

Abstract
Purpose
T cell dysfunction is a hallmark of GBM. While anergy and tolerance have been well characterized, T cell exhaustion remains relatively unexplored. Exhaustion, characterized in part by the upregulation of multiple immune checkpoints, is a known contributor to failures amidst immune checkpoint blockade, a strategy that has lacked success thus far in GBM. This study is among the first to examine, and credential as bona fide, exhaustion among T cells infiltrating human and murine GBM.
Experimental Design
Tumor-infiltrating and peripheral blood lymphocytes (TIL, PBL) were isolated from patients with GBM. Levels of exhaustion-associated inhibitory receptors and post-stimulation levels of the cytokines IFN-γ, TNF-α, and IL-2 were assessed by flow cytometry. T cell receptor (TCR) Vβ chain expansion was also assessed in TIL and PBL. Similar analysis was extended to TIL isolated from intracranial and subcutaneous immunocompetent murine models of glioma, breast, lung, and melanoma cancers.
Results
Our data reveal that GBM elicits a particularly severe T cell exhaustion signature among infiltrating T cells characterized by: 1) prominent upregulation of multiple immune checkpoints; 2) stereotyped T cell transcriptional programs matching classical virus-induced exhaustion; and 3) notable T cell hypo-responsiveness in tumor-specific T cells. Exhaustion signatures differ predictably with tumor identity, but remain stable across manipulated tumor locations.
Discussion
Distinct cancers possess similarly distinct mechanisms for exhausting T cells. The poor TIL function and severe exhaustion observed in GBM highlights the need to better understand this tumor-imposed mode of T cell dysfunction in order to formulate effective immunotherapeutic strategies targeting GBM.
Keywords: Glioblastoma, Immunotherapy, T Cell Exhaustion, Immune Checkpoint, PD-1
INTRODUCTION
Glioblastoma (GBM) is the most common and feared malignant primary brain tumor, persisting as one of few cancers that remain universally lethal. Despite improvements in standard of care, median survival remains at just 15–17 months (1). Immunotherapies continue to yield promise, but the efficacy needed to garner an FDA approval has remained wanting. Success has been limited by marked tumor heterogeneity (2), poor immune infiltration (3), and by the tumor’s notably potent capacities for subverting local and systemic immune responses. Regarding the latter, it is the T cells required for effective antitumor responses that are particularly victimized (4–6). Their plight is reflected in patient lymphopenia (7), as well as various characterized forms of classical T cell dysfunction, such as anergy (8,9) and regulatory T cell-imposed tolerance (10–13). Countering such dysfunction has not yet reliably yielded sustained and effective T cell activity within the tumor, however, raising concerns for additional unexplored tumor-imposed states of T cell dysfunction, such as exhaustion. Despite sporadic studies suggesting T cell phenotypes that might signal exhaustion, few formal characterizations of T cell exhaustion in patients or mice with GBM have been undertaken.
T cell exhaustion is a hyporesponsive state resulting from repeated or prolonged antigenic exposure under suboptimal conditions (14). Initially discovered in the context of chronic lymphocytic choriomeningitis virus (LCMV) infection (14,15), it is now known to contribute to disrupted T cell function in cancer as well (16). Exhaustion represents a specific transcriptional program in T cells that is often characterized phenotypically by the increased expression of multiple co-inhibitory receptors, many of which constitute classical or alternative immune checkpoints (17). Known receptors include PD-1, CTLA-4, TIM-3, LAG-3, BTLA, 2B4, CD160, CD39, VISTA, and TIGIT. Antagonizing or blocking PD-1 and CTLA-4 (the classical immune checkpoints) are well-recognized FDA-approved anti-cancer strategies aimed at improving T cell function in multiple malignancies, including melanoma and non-small cell lung carcinoma (NSCLC). To date, such strategies have shown only limited efficacy in GBM, creating the need to better understand treatment failures. One mechanism conferring resistance to classical checkpoint blockade has been the upregulation of alternative immune checkpoints such as TIM-3 and LAG-3(18), signifying that T cell exhaustion might play a substantial role in restraining therapeutic enhancements to T cell function in GBM. To date, few thorough investigations of classical and alternative immune checkpoint co-expression within GBM have been conducted. Likewise, the prominence of exhaustion amongst tumor-infiltrating lymphocytes (TIL) and its contribution to local T cell dysfunction in GBM is poorly described.
We report here that a substantial proportion of TIL derived from human GBM express multiple co-inhibitory immune checkpoints. These findings are recapitulated in two different murine models of glioma, where the mounting expression of PD-1, Tim-3, and Lag-3 correspond to diminished TIL responsiveness. TIL from glioma demonstrate significant dysfunction, and their transcriptional signatures and epigenetic profiles match that of traditional bona fide T cell exhaustion, as described in viral models. Interestingly, in our murine tumor models, patterns of TIL exhaustion prove to be tumor type-specific, with gliomas, lung carcinoma, breast carcinoma, and melanoma all eliciting characteristic, yet distinct exhaustion signatures that do not vary when tumor site is modified, including when each tumor is placed intracranially. Likewise, independent of location, the exhaustion signature and corresponding TIL dysfunction appear to be particularly severe amongst T cells infiltrating gliomas, highlighting a significant contribution for exhaustion to T cell dysfunction within these tumors.
MATERIALS AND METHODS
Patient samples
All studies were conducted with approval from the Massachusetts General Hospital Cancer Center Institutional Review Board or the Duke Cancer Center Institutional Review Board. All studies were conducted in accordance with recognized ethical guidelines (U.S. Common Rule, 45 CFR 46, 21 CFR 50, 21 CFR 56, 21 CFR 312, 21 CFR 812, and 45 CFR 164.508–514). with 21 treatment-naïve GBM patients undergoing primary surgical resection of intracranial GBM were included in the prospective collection of whole blood and tumor tissue. 15 healthy age-matched controls were included in the prospective collection of whole blood. Informed consent was obtained from all subjects. Blood specimens were collected into EDTA-containing tubes. All blood and tumor specimens were stored at room temperature and processed within 12 hours. All samples were labeled directly with antibodies for use in flow cytometry, and red blood cells subsequently lysed using eBioscience RBC lysis buffer (eBioscience, San Diego, CA). Cells were washed, fixed and analyzed on an LSRII FORTESSA flow cytometer (BD Bioscience, San Jose, CA).
Mice
Female C57BL/6 and VM-Dk mice were used at 6–12 weeks of age. C57BL/6 mice were purchased from Charles River Laboratories. VM-Dk mice were bred and maintained as a colony at Duke University. Animals were maintained under specific pathogen-free conditions at Cancer Center Isolation Facility (CCIF) of Duke University Medical Center. The Institutional Animal Care and Use Committee approved all experimental procedures.
Cell lines
Cell lines studied included SMA-560 malignant glioma, CT2A malignant glioma, E0771 breast medullary adenocarcinoma, B16F10 melanoma, and Lewis Lung Carcinoma (LLC). SMA-560 cells are syngeneic on the VM-Dk background, while all others are syngeneic in C57BL/6 mice. The SMA-560 cell line is derived from a spontaneous malignant glioma that originally arose on the VM-Dk background. Tumors have low S-100 expression and high glial fibrillary acid protein (GFAP) expression, and are most representative of anaplastic astrocytoma (19). The CT2A cell line is derived from a chemically induced tumor with 20-methylcholanthrene on the C57BL/6 background, and accurately reflects several characteristics of GBM including intra-tumoral heterogeneity, as well as radio- and chemo-resistance (20). SMA-560, CT2A, B16F10, and LLC cells were grown in vitro in Dulbecco’s Modified Eagle Medium (DMEM) with 2 mM 1-glutamine and 4.5 mg/mL glucose (Gibco, Gaithersburg, MD) containing 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA). E0771 cells were grown in vitro in RPMI 1640 (Gibco) containing 10% fetal bovine serum plus 1% HEPES (Gibco). Cells were harvested in the logarithmic growth phase. For intracranial implantation, tumor cells in PBS were then mixed 1:1 with 3% methylcellulose and loaded into a 250 μL syringe (Hamilton, Reno, NV). The needle was positioned 2 mm to the right of the bregma and 4 mm below the surface of the skull at the coronal suture using a stereotactic frame. 1 × 104 SMA-560, CT2A, E0771, and LLC cells or 500 B16F10 cells were delivered in a total volume of 5 μL per mouse. For subcutaneous implantation, 5 × 105 SMA-560, CT2A, E0771, and LLC cells or 2.5 × 105 B16F10 cells were delivered in a total volume of 200 μL per mouse into the subcutaneous tissues of the left flank.
Tissue processing
Tumors were minced, incubated in 100 Units/mL collagenase IV (Sigma-Aldrich, St. Luis, MO) and 0.1mg/mL DNase I (Roche Diagnostics, Indianapolis, IN) in RPMI supplemented with 10% FBS for 20–30 minutes in a Stomacher ® machine set at normal speed, washed through 70um nylon cell strainers (Falcon; BD Biosciences) in PBS with 2% FBS. Cells were immediately stained and subsequently analyzed by flow cytometry.
Flow cytometry and cytokine detection
Human-specific antibodies were purchased from BD Biosciences (CD3: SP34-2; CD4: RPA-T4) or BioLegend (CD8: HIT8a; PD1: EH12.2H7; TIM-3: F38-2E2; LAG3-: 11C3C65; CTLA-4: BNI3; TIGIT: A15153G; CD39: A1; IL-2: MQ1-17H12; TNF-α: MAb11; IFN-γ: 4S.B3 (San Diego, CA). Mouse-specific antibodies were purchased from BD Biosciences (CD3: 145-2C11; CD4: RM4-5; CD8: 53-6.7; PD1: J43; Lag-3: C9B7W; TIGIT: 1G9; CD244.2: 2B4; CD160: CNX46-3; CTLA4: UC10-4F10) or Biolegend (IFN-γ: XMG1.2; IL-2: JES6-5H4; TNF-α: MP6-XT22; Tim-3: RMT3-23; CD39: Duha59; BTLA: 8F4). Appropriate isotype controls were used when applicable. LIVE/DEAD ® Fixable Yellow Dead Cell Stain Kit (ThermoFischer) was used to exclude dead cells. Intracellular staining was performed using the eBioscience Fixation and Permeabilization Buffer kit. For intracellular cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA, 50nM) and ionomycin (500 nM) for 4–6h at 37°C, 5% CO2 in the presence of 1 ug/ml brefeldin A (BD biosciences). Surface staining was performed and cells were fixed and permeabilized with the BD Cytofix/Cytoperm kit and stained for IFN-γ, TNF-α, and IL-2. Tetramers for mODC1 were generated and used as described previously (21).
Analysis of T Cell Receptor Vβ Repertoire
5 patient TIL and PBMC and 5 control PBMC were isolated as described above. The repertoire was assessed using the IOTest ® Beta Mark TCR V beta Repertoire Kit (Cat: IM3497 Beckman Coulter, Brea, CA) according to manufacturer’s instructions.
Gene Expression Analysis
When moribund, tumor-bearing animals were sacrificed and T cells were isolated and sorted from tumors and spleens based on the following markers: Live, CD45+, CD3+, CD8+. CD8+ T cells isolated from tumor-draining cervical lymph nodes were additionally stained and sorted for CD44+. RNA was extracted using the Qiagen (Hilden, Germany) RNAeasy Mini Kit (Cat No: 74104) and DNase treated using Qiagen RNase-Free DNase Set (Cat No: 79254). RNA concentration and purity was determined with NanoDrop and RNA integrity with Agilent Bioanalyzer. cDNA was synthesized and hybridized to the Affymetrix (Santa Clara, CA) Clariom S Mouse Array.
PD-1 Methylation Analysis
Genomic DNA was isolated from sorted CD8+ T cells (as described above) from tumors, spleens, or tumor-draining cervical lymph nodes using the Quick-DNA Universal Kit Cat No: D4068 (Zymo Research, Irvine CA)) according to manufacturer’s instructions, and <1μg of DNA was subjected to bisulfite conversion using the EZ DNA Methylation Kit Cat No: D5001 (Zymo Research). The bisulfite converted DNA was then amplified for pyrosequencing analysis using the PyroMark PCR Kit (Qiagen, Cat No: 978703). Primers were selected for the CR-B site covering 4 CpG sites at the PD-1 promoter region as previously published (22). Pyrosequencing was performed on amplified bisulfite converted DNA using PyroMark Gold Q96 Reagent (Qiagen, Cat No: 972807).
Statistical Analysis
For human studies, the sample size of 21 patients and 10 controls was chosen so that a two-tailed t-test comparing groups has 90% power to detect a difference that is 1.1 times the standard deviation of the outcome variable in each group. For animal studies, sample sizes were chosen based on historical experience and were variable based on numbers of surviving mice available at experimental timepoints or technical limitations. For statistical comparisons, unpaired t-tests were generally used to compare groups. Analyses were not adjusted for multiple testing.
RESULTS
T cells infiltrating human glioblastoma express multiple immune checkpoints and exhibit impaired function
The checkpoint molecules PD-1, CTLA-4, TIM-3, LAG-3, CD160, 2B4, TIGIT, CD39, and BTLA have been identified as co-inhibitory regulators of T cell effector function during chronic viral infection and within tumors, limiting the scope and duration of T cell responses (17,23). Furthermore, their expression on T cells can frequently signal an exhausted state. To begin examining the contribution of exhaustion to T cell dysfunction in GBM, we used flow cytometry to phenotypically analyze tumor-infiltrating lymphocytes (TIL) and peripheral blood mononuclear cells (PBMC) isolated from treatment-naïve patients with GBM enrolled in a specimen collection protocol. PBMC from healthy controls were isolated for comparison. 21 patients were enrolled in total (patient and control characteristics listed in Supplementary Table 1).
We found that PD-1, LAG-3, TIGIT, and CD39, in particular, were highly expressed on patient CD8+ TIL (Fig 1A and B) (gating strategy depicted in Supplementary Fig 1A). Notably, PD-1 was especially prominent, being found on up to 96% of tumor-infiltrating CD8+ T cells, with most of these in turn being PD-1hi. Although the percentage of TIM-3+ CD8+ T cells was not statistically significantly greater among TIL than patient or control PBMC (p=0.059, TIM-3 was present on >20% of TIL in several patients, and the median fluorescence intensity (MFI) of TIM-3 was significantly greater on TIL than in control blood (Supplementary Fig 1B). The majority of immune checkpoints studied were present to a greater extent among TIL than among either patient or control PBMC. An exception was BTLA, which was lower among TIL, consistent with studies showing that BTLA is more highly expressed on naïve T cells and becomes downregulated following antigenic exposure (24). Regarding peripheral blood, patient and control PBMC did not differ substantially from each other with respect to checkpoint expression.
Figure 1. Elevated expression of checkpoint molecules and decreased cytokine production among TIL in human GBM.
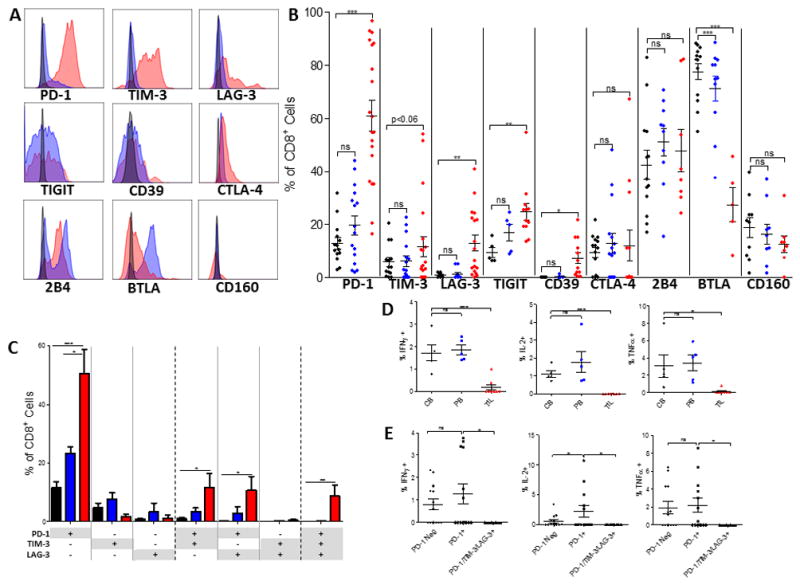
A. Representative histograms of checkpoint molecule expression on CD8+ T cells where red represents patient TIL, blue represents patient blood, and black represents relevant isotype control. CTLA-4 is an intracellular stain. B. Frequency of checkpoint molecule expression on CD8+ T cells isolated from TIL (PD-1, TIM-3 n=20; LAG-3 n=18; CTLA-4 n=13; CD39, TIGIT n=12; CD160, 2B4 n=8; BTLA n=5), patient blood (PD-1, TIM-3 n=16; CTLA-4 n=15; 2B4, BTLA n=11; LAG-3 n=12; CD160 n=9; TIGIT, CD39 n=5), or control blood (PD-1, TIM-3, CTLA-4 n=14; LAG-3 n=10; 2B4, BTLA n=13; CD160 n=10; TIGIT, CD39 n=5). C. Boolean gating was performed on CD8+ T cells isolated from control and patient blood and patient tumors to determine co-expression of PD-1, TIM-3, and LAG-3. Sample numbers: control blood (n=9), patient blood (n=6), and patient TIL (n=5). Bar graphs represent mean +/− SEM. D. Post-stimulation with PMA/Ionomycin intracellular staining was performed on patient TIL, blood and control blood for IFN-γ, IL-2, and TNF-α. E. Boolean gating was employed to determine percentage of cells producing cytokines among T cells not expressing PD-1, PD-1 single positive, or PD-1/TIM-3/LAG-3 triple positive cells. B–D: red represents patient TIL, blue represents patient blood, and black represents control blood. Throughout figure *p<0.05; **p<0.01; ***p<0.0001 by unpaired t-test between control and patient samples.
There was no difference in the percentage of CD8+ T cells expressing 2B4, CD160, or CTLA-4 across tissue types or between patients and controls. MFI data for all checkpoint molecules are available in Supplementary Fig 1B. Upon further characterization of T cells infiltrating human GBM, we found that tumor-infiltrating CD8+ T cells demonstrated a predominantly effector memory T cell phenotype (CD45RA-CD62L-) as opposed to naive (CD45RA+CD62L+) (Supplementary Fig 2), reflecting prior antigenic exposure.
While high levels of PD-1 expression on effector memory T cells can signal either activation or exhaustion, the mounting expression of additional alternative immune checkpoints frequently reflects the hierarchical loss of effector function, favoring the presence of T cell exhaustion (17). Therefore, we evaluated the degree of co-expression between PD-1 and the common alternative immune checkpoints TIM-3 and LAG-3 on CD8+ T cells from both tumors and blood. Boolean gating was employed to analyze the following T cell populations: CD8+ T cells that were positive for only one marker (e.g. PD-1+TIM-3-LAG-3-), positive for two markers (e.g. PD-1+TIM-3+LAG-3-), or triply positive (PD-1+TIM-3+LAG-3+). Higher percentages of CD8+ TIL in human GBM expressed either PD-1 alone, PD-1 in combination with TIM-3 or LAG-3, or all three markers, in comparison with CD8+ T cells isolated from either patient or control blood (Fig 1C). Single expression of TIM-3 or LAG-3 was not common in either TIL or blood. These findings reveal that both TIM-3 and LAG-3 expression are almost universally accompanied by PD-1 expression (and frequently by one another as well), with such co-expression suggesting a dysfunctional state.
We next assessed cytokine production of TIL and PBMC to determine whether the high immune checkpoint expression in TIL predicted T cell hyporesponsiveness. Following stimulation with PMA and ionomycin, intracellular IFN-γ, IL-2, and TNF-α were assessed by flow cytometry. There was no difference in cytokine production between patient and control PBMC, while TIL produced significantly less IFN-γ, IL-2, and TNF-α than control PBMC (Fig 1D). Within patient samples, cells that were PD-1 negative or PD-1 single positive produced comparable amounts of IFN-γ and TNF-α, while cells that were PD-1 single positive produced more IL-2 than PD-1 negative cells (Fig 1E), highlighting that high levels of PD-1 alone may represent a state of activation rather than exhaustion. Mounting expression of the alterative immune checkpoints TIM-3 and LAG-3 resulted in loss of function: triply positive PD-1+TIM-3+LAG-3+ T cells were unable to produce IFN-γ, IL-2, or TNF-α (Fig 1E).
Multiple immune checkpoint expression on TIL is recapitulated in murine models of malignant glioma
To examine the degree to which these findings might be recapitulated in mice, we employed two common immunologically relevant orthotopic models of murine malignant glioma: SMA-560 and CT2A. Beginning with the CT2A model, we found that PD-1 was expressed by up to 95% of CD8+ TIL (Fig 2A), similar to human GBM TIL. Tim-3, Lag-3, CTLA-4, 2B4, CD160, CD39, and TIGIT expression were also notably prevalent, and appeared to be more common than among GBM patient TIL, although no statistical comparisons across species were made.
Figure 2. Expression of multiple checkpoint molecules among TIL in CT2A and SMA-560 murine glioma models.
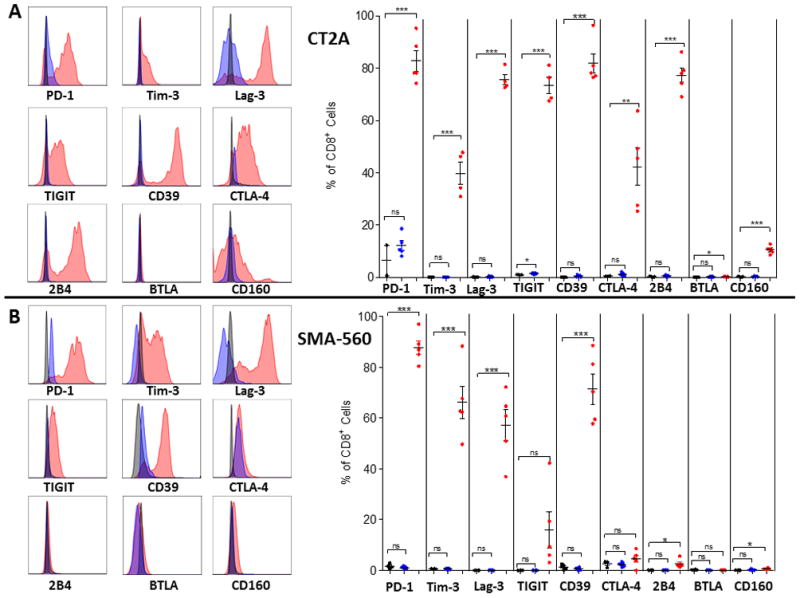
Tumors and blood were harvested when mice were moribund at day 21 post intracranial tumor implantation (n=5) for CT2A (A) or SMA-560 (B) tumor models. Representative histograms of checkpoint molecule expression on CD8+ T cells are shown where red represents TIL, blue represents tumor-bearing blood, and black represents relevant isotype control. For graphs, red represents TIL, blue represents tumor-bearing blood, and black represents naïve blood. Significance was determined using unpaired t-test between control and tumor-bearing samples. *p<0.05; ***p<.001.
CT2A TIL isolated from SMA-560 tumors likewise demonstrated increased expression of multiple immune checkpoints (particularly PD-1, Tim-3, Lag-3, and CD39), but consistently demonstrated less prominent upregulation of CTLA-4, 2B4, CD160, and TIGIT than what was found for CT2A (Fig 2A–B). Both the CT2A and SMA-560 models demonstrated a substantial proportion of TIL that were triply positive for PD-1, Tim-3, and Lag-3: upon Boolean analysis, a mean of 40% and 46% of infiltrating CD8+ T cells in CT2A and SMA-560, respectively, expressed all three immune checkpoints (Supplementary Fig 3). Similarly, in both murine models, we found that PD-1 was expressed on the majority of CD8+ TIL, while Tim-3 and Lag-3 were co-expressed with PD-1 rather than present on their own, akin to the observation in human GBM specimens.
TIL dysfunction, transcriptional signatures, and epigenetic modifications are consistent with bona fide T cell exhaustion
To determine whether glioma TIL demonstrate decreased functional capacity compared to lymphocytes isolated from other compartments in tumor-bearing and naïve animals, we harvested lymphocytes from spleens, tumor-draining (TD) cervical lymph nodes (LN), and tumors at day 18 following intracranial (IC) implantation with CT2A. Lymphocytes were also harvested from spleens and LN of age-matched naïve animals. As might be anticipated, CD8+ TIL expressed greater numbers and levels of immune checkpoints than CD8+ lymphocytes isolated from spleens or TDLN (Fig 3A). Notably, neither spleens nor LN from tumor-bearing animals demonstrated appreciable differences in the percentages of single-positive PD-1, Tim-3, or Lag-3 expressing T cells when compared to the same lymphoid organs from naïve animals, further revealing such expression patterns as concentrated amongst TIL. Accompanying the phenotypic variations across TIL, spleen, and LN, however, were significant differences in T cell function across the compartments. Following stimulation with PMA and ionomycin, intracellular IFN-γ, IL-2, and TNF-α were assessed by flow cytometry. Significantly fewer TIL were positive for IFN-γ, IL-2, and TNF-α than lymphocytes isolated from either spleens or LN, signaling a hypo-responsive state amongst TIL (Fig 3B). The MFIs of IFN-γ, IL-2, and TNF-α were also significantly lower amongst TIL than among T cells from other lymphoid compartments (Supplementary Fig 4A). Representative dot plots are shown in Supplementary Fig 4B. CD8+ TIL staining positively for IFN-γ, IL-2, or TNF-α were not uncommonly found to be PD-1 single positive (consistent with PD-1’s ambiguous identity as an activation marker). TIL triply positive for PD-1, Tim-3, and Lag-3 produced significantly less IL-2 than PD-1 singly positive TIL, and triply positive TIL trended towards decreased TNF-α production, though a significant difference was not seen. T cells producing IFN-γ were not found amongst triply positive TIL, indicating that mounting expression of alternative immune checkpoints corresponded to worsening function, particularly with regard to IFN-γ and IL-2 production (Fig 3C).
Figure 3. TIL isolated from murine gliomas demonstrate impaired function.
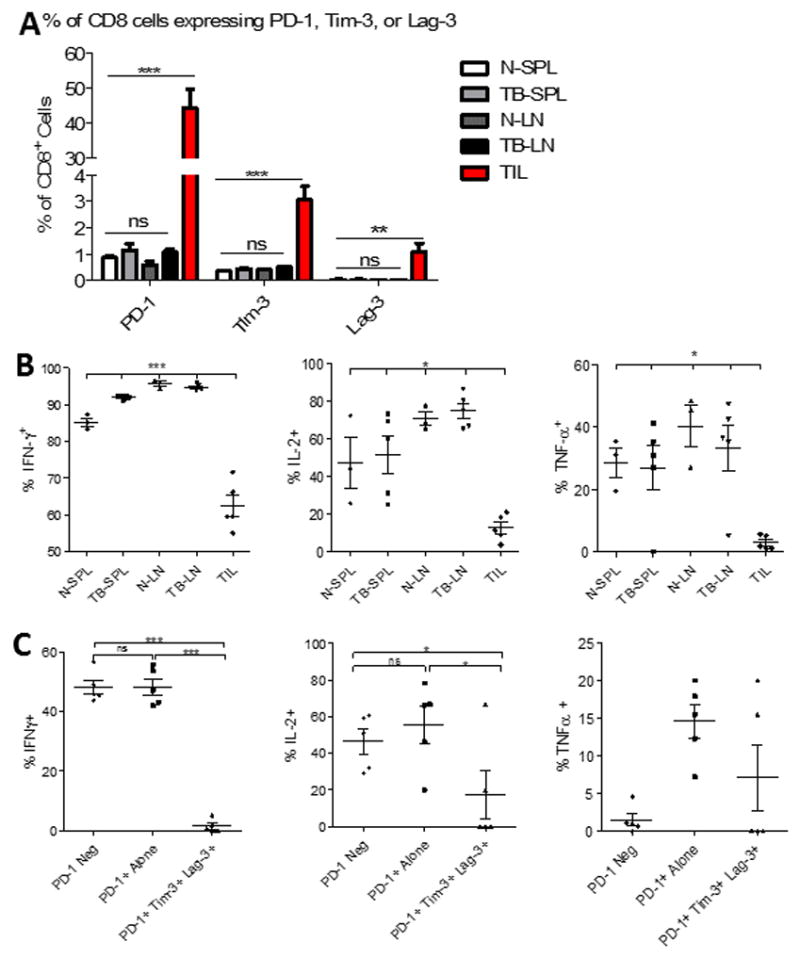
A. The percentage of CD8+ T cells expressing either PD-1, Tim-3, or Lag-3 alone was compared across immunologic compartments. T cells were isolated from the spleens (SPL) and cervical lymph nodes (LN) from either CT2A tumor-bearing (TB) (n=5) or naïve (N) (n=3) mice, as well as from tumors of TB mice (TIL). B. The capacity for CD8+ T cells isolated from the same sites to express IFN-γ, IL-2, and TNF-α, upon stimulation with PMA/ionomycin was compared. C. Boolean gating was employed to determine percentage of cells producing cytokines among TIL not expressing PD-1, PD-1 single positive, or PD-1/TIM-3/LAG-3 triple positive cells. Statistical significance was assessed via unpaired t-test between control and tumor-bearing samples. *p<0.05; **p<0.01; ***p<0.0001 throughout figure.
To further assess glioma TIL for features of bona fide T cell exhaustion, as described in models of chronic viral infection, we compared the gene-expression profiles of CD8+ CT2A TIL to those of naïve (CD44-) CD8+ T cells isolated from spleens, or to activated (CD44+) CD8+ T cells isolated from TDLN using the murine Clariom S microarray transcriptome-level platform (Affymetrix). The gene expression profile of TIL differed significantly from the profile of either naïve or effector T cells (Fig 4A). To determine whether this molecular signature was consistent with T cell exhaustion, we used gene-set enrichment analysis to compare the transcriptional profile of CT2A TIL with previously identified gene sets from virus-specific exhausted CD8+ T cells isolated from chronic LCMV clone 13 infections (31). We found enrichment for exhaustion genes and pathways in CT2A TIL that statistically matched patterns observed in virally-induced exhaustion (Fig 4B).
Figure 4. TIL demonstrate molecular signatures and epigenetic profiles consistent with T cell exhaustion.
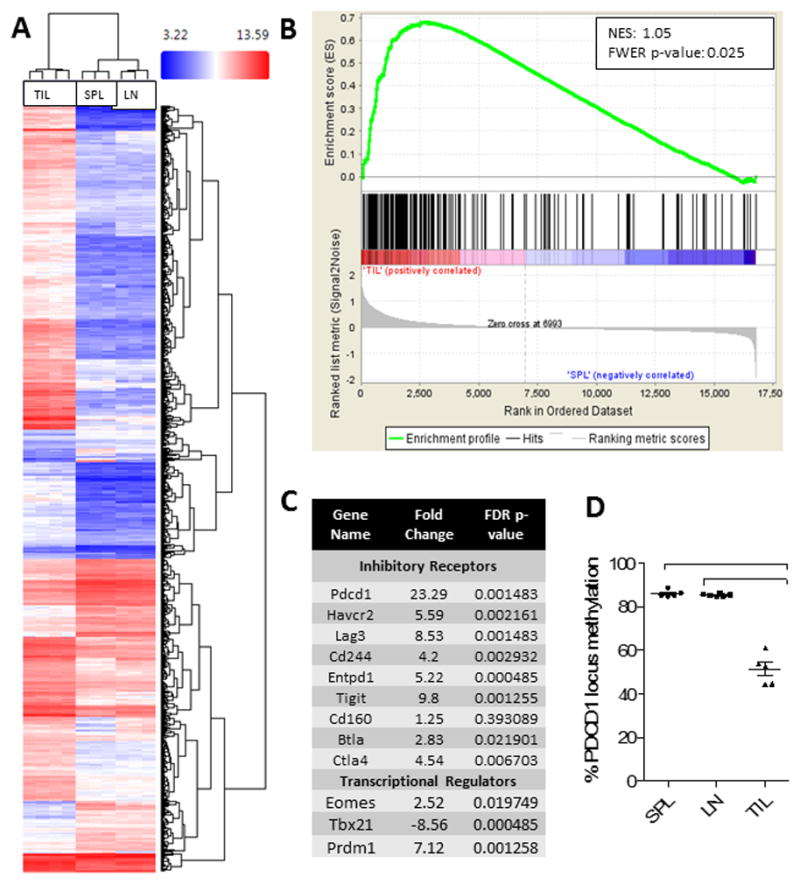
A. A hierarchical cluster of gene expression of CD8+ T cells isolated from tumors (TIL), spleens (SPL), or tumor-draining cervical lymph nodes (LN). Microarray analysis was performed using the Affymetrix Clariom S Array Platform and clustering was performed on the 1071 genes which were screened from 22701 total. Comparisons were made via ANOVA; p-value <0.001 and FDR p-value <0.001 (all conditions). B. Gene Set Enrichment Analysis (GSEA) was performed with the software GSEA v2.2.3 downloaded from the Broad Institute. Gene sets assessed included GSE30962, publically available through the NCBI on Gene Expression Omnibus. NES = normalized enrichment score. C. Table demonstrating representative transcripts over- or under-represented in TIL compared to SPL. D. Analysis of methylation at the PD-1 promoter (Pdcd1) in CD8+ T cells from TIL, SPL, or LN via ANOVA with repeated measures followed by Bonferroni post-test ***p<0.0001.
T cell exhaustion during chronic viral infection is associated with the loss of TbethiPD-1int T cells and the accumulation of terminally differentiated EomeshiPD-1hi exhausted T cells (32). Likewise, our data show that GBM TIL demonstrate elevated Eomes and decreased Tbet levels (Fig 4C). Blimp-1, a transcription factor associated with T cell exhaustion in chronic viral infections (33), is also elevated among CT2A TIL. As additional confirmation, we examined the methylation status of the PD-1 (pdcd1) gene locus. Chronic viral infection has been show to enforce demethylation of the PD-1 locus in antigen-specific T cells (34). We found that the PD-1 locus is significantly demethylated in glioma TIL compared to naïve and effector T cells, consistent with expectations for T cell exhaustion (Fig 4D).
T cell exhaustion arises preferentially amidst tumor-specific T cells
To determine whether T cell exhaustion arises moreso amidst tumor-specific T cells, we utilized the SMA-560 glioma model. The SMA-560 glioma model expresses the recently characterized ODC1-Q129L mutation, which results in a functionally immunogenic endogenous neoantigen (21). As such, we were able to detect neoantigen-specific T cells within IC SMA-560 gliomas using a newly developed ODC1 tetramer. In so doing, we found that ODC1 tetramer-positive T cells contained higher proportions of triply positive PD-1+Tim-3+Lag-3+ T cells, and were less functional upon stimulation than their tetramer-negative counterparts (Fig 5A).
Figure 5. T cell exhaustion arises preferentially amongst tumor-specific T cells.
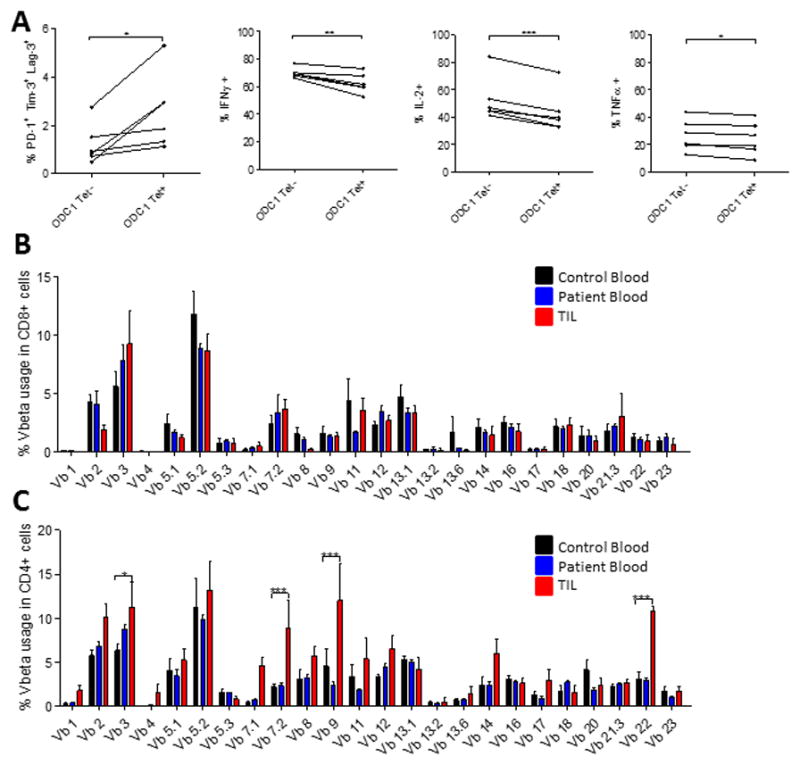
A. 5 mice were implanted with SMA-560 tumors IC. Tumors were harvested when mice were moribund and TIL isolated. TIL were stimulated with the ODC-1 peptide for 6 hours, stained with the ODC-1 tetramer conjugated to APC, several immune checkpoints and intracellular stain for cytokines was performed. Significance was assessed via paired t-test between tetramer positive and negative cells. Vbeta analysis was performed on CD8+ (B) and CD4+ (C) T cells isolated from human GBM TIL (n=5), patient blood (n=5), or control blood (n=5). Significance was assessed using a two-way ANOVA to assess for interaction between Vb and sample type, followed by Bonferroni post-tests between patient and control samples. *p<0.05; **p<0.01; ***p<0.001.
We next sought to determine whether T cell exhaustion likewise preferentially arises in tumor-specific T cells in human GBM. Given the inability to reliably detect and compare neoantigen-specific T cells across heterogeneous patient specimens, we assessed T cell clonal expansion as a surrogate measure for antigen-specificity, with the goal of phenotypically characterizing any expanded clones. We analyzed the T cell receptor (TCR) Vβ chains of 5 matched patient TIL and PBMC samples, and 5 control PBMC samples. Surprisingly, human GBM-infiltrating CD8+ T cells exhibited no detectable clonal expansion, precluding phenotypic characterization of expanded antigen-specific clones (Fig 5B). This lack of clonal expansion suggests either ineffectual tumor antigen presentation or the onset of functional defects amidst antigen-specific CD8+ T cells immediately upon tumor infiltration. The latter possibility appears more likely, as the limited expansion of CD8+ T cells contrasted starkly with ample clonal expansion amidst infiltrating CD4+ T cells (Fig 5C). Furthermore, we demonstrated that while CD4+ T cells express multiple immune checkpoints (Supplementary Fig 5A), they remain functional, producing equal amounts of IFN-γ as patient and control blood when stimulated (Supplementary Fig 5B).
T cell exhaustion signatures reflect tumor type rather than location and are particularly severe amongst malignant glioma
In prior experiments (Figs 2A, 2B), we revealed that the SMA-560 and CT2A glioma models yield consistent yet characteristically distinct exhaustion signatures among infiltrating T cells. This suggested that varying tumor types might elicit reproducibly distinct patterns of exhaustion. To determine whether the intracranial environment might also contribute to the glioma-induced T cell exhaustion signatures observed, we implanted CT2A and SMA-560 gliomas both IC and subcutaneously (SQ) and analyzed TIL at late stages of tumor growth. The phenotypic T cell exhaustion signature remained constant for each glioma model, independent of the tumor’s location in the brain versus the flank (Fig 6A).
Figure 6. T cell exhaustion signatures reflect tumor histology rather than intracranial location and are particularly severe among malignant glioma.
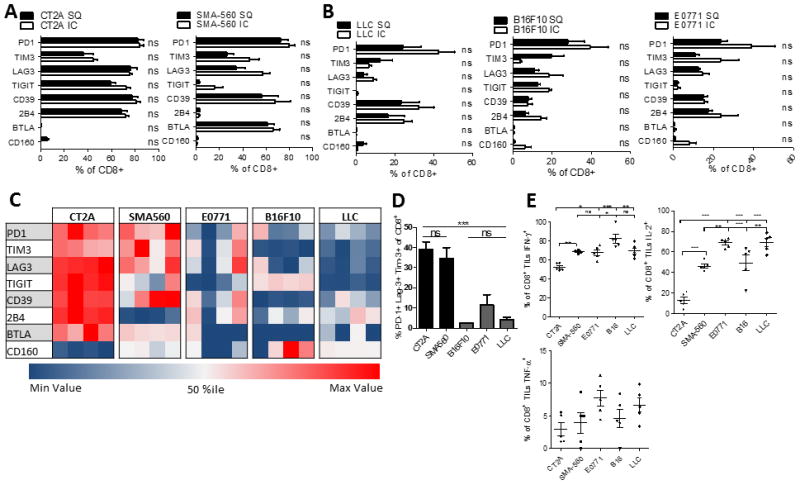
A. The percentage of CD8+ T cells expressing immune checkpoints within either intracranial (IC) or subcutaneous (SC) CT2A and SMA-560 glioma models. TIL were isolated when mice bearing IC tumors (n=5) were moribund or when SC tumors (n=5) reached 20mm at the largest diameter. B. The percentage of CD8+ T cells expressing various immune checkpoints among IC or SC B16F10 (melanoma), E0771 (breast) and LLC (lung). C. Heat map representing the % of CD8+ TIL expressing immune checkpoints across five different models of IC malignancies (n=4). Each row represents one immune checkpoint with associated percentiles. Each column represents an individual mouse. D. The percentage of CD8+ TIL co-expressing PD-1, Tim-3, and Lag-3 across five different models of intracranial malignancies. Differences were assessed via one-way ANOVA with Tukey’s post hoc test. E. The percentage of CD8+ TIL staining for IFN-γ, IL-2, and TNF-α. *p<0.05; **p<0.01; ***p<0.0001.
We then determined whether infiltrating T cell exhaustion signatures differ substantially across gliomas or other solid tumors commonly metastatic to the brain. To this end, we first validated IC engraftment of melanoma (B16F10), breast (E0771), and lung (LLC) carcinoma cell lines (Supplementary Fig 6A). Subsequently, we implanted B16F10, E0771, and LLC either IC or SQ into syngeneic C57BL/6 mice, harvested tumors when mice were moribund, and determined the TIL exhaustion patterns. Once again, each tumor yielded a characteristic and distinct TIL exhaustion signature that did not vary with intracranial versus peripheral location (Fig 6B). Representative staining of PD-1 across tumor models is shown in Supplementary Fig 6B.
Among tumor types, the SMA-560 and CT2A malignant gliomas each yielded TIL with comparatively severe phenotypic exhaustion patterns (Fig 6C). This included significantly increased numbers of TIL that were triply PD-1+Tim-3+Lag-3+ (Fig 6D). Correspondingly, TIL isolated from gliomas exhibited the poorest function, with fewer cells producing IL-2 than TIL isolated from non-glioma tumor models (Fig 6E) (MFI data shown in Supplementary Fig 6C). CT2A gliomas, in particular, harbored the least functional T cells, exhibiting significantly impaired IFN-γ and IL-2 production compared to TIL from SMA-560 gliomas or other tumor models (Fig 6E). Likewise, the greater functional impairment corresponded to higher levels of the alternative immune checkpoints TIGIT, CD39, and 2B4 on T cells, compared to those of the SMA-560 glioma model (Fig 6C). Importantly, the severity of TIL exhaustion patterns did not appear to correspond to levels of T cell infiltration across tumor types: CT2A and LLC, despite very different exhaustion signatures, both demonstrated low T cell infiltration (“cold” tumors), while E0771, SMA-560, and E0771 all demonstrated greater degrees of infiltration (Supplementary Fig 7). The degree of T cell infiltration may not influence T cell exhaustion profiles, suggesting that the association of T cell infiltration to treatment response to checkpoint blockade may not be associated with degree of T cell exhaustion (35). Taken together, these findings suggest that exhaustion patterns among tumor-infiltrating T cells are 1) determined by tumor-intrinsic factors and not environment; 2) strongly influence infiltrating T cell function; and 3) vary in severity across and reflect tumor type, with gliomas instigating particularly severe exhaustion.
DISCUSSION
Immune checkpoint blockade has garnered significant interest in recent years following dramatic successes against a variety of solid tumors (36–40) and resultant FDA approval as an immunotherapeutic strategy for multiple malignancies. The efficacy of classical checkpoint blockade (specifically anti-PD-1 and anti-CTLA-4) may be hindered, however, by the emergence of a number of secondary or alternative checkpoints on T cells (18), including TIM-3, LAG-3, BTLA, 2B4, CD160, CD39, and TIGIT (41–43). Mounting expression of these alternative checkpoints on T cells provides additional avenues for T cell shutdown following activation, but also serves to signal T cell transition into a dysfunctional, exhausted state (43,44). Such exhaustion may be reversible in earlier stages, but can rapidly progress to a late phase in which function declines beyond rescue (45,46).
T cell dysfunction has long been a hallmark of GBM, as we, and others, have highlighted (4–6). Classically, such dysfunction can be categorized under the labels of senescence, ignorance, anergy, tolerance, and exhaustion (47). Anergy and tolerance are well characterized in GBM, but this study suggests exhaustion as a major contributor to the failure of T cells that otherwise successfully arrive at tumor and initially become activated. Whereas much focus to date has been on activating T cells and ensuring their access to tumors situated within the confines of the brain, we demonstrate that those T cells successfully arriving tumor are still rendered hypo-responsive by the tumor, with exhaustion revealed as a predominant and underappreciated mode of dysfunction. Accordingly, we are among the first to thoroughly characterize immune checkpoint expression on GBM patient CD8+ TIL, and to subsequently credential such expression as bona fide exhaustion by performing functional and transcriptome analyses. Our study highlights that phenotypic expression of checkpoints may not accurately reflect an exhausted state: we show that CD4+ TIL remain functional despite expression of multiple immune checkpoints, and that CD8+ T cells expressing PD-1 alone remain functional. In light of these findings, we demonstrate that T cell functional status and molecular signatures remain important confirmations of true T cell exhaustion.
Prevalent amongst TIL in both patients and mice with GBM are T cells co-expressing PD-1 in conjunction with the alternative inhibitory immune checkpoints, TIM-3 and LAG-3. This co-expression appears to specifically mark TIL as hypofunctional in our study and suggests the importance of TIM-3 and LAG-3 as immune checkpoint targets of import in GBM. Preclinical work has indeed highlighted a possible synergy between PD-1, Tim-3, and Lag-3 blockade in mice with GBM (48). Monoclonal antibodies blocking human TIM-3 and LAG-3 are in early phase clinical trials in GBM and other solid tumors. A phase I clinical trial of anti-LAG-3 alone or in combination with anti-PD-1 is currently recruiting GBM patients (NCT02658981), while a phase I clinical trial of anti-TIM-3 alone or in combination with anti-PD-1 is currently recruiting patients with advanced solid tumors (NCT02817633). The results of these trials will help to assess whether combinatorial checkpoint blockade in GBM might increase the likelihood of functional rescue for T cells infiltrating these tumors. Data from these trials will be highly anticipated, given the recent announcements regarding the failure of PD-1 blockade monotherapy in a phase III trial in GBM.
Checkpoint blockade, as with all immunotherapies, encounters multiple challenges in GBM, including considerable intratumoral heterogeneity (2,49,50), poor immunogenicity (51,52), low mutational burden (53), poor T cell infiltration (54), and substantial tumor-elicited T cell dysfunction (10). Each of these challenges may need to be addressed before we can anticipate the ingress of prodigious immunotherapeutic success in GBM. GBM has frequently posed more difficulty than other cancers, evident in what remains a 100% mortality rate, despite what is essentially a local disease with only exceedingly rare cases of metastasis. Yet, despite its exclusively local progression, GBM elicits severe immune dysfunction, both locally and systemically. Here, we demonstrate that GBM also elicits a uniquely severe T cell exhaustion signature compared to other tumor types. This finding is manifested in the increased expression of multiple co-inhibitory immune checkpoints and the decreased functional capacity of glioma-infiltrating T cells in comparison to other T cells infiltrating other models of intracranial malignancy. The mounting expression of alternative immune checkpoints may indicate a state of terminal exhaustion that cannot be reversed by traditional checkpoint blockade alone.
Importantly, different tumor histologies (and even different glioma models) in our studies elicited differing exhaustion signatures among infiltrating T cells, regardless of where in the body these tumors were introduced. These results suggest that different tumors might induce reproducibly distinct patterns of exhaustion. They also therefore suggest that the mechanisms whereby tumors induce exhaustion amongst T cells may not be universal, and may represent a “convergent evolution” of sorts. These findings, however, may also begin to provide the foundation for personalized and rationally designed alternative immune checkpoint blockade: checkpoint blockade strategies successful in one cancer type may not be appropriate in others, and may ultimately require thorough characterization of the dysfunction elicited among a given patient’s TIL.
Lastly, the lack of influence of tumor location on TIL exhaustion signature suggests a limited role for CNS-specific factors in the programming of the T cell exhaustion, a point of optimism for those looking to thwart T cell exhaustion within the brain. Such strategies, as applied to primary peripheral tumors metastasizing to the brain, may prove equally successful against brain metastases or primary brain tumors, once afforded appropriate CNS access. Future studies will need to examine factors influencing tumor-specific induction of T cell exhaustion, as well as the capacities for T cell rescue with rationally designed immune checkpoint blockade or additional strategies.
Supplementary Material
Statement of Translational Relevance.
Primary malignant brain neoplasms are responsible for over 15,000 deaths annually in the United States. Although several immunotherapeutic strategies have shown remarkable success and are FDA-approved in other malignancies, immunotherapies for glioblastoma (GBM) have shown limited success in clinical trials. Immunotherapies are potentially limited in GBM by several factors, including tumor heterogeneity, low tumor mutational burden, low levels of T cell infiltration, and GBM’s potent immunosuppressive capabilities. The latter most prominently results in profound T cell dysfunction, damaging the efficacy of immune-based platforms. Much of the immunotherapeutic focus has been on activating T cells and ensuring their access to tumors situated within the confines of the brain. We demonstrate, however, that those T cells successfully arriving at the tumor are still rendered hypo-responsive by the tumor, with exhaustion revealed as a significant mode of dysfunction. Notably, we find that GBM elicits a particularly severe exhaustion signature among infiltrating T cells, characterized by prominent upregulation of multiple immune checkpoints and stereotyped exhaustion transcriptional signatures. The severity of the observed T cell exhaustion suggests tumor-imposed dysfunction that might not be reversed with immune checkpoint blockade alone, highlighting the new and urgent need to address the underlying mechanisms contributing to tumor-imposed exhaustion in order to formulate effective immunotherapies targeting GBM.
Acknowledgments
Financial Support: The work was supported in part by the National Institutes of Health Duke Brain SPORE Developmental Research Program (P.E.F.), the Medical Scientist Training Program at Duke University School of Medicine (K.W.), the NIH grant K08NS092912 (G.P.D.), the American Cancer Society-Institutional Research grant (G.P.D.), and the Physician-Scientist Training Program at Washington University School of Medicine (T.M.J.).
Abbreviations
- GBM
glioblastoma
- TIL
tumor-infiltrating lymphocytes
Footnotes
Conflicts of Interest: Glenn Dranoff is currently an employee and stock-holder at Novartis, Inc.
All authors: read, revised, and approved the final manuscript
Statement of Author Contributions:
K.W., S.K., C.J., A.G., and P.E.F. obtained and/or analyzed clinical data
K.W., P.C., K.R., H.K., C.D., H.F., A.E., X.C., L.H., G.D., K.W.H., T.M.J., G.P.D., and P.E.F. designed, carried out, and/or analyzed all in vivo experiments
P.H. provided statistical support
K.W., A.G., and K.W.H., and P.F. wrote the manuscript
References
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. Journal of neuroimmunology. 2013;264(1–2):71–83. doi: 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. Journal of neuroimmunology. 1999;100(1–2):216–32. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 5.Dunn GP, Fecci PE, Curry WT. Cancer immunoediting in malignant glioma. Neurosurgery. 2012;71(2):201–22. doi: 10.1227/NEU.0b013e31824f840d. discussion 22–3. [DOI] [PubMed] [Google Scholar]
- 6.Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(22):5620–9. doi: 10.1158/1078-0432.CCR-14-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clinical and experimental immunology. 1977;29(1):61–6. [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott LH, Brooks WH, Roszman TL. Cytokinetic basis for the impaired activation of lymphocytes from patients with primary intracranial tumors. Journal of immunology. 1984;132(3):1208–15. [PubMed] [Google Scholar]
- 9.Ausiello CM, Palma C, Maleci A, Spagnoli GC, Amici C, Antonelli G, et al. Cell mediated cytotoxicity and cytokine production in peripheral blood mononuclear cells of glioma patients. European journal of cancer. 1991;27(5):646–50. doi: 10.1016/0277-5379(91)90235-6. [DOI] [PubMed] [Google Scholar]
- 10.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer research. 2006;66(6):3294–302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 11.El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-oncology. 2006;8(3):234–43. doi: 10.1215/15228517-2006-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Andaloussi A, Han Y, Lesniak MS. Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. Journal of neurosurgery. 2006;105(3):430–7. doi: 10.3171/jns.2006.105.3.430. [DOI] [PubMed] [Google Scholar]
- 13.Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(14 Pt 1):4294–305. doi: 10.1158/1078-0432.CCR-06-0053. [DOI] [PubMed] [Google Scholar]
- 14.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of virology. 2003;77(8):4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. The Journal of experimental medicine. 1998;188(12):2205–13. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nature medicine. 1999;5(6):677–85. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 17.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature communications. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serano RD, Pegram CN, Bigner DD. Tumorigenic cell culture lines from a spontaneous VM/Dk murine astrocytoma (SMA) Acta Neuropathol. 1980;51(1):53–64. doi: 10.1007/BF00688850. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Murillo R, Martinez A. Standardization of an orthotopic mouse brain tumor model following transplantation of CT-2A astrocytoma cells. Histol Histopathol. 2007;22(12):1309–26. doi: 10.14670/HH-22.1309. [DOI] [PubMed] [Google Scholar]
- 21.Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, et al. Endogenous Neoantigen-Specific CD8 T Cells Identified in Two Glioblastoma Models Using a Cancer Immunogenomics Approach. Cancer immunology research. 2016;4(12):1007–15. doi: 10.1158/2326-6066.CIR-16-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPherson RC, Konkel JE, Prendergast CT, Thomson JP, Ottaviano R, Leech MD, et al. Epigenetic modification of the PD-1 (Pdcd1) promoter in effector CD4(+) T cells tolerized by peptide immunotherapy. Elife. 2014:3. doi: 10.7554/eLife.03416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. The Journal of clinical investigation. 2011;121(6):2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. The Journal of clinical investigation. 2010;120(1):157–67. doi: 10.1172/JCI40070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Maaten L, Hinton G. Visualizing Data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–605. [Google Scholar]
- 26.Grimes ML, Lee WJ, van der Maaten L, Shannon P. Wrangling phosphoproteomic data to elucidate cancer signaling pathways. PloS one. 2013;8(1):e52884. doi: 10.1371/journal.pone.0052884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang B, Zhao W, Li H, Chen Y, Tian H, Li L, et al. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol Immunother. 2016;65(3):305–14. doi: 10.1007/s00262-016-1799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nature reviews Immunology. 2011;11(3):201–12. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90(4):1600–10. [PubMed] [Google Scholar]
- 30.Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS pathogens. 2015;11(10):e1005177. doi: 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.West EE, Youngblood B, Tan WG, Jin HT, Araki K, Alexe G, et al. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity. 2011;35(2):285–98. doi: 10.1016/j.immuni.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220–5. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, et al. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31(2):309–20. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35(3):400–12. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell. 2016;30(3):500. doi: 10.1016/j.ccell.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015;373(13):1270–1. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 37.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. The New England journal of medicine. 2015;372(26):2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 38.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. The New England journal of medicine. 2015;373(19):1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. The New England journal of medicine. 2015;372(21):2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 40.Ferris RL, Blumenschein G, Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. The New England journal of medicine. 2016;375(19):1856–67. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. The Journal of clinical investigation. 2015;125(5):2046–58. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature immunology. 2009;10(1):29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer research. 2012;72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. The Journal of clinical investigation. 2015;125(11):4053–62. doi: 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. Journal of virology. 2012;86(15):8161–70. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature reviews Immunology. 2015;15(8):486–99. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirzaei R, Sarkar S, Yong VW. T Cell Exhaustion in Glioblastoma: Intricacies of Immune Checkpoints. Trends in immunology. 2017;38(2):104–15. doi: 10.1016/j.it.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-15-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(3):851–6. doi: 10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T, Park DM. The evidence of glioblastoma heterogeneity. Scientific reports. 2015;5:7979. doi: 10.1038/srep07979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest. 2005;85(3):328–41. doi: 10.1038/labinvest.3700233. [DOI] [PubMed] [Google Scholar]
- 52.Yeung JT, Hamilton RL, Ohnishi K, Ikeura M, Potter DM, Nikiforova MN, et al. LOH in the HLA class I region at 6p21 is associated with shorter survival in newly diagnosed adult glioblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(7):1816–26. doi: 10.1158/1078-0432.CCR-12-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(13):4296–308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.