

Abstract
Background:
White matter hyperintensities (WMH) on magnetic resonance imaging have been postulated to be a core feature of Alzheimer’s disease. Clinicopathological studies are needed to elucidate and confirm this possibility.
Objective:
This study examined: 1) the association between ante-mortem WMH and autopsy-confirmed Alzheimer’s disease neuropathology (ADNP), 2) the relationship between WMH and dementia in participants with ADNP, and 3) the relationships among cerebrovascular disease, WMH and ADNP.
Methods:
The sample included 82 participants from the National Alzheimer’s Coordinating Center’s Data Sets who had quantitated volume of WMH from ante-mortem FLAIR MRI and available neuropathological data. The Clinical Dementia Rating (CDR) scale (from MRI visit) operationalized dementia status. ADNP+ was defined by moderate to frequent neuritic plaques and Braak stage III-VI at autopsy. Cerebrovascular disease neuropathology included infarcts or lacunes, microinfarcts, arteriolosclerosis, atherosclerosis, and cerebral amyloid angiopathy.
Results:
60/82 participants were ADNP+. Greater volume of WMH predicted increased odds for ADNP (p=0.037). In ADNP+ participants, greater WMH corresponded with increased odds for dementia (CDR≥1; p=0.038). WMH predicted cerebral amyloid angiopathy, microinfarcts, infarcts and lacunes (p’s<0.04). ADNP+ participants were more likely to have moderate-severe arteriolosclerosis and cerebral amyloid angiopathy compared to ADNP− participants (p’s<0.04).
Conclusions:
This study found a direct association between total volume of WMH and increased odds for having ADNP. In patients with Alzheimer’s disease, FLAIR MRI WMH may be able to provide key insight into disease severity and progression. The association between WMH and ADNP may be explained by underlying cerebrovascular disease.
Keywords: Alzheimer’s disease, white matter hyperintensities, Alzheimer’s disease neuropathology, cerebrovascular disease, dementia, magnetic resonance imaging
INTRODUCTION
Great strides have been made in the accurate in vivo diagnosis of Alzheimer’s disease (AD) partially due to advances in biomarkers that can detect AD-specific neuropathology, such as positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) protein analysis of amyloid and tau [1–7]. Biomarkers not directly related to amyloid or tau also play a valuable role in the detection and monitoring of AD severity and progression and are known as downstream biomarkers [1,8]. For example, patterns of volume loss (e.g., hippocampal atrophy) on structural magnetic resonance imaging (MRI) have long been used to assist in the differential diagnosis of AD dementia. Atrophy, however, is not specific to AD-related neuropathology, but is a general marker of neurodegeneration and is therefore considered a downstream biomarker of AD [1].
White matter hyperintensities (WMH) are MRI markers of non-specific pathologies that have long been a target of clinical research into cognitive impairment and AD. WMH refer to regions in the white matter that appear hyperintense on T2 fluid attenuated inversion recovery (FLAIR) sequences. The etiologies of WMH are multifaceted (e.g., gliosis, axonal loss) [9–15], but WMH accompany aging and cardiovascular disease and are often presumed to be of vascular origin and reflect small vessel cerebrovascular disease (CBVD) [9,16–21]. International consensus-based guidelines emphasize the pathologies underlying WMH (along with cerebral amyloid angiopathy [CAA], microbleeds, microinfarcts, among others) as mechanisms of vascular cognitive impairment (VCI) [22].
WMH predict accelerated cognitive decline and increased risk for AD dementia [23–29] and MRI WMH may be a downstream biomarker for AD. A recent study proposed that the underlying pathologies of WMH are a core feature of AD, based on findings that participants with autosomal-dominant AD had increased burden of WMH long before clinical symptom onset and WMH predicted CSF beta-amyloid levels in mutation carriers only [30]. WMH have been shown to predict increased CSF total tau and progressive medial temporal lobe atrophy in AD [31,32], as well as PET cortical amyloid uptake in older adults [16,33] and individuals with AD dementia [34], even more so than traditional AD neuroimaging and cognitive biomarkers [33]. The spatial distribution of WMH in AD has also been shown to be distinct from that found in “normal aging” [35–39], suggesting that WMH may have some degree of specificity to AD. The relationship between WMH and AD is often attributed to WMH being a marker of CBVD. CBVD (e.g., arteriosclerosis, CAA) and AD neuropathology are highly comorbid [40–45] and CBVD may contribute to the pathogenesis of AD [46–53]. However, CBVD (and resulting MRI WMH) can also be white matter sequelae of AD neuropathology (ADNP) [15], and there remains a debate on whether CBVD in AD is additive or synergistic.
Although considerable evidence has linked WMH to in vivo AD biomarkers, correlation of ante-mortem MRI WMH with postmortem AD neuropathology (ADNP) is necessary to validate MRI WMH as a potential biomarker for AD. Relatively few studies have investigated the clinicopathological relationship between WMH and ADNP. Moghekar et al. found that greater severity of visually-rated WMH was associated with more severe ADNP, based on a higher Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic plaque score and Braak stage in 50 participants from the Baltimore Longitudinal Study of Aging Autopsy Program; WMH were also associated with a diagnosis of dementia [54]. Ante-mortem accumulation of WMH over time also predicted a higher Braak stage in an advanced age autopsy sample of 66 individuals (mean age at death = 95) [55]. A recent ex vivo study further linked parietal white matter lesions with demyelination and axonal loss (and not ischemic injury) in participants with AD potentially reflecting Wallerian degeneration subsequent to cortical AD pathology; cortical phosphorylated tau (p-tau) pathology further correlated with white matter lesion severity [15]. Past work has found an inverse relationship between quantitatively measured WMH and Braak stage [21], and research from the Ischemic Vascular Dementia Program Project found no association between quantitated WMH and ADNP [56].
Overall, the association between ante-mortem WMH and postmortem ADNP remains inconclusive due to mixed findings from the few clinicopathological studies that have been conducted, possibly due to failures and/or inconsistences in accounting for apolipoprotein E (APOE) ε4 allele status, lack of inclusion of a comparison group without ADNP, and use of visual rating scales of WMH instead of automated quantification. In addition, the majority of studies examining WMH and clinical status in AD dementia have been in vivo and without autopsy confirmation. The interactions among WMH, CBVD neuropathology, and ADNP also remains poorly understood. To address these knowledge gaps, this study investigated the association between estimated volume of T2 FLAIR WMH (using a Bayesian probability structure) and odds for ADNP in an autopsy sample of 82 participants with and without ADNP from the National Alzheimer’s Coordinating Center (NACC) Uniform Data Set (UDS) and Neuropathology Data Set (NDS). The association between WMH and dementia status (defined by a Clinical Dementia Rating [CDR] scale score ⩾1) in participants with autopsy-confirmed ADNP was tested. The relationships among WMH, CBVD neuropathology, and ADNP were also examined.
METHODS
Participants and Design
The sample included 82 deceased participants from the NACC-UDS and NACC-NDS (see Table 1). The NACC was established by the National Institute on Aging (NIA) in 1999 to promote AD research and is a publicly available database of clinical and neuropathological data gathered from approximately 30 NIA-funded Alzheimer’s Disease Centers (ADCs) across the United States. Since 2005, ADCs have contributed standardized cognitive, behavioral, and functional participant data each year to a common database, known as the NACC-UDS. A full description of NACC-UDS is provided elsewhere [57–59]. Participants complete UDS visits on an annual basis and each participant in this sample completed between 1 and 10 visits (mean = 3.85, SD = 2.00). In 2011, the NIA appointed an Imaging Subcommittee of the ADCs Clinical Task Force, and ADCs across the U.S. began to voluntarily submit structural MRI sequences on participants enrolled in NACC-UDS in 2013. There are currently 19 sites that contribute MRI data to NACC. A subset of UDS participants also agree to postmortem brain donation and neuropathological examination to form the NACC-NDS [50,51,57]. The NACC databases are approved by the University of Washington Institutional Review Board, and informed consent from participants was obtained at the individual ADCs that contribute data to NACC.
Table 1.
Demographic and clinical features among Alzheimer’s disease neuropathology positive and negative participants
| Characteristica | ADNP+ | ADNP− | p |
|---|---|---|---|
| Number of subjects | 60 | 22 | NA |
| Age at initial visit (years), mean (SD) | 79.4 (7.8) | 78.7 (8.9) | 0.74 |
| Age at MRI visit (years), mean (SD) | 80.7 (7.8) | 80.3 (10.0) | 0.85 |
| Age at death (years), mean (SD) | 83.8 (7.9) | 83.6 (9.1) | 0.92 |
| Male, n (%) | 33 (55.0%) | 12 (54.6%) | 0.97 |
| Education (years), mean (SD) | 14.1 (3.2) | 13.1 (4.7) | 0.30 |
| Non-white race, n (%) | 9 (15.0%) | 3 (13.6%) | 0.50 |
| Diagnosis at visit closest to MRI, n (%) | |||
| Normal cognition | 4 (6.7%) | 8 (36.4%) | 0.003 |
| Cognitively impaired, not MCI | 0 (0%) | 1 (4.5%) | 0.60 |
| MCI | 11 (18.3%) | 7 (31.8%) | 0.31 |
| Dementia | 45 (75.0%) | 6 (27.3%) | 0.0002 |
| Global CDR at Visit closest to MRI, mean (SD) | 1.12 (0.76) | 0.75 (0.81) | 0.008 |
| Diagnosis at final study visit, n (%) | |||
| Normal cognition | 3 (5.0%) | 8 (36.4%) | 0.0009 |
| Cognitively impaired, not MCI | 0 (0.0%) | 1 (4.5%) | 0.60 |
| MCI | 6 (10.0%) | 6 (27.3%) | 0.050 |
| Dementia | 51 (85.0%) | 7 (31.8%) | < 0.0001 |
| Global CDR at final study visit, mean (SD) | 1.62 (0.91) | 0.86 (0.90) | 0.002 |
| ≥ 1 APOE e4 allele, n (%) | 32 (56.1%) | 2 (9.1%) | 0.001 |
| Time between MRI and death (years), n (%) | 3.5 (2.4) | 3.8 (2.6) | 0.71 |
Abbreviations: SD = standard deviation; ADNP = Alzheimer’s disease neuropathology; MRI = magnetic resonance imaging; CDR = Clinical Dementia Rating; MCI = mild cognitive impairment; APOE = apolipoprotein E; NA = not applicable
Missing data (ADNP+, ADNP−): APOE genotype (n=3, n=0); Education (n=2, n=0)
A formal data request was submitted to NACC for this study. The NACC-UDS and NDS data that were queried are described below. The sample was restricted to participants who had total volume of T2 FLAIR WMH from their most recent UDS visit quantitated (see White Matter Hyperintensities section) as well as those who agreed to brain donation and had available neuropathological data.
Neuropathology
Neuropathological data are collected via a standardized Neuropathology Form and Coding Guidebook. Refer to Beekly et al. (2004) [57] for a description of the NACC-NDS. The NACC-NDS has been revised several times since it began in 2002, with the most recent and substantial revision occurring in 2014 (version 10 of the Neuropathology Data Form). Version 10 includes a much more granular and refined assessment of CBVD neuropathology but there is limited amount of data collected to date. Therefore, this study used CBVD neuropathology variables that have been harmonized across versions 1 thru 10 of the NACC-NDS. There are indeed differences in the data elements and data coding between versions 1–9 and version 10. To resolve these discrepancies, NACC recoded and/or created derived variables to harmonize the data across versions 1–9 and 10. The following NACC neuropathology variables were used: NACCNEUR (CERAD score), NACCBRAA (Braak staging), NACCAMY (CAA), NACCARTE (arteriolosclerosis), NACCAVAS (atherosclerosis), NACCINF (infarcts and lacunes), and NACCMICR (microinfarcts). Neuropathologists from the ADCs make diagnoses and severity ratings based on methods and protocols put forth in the NACC Neuropathology Diagnosis Coding Guidebook.
The NACCNEUR and NACCBRAA variables were used to compute a binary ADNP+ or ADNP− variable. NACCNEUR (CERAD score) refers to the density of neocortical plaques and is rated on a 0 (no neuritic plaques) to 3 (frequent neuritic plaques) scale. The NACC Neuropathology Diagnosis Guidebook defines neuritic plaques to be “plaques with argyrophilic, thioflavin-S-positive or tau-positive dystrophic neurites with or without dense amyloid cores.” NACCBRAA is the Braak staging of neurofibrillary degeneration and is rated on a 0 (no AD neurofibrillary degeneration) to 6 (widespread neurofibrillary degeneration that has progressed to the primary cortices) scale. The NACC Neuropathology Diagnosis Coding Guidebook instructs neuropathologists from each ADC that a number of stains are acceptable to stain for beta-amyloid (e.g., immunohistochemistry [preferred], thioflavin-S, silver histochemical stains) and tau (e.g., Gallyas stains, tau immunostains, other silver stains). The criteria for an individual to be classified as ADNP+ was evidence of both moderate-severe neurofibrillary tangles, as determined by having a Braak stage of III or higher, and a moderate-frequent density of neocortical neuritic plaques, based on CERAD staging criteria (i.e., score of 2 or 3). Similar operationalization of ADNP has been previously used [50,51]. Participants with moderate to frequent neuritic plaques and Braak stage 0-II, or none or sparse neuritic plaques and Braak stage III-VI were not included in this study to limit ambiguity regarding the presence of ADNP and more clearly delineate the ADNP+ and ADNP− groups.
The neuropathological evaluation also involves qualitative and subjective ratings of the presence and severity of several vascular pathologies, including atherosclerosis of the circle of Willis (NACCAVAS), arteriolosclerosis (NACCARTE), and CAA (NACCAMY). These are all rated on the following scale: 0=none, 1=mild, 2=moderate, 3=severe. Atherosclerosis is defined by intimal and medial fibrofatty atheromatous plaques in the large arteries of the circle of Willis. Arteriolosclerosis is defined by concentric hyaline thickening of the media arterioles. Using special stains for amyloid (e.g., Congo red, thioflavin-S or beta-amyloid immunostaining) and adapted guidelines [60,61], the presence and severity of global CAA burden is determined. Version 10 of the NACC Neuropathology Diagnosis Codebook describes CAA as follows: Mild CAA (score of 1) is defined as scattered positivity in parenchymal and/or leptomeningeal vessels, possibly in only one brain area; moderate CAA (score of 2) refers to intense positivity in many parenchymal and/or leptomeningeal vessels; and severe CAA (score of 3) involves widespread (more than one brain area of intensive positivity in parenchymal and leptomeningeal vessels. The presence or absence (0 = no, 1 = yes) of infarcts or lacunes (NACCINF) and microinfarcts (NACCMICR) were also evaluated. For infarcts and lacunes, the NACCINF variable combines data on large cerebral artery infarcts, lacunes, and gross infarcts across Neuropathology Form versions 1 thru 10. For microinfarcts, NACCMICR combines the presence of microinfarcts across the Neuropathology Form versions and does not distinguish between acute versus old microinfarcts, or identify the location of microinfarcts, as these only began to be assessed in version 10. NACCMICR is considered a “flag” for the presence of microinfarcts.
White Matter Hyperintensities
A subset of ADCs contributed T1 and T2 FLAIR scan files to the NACC. Participants’ most recent UDS visit MRI was used. There were no significant differences between the ADNP groups in terms of time between MRI and death (see Table 1). Total volume of FLAIR WMH for NACC is calculated by the Imaging of Dementia & Aging (IDeA) Lab at the University of California Davis (Director: Charles DeCarli, M.D.). Methods for WMH estimation include those used for the Alzheimer’s Disease Neuroimaging Initiative-II [62]. For a description of methods for WMH estimation, see https://www.alz.washington.edu/WEB/adni_proto.pdf. The FLAIR is transformed to the T1 image using linear image registration (FLIRT from the FSL toolbox). Inhomogeneity correction of the co-registered FLAIR and T1 is performed using a histogram normalization method [63]. The T1 scan is aligned to a common template atlas and WMH are estimated using a Bayesian probability structure [62] based on previously developed WMH probability maps of more than 700 individuals with semi-automatic detection of WMH followed by manual editing. Likelihood estimates of the native image are calculated and all segmentation is performed in standard space to generate probability likelihood values of WMH at each white matter voxel. A threshold of 3.5 standard deviations above the mean is applied to the probabilities to result in a binary WMH mask. The segmented WMH are transformed to native space and summary volume of WMH (in cm3) is calculated. Gray and white matter were also segmented using an Expectation-Maximization algorithm [64] and were summed to create a total brain volume (TBV) composite. TBV was included as a covariate to control for individual differences in the amount of brain tissue present, which would influence the volume of WMH. Adjustment for TBV was also conducted to determine whether WMH are associated with ADNP above and beyond a general MRI marker of volume loss related to AD.
Clinical Status
The global rating from the CDR scale from the UDS visit closest in time to the MRI was used to assess dementia status in the sample [65,66]. The CDR is used to stage dementia severity through assessment of memory, orientation, judgment/problem-solving, community affairs, home and hobbies, and personal care. Each domain is scored on 0 to 3 scale and an algorithm is used to calculate a global rating of impairment severity designated as: 0 (no dementia), 0.5 (questionable dementia or mild cognitive impairment [MCI]), 1.0 (mild dementia), 2.0 (moderate dementia), or 3.0 (severe dementia). Typically, a CDR of 0 is consistent with no impairment and a CDR of 0.5 reflects MCI [65].
Demographic, Clinical, and Genotype Characteristics
Demographic and medical characteristics are ascertained during annual UDS clinical evaluations. To characterize the in vivo vascular status of the sample, a modified Framingham Stroke Risk Profile (mFSRP) was calculated based on data (i.e., age, systolic blood pressure, and history of diabetes, cigarette smoking, cardiovascular disease [myocardial infarction, congestive heart failure, angina], and atrial fibrillation) from participant’s most recent UDS visit [67]. Left ventricular hypertrophy is a component of the FSRP, but this data was not available from NACC and thus was not included in the calculation of the FSRP for this sample. Biofluid samples are collected by the individual ADCs to determine APOE ε4 allele status, which was dichotomized into ε4 carriers vs. non-carriers.
Statistical Analysis
Primary study hypotheses were evaluated through a series of logistic regression models; all analyses were conducted using the “rms” package for R version 3.1.1. For all models, APOE ε4 carrier status, age, and TBV were selected a priori to be included as covariates and were evaluated in a single model simultaneous with volume of WMH. Volume of WMH served as a predictor variable in all analyses and not as an outcome. All predictor variables included were from the time of the MRI, with the exception of age. Age at death was included as covariate for all analyses with neuropathological outcomes as the dependent variable. Age at the time of the MRI was used for analyses examining CDR, given the CDR score closest in time to the MRI served as the dependent variable. Binomial logistic regression models examined the relationship between volume of WMH and odds for ADNP (1 = presence; 0 = absence). In ADNP+ participants, a binomial logistic regression analysis examined the relationship between WMH and odds for dementia (i.e., Global CDR <1 versus ⩾ 1) and an ordinal logistic regression model examined the association between WMH and dementia severity (i.e., Global CDR scores of 1, 2, and 3). Binomial logistic regression models were used to test the association between WMH and presence of CBVD, including microinfarcts, infarcts/lacunes, arteriolosclerosis, atherosclerosis, and CAA. Atherosclerosis, arteriolosclerosis, and CAA were all evaluated on a rating scale with options ranging from 0 (none) to 3 (severe). However, these three variables were dichotomized into moderate-severe and none-mild for analyses due to small sample sizes for some ratings and to separate the sample into groups of individuals with a pathological presence vs. absence of these variables. Finally, the concordance between the presence of vascular pathologies in the ADNP+ and ADNP− groups was evaluated using chi-square tests of independence.
RESULTS
Clinical and Neuropathological Characteristics
Sixty participants met criteria for ADNP+ and the 22 remaining participants were ADNP−. Demographics and medical characteristics for the ADNP+ and ADNP− groups are presented in Tables 1–2. There were no differences between the groups on age, race, education, or ante-mortem cardiovascular status, with the exception of diabetes (a greater proportion of ADNP− participants had diabetes). ADNP+ participants were more likely to be carriers of the APOE ε4 allele, have a higher Global CDR score, and have a clinical diagnosis of AD. Among the 25 individuals with a Braak stage of III or IV, 12% (3/25) received a clinical diagnosis of normal cognition, 16% (4/25) had a diagnosis of MCI, and 72% (18/25) had a diagnosis of dementia. In contrast, 94% of individuals (33/35) with a Braak stage of V or VI had a diagnosis of dementia and the remaining two participants received a diagnosis of MCI. A similar pattern emerged with regard to CERAD staging. 68% of individuals (17/25) with a CERAD staging score of 2 had dementia at their final study visit; 24% (6/25) had MCI and 8% (2/25) were diagnosed with normal cognition. Amongst the 35 individuals with a CERAD staging score of 3, 97% (34/35) received a diagnosis of dementia, with the final individual receiving a diagnosis of normal cognition.
Table 2.
Medical status of Alzheimer’s disease neuropathology positive and negative individuals at final study visit
| Characteristica | ADNP+ | ADNP− | p |
|---|---|---|---|
| Framingham Stroke Risk Profile, mean (SD) | 13.5 (5.1) | 15.1 (5.4) | 0.21 |
| Systolic blood pressure, mean (SD)b | 133.2 (18.7) | 138.6 (25.4) | 0.31 |
| Diastolic blood pressure, mean (SD)b | 69.1 (10.4) | 68.7 (11.2) | 0.89 |
| Diabetes (recent or remote), n (%) | 6 (10.0%) | 7 (31.8%) | 0.02 |
| Reported smoking within 30 days, n (%) | 2 (3.5%) | 1 (4.8%) | 0.80 |
| Cardiovascular disease (recent or remote), n (%) | 14 (23.7%) | 7 (33.3%) | 0.39 |
| Atrial fibrillation (recent or remote), n (%) | 8 (13.3%) | 4 (19.1%) | 0.53 |
| Hypertension (recent or remote), n (%) | 41 (71.9%) | 15 (71.4%) | 0.97 |
| Stroke (recent or remote), n (%) | 11 (19.6%) | 2 (9.5%) | 0.30 |
| Antihypertensive use at any visit, n (%) | 42 (70.0%) | 16 (72.7%) | 0.81 |
| Antilipid use at any visit, n (%) | 26 (43.3%) | 9 (40.9%) | 0.84 |
Abbreviations: SD = standard deviation; ADNP = Alzheimer’s disease neuropathology
Missing data (ADNP+, ADNP−): systolic blood pressure (n=1, n=1); diastolic blood pressure (n=1, n=1); cigarettes (n=1, n=1); cardiovascular disease (n=1, n=1); stroke (n=4, n=1); hypertension (n=3, n=1); atrial fibrillation (n=0, n=1)
11 ADNP+ and 3 ADNP− participants did not have a blood pressure reading at last visit; used blood pressure measurement closest to the last visit before death
WMH, Dementia, and ADNP
A logistic regression model (Table 3) demonstrated that greater volume of WMH was associated with increased odds for ADNP (odds ratio [OR] = 1.04 per cm3, p = 0.037), after controlling for APOE ε4 carrier status, age at death, and TBV. Individuals at the 75th percentile of WMH in the sample were 141% more likely to have ADNP compared to the 25th percentile. Figure 1 shows an exemplary T2 FLAIR sequence of a participant with ADNP who had high WMH burden. In ADNP+ participants, logistic regression model controlling for APOE ε4 carrier status, age, and TBV, demonstrated that increased volume of WMH was associated with increased odds for the presence of dementia (i.e., CDR ≥ 1) (OR = 1.05, p = 0.038). In an ordinal logistic regression model among ADNP+ individuals with dementia (i.e., CDR ≥ 1), higher volumes of WMH were associated with increased odds of having more severe dementia, as reflected by the CDR score (OR = 1.06, p’s < 0.04). See Table 4 for a summary of the CDR results. Three ADNP+ participants did not have APOE ε4 allele data available and were excluded from these analyses.
Table 3.
Results from logistic regression model examinining WMH and Alzheimer’s disease neuropathology
| Predicting ADNP: R2 = 0.40; AUC = 0.84 | ||||
|---|---|---|---|---|
| B | Wald Z | p | OR (95 % CI) | |
| Intercept | −13.05 | −2.19 | 0.028 | -- |
| APOE ε4 carrier | 3.39 | 3.75 | < 0.001 | 29.57 (5.04–173.37) |
| Age at death | 0.07 | 1.56 | 0.120 | 1.07 (0.98–1.16) |
| Total brain volume | 0.01 | 2.12 | 0.034 | 1.01 (1.00–1.01) |
| WMH volume | 0.04 | 2.09 | 0.037 | 1.04 (1.00–1.08) |
OR = Odds ratio per unit increase in the predictor. Total brain and WMH volume was measured in cm3.
Figure 1.
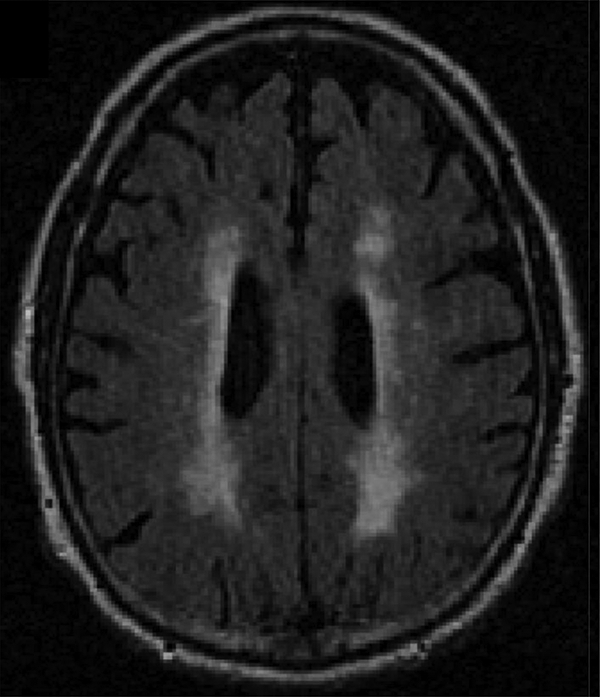
Exemplary White Matter Hyperintensity Burden in a Participant with Alzheimer’s Disease Neuropathology. The below is a T2 FLAIR sequence from a selected participant who had Alzheimer’s disease neuropathology. Participant was selected to exemplify the pattern and high burden of WMH in AD.
Table 4.
Results from binomial and ordinal logistic regression models examining WMH and Global Clinical Dementia Rating (CDR) score from MRI visit
| Model 1: CDR < 1 or ⩾ 1 | Model 2: CDR 1, 2, and 3 | |||||||
|---|---|---|---|---|---|---|---|---|
| B | Wald Z | p | OR (95% CI) | B | Wald Z | p | OR (95% CI) | |
| Constant 1* | 4.00 | 0.50 | 0.62 | -- | −0.88 | −0.09 | 0.93 | -- |
| Constant 2* | -- | -- | -- | -- | −2.78 | −0.30 | 0.77 | -- |
| APOE ε4 carrier | 2.43 | 2.88 | 0.004 | 11.35 (2.17–59.45) | 1.64 | 1.30 | 0.19 | 5.15 (0.43–61.20) |
| Age at MRI visit | −0.09 | −1.35 | 0.18 | 0.92 (0.81–1.04) | 0.05 | 0.72 | 0.47 | 1.05 (0.92–1.21) |
| Total brain volume | 0.00 | 0.47 | 0.64 | 1.00 (0.99–1.01) | −0.01 | −1.35 | 0.18 | 0.99 (0.98–1.00) |
| WMH volume | 0.05 | 2.07 | 0.038 | 1.05 (1.00–1.10) | 0.06 | 2.05 | 0.040 | 1.06 (1.00–1.12) |
In both models, greater volume of white matter hyperintensities predicted increased odds for having a higher CDR score. Only ADNP+ participants were included in analyses. Total model fit statistics: R2 = 0.35 and AUC = 0.79 for Model 1; R2 = 0.26 and AUC = 0.71 for Model 2. OR = Odds Ratio.
In Model 2, Constants 1 and 2 were applied for CDR values ⩾ 2 and 3, respectively. Model 1 was a logistic regression model with only one intercept.
WMH and CBVD Neuropathology
As shown in Table 5, unadjusted chi-square tests showed that ADNP+ participants were more likely to have moderate-severe arteriolosclerosis (p = 0.035) and CAA (p = 0.013), compared to the ADNP− participants. No participant in the sample had moderate-severe CAA without comorbid ADNP. There were no significant between ADNP group differences for the presence of microinfarcts, infarcts or lacunes, and moderate-severe atherosclerosis.
Table 5.
Cerebrovascular neuropathology among Alzheimer’s disease neuropathology positive and negative individuals
| Neuropathology characteristica | ADNP+ | ADNP− | pb |
|---|---|---|---|
| Infarct or lacune, n (%) | 18 (30.0%) | 9 (40.9%) | 0.505 |
| Microinfarct, n (%) | 27 (45.0%) | 10 (45.5%) | 1.000 |
| Arteriolosclerosis, n (%) moderate-severe | 34 (57%) | 6 (27%) | 0.035 |
| Atherosclerosis, n (%) moderate-severe | 24 (41%) | 8 (38%) | 0.997 |
| Cerebral amyloid angiopathy, n (%) moderate-severe |
17 (28%) | 0 | 0.013 |
Abbreviations: SD = standard deviation; ADNP = Alzheimer’s disease neuropathology
Missing data (ADNP+, ADNP−): Atherosclerosis (n=2, n=1)
Significant values were calculated using chi-square tests of independence
A multiple linear regression model controlling for age, APOE ε4 carrier status, and TBV showed volume of WMH was not significantly associated with mFSRP scores at the time of the MRI scan (p = 0.11). Binomial logistic regression models (Table 6) demonstrated that increased volume of WMH was associated with increased odds for moderate-severe CAA (OR = 1.04, p = 0.032), microinfarcts (OR = 1.03, p = 0.029), and infarcts or lacunes (OR = 1.04, p = 0.007). There were no significant relationships between WMH and moderate-severe levels of atherosclerosis or arteriolosclerosis. Given the relationship between WMH and CAA, and CAA and ADNP, sensitivity analyses were conducted to examine the effect of WMH on ADNP with CAA included as a covariate. When moderate-severe CAA was simultaneously included in a model predicting ADNP, the coefficient for WMH was of a similar magnitude to the model presented in Table 3 (B = 0.04, OR = 1.04), but there was no longer statistical significance (Wald Z = 1.71, p = 0.09).
Table 6.
Results from logistic regression models examining WMH in the prediction of cerebrovascular neuropathology
| 1. Atherosclerosis (n = 31) | 2. Arteriolosclerosis (n = 39) | 3. Amyloid angiopathy (n = 16) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| B | p | OR (95% CI) | B | p | OR (95% CI) | B | p | OR (95% CI) | |
| Intercept | −7.91 | 0.15 | -- | −13.46 | 0.01 | -- | −10.19 | 0.11 | -- |
| APOE ε4 carrier | 0.30 | 0.58 | 1.35 (0.47–3.86) | 1.51 | 0.009 | 4.54 (1.46–14.11) | 1.25 | 0.07 | 3.49 (0.89–13.70) |
| Age at death | 0.11 | 0.014 | 1.12 (1.02–1.22) | 0.12 | 0.004 | 1.13 (1.04–1.23) | 0.03 | 0.52 | 1.03 (0.94–1.13) |
| Total brain volume | −0.00 | 0.39 | 1.00 (0.99–1.00) | 0.00 | 0.41 | 1.00 (1.00–1.01) | 0.00 | 0.15 | 1.00 (1.00–1.01) |
| WMH volume | 0.01 | 0.37 | 1.01 (0.99–1.03) | 0.02 | 0.21 | 1.02 (0.99–1.05) | 0.04 | 0.032 | 1.04 (1.00–1.08) |
| 4. Microinfarcts (n = 36) | 5. Infarcts/lacunes (n = 26) | ||||||||
| B | p | OR (95% CI) | B | p | OR (95% CI) | ||||
| Intercept | −7.91 | 0.12 | -- | −5.47 | 0.31 | -- | |||
| APOE ε4 carrier | −0.11 | 0.84 | 0.90 (0.32–2.53) | −0.07 | 0.91 | 0.93 (0.29–2.95) | |||
| Age at death | 0.07 | 0.07 | 1.07 (0.99–1.15) | 0.06 | 0.18 | 1.06 (0.98–1.15) | |||
| Total brain volume | 0.00 | 0.62 | 1.00 (1.00–1.01) | −0.00 | 0.71 | 1.00 (0.99–1.00) | |||
| WMH volume | 0.03 | 0.029 | 1.03 (1.00–1.06) | 0.04 | 0.007 | 1.04 (1.01–1.08) | |||
Total model fits are as follows: 1. R2 = 0.24, AUC = 0.74; 2. R2 = 0.26, AUC = 0.75; 3. R2 = 0.18, AUC = 0.73; 4. R2 = 0.20, AUC = 0.72; 5. R2 = 0.26, AUC = 0.77
DISCUSSION
In this autopsy sample of 82 participants from the NACC-UDS and NACC-NDS, greater volume of WMH was associated with increased odds for ADNP, after controlling for age APOE e4 carrier status, and TBV. Greater burden of WMH also corresponded to increased odds for dementia, as well as worse dementia severity in participants with autopsy-confirmed ADNP. WMH were further associated with increased odds for having CBVD neuropathology (i.e., microinfarcts, infarcts or lacunes, CAA), and ADNP+ participants were more likely to have CBVD neuropathology (i.e., arteriolosclerosis and CAA) compared to ADNP− participants. Overall, the current results provide clinicopathological evidence for a direct relationship between ante-mortem FLAIR WMH and ADNP, which may partially be a function of underlying CBVD neuropathology, such as CAA.
The few ex vivo studies that have examined the association between ante-mortem WMH and ADNP have reported mixed findings, including positive [54], inverse [21], and no significant associations [56]. Reasons for the inconsistent findings could be related to sample differences in the presence and severity of ADNP and comorbid pathologies, methods used to quantitate WMH (e.g., use of visual rating scales), the lack of an ADNP− comparison group, and/or a failure to control for key confounding variables that contribute to ADNP (e.g., APOE ε4 carrier status). The current study addresses all of these limitations and included a clinically and neuropathologically enriched sample of participants with clinical AD with advanced ADNP, i.e., Braak stage greater than III and a CERAD score of 2 or 3. Furthermore, this study used a Bayesian probability structure to quantitatively estimate volume of FLAIR WMH and thus extends previous research that linked visually-rated WMH with ADNP [54]. The present study showed that WMH increased odds for dementia (and ADNP) in participants with autopsy-confirmed ADNP above and beyond the contributions of APOE ε4 carrier status. These findings extend in vivo studies that associate WMH with accelerated cognitive decline and brain atrophy, and increased risk for AD dementia [23–28,31,32,68], but were without autopsy confirmation of underlying ADNP. Taken together, the present findings corroborate recent research studies that propose WMH to be a contributor to the pathogenesis of AD [30,35].
In the present study, greater volume of ante-mortem WMH predicted increased odds for ADNP and dementia, suggesting that the pathologies of WMH may promote AD-related neurodegeneration. Previous studies that have examined the association between WMH and CSF total tau in AD dementia also provide evidence that WMH promote tau pathology and exacerbate the effects of tau on clinical status; additionally, the relationship between WMH and CSF total tau may be independent of beta-amyloid [32,69]. WMH are typically interpreted to be MRI markers of small vessel CBVD, which may explain their association with ADNP. In this sample, greater volume of ante-mortem WMH indeed corresponded to multiple CBVD pathologies (i.e., microinfarcts, infarcts/lacunes, and moderate-severe CAA) and ADNP+ participants had worse comorbid arteriolosclerosis and CAA, compared to ADNP−participants. The pathways by which CBVD may influence or interact with the development or progression of ADNP likely involve cerebral blood flow dysregulation. CBVD can lead to cerebral hypoperfusion that could result in degradation of the neurovascular unit and subsequent deposition of beta-amyloid and tau, eventually leading to dementia (this vascular pathway has been the focus of several literature reviews [70–72]). The relationship between cerebral blood flow and ADNP remains inconclusive, given a recent study that found no association between cerebral blood flow and cortical uptake of amyloid (flutemetamol) or tau (AV-1451) tracers on PET imaging in 11 middle-aged to elderly patients with unilateral occlusion of precerebral arteries [73].
In the setting of AD, WMH may be indicative of AD pathology and not small vessel CBVD [15,74]. For instance, parietal white matter lesions in AD have been shown to reflect axonal loss and demyelination, and not ischemic disease; the authors hypothesized this to be a manifestation of Wallerian degeneration triggered by cortical p-tau pathology [15]. Overall, the associations among WMH, CBVD, and ADNP seem to involve a complex combination of neurovascular and non-neurovascular pathways. Continued research on the exact role that CBVD plays in the pathogenesis of AD is much needed, given vascular health (e.g., cerebral blood flow) may be modifiable through pharmacological and/or behavioral interventions [75,76].
Interestingly, CAA was associated with both ADNP and WMH in this sample. Ante-mortem WMH have been previously linked with more severe CAA neuropathology [77], and in vivo studies have shown MRI WMH are associated with CAA [18,78]. A previous study that induced hyperhomocysteinemia (a risk factor for stroke) in APP/PS1 transgenic mice found that congophilic amyloid deposition was reduced in the parenchyma, but increased in the vasculature, i.e., CAA [79]. It is thus possible that CBVD may shift the deposition of amyloid to the vasculature in AD.
WMH are often found in the setting of cardiovascular disease, which is a known risk factor for AD dementia [80]. In this sample, WMH were not associated with the mFSRP or atherosclerosis, consistent with findings from another autopsy sample on this topic [21]. ADNP+ participants also did not exhibit greater rates of cardiovascular disease or atherosclerosis compared to ADNP− participants. Autopsy studies on aging include highly selective samples and thus limits their generalizability, i.e., there are distinct mechanisms that drive an observed autopsy sample. For this study, those with the most severe cardiovascular disease (and CBVD) may have been more likely to be excluded due to attrition (e.g., premature death). In this sample, WMH are therefore more likely related to age-related microvascular changes (e.g., arteriosclerosis) [21,81,82] and/or MRI manifestations of ADNP (as discussed above) rather than large vessel pathologies associated with cardiovascular disease. Cause of death would shed further insight into the cardiovascular status, however, such data are not available from the NACC-NDS.
The present study has several limitations. Although the sample size was relatively large for a study that includes ante-mortem neuroimaging and neuropathological data, larger clinicopathological studies are needed to replicate the current findings. The cross-sectional design and time between MRI and autopsy are additional limitations. Prospective clinicopathological studies that include multiple ante-mortem MRIs will clarify whether WMH are a cause or consequence of ADNP. Version 10 of the Neuropathology Data form includes a more granular assessment of CBVD neuropathology but was not implemented until 2014 and therefore we used vascular pathology variables that have been harmonized across versions 1–10. This was done in order to maximize our sample size to examine the clinicopathological relationship between WMH with CBVD neuropathology and ADNP. As a consequence, however, there are limitations associated with some of the CBVD neuropathology variables; for example, the location of microinfarcts was not recorded in versions 1–9 (but now is for Version 10). Another limitation is that there are slight variations across the different versions and NACC sites in the staining methods and techniques used. The NACC-NDS version 10 and other more refined alternative neuropathological methods for assessing CBVD neuropathology, such as those for CAA [83], will clarify the precise relationships among WMH, CBVD, and ADNP.
The spatial distribution of WMH (in contrast to total volume) appears to be a more sensitive predictor of AD dementia [35–39] and the relationship between WMH location and ADNP should be explored [8]. For example, WMH in the frontal, temporal, and parietal lobes appear to be more likely to indicate underlying AD pathology [36]. Diffusion tensor imaging (DTI) may detect white matter injury before it appears as WMH on FLAIR and may be more sensitive to the detection of ADNP. Ante-mortem DTI metrics of lower fractional anisotropy and higher mean diffusivity in medial temporal limbic and medial parietal white matter fiber paths were recently found to be associated with high neurofibrillary tangle pathology (Braak IV-VI) [84]. Lastly, the odds ratio for the association between WMH and dementia was 1.05, meaning that each additional cm3 of WMH volume corresponded to a 5% increased probability that the participant would have dementia. Similar effect sizes were observed for the other relationships examined. A cm3 voxel is rather large and therefore the clinical meaningfulness of the present findings may be limited.
CONCLUSIONS
In this autopsy sample of participants from the NACC-UDS and NDS, there was a direct association between greater ante-mortem T2 FLAIR WMH and increased odds for ADNP. In participants with autopsy-confirmed ADNP, FLAIR WMH corresponded to greater odds for the presence of dementia, as well as more severe dementia. This study provides additional evidence for the contribution of CBVD in the pathogenesis of AD. MRI WMH may be an important marker of disease severity and progression in AD.
ACKNOWLEDGMENTS
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI David Teplow, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), and P50 AG005681 (PI John Morris, MD). This work was supported by grants from the NIH (P30 AG013846; R01 NS078337; R56 9500304025; U01 NS093334, U01NS086659–01; 1F32NS096803–01; K23AG046377; RF1AG05416). This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through BU-CTSI Grant Number 1UL1TR001430. The content is the responsibility of the authors and does not necessarily represent the official views of the NIH. The funding sources provided data and salary support for some of the authors. However, these funding sources did not play any role in the study design, analysis and interpretation of data, in the writing of the report, or in the decision to submit the article for publication.
Footnotes
CONFLICT OF INTEREST
RAS is a paid consultant to Eli Lilly (Indianapolis, IN, USA), Avanir Pharmaceuticals (Aliso Viejo, CA), and Biogen (Cambridge, MA, USA). He is a member of the Board of Directors of King-Devick Technologies, Inc. (Chicago, IL, USA), and he receives royalties for published neuropsychological tests from Psychological Assessment Resources, Inc. (Lutz, FL, USA). CD is a paid consultant to Novartis Pharmaceuticals (Basel, Switzerland). For the remaining authors, there are no conflicts of interest to declare.
REFERENCES
- 1.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B (2016) A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N, Sperling R. (2016) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79, 110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang JH, Korecka M, Figurski MJ, Toledo JB, Blennow K, Zetterberg H, Waligorska T, Brylska M, Fields L, Shah N, Soares H, Dean RA, Vanderstichele H, Petersen RC, Aisen PS, Saykin AJ, Weiner MW, Trojanowski JQ, Shaw LM, Alzheimer’s Disease Neuroimaging I. (2015) The Alzheimer’s Disease Neuroimaging Initiative 2 Biomarker Core: A review of progress and plans. Alzheimers Dement 11, 772–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, Holtta M, Rosen C, Olsson C, Strobel G, Wu E, Dakin K, Petzold M, Blennow K, Zetterberg H. (2016) CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 15, 673–84. [DOI] [PubMed] [Google Scholar]
- 5.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Cedarbaum J, Green RC, Harvey D, Jack CR, Jagust W, Luthman J, Morris JC, Petersen RC, Saykin AJ, Shaw L, Shen L, Schwarz A, Toga AW, Trojanowski JQ, Alzheimer’s Disease Neuroimaging I. (2015) 2014 Update of the Alzheimer’s Disease Neuroimaging Initiative: A review of papers published since its inception. Alzheimers Dement 11, e1–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan TK, Alkon DL. (2015) Alzheimer’s Disease Cerebrospinal Fluid and Neuroimaging Biomarkers: Diagnostic Accuracy and Relationship to Drug Efficacy. J Alzheimers Dis 46, 817–36. [DOI] [PubMed] [Google Scholar]
- 7.Palmqvist S, Zetterberg H, Mattsson N, Johansson P, Alzheimer’s Disease Neuroimaging I, Minthon L, Blennow K, Olsson M, Hansson O, Swedish Bio FSG. (2015) Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 85, 1240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R, Cappa S, Crutch S, Engelborghs S, Frisoni GB, Fox NC, Galasko D, Habert MO, Jicha GA, Nordberg A, Pasquier F, Rabinovici G, Robert P, Rowe C, Salloway S, Sarazin M, Epelbaum S, de Souza LC, Vellas B, Visser PJ, Schneider L, Stern Y, Scheltens P, Cummings JL. (2014) Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 13, 614–29. [DOI] [PubMed] [Google Scholar]
- 9.Dallaire-Theroux C, Callahan BL, Potvin O, Saikali S, Duchesne S. (2017) Radiological-Pathological Correlation in Alzheimer’s Disease: Systematic Review of Antemortem Magnetic Resonance Imaging Findings. J Alzheimers Dis 57, 575–601. [DOI] [PubMed] [Google Scholar]
- 10.Englund E (1998) Neuropathology of white matter changes in Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord 9 Suppl 1, 6–12. [DOI] [PubMed] [Google Scholar]
- 11.Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, Radner H, Lechner H. (1993) Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology 43, 1683–9. [DOI] [PubMed] [Google Scholar]
- 12.Thomas AJ, O’Brien JT, Davis S, Ballard C, Barber R, Kalaria RN, Perry RH. (2002) Ischemic basis for deep white matter hyperintensities in major depression: a neuropathological study. Arch Gen Psychiatry 59, 785–92. [DOI] [PubMed] [Google Scholar]
- 13.Udaka F, Sawada H, Kameyama M. (2002) White matter lesions and dementia: MRI-pathological correlation. Ann N Y Acad Sci 977, 411–5. [DOI] [PubMed] [Google Scholar]
- 14.Wardlaw JM, Valdes Hernandez MC, Munoz-Maniega S. (2015) What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 4, 001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, Colloby SJ, Dey M, Martin-Ruiz C, Taylor JP, Thomas AJ, McKeith IG, De Carli C, Attems J. (2017) Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brickman AM, Guzman VA, Gonzalez-Castellon M, Razlighi Q, Gu Y, Narkhede A, Janicki S, Ichise M, Stern Y, Manly JJ, Schupf N, Marshall RS. (2015) Cerebral autoregulation, beta amyloid, and white matter hyperintensities are interrelated. Neurosci Lett 592, 54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garde E, Mortensen EL, Krabbe K, Rostrup E, Larsson HB. (2000) Relation between age-related decline in intelligence and cerebral white-matter hyperintensities in healthy octogenarians: a longitudinal study. Lancet 356, 628–34. [DOI] [PubMed] [Google Scholar]
- 18.Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, Killiany RJ, Blacker D, Albert MS, Guttmann CR, Greenberg SM. (2008) Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 39, 1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SC, Markus HS. (2002) Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology 59, 321–6. [DOI] [PubMed] [Google Scholar]
- 20.Wolters FJ, Zonneveld HI, Hofman A, van der Lugt A, Koudstaal PJ, Vernooij MW, Ikram MA, Heart-Brain Connection Collaborative Research G. (2017) Cerebral Perfusion and the Risk of Dementia: A Population-Based Study. Circulation. [DOI] [PubMed] [Google Scholar]
- 21.Shim YS, Yang DW, Roe CM, Coats MA, Benzinger TL, Xiong C, Galvin JE, Cairns NJ, Morris JC. (2015) Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dement Geriatr Cogn Disord 39, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O’Brien JT, Barkhof F, Benavente OR, Black SE, Brayne C, Breteler M, Chabriat H, Decarli C, de Leeuw FE, Doubal F, Duering M, Fox NC, Greenberg S, Hachinski V, Kilimann I, Mok V, Oostenbrugge R, Pantoni L, Speck O, Stephan BC, Teipel S, Viswanathan A, Werring D, Chen C, Smith C, van Buchem M, Norrving B, Gorelick PB, Dichgans M, nEuroimaging STfRVco. (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12, 822–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bilello M, Doshi J, Nabavizadeh SA, Toledo JB, Erus G, Xie SX, Trojanowski JQ, Han X, Davatzikos C. (2015) Correlating Cognitive Decline with White Matter Lesion and Brain Atrophy Magnetic Resonance Imaging Measurements in Alzheimer’s Disease. J Alzheimers Dis 48, 987–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brickman AM, Muraskin J, Zimmerman ME. (2009) Structural neuroimaging in Alzheimer’s disease: do white matter hyperintensities matter? Dialogues Clin Neurosci 11, 181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brickman AM, Zahodne LB, Guzman VA, Narkhede A, Meier IB, Griffith EY, Provenzano FA, Schupf N, Manly JJ, Stern Y, Luchsinger JA, Mayeux R. (2015) Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer’s disease incidence. Neurobiol Aging 36, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carmichael O, Schwarz C, Drucker D, Fletcher E, Harvey D, Beckett L, Jack CR Jr., Weiner M, DeCarli C, Alzheimer’s Disease Neuroimaging I. (2010) Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol 67, 1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kloppenborg RP, Nederkoorn PJ, Geerlings MI, van den Berg E. (2014) Presence and progression of white matter hyperintensities and cognition: a meta-analysis. Neurology 82, 2127–38. [DOI] [PubMed] [Google Scholar]
- 28.Maillard P, Carmichael O, Fletcher E, Reed B, Mungas D, DeCarli C. (2012) Coevolution of white matter hyperintensities and cognition in the elderly. Neurology 79, 442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kertesz A, Polk M, Carr T. (1990) Cognition and white matter changes on magnetic resonance imaging in dementia. Arch Neurol 47, 387–91. [DOI] [PubMed] [Google Scholar]
- 30.Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, Marcus DS, Fagan AM, Goate A, Fox NC, Cairns NJ, Holtzman DM, Buckles V, Ghetti B, McDade E, Martins RN, Saykin AJ, Masters CL, Ringman JM, Ryan NS, Forster S, Laske C, Schofield PR, Sperling RA, Salloway S, Correia S, Jack C, Jr., Weiner M, Bateman RJ, Morris JC, Mayeux R, Brickman AM, Dominantly Inherited Alzheimer N. (2016) White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol 79, 929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiford CM, Manning EN, Bartlett JW, Cash DM, Malone IB, Ridgway GR, Lehmann M, Leung KK, Sudre CH, Ourselin S, Biessels GJ, Carmichael OT, Fox NC, Cardoso MJ, Barnes J, Alzheimer’s Disease Neuroimaging I. (2017) White matter hyperintensities are associated with disproportionate progressive hippocampal atrophy. Hippocampus 27, 249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tosto G, Zimmerman ME, Hamilton JL, Carmichael OT, Brickman AM, Alzheimer’s Disease Neuroimaging I. (2015) The effect of white matter hyperintensities on neurodegeneration in mild cognitive impairment. Alzheimers Dement 11, 1510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kandel BM, Avants BB, Gee JC, McMillan CT, Erus G, Doshi J, Davatzikos C, Wolk DA. (2016) White matter hyperintensities are more highly associated with preclinical Alzheimer’s disease than imaging and cognitive markers of neurodegeneration. Alzheimers Dement (Amst) 4, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimmer T, Faust M, Auer F, Alexopoulos P, Forstl H, Henriksen G, Perneczky R, Sorg C, Yousefi BH, Drzezga A, Kurz A. (2012) White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol Aging 33, 2766–73. [DOI] [PubMed] [Google Scholar]
- 35.Lindemer ER, Greve DN, Fischl B, Augustinack JC, Salat DH, Alzheimer’s Disease Neuroimaging I. (2017) Differential Regional Distribution of Juxtacortical White Matter Signal Abnormalities in Aging and Alzheimer’s Disease. J Alzheimers Dis 57, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindemer ER, Greve DN, Fischl BR, Augustinack JC, Salat DH. (2017) Regional staging of white matter signal abnormalities in aging and Alzheimer’s disease. Neuroimage Clin 14, 156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee DY, Fletcher E, Martinez O, Ortega M, Zozulya N, Kim J, Tran J, Buonocore M, Carmichael O, DeCarli C. (2009) Regional pattern of white matter microstructural changes in normal aging, MCI, and AD. Neurology 73, 1722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee DY, Fletcher E, Martinez O, Zozulya N, Kim J, Tran J, Buonocore M, Carmichael O, DeCarli C. (2010) Vascular and degenerative processes differentially affect regional interhemispheric connections in normal aging, mild cognitive impairment, and Alzheimer disease. Stroke 41, 1791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshita M, Fletcher E, Harvey D, Ortega M, Martinez O, Mungas DM, Reed BR, DeCarli CS. (2006) Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology 67, 2192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brun A, Englund E. (1986) A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol 19, 253–62. [DOI] [PubMed] [Google Scholar]
- 41.Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, Maurage CA, Kalaria RN. (2012) Staging and natural history of cerebrovascular pathology in dementia. Neurology 78, 1043–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, Oakley AE, Englund E, O’Brien JT, Ince PG, Kalaria RN. (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 119, 579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalaria R (2002) Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci 203–204, 29–34. [DOI] [PubMed] [Google Scholar]
- 44.Kalaria RN, Ballard C. (1999) Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord 13 Suppl 3, S115–23. [DOI] [PubMed] [Google Scholar]
- 45.Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN. (1996) Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol 148, 2083–95. [PMC free article] [PubMed] [Google Scholar]
- 46.Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, Obrien RJ. (2010) Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol 68, 231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Monsell SE, Kukull WA, Trojanowski JQ. (2013) Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 136, 2697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tolppanen AM, Solomon A, Soininen H, Kivipelto M. (2012) Midlife vascular risk factors and Alzheimer’s disease: evidence from epidemiological studies. J Alzheimers Dis 32, 531–40. [DOI] [PubMed] [Google Scholar]
- 49.Zlokovic BV. (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12, 723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alosco ML, Duskin J, Besser LM, Martin B, Chaisson CE, Gunstad J, Kowall NW, McKee AC, Stern RA, Tripodis Y. (2017) Modeling the Relationships Among Late-Life Body Mass Index, Cerebrovascular Disease, and Alzheimer’s Disease Neuropathology in an Autopsy Sample of 1,421 Subjects from the National Alzheimer’s Coordinating Center Data Set. J Alzheimers Dis 57, 953–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Besser LM, Alosco ML, Ramirez Gomez L, Zhou XH, McKee AC, Stern RA, Gunstad J, Schneider JA, Chui H, Kukull WA. (2016) Late-Life Vascular Risk Factors and Alzheimer Disease Neuropathology in Individuals with Normal Cognition. J Neuropathol Exp Neurol 75, 955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Honig LS, Kukull W, Mayeux R. (2005) Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center. Neurology 64, 494–500. [DOI] [PubMed] [Google Scholar]
- 53.Santos CY, Snyder PJ, Wu WC, Zhang M, Echeverria A, Alber J. (2017) Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimers Dement (Amst) 7, 69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moghekar A, Kraut M, Elkins W, Troncoso J, Zonderman AB, Resnick SM, O’Brien RJ. (2012) Cerebral white matter disease is associated with Alzheimer pathology in a prospective cohort. Alzheimers Dement 8, S71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Erten-Lyons D, Woltjer R, Kaye J, Mattek N, Dodge HH, Green S, Tran H, Howieson DB, Wild K, Silbert LC. (2013) Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology 81, 977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, Weiner MW, Ellis WG, Zarow C, Mungas D, Reed BR, Kramer JH, Schuff N, DeCarli C, Chui HC. (2008) Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 63, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, Kukull WA, Centers NI-AsD. (2004) The National Alzheimer’s Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord 18, 270–7. [PubMed] [Google Scholar]
- 58.Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA. (2006) The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20, 210–6. [DOI] [PubMed] [Google Scholar]
- 59.Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, Cummings J, DeCarli C, Foster NL, Galasko D, Peskind E, Dietrich W, Beekly DL, Kukull WA, Morris JC. (2009) The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 23, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olichney JM, Hansen LA, Lee JH, Hofstetter CR, Katzman R, Thal LJ. (2000) Relationship between severe amyloid angiopathy, apolipoprotein E genotype, and vascular lesions in Alzheimer’s disease. Ann N Y Acad Sci 903, 138–43. [DOI] [PubMed] [Google Scholar]
- 61.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr., (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30, 637–49. [DOI] [PubMed] [Google Scholar]
- 62.DeCarli C, Miller BL, Swan GE, Reed T, Wolf PA, Garner J, Jack L, Carmelli D. (1999) Predictors of brain morphology for the men of the NHLBI twin study. Stroke 30, 529–36. [DOI] [PubMed] [Google Scholar]
- 63.DeCarli C, Murphy DG, Teichberg D, Campbell G, Sobering GS. (1996) Local histogram correction of MRI spatially dependent image pixel intensity nonuniformity. J Magn Reson Imaging 6, 519–28. [DOI] [PubMed] [Google Scholar]
- 64.Rajapakse JC, Giedd JN, DeCarli C, Snell JW, McLaughlin A, Vauss YC, Krain AL, Hamburger S, Rapoport JL. (1996) A technique for single-channel MR brain tissue segmentation: application to a pediatric sample. Magn Reson Imaging 14, 1053–65. [DOI] [PubMed] [Google Scholar]
- 65.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. (1982) A new clinical scale for the staging of dementia. Br J Psychiatry 140, 566–72. [DOI] [PubMed] [Google Scholar]
- 66.Morris JC. (1993) The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–4. [DOI] [PubMed] [Google Scholar]
- 67.D’Agostino RB, Wolf PA, Belanger AJ, Kannel WB. (1994) Stroke risk profile: adjustment for antihypertensive medication. The Framingham Study. Stroke 25, 40–3. [DOI] [PubMed] [Google Scholar]
- 68.Barnes J, Carmichael OT, Leung KK, Schwarz C, Ridgway GR, Bartlett JW, Malone IB, Schott JM, Rossor MN, Biessels GJ, DeCarli C, Fox NC, Alzheimer’s Disease Neuroimaging I. (2013) Vascular and Alzheimer’s disease markers independently predict brain atrophy rate in Alzheimer’s Disease Neuroimaging Initiative controls. Neurobiol Aging 34, 1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bos I, Verhey FR, Ramakers I, Jacobs HIL, Soininen H, Freund-Levi Y, Hampel H, Tsolaki M, Wallin AK, van Buchem MA, Oleksik A, Verbeek MM, Olde Rikkert M, van der Flier WM, Scheltens P, Aalten P, Visser PJ, Vos SJB. (2017) Cerebrovascular and amyloid pathology in predementia stages: the relationship with neurodegeneration and cognitive decline. Alzheimers Res Ther 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de la Torre JC. (2002) Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33, 1152–62. [DOI] [PubMed] [Google Scholar]
- 71.Kalaria RN, Akinyemi R, Ihara M. (2012) Does vascular pathology contribute to Alzheimer changes? J Neurol Sci 322, 141–7. [DOI] [PubMed] [Google Scholar]
- 72.Hays CC, Zlatar ZZ, Wierenga CE. (2016) The Utility of Cerebral Blood Flow as a Biomarker of Preclinical Alzheimer’s Disease. Cell Mol Neurobiol 36, 167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hansson O, Palmqvist S, Ljung H, Cronberg T, van Westen D, Smith R. (2018) Cerebral hypoperfusion is not associated with an increase in amyloid beta pathology in middle-aged or elderly people. Alzheimers Dement 14, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McAleese KE, Firbank M, Dey M, Colloby SJ, Walker L, Johnson M, Beverley JR, Taylor JP, Thomas AJ, O’Brien JT, Attems J. (2015) Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol Commun 3, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hajjar I, Hart M, Chen YL, Mack W, Novak V, H CC, Lipsitz L. (2013) Antihypertensive therapy and cerebral hemodynamics in executive mild cognitive impairment: results of a pilot randomized clinical trial. J Am Geriatr Soc 61, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacIntosh BJ, Swardfager W, Crane DE, Ranepura N, Saleem M, Oh PI, Stefanovic B, Herrmann N, Lanctot KL. (2014) Cardiopulmonary fitness correlates with regional cerebral grey matter perfusion and density in men with coronary artery disease. PLoS One 9, e91251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Charidimou A, Martinez-Ramirez S, Reijmer YD, Oliveira-Filho J, Lauer A, Roongpiboonsopit D, Frosch M, Vashkevich A, Ayres A, Rosand J, Gurol ME, Greenberg SM, Viswanathan A. (2016) Total Magnetic Resonance Imaging Burden of Small Vessel Disease in Cerebral Amyloid Angiopathy: An Imaging-Pathologic Study of Concept Validation. JAMA Neurol 73, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marnane M, Al-Jawadi OO, Mortazavi S, Pogorzelec KJ, Wang BW, Feldman HH, Hsiung GY, Alzheimer’s Disease Neuroimaging I. (2016) Periventricular hyperintensities are associated with elevated cerebral amyloid. Neurology 86, 535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sudduth TL, Weekman EM, Brothers HM, Braun K, Wilcock DM. (2014) beta-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res Ther 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qiu C, Winblad B, Marengoni A, Klarin I, Fastbom J, Fratiglioni L. (2006) Heart failure and risk of dementia and Alzheimer disease: a population-based cohort study. Arch Intern Med 166, 1003–8. [DOI] [PubMed] [Google Scholar]
- 81.Leoni RF, Oliveira IA, Pontes-Neto OM, Santos AC, Leite JP. (2017) Cerebral blood flow and vasoreactivity in aging: an arterial spin labeling study. Braz J Med Biol Res 50, e5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spangler KM, Challa VR, Moody DM, Bell MA. (1994) Arteriolar tortuosity of the white matter in aging and hypertension. A microradiographic study. J Neuropathol Exp Neurol 53, 22–6. [DOI] [PubMed] [Google Scholar]
- 83.Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, Yamada M, McCarron M, Minett T, Matthews F, Greenberg S, Mann D, Kehoe PG. (2014) Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 3, 19–32. [PMC free article] [PubMed] [Google Scholar]
- 84.Kantarci K, Murray ME, Schwarz CG, Reid R, Przybelski, SA, Lesnick T, Zuk SM, & Raman MR. (2017) White matter integrity on DTI and the pathologic staging of Alzheimer’s disease. Neurobiol Aging. [DOI] [PMC free article] [PubMed] [Google Scholar]