

Abstract
A primary cilium is found on most mammalian cells, where it acts as a cellular antenna for the reception of both mechanical and chemical signals. A variety of diseases are associated with defective ciliogenesis, reflecting the ubiquity of the function of cilia and the number of proteins required for their assembly. Proper cilia length is necessary for cilia signaling and is regulated through a poorly understood balance of assembly and disassembly rates. FHDC1 is a unique member of the formin family of cytoskeletal regulatory proteins. Overexpression of FHDC1 induces F-actin accumulation and microtubule stabilization and acetylation. We find that overexpression of FHDC1 also has profound effects on ciliogenesis; in most cells FHDC1 overexpression blocks cilia assembly, but the cilia that are present are immensely elongated. FHDC1-induced cilia growth requires the FHDC1 FH2 and microtubule-binding domain and results from F-actin–dependent inhibition of cilia disassembly. FHDC1 depletion, or treatment with a pan-formin inhibitor, inhibits cilia assembly and induces cilia resorption. Endogenous FHDC1 protein localizes to cytoplasmic microtubules converging on the base of the cilia, and we identify the subdistal appendage protein Cep170 as an FHDC1 interacting protein. Our results suggest that FHDC1 plays a role in coordinating cytoskeletal dynamics during normal cilia assembly.
INTRODUCTION
A nonmotile primary cilium is found on nearly every mammalian cell type, where it serves as a cellular signaling hub responding to both mechanical and chemical stimuli from the surrounding environment. Defective cilia assembly causes a spectrum of diseases termed “ciliopathies.” These include polycystic kidney disease, nephronophthisis, Joubert syndrome, Bardet–Biedel syndrome, and Meckel–Gruber syndrome. Cilia defects have also been associated with cancer progression and diabetes. The wide array of cilia-associated diseases are thought to reflect the sheer number of proteins that are required to orchestrate normal cilia assembly (Reiter and Leroux, 2017).
Cilia are typically assembled in quiescent G0 cells, but may also form transiently during G1 before being disassembled again in S or G2 (Tucker et al., 1979). Cilia formation initiates from the basal body, which itself serves as a template for the assembly of axonemal microtubules. The basal body is anchored in place at the cell periphery by the distal (DA, a.k.a transition fibers) and subdistal appendages (SDA, a.k.a. basal feet) of the mother centriole as it transitions into the basal body. The SDA anchor the basal body to the actin and microtubule networks and the SDA and DA together form the transition zone (TZ). The TZ, SDA, and DA together form a docking platform for delivery of vesicles from both the Golgi and the endosome recycling compartment that are needed to initiate ciliogenesis (Sanchez and Dynlacht, 2016). The TZ is also required as a launch pad for vesicle delivery from the basal body into the gated domain of the growing cilia. Defects in SDA assembly are associated with a number of ciliopathies (Ishikawa et al., 2005; Chih et al., 2012; Thauvin-Robinet et al., 2014; Veleri et al., 2014) and anchoring of the SDA complex to the microtubule network requires the SDA protein Cep170 and the MT-binding protein EB1 (Guarguaglini et al., 2005; Schroder et al., 2007; Mazo et al., 2016; Huang et al., 2017).
Entry into and exit from the cilia are tightly regulated and mediated by the intraflagellar transport machinery. Anterograde trafficking delivers cargo to the cilia tip, is kinesin-dependent, and involves the IFT-B complex; retrograde trafficking requires cytoplasmic dynein2 and the IFT-A complex. IFT-B is required for delivery of tubulin subunits and other building blocks to the growing cilia tip. Retrograde IFT-A trafficking is also required for cilia assembly, although its role is not well understood. Cilia length is determined by the balance of the assembly rate and the intrinsic disassembly rate of the axonemal microtubules. Axonemal microtubules undergo extensive posttranslational modifications and it is thought that this must play some role in governing cilia assembly and microtubule stabilization (Gaertig and Wloga, 2008; Magiera and Janke, 2014). Cilia assembly is also responsive to changes in actin dynamics, with both positive and negative roles assigned to actin filaments during primary cilia assembly (Avasthi et al., 2014; Kim et al., 2015; Nager et al., 2017; Phua et al., 2017; Yeyati et al., 2017). Many actin regulatory proteins are localized to the cilia, and the centrosome itself acts as both an actin- and a microtubule-organizing centre (Farina et al., 2016; Kohli et al., 2017).
FHDC1 (a.k.a. INF1) is a unique member of the formin family of cytoskeletal remodeling proteins. FHDC1 contains an N-terminal Formin Homology 1 (FH1)–Formin Homology 2 (FH2) cytoskeletal regulatory unit, with which it is able to regulate both actin and microtubule dynamics (Young et al., 2008; Thurston et al., 2012). Unlike other formins, FHDC1 also contains a distinct C-terminal microtubule-binding domain (MTBD). The endogenous FHDC1 protein is found primarily in association with cytoplasmic microtubules and has an apparent affinity for the acetylated microtubule network (Young et al., 2008). Overexpression of FHDC1 induces microtubule acetylation in quiescent cells (Young et al., 2008; Thurston et al., 2012). It also induces dispersion of the Golgi ribbon, and this effect is mediated by the Golgi-derived microtubule network and is not dependent on centrosome-derived microtubules (Copeland et al., 2016). We show here that FHDC1 overexpression also has profound effects on cilia assembly, either blocking ciliogenesis or inducing extensive cilia elongation. Conversely, FHDC1 depletion or formin inhibition inhibits cilia assembly and reduces cilia length. The overexpression effects of FHDC1 are F-actin dependent and require both the intact FH2 and MTBD of FHDC1. FHDC1 apparently acts at the centrosome/basal body to affect cilia assembly and, consistent with this, we identified the subdistal appendage protein Cep170 as an FHDC1-interacting protein.
RESULTS
We reported previously that overexpression of FHDC1 induces stabilization and extensive acetylation of the cytoplasmic microtubule network (Young et al., 2008; Thurston et al., 2012). In addition to stable cytoplasmic microtubules, the axonemal microtubules of the primary cilia are also distinguished by microtubule acetylation (Piperno and Fuller, 1985). Thus, we wanted to determine whether FHDC1 also affects cilia formation, and to do this we first examined the effects of transient FHDC1 overexpression on cilia assembly in NIH 3T3 fibroblasts. The cells were transiently transfected with plasmids encoding an epitope-tagged derivative of FHDC1 and induced to assemble primary cilia by incubation in low-serum media. After 48 h, the cells were fixed and stained for acetylated α-tubulin to detect cilia and for the flag-tag on FHDC1. mCherry-overexpressing cells served as controls. Under these conditions, nearly 90% of serum-starved control cells generated primary cilia, and the cilia had an average length of 2.9 μm (Figure 1, A, D, and E). In contrast, overexpression of FHDC1 had two major effects on cilia assembly (Figure 1). In roughly 75% of the transfected cells, FHDC1 overexpression blocked cilia assembly (Figure 1B) while still inducing acetylation of cytoplasmic microtubules. In contrast, the remaining 25% of transfected cells had hugely elongated primary cilia characterized by numerous kinks and bends (Figure 1, C, D, and G). These elongated cilia had an average length of 15 μm and varied greatly in size, ranging from 4 μm to more than 40 μm in length (Figure 1, C, E, and G). This effect is unique to FHDC1; overexpression of constitutively active derivatives of 12 other mammalian formins had no significant effect on ciliogenesis (Supplemental Figure S1). We confirmed the identity of the elongated cilia induced by FHDC1 overexpression using the cilia membrane marker Arl13b. GFP-Arl13b (Larkins et al., 2011) alone or coexpressed with FHDC1 was used to mark cilia in serum-starved NIH 3T3s. GFP-Arl13b was clearly localized to cilia in control (Figure 1F) and FHDC1-overexpressing cells (Figure 1G), confirming that these elongated structures indeed represent protrusive primary cilia.
FIGURE 1:

FHDC1 expression induces cilia elongation. NIH 3T3 fibroblasts were transfected with plasmids encoding flag-tagged FHDC1 (red) or mCherryFP (red) as a control. Cilia formation was induced by incubation in low serum media. At 48 h posttransfection the cells were fixed and primary cilium formation was assessed by immunofluorescence using an anti-acetylated tubulin antibody (green). (A) The majority of mCherry-expressing control cells generated a short primary cilium. (B) The majority of FHDC1-expressing cell formed an extensive cytoplasmic network of acetylated microtubules, but did not produce a primary cilium. (C) Approximately 20% of FHDC1-expressing cells generated an elongated primary cilium that was often kinked or bent. (D) Quantification of data shown in A–C. N = 5, >100 cells counted per sample. Error bars = SEM. (E) Quantification of cilia length for data shown in A–C. Cilia from mCherry-expressing controls cells have an average length of 2.9 μm (red bar). Cilia from FHDC1-expressing cells range broadly in size with an average length of 15 μm. (F) GFP-Arl13b was expressed in control cells by transient transfection. The transfected cells were fixed 48 h later and the effects on ciliogenesis were assessed by immunofluorescence. GFP-Arl13b was clearly localized to the primary cilia. (G) Flag-tagged FHDC1 (white) was coexpressed with GFP-Arl13b (green) by transient transfection and effects on ciliogenesis were assessed as in A. In all ciliated cells the exogenous FHDC1 has clearly accumulated along the length of the primary cilia and colocalized with GFP-Arl13b.
We noted that overexpressed FHDC1 protein was present along the length of the cilia (Figure 1, C and G), and we wanted to determine whether this was specific to FHDC1 or whether passage through the cilia gate had been generally affected. To address this, we coexpressed GFP with FHDC1 to see whether both proteins were similarly recruited to the cilia. Following transfection, cilia formation was induced by serum starvation as before and recruitment to the cilia was assessed by immunofluorescence. Cilia formed in the majority of control cells expressing GFP alone, and GFP was not obviously recruited to the cilia. Similarly, when coexpressed with FHDC1, GFP did not obviously accumulate in the cilia. while FHDC1 was very clearly localized there (Supplemental Figure S1, F and G).
Given these results, we wanted to determine whether the endogenous FHDC1 protein also localizes to the cilium. As before, cilia assembly was induced in NIH 3T3 cells by serum starvation and the cells were fixed 48 h later. Subcellular localization of FHDC1 was determined by immunofluorescence using anti-FHDC1 antibody (Young et al., 2008); cilia were detected as above. In ciliated cells, FHDC1 protein was predominantly localized to a network of cytoplasmic microtubules that converged on the base of the cilia (Figure 2). We did not observe any obvious localization of endogenous FHDC1 along the axoneme, although given the small volume of the cilium, any protein present may be below the limits of detection (Nachury, 2014).
FIGURE 2:

Endogenous FHDC1 is enriched at a cluster of microtubules converging on the basal body. (A–C) NIH 3T3 fibroblasts were incubated in low-serum media to induce cilia assembly. Serum-starved cells were fixed after 48 h and stained for acetylated microtubules (green) and endogenous FHDC1 (red). The endogenous FHDC1 protein is present on cytoplasmic microtubules that come together at the bases of the cilia.
Cilia assembly is affected by perturbations to both actin and microtubule networks (Sharma et al., 2011; Avasthi et al., 2014), and FHDC1 directly affects both actin and microtubule dynamics via its N-terminal FH2 domain and C-terminal MTBD (Young et al., 2008; Thurston et al., 2012). To determine which aspect of FHDC1 activity might be affecting ciliogenesis, a series of FHDC1 derivatives selectivity defective for either FH2 or MTBD activity were overexpressed in NIH 3T3 cells and their effects on primary cilia assembly assessed by immunofluorescence. FHDC1.I180A converts a conserved residue in the FH2 domain from isoleucine to alanine and blocks the ability of FH2 to bind the barbed end of F-actin (Xu et al., 2004; Thurston et al., 2012). Expression of this derivative had only minor effects on the number of ciliated cells, while it induced a significant increase in average cilia length (Figure 3, B, G, and H). This increase was not as pronounced as with wild-type FHDC1 and did not result in such extreme variations in length. Similar results were obtained with overexpression of a C-terminal derivative of FHDC1 (486C), containing the intact MTBD, but with FH1 and FH2 deleted (Figure 3, E, G, and H). In contrast, expression of the MTBD-deleted derivative 958N had no effect on the number of cells with cilia or on cilia length (Figure 3. C, G, and H). Similar results were obtained with a derivative consisting of the isolated FH1–FH2 unit (485N). This derivative had only minor effects on the number of cells with cilia; however, unlike the larger 958N derivative, it caused a small, but significant, reduction in average cilia length. We confirmed the expected effect of each derivative on F-actin assembly by phalloidin staining (Supplemental Figure S2, A–F), which suggests that FHDC1-induced F-actin accumulation is not itself sufficient to inhibit cilia assembly. This analysis also suggests that the MTBD may serve as a cilia localization domain for FHDC1 (Figure 3; Supplemental Figure S2). Thus FHDC1 overexpression has two distinct effects on cilia assembly: FH2- and MTBD-dependent inhibition of cilia assembly with immense elongation when cilia are present (as seen with full-length FHDC1) or MTBD-dependent increase in average cilia length (as seen with I180A and 486C).
FIGURE 3:

FHDC1-induced cilia elongation is FH2- and MTBD-dependent. A series of FHDC1 derivatives were expressed in NIH 3T3s by transient transfection. Cilia formation was induced by incubation in low-serum media, the transfected cells were fixed 48 h later ,and the effects on ciliogenesis were assessed by immunofluorescence using an anti-acetylated tubulin antibody (green). FHDC1 expression was detected by virtue of the N-terminal flag epitope tag (red). (A) Expression of full-length FHDC1 inhibits primary cilia formation, but when present the cilia are greatly elongated. (B) FHDC1.I180A-expressing cells assemble primary cilia in most cases. The cilia are longer than control cilia, but do not display the same variation in length induced by FHDC1. (C) Expression of FHDC1.958N had no effect on the number of ciliated cells nor on cilia length. (D) Expression of FHDC1.485N did not affect the number of cells with cilia; however, the cilia were noticeably shorter than on controls. (E) Expression of FHDC1.486C had effects on cilia number and length similar to those of expression of FHDC1.I180A. (F) Schematic of FHDC1 derivatives. Boxes 1 through 5 indicate regions of homology conserved between FHDC1 orthologs. Boxes 4 and 5 comprise the MTBD. (G, H) Quantification of data shown in A–E. N = 3, >100 cells counted per sample. Error bars = SEM. *indicates significant difference from mCherry-expressing controls, p < 0.01.
The effects of the FHDC1 deletion derivatives on ciliogenesis were reminiscent of our previous observations regarding the effects of FHDC1 on assembly of the Golgi ribbon (Copeland et al., 2016; see Supplemental Figure S2, G–K) and a close juxtaposition of the Golgi and basal body are thought to be required for normal cilia assembly (Knodler et al., 2010; Asante et al., 2013). This raises the possibility that inhibition of cilia assembly is a consequence of FHDC1-induced Golgi dispersion. In an effort to separate these two effects, we generated a series of C-terminal FHDC1 truncations that progressively deleted the MTBD. These deletion derivatives were assessed for their effects on Golgi dispersion and cilia assembly by immunofluorescence. Full-length FHDC1 and the truncation mutants 1004N and 978N all efficiently induced Golgi dispersion in the majority of transfected cells (Figure 4, A–D). These derivatives differed, however, in their effects on ciliogenesis. Overexpression of 1004N had intermediate effects on both the number of ciliated cells and the extent of cilia elongation (Figure 4E). In contrast, 978N had no obvious effect on either cilia length or the number of ciliated cells (Figure 4, C, E, and F). Thus, the ability to inhibit cilia assembly or induce cilia elongation seem linked, while the effects of FHDC1 on Golgi dispersion and cilia assembly are not.
FIGURE 4:

FHDC1-induced Golgi dispersion does not inhibit ciliogenesis. Full-length FHDC1 and a series of FHDC1 C-terminal–deletion derivatives (red) were expressed in NIH 3T3 cells and the effects on Golgi dispersion and ciliogenesis were assessed by immunofluorescence. Primary cilia were detected with anti-acetylated tubulin antibody (white) and the Golgi was detected via coexpression of the Golgi marker GFP-GalT (green). (A) Expression of full-length FHDC1 induces Golgi dispersion in essentially all transfected cells. Cells with clearly dispersed Golgi were still able to assemble greatly elongated primary cilia. (B) Expression of FHDC1.1004N (codons 1–1004) induces Golgi dispersion in nearly all cells. Primary cilia were present in more than half of 1004N-expressing cells. (C) Expression of FHDC1.978N (codons 1–978) induced Golgi dispersion in the majority of transfected cells and had little effect on cilia length or number. (D–F) Quantification of data shown in A–C. N = 3, >100 cells counted per experiment, error bars = SEM. Red bar indicates mean cilia length. *indicates significant difference from FHDC1-expressing cells, p < 0.001.
The effects of FHDC1 on cilia length and number are FH2-dependent and it has been suggested recently that axonemal F-actin polymerization induces ciliary decapitation and disassembly (Phua et al., 2017). To determine whether FHDC1 induces accumulation of axonemal F-actin, and whether this causes FHDC1-induced inhibition of cilia assembly, we compared the effects of FHDC1 and FHDC1.I180A overexpression on ciliary F-actin accumulation (Figure 5; Supplemental Figure S3). As before, both FHDC1 and I180A were recruited to the elongated cilia (Figure 5, B–D, and Supplemental Figure 3, B and C). In I180A-expressing cells there is no detectable phalloidin staining in the cilia, consistent with the I180A derivative being unable to regulate actin dynamics (Figure 5B; Supplemental Figure S3B). In FHDC1-expressing cells, F-actin is clearly present along the length of essentially all elongated cilia (Figure 5, C and D; Supplemental Figure S3C). This would suggest that F-actin induced decapitation is not related to the inhibition of cilia assembly by FHDC1. Indeed, it seems that in this context axonemal F-actin is associated with cilia elongation instead. To test this directly, we treated cells overexpressing mCherry, FHDC1, and FHDC1.I180A with the actin-depolymerizing drug latrunculin B. Vehicle-treated cells served as controls (Figure 6). LatB treatment was sufficient to induce depolymerization of F-actin in all cells (Figure 6, A and B). LatB treatment did not affect cilia length in mCherry- and I180A-overexpressing cells. In FHDC1-expressing cells, however, LatB treatment caused a significant reduction in cilia length and reduced the variation in length as well (Figure 6, B and C), essentially phenocopying the effects of I180A overexpression on cilia length. LatB treatment did not affect the number of ciliated cells over the course of the 2-h treatment (Figure 6D).
FIGURE 5:

F-actin is present in the cilia of FHDC1-overexpressing cells. FHDC1 and FHDC1.I180A were expressed in NIH 3T3 cells by transient transfection and the effects on F-actin accumulation (green) and cilia assembly (white) were assessed by immunofluorescence. mCherry was used as a control. (A) F-actin is not detected in cilia of mCherry-expressing cells. (B) FHDC1.I180A is recruited to the cilia. F-actin is not detected in cilia of FHDC1.I180A-overexpressing cells. (C, D) F-actin is readily detected by phalloidin staining in the elongated cilia of FHDC1-overexpressing cells.
FIGURE 6:

FHDC1-induced cilia elongation is F-actin dependent. mCherry, FHDC1, or FHDC1.I180A (red) were expressed in NIH 3T3 cells by transient transfection, and the transfected cells were serum-starved to induce cilia assembly. At 40 h after transfection the cells were treated with 2-μm latrunculin B (Lat B) for 2 h and the effects on cilia length and number were assessed by immunofluorescence. F-actin (green) was detected with Alexa488-phalloidin and acetylated microtubules (white) were detected with anti-acetyl-α-tubulin. For all samples, LatB treatment did not affect the number of ciliated cells. (A, B) LatB treatment did not affect the length of cilia in mCherry-expressing cells. LatB treatment significantly reduced the length of cilia in FHDC1-expressing cells. LatB had minimal effects on cilia length in FHDC1.I180A-expressing cells. (C, D) Quantification of data shown in A and B. N = 3, >100 cells counted per sample. Error bars = SEM. Red bar indicates average cilia length.
Increases in cilia length can be driven either by promoting assembly or by inhibiting disassembly (Sanchez and Dynlacht, 2016) and F-actin has been proposed to affect both these processes (Quarmby, 2014; Phua et al., 2017). To distinguish between these possibilities, we tested the effects of FHDC1 expression on serum-induced cilia disassembly (Pugacheva et al., 2007; Phua et al., 2017; Figure 7). mCherry, FHDC1, and FHDC1.I180A were expressed in NIH 3T3s by transient transfection and ciliogenesis induced by growth in low-serum media. After 48 h, cells were stimulated with 10% serum to induce cilia disassembly and fixed 2, 3, and 4 h after serum stimulation. In Cherry and I180A cells, an obvious reduction in the number of ciliated cells was observed 2 h after serum stimulation (Figure 7A). This effect continued at 3 h and began to recover at 4 h, presumably as cilia assembly was reinitiated in G1. In contrast, the number of ciliated cells in the FHDC1 sample remained constant with no effect at 2 h and only a slight decrease at 3 and 4 h (Figure 7A), while the cilia that were present actually increased in size (Figure 7B). These results suggest that FHDC1 affects cilia length via two mechanisms: FH2- and MTBD-dependent inhibition of disassembly and MTBD-dependent promotion of assembly. It also suggests that FHDC1-induced inhibition of cilia disassembly is F-actin–dependent.
FIGURE 7:
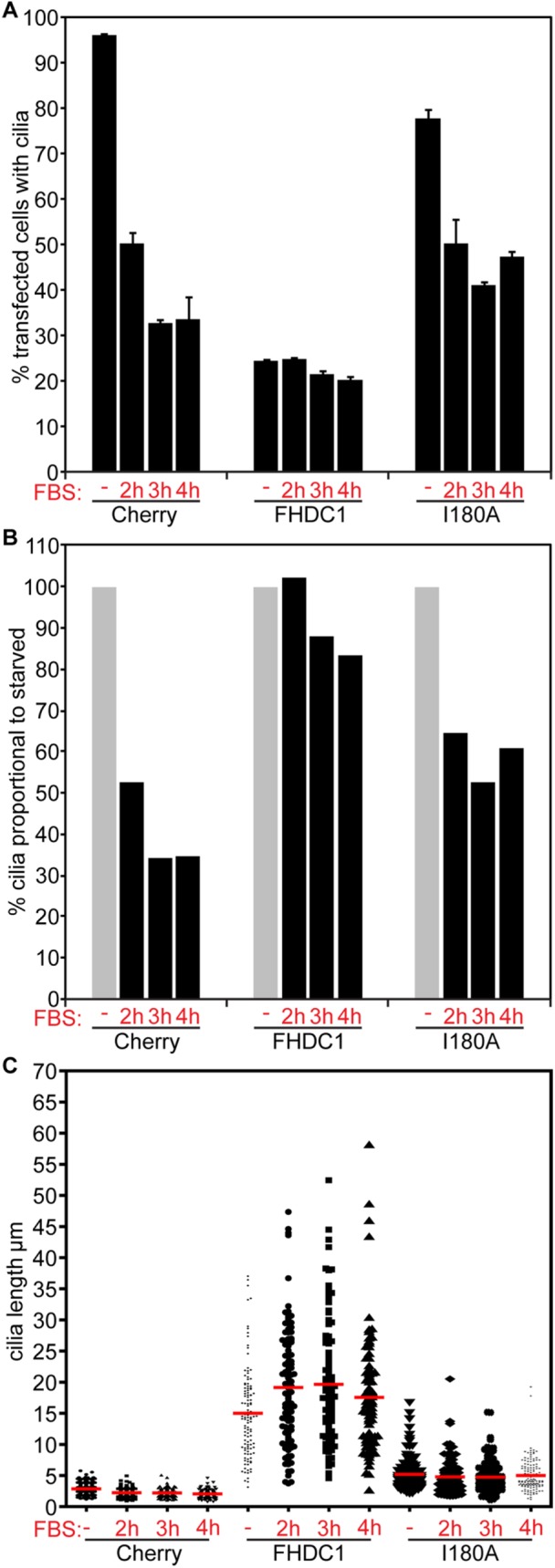
FHDC1 expression inhibits cilia disassembly. mCherry, FHDC1, or FHDC1.I180A was expressed in NIH 3T3 cells by transient transfection and the transfected cells were serum-starved to induce cilia assembly. At 48 h after transfection the cells were stimulated with complete medium for 2, 3, or 4 h and the effects on ciliogenesis were assessed by immunofluorescence. (A) Serum stimulation rapidly induced cilia disassembly in mCherry- and I180A-expressing cells. The number of FHDC1-expressing cells with cilia was not affected by serum stimulation. N = 3, >100 cells counted per experiment. Error bars = SEM. (B) The data in A were replotted as percentages of the unstimulated values to facilitate comparison of the relative effects of FHDC1 and FHDC1.I180A on cilia resorption. (C) Quantification of cilia length for the data shown in A. N = 3, >100 cilia measured per sample.
Our results show that excess FHDC1 protein disrupts the normal mechanisms governing cilia assembly; they do not, however, address the requirement for FHDC1 in this process. We first addressed the general need for formin activity during ciliogenesis using the pan-formin inhibitor smiFH2 (Rizvi et al., 2009). Serum-starved NIH 3T3s were treated with 5 or 10 μm smiFH2 for 30 or 60 min prior to fixation (Figure 8). SmiFH2 treatment resulted in a dose-dependent reduction both in the number of cells with cilia and in average cilia length (Figure 8, D and E), suggesting that formin activity is required to maintain cilia in these cells. We next turned to the CRISPR/Cas9 system to attempt to knock out FHDC1 in our cells (see Materials and Methods for details). Despite extensive efforts, we were unable to obtain complete FHDC1 knockout clones, but we were able to isolate two clonal lines with notably reduced levels of FHDC1 protein, as determined by immunoblot (Figure 8F). Both behaved similarly in preliminary experiments and one line was selected for further analysis. The partial FHDC1 knockout cells exhibited defective cilia assembly and aberrant regulation of cilia length, as assessed by immunofluorescence. We also noted that the basal body was often adrift in these cells and away from its normal perinuclear position (Figure 8, G–J). We used small interfering RNA (siRNA) to further deplete FHDC1 expression in the wild-type and CRISPR cell lines (Copeland et al., 2016). This was sufficient to significantly reduce FHDC1 expression in wild-type cells and essentially eliminate it in the partial knockout cell-line (Figure 8H). In both cases knockdown of FHDC1 greatly reduced both cilia length (Figure 8I) and the number of ciliated cells (Figure 8J). We also noted apparent defective centrosome assembly in the FHDC1-depleted cells where the majority of cells lack distinct puncta of perinuclear γ-tubulin (Supplemental Figure S3). Together this suggests that FHDC1 participates in the normal assembly of cilia in these cells.
FIGURE 8:

FHDC1 is required for normal cilia assembly. (A–C) NIH 3T3 cells were maintained in low-serum media for 48 h to induce cilia formation. F-actin was detected with phalloidin (green) and cilia were detected with anti-acetylated tubulin (red). Cells treated for 60 min with increasing amounts of smiFH2 lost their cilia in a dose-dependent manner. (D, E) Cells were treated with 0, 5, or 10 μM smiFH2 for 30 and 60 min. Cilia assembly was assessed as in A–C. Both cilia length and the number of cells with cilia were obviously decreased after 30 min of smiFH2 treatment and more so after 60 min. Control cells treated with DMSO (vehicle) maintained their cilia. N = 3, >100 cells counted per experiment. Error bars = SEM. (F) Isolation of two CRISPR lines with diminished levels of FHDC1 expression as determined by immunoblotting. (G) Depletion of FHDC1 results in defective cilia assembly. Partial FHDC1 knockout cells, with diminished levels of FHDC1 expression, are defective for cilia length regulation. These cells frequently exhibit aberrant cilia localization away from the perinuclear region. (H–K) siRNA-mediated knockdown was used to further deplete FHDC1 expression in the partial knockout cell line. NC = negative control; k/d = FHDC1 knockdown. (H–J) Depletion of FHDC1 expression decreases cilia length and reduces the number of ciliated cells following serum starvation. N = 3, >100 cells counted per experiment. Error bars = SEM. * indicates p < 0.0001. (K) FHDC1 was essentially undetectable following siRNA transfection of the partial knockout cells as determined by immunoblotting.
In an effort to shed light on how FHDC1 is affecting cilia assembly, we used BioID (Roux et al., 2012, 2013) to identify potential FHDC1 interacting proteins. A BirA*–FHDC1 fusion derivative was generated, and we characterized its effect on cilia assembly and Golgi dispersion in NIH 3T3 fibroblasts. Overexpression of BirA*–FHDC1 had similar effects on cilia assembly and Golgi dispersion as observed with other full-length FHDC1 derivatives (Supplemental Figure S4, A–C). Following treatment with exogenous biotin, BirA*–FHDC1 expression induced biotinylation of cytoplasmic microtubules and the primary cilium, as well as distinct puncta present either at the base of the cilia or at sites where cytoplasmic microtubules converge (Figure 9, A and B). These puncta are seen in both ciliated and nonciliated cells and colocalize with γ-tubulin staining (Figure 9B), suggesting that they may represent the centrosome or basal body. Surprisingly, these puncta were often widely separated within the cell. Using the BirA*–FHDC1 derivative, a BioID interactome screen was carried out in HEK293T cells (see Materials and Methods for details). We identified a number of putative FHDC1-interacting proteins that are known to affect cilia assembly (Cep170, EB1, Snap29, Arl2; Guarguaglini et al., 2005; Schroder et al., 2007; Davidson et al., 2013; Lu et al., 2015; Huang et al., 2017) and focused on Cep170 for further study.
FIGURE 9:

Cep170 is an FHDC1-interacting protein. BirA*-tagged FHDC1 was expressed in NIH 3T3 cells by transient transfection. Ciliogenesis was induced as before in low-serum media. BirA*-FHDC1 (red) was detected by virtue of the encoded myc-tag. (A) BirA*-FHDC1 is recruited to the elongated cilia and, following exogenous biotin treatment, induces biotinylation of cytoplasmic microtubules (green) and the cilia (acetylated tubulin, white). Specific punctate structures within the cytoplasmic microtubule network were also labeled (arrows). Biotinylated proteins were detected with Alexa488-streptavidin. (B) BirA*-FHDC1 (red) induces biotinylation of puncta (green) that colocalize with γ-tubulin puncta detected with anti–γ-tubulin (white). (C) BirA*-FHDC1 induces biotinylation of endogenous Cep170. As in B, BirA*-FHDC1 was expressed by transient transfection and its ability to induce biotinylation of the endogenous Cep170 was assessed by IB. myc-FHDC1 and FMNL2-BirA* were included as negative controls. Total biotinylated proteins were isolated from each sample using streptavidin–agarose beads. The eluted proteins were immunoblotted using the indicated antibodies. Endogenous Cep170 is only present in the pool of biotinylated proteins induced by BirA*-FHDC1 (arrow).
Cep170 is a subdistal appendage protein originally identified via its interaction with PLK1; it is found only at the mother centriole/basal body and is needed to connect mature centrioles to the cytoplasmic microtubule network (Guarguaglini et al., 2005; Huang et al., 2017). We first confirmed the FHDC1 interaction with Cep170 using a directed version of BioID. BirA*–FHDC1 was coexpressed with GFP–Cep170 or GFP alone. Following biotin treatment, the cells were lysed and GFP–Cep170 or the GFP control was immunoprecipitated using GFP–Trap. The input and eluate samples were immunoblotted and the resultant blots probed with streptavidin-HRP to detect the biotin tag. Coexpression of BirA*–FHDC1 induced biotinylation of GFP–Cep170, but not GFP alone (Supplemental Figure S4D). Using a similar approach, we confirmed that BirA*–FHDC1 was also able to label the endogenous Cep170 protein. In this case, myc-tagged FHDC1 and a BirA*-tagged derivative of the formin FMNL2 (FMNL2–BirA*) were used as controls. Each derivative was expressed in NIH 3T3 cells, lysates were prepared from the biotin-treated cells, and the biotinylated proteins were isolated from each using streptavidin beads. The streptavidin-bound proteins were eluted, immunoblotted, and probed for the presence of endogenous Cep170. Cep170 was present only in the BirA*–FHDC1 lysate and was not detected in either myc–FHDC1 or FMNL2—BirA* lysates (Figure 9E), consistent with a specific interaction between FHDC1 and the endogenous Cep170 protein.
As noted above, in cells expressing BirA*–FHDC1, we frequently detected two or more well-separated biotinylated puncta that colocalized with γ-tubulin (Figure 9, A and B). These puncta likely represent the centrosome and suggest that the centriole cycle may be disrupted in these cells (Kong et al., 2014). To determine whether FHDC1 can induce defects in centriole maturation, we compared the effects of FHDC1 and FHDC1.I180A expression on centrosome morphology. GFP–Cep170 was used as a marker for the basal body/mother centriole and γ-tubulin as a marker for the centrosome. mCherry expression was used as an additional control. In quiescent cells, only one Cep170 punctum should be observed in close association with the basal body (Guarguaglini et al., 2005). As expected, in cells overexpressing mCherry or FHDC1.I180A, only one GFP–Cep170 punctum was observed, and this colocalized with the γ-tubulin marker (Figure 10, A, D, and E). In contrast, two, and sometimes three, GFP–Cep170 puncta were detected in the majority of FHDC1-transfected cells (Figure 10, B, C, and E). We noted that the Cep170/γ-tubulin puncta were often widely separated (Figure 10, B and C) and quantified this effect using γ-tubulin alone as a marker (Figure 10, F–J). In Cherry and FHDC1.I180A cells, two visible γ-tubulin puncta could often be detected in close apposition, typically <0.8 μm apart. We designated centrioles as “separated” if they were separated by more than twice this distance. Using this criterion, the majority of γ-tubulin puncta were widely separated in FHDC1-overexpressing cells, and three and sometimes four puncta were observed. Similar results were obtained using the centriole marker centrin2 and the SDA marker ODF2/cenexin (Supplemental Figure S6). Taken together, these observations are indicative of disruption of the normal centriole maturation cycle and similar to what is seen when PLK1 signaling is defective (Kong et al., 2014).
FIGURE 10:

FHDC1 overexpression disrupts the centriole maturation cycle. FHDC1 or FHDC1.I180A was expressed by transient transfection, and the effects on centrosome morphology and Cep170 localization were assessed by immunofluorescence. (A) mCherry (red) expression does not affect γ-tubulin staining (white), nor does it affect the number of Cep170 puncta detected with anti-Cep170 antibody (green). (B, C) FHDC1 overexpression induces the accumulation of supernumerary Cep170 puncta in both ciliated and nonciliated cells. (D) FHDC1.I180A overexpression does not affect centrosome morphology. (E) Quantification of data shown in A–D. N = 3, >100 cells counted per sample. Error bars = SEM. (F) mCherry (red) expression does not affect centriole number nor the distance between centrioles as detected by γ-tubulin staining (green). (G, H) FHDC1 (red) overexpression induces centriole separation and supernumerary centrioles in both ciliated and nonciliated cells. (I) FHDC1.I180A (red) overexpression does not affect centriole number or their separation morphology. (J) Quantification of data shown in F–I. N = 3, >100 cells counted per sample. Error bars = SEM.
If these effects of FHDC1 on the centriole cycle are related to its effects on cilia assembly, then they too should be F-actin–dependent. To test this proposal, cells expressing FHDC1, FHDC1.I180A, or mCherry were treated with LatB and the effects on centrosome morphology were compared with those in cells treated with vehicle alone. Prior to LatB treatment, two closely juxtaposed centrioles were present in mCherry- and FHDC1.I180A-expressing cells, while the centrioles in FHDC1-overexpressing cells were well separated (Figure 11, A–C, left panels). Following 2 h of LatB treatment, there was no change in centriole/centrosome morphology in mCherry and FHDC1.I180A cells (Figure 11, A–D). In FHDC1-transfected cells, however, LatB treatment was sufficient to reverse FHDC1-induced centriole separation (Figure 11, A–D).
FIGURE 11:

FHDC1-induced centriole separation is F-actin dependent. mCherry, FHDC1, or FHDC1.I180A was expressed in NIH 3T3 cells by transient transfection, and the effects on centriole separation were assessed by IF. (A) Overexpression of mCherry (red) does not affect centriole (γ-tubulin, green) separation, and centriole localization is not affected by 2 h of treatment with the actin-disrupting drug LatB. (B) Overexpression of FHDC1.I180A (red) does not affect centriole separation, and centriole localization is not affected by 2 h of treatment with the actin-disrupting drug LatB. (C) Overexpression of FHDC1 induces centriole separation and this effect is reversed following treatment with LatB. (D) Quantification of data shown in A–C. N = 3, >100 cells counted per sample. Error bars = SEM.
Our results suggest that at least some of the effects of FHDC1 on cilia assembly are being mediated by its effects on the subdistal appendages and the mother centriole/basal body. Cep170 recruits PLK1 to the mother centriole (Guarguaglini et al., 2005), where the kinase plays multiple regulatory roles (Nigg and Holland, 2018). In cycling cells, PLK1 promotes centriole duplication and mother centriole maturation and PLK1 is required for cilia assembly as cells enter G0 and quiescence (Kong et al., 2014). Later, PLK1 promotes cilia disassembly at the G0/G1 transition by activating the inhibitory kinesin Kif2A which blocks microtubule assembly at the cilia tip (Wang et al., 2013; Miyamoto et al., 2015). To test whether there is a connection between FHDC1 and PLK1 signaling, we coexpressed FHDC1 with wild-type (WT), kinase-dead (K82R), and constitutively active (T210D) PLK1 derivatives. As expected, kinase-dead PLK1 did not affect FHDC1-induced inhibition of cilia assembly (Figure 12, E and G), nor did it rescue FHDC1-induced cilia elongation (Figure 12, E and H). Similarly, WT PLK1 had only modest effects on FHDC1-induced inhibition of cilia assembly and cilia elongation (Figure 12, D, G, and H). Expression of the constitutively active T210D derivative, however, was able to rescue both of these effects; repressing FHDC1-induced cilia elongation and more than doubling the number of FHDC1-expressing cells with cilia (Figure 12, F–H). Coexpression of the PLK1 derivatives did not affect cilia assembly in control mCherry-expressing cells (Figure 12, A–C, G, and H).
FIGURE 12:

FHDC1-induced effects on cilia assembly are rescued by expression of a constitutively active derivative of PLK1. (A–C) mCherry was coexpressed in NIH 3T3 cells with wild-type PLK1 or kinase-dead (K82R) or constitutively active (T210D) PLK1 derivatives (cyan), and the effects on ciliogenesis were assessed by immunofluorescence. None of the coexpressed PLK1 derivatives have substantial effects on ciliogenesis in mCherry control ceclls. (D–F) FHDC1 was coexpressed with wild-type PLK1 or kinase-dead (K82R) or constitutively active (T210D) PLK1 (cyan), and the effects on ciliogenesis were assessed by IF. Coexpression of PLK1.T210D suppresses the effects of FHDC1 on cilia length and the number of ciliated cells. (G, H) Quantification of data shown in A–F. N = 3, >100 cells counted per sample. Error bars = SEM. *indicates p < 0.005’, **indicates p < 0.0001.
DISCUSSION
We show here that formin activity is required in NIH 3T3 fibroblasts for the normal assembly of primary cilia and that the formin FHDC1 is of particular importance. Treatment with the pan-formin inhibitor smiFH2 caused a dose-dependent reduction in both cilia length and the number of cells that form cilia. Depletion of FHDC1 had similar effects, inhibiting cilia assembly and causing a reduction in cilia length. In the reverse situation, FHDC1 overexpression blocked cilia formation in the majority of cells, while inducing massive elongation in cells where cilia were assembled. Indeed, the longest cilia we have measured are in excess of 100 μm. This elongation likely arises from two distinct mechanisms. The first is MTBD-, FH2-, and F-actin-dependent and inhibits cilia disassembly, resulting in a continuous distribution of elongated cilia length. The two effects of FHDC1 on cilia seem linked, suggesting that elongation and inhibition are mediated by a common target. Our results suggest that this target is likely Cep170 and the subdistal appendages of the centrosome/basal body. FHDC1 also affects cilia length via a second mechanism that is solely MTBD-dependent and likely results from promotion of assembly via stabilization of axonemal microtubules.
Three likely targets for FHDC1-induced effects on cilia assembly initially presented themselves: the Golgi ribbon, F-actin assembly, and the centrosome. Our results, however, allowed us to eliminate the first two. First, there is no correlation between the effects of FHDC1 on Golgi ribbon assembly and its effects on cilia: expression of FHDC1 derivatives that do not affect ciliogenesis still induce Golgi dispersion and FHDC1-overexpressing cells with dispersed Golgi still produce grossly elongated cilia. These findings also suggest that close apposition of the basal body to the Golgi ribbon is not essential for cilia assembly. Second, FHDC1-induced inhibition of cilia production is not a secondary consequence of F-actin accumulation. There are conflicting reports regarding the role of F-actin in cilia assembly, with both negative and positive effects attributed to actin filaments (Chaitin, 1991; Avasthi et al., 2014; Kim et al., 2015; Kohli et al., 2017; Nager et al., 2017; Phua et al., 2017; Yeyati et al., 2017). We find that formin-induced F-actin assembly alone is not sufficient to either block or promote cilia assembly, but that F-actin is necessary for FHDC1-induced cilia elongation and centriole separation. Indeed, F-actin is observed along the length of the elongated cilia in FHDC1-overexpressing cells. This observation suggests that F-actin accumulation in the cilia does not itself induce ciliary decapitation (Phua et al., 2017). Instead, we find that F-actin in this context is needed to promote elongation by inhibiting disassembly. FHDC1-induced F-actin also drives centriole separation, with both effects reliant upon targeting of FH2 activity to the basal body and/or cilia via the MTBD of FHDC1.
Separately from its effects on Golgi ribbon and F-actin assembly, our data point to a novel role for FHDC1 at the centrosome/basal body during ciliogenesis. The endogenous FHDC1 protein accumulates on a tangle of microtubules that converge on the basal body in ciliated cells, reminiscent of the stable microtubules reported to affect centrosome positioning during ciliogenesis in RPE1 cells (Pitaval et al., 2017). Depletion of FHDC1 disrupts normal centrosome morphology and, consistent with this, we identified Cep170, a component of the subdistal appendages, as an FHDC1-binding protein. Cep170 is needed to anchor the centrosome to the cytoplasmic microtubule network (Guarguaglini et al., 2005; Huang et al., 2017), and recent work suggests that Cep170 and the subdistal appendages play a critical role in both ciliogenesis and centrosome function in a cell type–dependent manner (Mazo et al., 2016; Huang et al., 2017). Overexpression of FHDC1 apparently has multiple effects on the centriole maturation/duplication cycle, causing the formation of supernumerary γ-tubulin, Centrin2, and cenexin foci. These foci are often well separated, but apparently remain tethered, as the centrioles are able to “reengage” in response to LatB treatment. The accumulation of multiple puncta positive for the SDA protein Cep170 or cenexin/ODF2 is also indicative of a maturation defect, as Cep170 is normally restricted to a single centriole per G0/G1 cell (Guarguaglini et al., 2005). Consistent with disruption of Cep170/SDA function, these centrioles are also no longer anchored to their normal perinuclear position (Veleri et al., 2014; Huang et al., 2017).
Cep170 was originally identified as a PLK1-binding protein, and centriole distancing, disengagement, and maturation are all regulated by PLK1 at distinct phases of the cell cycle (Nigg and Holland, 2018). It has been proposed that the subdistal appendages also act as a signaling platform for PLK1-dependent regulation of cilia disassembly (Miyamoto et al., 2015; Nigg and Holland, 2018). Thus, altering levels of FHDC1 protein expression likely disrupts a putative SDA-dependent PLK1 signaling scaffold, affecting cilia and centriole dynamics. This would be consistent with our observation that constitutively active PLK1 T210D mutant, but not wild-type PLK1, is able to rescue FHDC1-induced cilia elongation and inhibition of cilia assembly.
On the basis of our results, we propose that the effects of FHDC1 overexpression on ciliogenesis are largely mediated at the centriole/basal body. In this model, excess FHDC1 disrupts the normal maturation of the mother centriole and thereby inhibits cilia assembly in the majority of cells. In cells where cilia assembly has already begun, however, excess FHDC1 is unable to halt this process and instead disrupts the regulation of disassembly, resulting in runaway elongation. This model would account for the consistent proportion (roughly 20%) of ciliated cells overexpressing FHDC1, similar to the proportion of G1 ciliated cells found in nonsynchronized populations of NIH 3T3 fibroblasts (Tucker et al., 1979). We should note that additional effects of axonemal F-actin on the inhibition of cilia disassembly cannot be ruled out, but are difficult to separate from the F-actin–dependent effects of FHDC1 on the centrosome.
Our data suggest that the endogenous FHDC1 protein plays an important role at the subdistal appendages. As noted above, in ciliated cells, endogenous FHDC1 is localized to a tangle of microtubules at the base of the cilia. When FHDC1 activity is depleted, cilia assembly is inhibited, and this is apparently associated with disruption of the normal morphology and maturation of the centrosome. Cep170 depletion also disrupts anchoring of the centrosome to the cytoplasmic microtubule network (Guarguaglini et al., 2005; Huang et al., 2017), and we propose that the association of FHDC1 with Cep170 stabilizes this connection. Thus, early in cilia assembly, FHDC1 would assist in the maturation and positioning of the basal body, and then, once cilia assembly has initiated, it may also promote cilia elongation by inhibiting disassembly. This would be consistent with inhibition of disassembly by FHDC1 overexpression and might relate to previous observations of transient F-actin accumulation during the initial stages of ciliogenesis (Avasthi et al., 2014). Although we favor a model where FHDC1 acts primarily at the subdistal appendages, we cannot rule out additional roles for FHDC1 along the axonemal microtubules. Overexpressed FHDC1 is readily recruited to the cilia via its MTBD, and ciliary localization of overexpressed proteins is often indicative of their endogenous localization (Dishinger et al., 2010). While we were unable to detect the endogenous FHDC1 protein in the cilium, its exceptionally small volume greatly limits the ability to detect endogenous ciliary proteins (Nachury, 2014). Indeed, the kinked cilia and bulbous tips induced by FHDC1 overexpression are similar to the defects observed in Kif7-depleted cells (He et al., 2014) and may indicate an additional role for FHDC1 in the cilium itself.
In conclusion, we note that both FHDC1 and subdistal appendages are evolutionary innovations that have arisen in the vertebrate lineage (Young et al., 2008; Loncarek and Bettencourt-Dias, 2018). Previous observations suggest that there is cell-type specificity both in the regulation of cilia assembly and in the structure of the primary cilia (Choksi et al., 2014; Kong et al., 2014; Huang et al., 2017). FHDC1 is not ubiquitously expressed (Young et al., 2008), and hence its role in cilia assembly is by definition cell type–specific. Our results suggest that FHDC1 is one factor that contributes to the generation of diversity in both cilia structure and function in mammalian cells.
MATERIALS AND METHODS
Reagents and plasmids
Constitutively active formin derivatives and FHDC1 point mutant and deletion derivatives were previously described (Copeland et al., 2007, 2016; Vaillant et al., 2008; Young et al., 2008; Thurston et al., 2012). FHDC1 cDNA was subcloned into pcDNA3.1 mycBioID (Addgene #35700; Roux et al., 2012) using standard procedures. FHDC1 gRNA constructs were generated in PX459 (Addgene #62988) according to published protocols (Ran et al., 2013). EGFP-Gal-T (Addgene #11929) was a gift from Jennifer Lippincott-Schwartz, National Institutes of Health (Cole et al., 1996). Cer-C3-PLK1 (Addgene #68132), K82R (Addgene #68134), and T210D (Addgene #68133) were gifts from Catherine Lindon, University of Cambridge. EGFP-Centrin2 (Addgene #41147) was a gift from Erich Nigg, University of Basel (Kleylein-Sohn et al., 2007). EGFP-Cep170 (Addgene #41150) was a gift from Erich Nigg (Guarguaglini et al., 2005). pLVX-EGFP-C3-Cenexin (Addgene 73334) was a gift from Manuel Thery, CEA. The following antibodies were used in this study: rabbit anti-Cep170 (Guarguaglini et al., 2005), mouse anti–α-tubulin (Sigma, T5168), mouse anti-acetylated tubulin (clone 6-11B-1 Sigma T-6793), mouse anti-FLAG (Sigma, F7425), rabbit affinity–purified anti-FHDC1 (Young et al., 2008), Alexa Fluor 488 Phalloidin (Molecular Probes, A12379). streptavidin HRP, streptavidin488, and streptavidin beads. smiFH2 was obtained from Calbiochem (340316-62-3). Latrunculin B was from Sigma-Aldrich.
Cell culture, transfections, and treatments
NIH 3T3 fibroblasts were obtained from the American Type Culture Collection (ATCC) and cultured in DMEM (Wisent) supplemented with 10% DBS (ATCC) in 5% CO2 according to the supplied guidelines. Mycoplasma contamination was tested biweekly. Cilia assembly was induced by incubation in low-serum media (DMEM + 0.5% DBS) for 48 h. Transient transfections were performed using PEI as described previously (Young et al., 2008). Briefly, 1.5 μg total plasmid DNA was diluted in 50 μl of Optimem, 5 μl of 1 mg/ml PEI was added, and the mixture was incubated at room temperature for 25–30 min. The DNA/PEI mix was added to cells in 1 ml of Optimem and left for 5 h under normal culture conditions. At the end of 5 h the media were replaced with 2 ml of the appropriate culture media. siRNA-mediated knockdown was performed as previously described (Young et al., 2008). FHDC1 siRNA Duplex 1: 5′-GCUAUAGCACCAAAGAGAAAUUCCT-3′ and 5′-AGGAAUUUCUCUUUGGUGCUAUAGCAU-3′; Duplex 2: 5′-CCAUCGUAGAGGAUAUCUATT-3′ and 5′-UAGAUAUCCUCUACGAUGGAC-3′ (Young et al., 2008; Copeland et al., 2016).
CRISPR
Guide RNA (gRNA) targeting the first coding exon of mouse FHDC1 were designed using the http://crispr.mit.edu/ database. gRNA pair 1: 5′-CACCGCAATGGTCTTCTTGTCGATC-3′, 5′-AAACGATCGACAAGAAGACCATTGC-3′; pair 2: 5′-CACCGAACGAATGCGGAGCTTTTTC-3′, 5′-AAACGAAAAAGCTCCGCATTCGTTC-3′; pair 3: 5′-CACCGCATTTTCTTTATCGCTGGCC-3′, 5′-AAACGGCCAGCGATAAAGAAAATGC-3′. gRNA duplexes were subcloned into pX459 using standard procedures (Ran et al., 2013). The gRNA pX459 derivatives were cotransfected with pEF-mCherry as a transfection marker. The following day, transfected cells were bulk sorted for mCherry expression on a Beckman MoFlo AstriosEQ. Positive cells were treated with 2 μg/ml puromycin for 3 d and allowed to recover for 1 d, and individual live cells were sorted into 96-well plates based on side and forward scatter. Individual clones were expanded and screened for FHDC1 expression by immunoblotting with anti-FHDC1 antibody (Young et al., 2008). No complete FHDC1 knockouts were identified after more than 50 clones were screened, but clones with partial loss of expression were identified for gRNA pair 2. NIH 3T3 fibroblasts are tetraploid (Leibiger et al., 2013), which may account for the difficulty in isolating complete knockouts. Genomic DNA was harvested from one control isolate and two putative partial FHDC1 knockouts. The targeted genomic region was amplified by PCR, and the products were isolated by agarose gel electrophoresis. Amplification of genomic DNA from control cells yielded a single band of the expected size, 618 base pairs. Amplification of genomic DNA from clone 2.11 produced three amplicons corresponding to wild type (618 base pairs), a 2–base pair deletion at codon 102 (616 base pairs), and a large deletion of 206 base pairs at codon 36 (412 base pairs). The deletion at codon 102 induces a frameshift and results in termination 20 codons downstream. The larger deletion at codon 36 induces a frameshift and results in termination 14 codons downstream.
BioID interactome screen
An FHDC1 interactome was generated using the BioID system incorporating quantitative SILAC-based affinity purification as previously described SILAC (Trinkle-Mulcahy et al., 2008; Roux et al., 2012). Cells expressing myc-FHDC1 were encoded in media containing “light (L)” l-arginine and l-lysine (Sigma-Aldrich), while cells expressing birA*-FHDC1 were encoded in “heavy (H)” l-arginine 13C/15N and l-lysine 13C/15N (Cambridge Isotope Laboratories). Biotin (50 mM) was added to the media 24 h posttransfection and cells were cultured for an additional 24 h prior to lysis. Extracts were prepared in ice cold RIPA buffer, with sonication to ensure complete cell lysis, and cleared by centrifugation at 13,500 × rpm for 15 min at 4°C. Protein concentration was determined by BCA assay and equal amounts of each lysate were incubated separately with streptavidin agarose beads (Solulink) for 3 h at 4°C. The beads were combined and washed five times in low-salt RIPA buffer, and bound proteins were eluted by boiling for 5 min in 2% SDS supplemented with 30 mM biotin. Eluted proteins were separated by SDS–PAGE and trypsin-digested. An aliquot of the tryptic digest (prepared in 5% acetonitrile [ACN]/0.1% trifluoroacetic acid [TFA] in water) was analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) on an LTQ-Orbitrap mass spectrometer system (ThermoElectron) as previously described (Trinkle-Mulcahy et al., 2008). The resulting data set was filtered for known background contaminants (Lambert et al., 2015), and putative positives were prioritized based on the H:L ratio and gene function.
Immunofluorescence
Cells were prepared for immunofluorescence as described previously (Young et al., 2008). Briefly, cells cultured on acid-washed glass coverslips were fixed for 10 min directly in 4% paraformaldehyde freshly prepared in PHEM buffer (Schliwa and van Blerkom, 1981). Following fixation, the cells were permeabilized and blocked for 20 min in 0.3% Triton X-100, 5% donkey serum (DS) in 1 × PBS (phosphate-buffered saline). The coverslips were washed in 1 × PBS and incubated with the appropriate primary antibody in 0.03% Triton X-100, 5% DS in 1 × PBS for 1 h at room temperature. The coverslips were washed three times in 1 × PBS and then incubated with secondary antibody in the same solution for 1 h at room temperature. After being washed in 1 × PBS, the coverslips were mounted in Vectashield with DAPI (4′,6-diamidino-2-phenylindole) and sealed with nail polish. Primary cilia length was measured from 40× magnification micrographs using the curved measurement tool in Northern Eclipse. Statistical significance of changes in cilia length was evaluated using an unequal-variances t test, and significance of percent changes in the number of ciliated cells was evaluated using a two-tailed t test for percentages. Golgi dispersion was assessed as previously described (Copeland et al., 2016). Briefly, Golgi were considered “dispersed” if there was no obvious polarity to its position and >30 individual fragments were present. Fragmented Golgi that were polarized or nonpolarized ribbons were classed as “intermediate.” Statistical significance of percent change in cells with dispersed Golgi were evaluated using a two-tailed t test for percentages.
Microscopy
All microscopy was performed on a Zeiss AXIO Imager.Z1 with a Zeiss Apotome.2 structured illumination system for optical sectioning and using a 63× (NA 1.4) oil immersion lens or 40× (NA 0.75) dry objective and a Zeiss AxioCam HRm camera (60N-C 1″ 1.0 × 426114) controlled with AxioVision (Zeiss, release 4.8.2). Coverslips were mounted in Vectashield (Vector Labs) with or without DAPI. Cy5, Alexaflour 488, and 594 secondary antibodies were from Jackson Labs. Figures were prepared in Adobe Photoshop and Adobe Illustrator. Cilia lengths were measured using the measure tool of Northern Eclipse software.
Supplementary Material
Acknowledgments
We thank Tamara Caspary and Lynne Quarmby for their support, insight, and useful advice. We thank Bailey Paterson, Christine Tenneson, and Wojciech Kulacz for preliminary observations. Microscopy support was provided by the Cell Biology and Image Acquisition core facility, and flow cytometry was performed by the Flow Cytometry and Virometry core facility, both in the Faculty of Medicine, University of Ottawa. This work was initiated with support from Grant 102616 from the Canadian Institutes of Health Research and continued with support from Grant 05921/RGPIN/2016 from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
Abbreviations used:
- DA
distal appendage
- DAPI
4′,6-diamidino-2-phenylindole
- FH1
formin homology 1
- FH2
formin homology 2
- FHDC1
FH2 domain containing 1
- FMNL2
formin-like 2
- GFP
green fluorescent protein
- IFT-A
intraflagellar transport A
- IFT-B
intraflagellar transport B
- MTBD
microtubule-binding domain
- SDA
subdistal appendage
- TZ
transition zone
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-02-0088) on May 9, 2018.
REFERENCES
- Asante D, Maccarthy-Morrogh L, Townley AK, Weiss MA, Katayama K, Palmer KJ, Suzuki H, Westlake CJ, Stephens DJ. (2013). A role for the Golgi matrix protein giantin in ciliogenesis through control of the localization of dynein-2. J Cell Sci , 5189–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avasthi P, Onishi M, Karpiak J, Yamamoto R, Mackinder L, Jonikas MC, Sale WS, Shoichet B, Pringle JR, Marshall WF. (2014). Actin is required for IFT regulation in Chlamydomonas reinhardtii. Curr Biol , 2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitin MH. (1991). Actin filaments in the photoreceptor cilium of the rds mutant mouse. Exp Eye Res , 107–113. [DOI] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. (2012). A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol , 61–72. [DOI] [PubMed] [Google Scholar]
- Choksi SP, Lauter G, Swoboda P, Roy S. (2014). Switching on cilia: transcriptional networks regulating ciliogenesis. Development , 1427–1441. [DOI] [PubMed] [Google Scholar]
- Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J. (1996). Diffusional mobility of Golgi proteins in membranes of living cells. Science , 797–801. [DOI] [PubMed] [Google Scholar]
- Copeland SJ, Green BJ, Burchat S, Papalia GA, Banner D, Copeland JW. (2007). The diaphanous inhibitory domain/diaphanous autoregulatory domain interaction is able to mediate heterodimerization between mDia1 and mDia2. J Biol Chem , 30120–30130. [DOI] [PubMed] [Google Scholar]
- Copeland SJ, Thurston SF, Copeland JW. (2016). Actin- and microtubule-dependent regulation of Golgi morphology by FHDC1. Mol Biol Cell , 260–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AE, Schwarz N, Zelinger L, Stern-Schneider G, Shoemark A, Spitzbarth B, Gross M, Laxer U, Sosna J, Sergouniotis PI, et al (2013). Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet , 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishinger JF, Kee HL, Jenkins PM, Fan S, Hurd TW, Hammond JW, Truong YN, Margolis B, Martens JR, Verhey KJ. (2010). Ciliary entry of the kinesin-2 motor KIF17 is regulated by importin-beta2 and RanGTP. Nat Cell Biol , 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina F, Gaillard J, Guerin C, Coute Y, Sillibourne J, Blanchoin L, Thery M. (2016). The centrosome is an actin-organizing centre. Nat Cell Biol , 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertig J, Wloga D. (2008). Ciliary tubulin and its post-translational modifications. Curr Top Dev Biol , 83–113. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. (2005). The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol Biol Cell , 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Subramanian R, Bangs F, Omelchenko T, Liem KF, Jr, Kapoor TM, Anderson KV. (2014). The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat Cell Biol , 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Xia Y, Zhang D, Wang S, Bao Y, He R, Teng J, Chen J. (2017). Hierarchical assembly of centriole subdistal appendages via centrosome binding proteins CCDC120 and CCDC68. Nat Commun , 15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Kubo A, Tsukita S. (2005). Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat Cell Biol , 517–524. [DOI] [PubMed] [Google Scholar]
- Kim J, Jo H, Hong H, Kim MH, Kim JM, Lee JK, Heo WD. (2015). Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat Commun , 6781. [DOI] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. (2007). Plk4-induced centriole biogenesis in human cells. Dev Cell , 190–202. [DOI] [PubMed] [Google Scholar]
- Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W. (2010). Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA , 6346–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli P, Hohne M, Jungst C, Bertsch S, Ebert LK, Schauss AC, Benzing T, Rinschen MM, Schermer B. (2017). The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep , 1521–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, Farmer V, Shukla A, James J, Gruskin R, Kiriyama S, Loncarek J. (2014). Centriole maturation requires regulated Plk1 activity during two consecutive cell cycles. J Cell Biol , 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC. (2015). Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics , 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkins CE, Aviles GD, East MP, Kahn RA, Caspary T. (2011). Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell , 4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibiger C, Kosyakova N, Mkrtchyan H, Glei M, Trifonov V, Liehr T. (2013). First molecular cytogenetic high resolution characterization of the NIH 3T3 cell line by murine multicolor banding. J Histochem Cytochem , 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncarek J, Bettencourt-Dias M. (2018). Building the right centriole for each cell type. J Cell Biol , 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS, et al (2015). Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol , 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Janke C. (2014). Post-translational modifications of tubulin. Curr Biol , R351–R354. [DOI] [PubMed] [Google Scholar]
- Mazo G, Soplop N, Wang WJ, Uryu K, Tsou MB. (2016). Spatial control of primary ciliogenesis by subdistal appendages alters sensation-associated properties of cilia. Dev Cell , 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Hosoba K, Ochiai H, Royba E, Izumi H, Sakuma T, Yamamoto T, Dynlacht BD, Matsuura S. (2015). The microtubule-depolymerizing activity of a mitotic kinesin protein KIF2A drives primary cilia disassembly coupled with cell proliferation. Cell Rep , 664–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV. (2014). How do cilia organize signalling cascades? Philos Trans R Soc Lond B Biol Sci 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nager AR, Goldstein JS, Herranz-Perez V, Portran D, Ye F, Garcia-Verdugo JM, Nachury MV. (2017). An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell , 252–263 e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Holland AJ. (2018). Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol , 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phua SC, Chiba S, Suzuki M, Su E, Roberson EC, Pusapati GV, Setou M, Rohatgi R, Reiter JF, Ikegami K, et al (2017). Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell , 264–279 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Fuller MT. (1985). Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J Cell Biol , 2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitaval A, Senger F, Letort G, Gidrol X, Guyon L, Sillibourne J, Thery M. (2017). Microtubule stabilization drives 3D centrosome migration to initiate primary ciliogenesis. J Cell Biol , 3713–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell , 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarmby L. (2014). Cilia assembly: a role for F-actin in IFT recruitment. Curr Biol , R796–R798. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc , 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Leroux MR. (2017). Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol , 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, Gardel ML, Kozmin SA, Kovar DR. (2009). Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem Biol , 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Burke B. (2013). BioID: a screen for protein–protein interactions. Curr Protoc Protein Sci , Unit 19 23. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol , 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Dynlacht BD. (2016). Cilium assembly and disassembly. Nat Cell Biol , 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliwa M, van Blerkom J. (1981). Structural interaction of cytoskeletal components. J Cell Biol , 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder JM, Schneider L, Christensen ST, Pedersen LB. (2007). EB1 is required for primary cilia assembly in fibroblasts. Curr Biol , 1134–1139. [DOI] [PubMed] [Google Scholar]
- Sharma N, Kosan ZA, Stallworth JE, Berbari NF, Yoder BK. (2011). Soluble levels of cytosolic tubulin regulate ciliary length control. Mol Biol Cell , 806–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Perez V, Shida T, Franco B, Jego L, Ye F, Pasquier L, Loget P, et al (2014). The oral–facial–digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet , 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston SF, Kulacz WA, Shaikh S, Lee JM, Copeland JW. (2012). The ability to induce microtubule acetylation is a general feature of formin proteins. PLoS One , e48041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, Vandermoere F, Morrice NA, Swift S, Rothbauer U, Leonhardt H, et al (2008). Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol , 223–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RW, Pardee AB, Fujiwara K. (1979). Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell , 527–535. [DOI] [PubMed] [Google Scholar]
- Vaillant DC, Copeland SJ, Davis C, Thurston SF, Abdennur N, Copeland JW. (2008). Interaction of the N- and C-terminal autoregulatory domains of FRL2 does not inhibit FRL2 activity. J Biol Chem , 33750–33762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veleri S, Manjunath SH, Fariss RN, May-Simera H, Brooks M, Foskett TA, Gao C, Longo TA, Liu P, Nagashima K, et al (2014). Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat Commun , 4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Chen Q, Zhang X, Zhang B, Zhuo X, Liu J, Jiang Q, Zhang C. (2013). PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. J Cell Sci , 1355–1365. [DOI] [PubMed] [Google Scholar]
- Xu Y, Moseley JB, Sagot I, Poy F, Pellman D, Goode BL, Eck MJ. (2004). Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell , 711–723. [DOI] [PubMed] [Google Scholar]
- Yeyati PL, Schiller R, Mali G, Kasioulis I, Kawamura A, Adams IR, Playfoot C, Gilbert N, van Heyningen V, Wills J, et al (2017). KDM3A coordinates actin dynamics with intraflagellar transport to regulate cilia stability. J Cell Biol , 999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KG, Thurston SF, Copeland S, Smallwood C, Copeland JW. (2008). INF1 is a novel microtubule-associated formin. Mol Biol Cell , 5168–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.