

Abstract
Inositol trisphosphate receptors (IP3R) are ubiquitous Ca2+-permeable channels that mediate release of Ca2+ from the endoplasmic reticulum to regulate numerous processes including cell division, cell death, differentiation and fertilization. IP3R is activated by both IP3 and its permeant ion Ca2+. At high concentrations, however, Ca2+ inhibits activity ensuring precise spatiotemporal control over intracellular Ca2+. Despite extensive characterization of IP3R, the mechanisms by which these molecules control channel gating have remained elusive. Here, we present structures of full-length human type 3 IP3R in ligand-bound and ligand-free states. Multiple IP3-bound structures demonstrate that the large cytoplasmic domain provides a platform for propagation of long-range conformational changes to the ion conduction gate. Structures in the presence of Ca2+ reveal two Ca2+ binding sites that induce the disruption of numerous interactions between subunits, thereby inhibiting IP3R. These structures thus begin to provide a mechanistic basis for understanding the regulation of IP3R.
Introduction
Inositol trisphosphate receptors (IP3R) are a family of large tetrameric Ca2+-permeable cation channels predominantly expressed in the endoplasmic reticulum (ER)1–5. Three IP3R subtypes exist in humans that share between 64 and 70% sequence identity but differ in their tissue expression and subcellular localization, resulting in homo- and heterotetrameric channels that perform a variety of physiological roles6,7. Activation of IP3R by cytoplasmic Ca2+ and the second messenger inositol 1,4,5-trisphosphate (IP3) allows Ca2+ to flow from the ER into the cytoplasm2,8–11. The dual regulation of IP3R by Ca2+ and IP3 integrates extracellular signaling pathways that lead to the generation of cytoplasmic IP3 with intracellular Ca2+ signaling to jointly regulate the activity of numerous cellular processes including fertilization, differentiation, cell division and cell death12–14. Underscoring the essential role of these channels in diverse cell signaling pathways, the dysregulation of IP3R is linked to cardiac disease, cancer, neurological disorders and other pathologies6,15–19.
Electrophysiological and optical recordings of IP3R activity in a variety of contexts have demonstrated that IP3R displays a biphasic response to cytoplasmic Ca2+ (refs9,18,20–22). While low concentrations of cytoplasmic Ca2+ activate IP3R, high concentrations of Ca2+ inhibit channel activity. The biphasic regulation of IP3R by its permeant ion ensures precise spatiotemporal control of Ca2+ signaling. Depending on the input stimuli, IP3R can generate cell-wide Ca2+ spikes or oscillatory waves as well as more localized Ca2+ “blips” or “puffs”23–25. Proper signal encoding requires Ca2+-dependent inhibition as well as Ca2+- and IP3-dependent activation.
Despite intensive investigation into the regulation of IP3R by Ca2+, the mechanisms of Ca2+-driven modulation remain open questions26–29. FRET experiments, partial proteolysis experiments, and negative-stain electron microscopic images suggest that Ca2+ stimulates large conformational changes, though the molecular details of these changes are unknown30,31. Analysis of single channel recordings suggest a high affinity activating Ca2+ site and a low affinity inhibiting Ca2+ site, though the residues that comprise these sites have yet to be identified32. In contrast, the molecular details of IP3 binding to IP3R have been better characterized. Mutational analysis identified the first ~600 residues of IP3R as the IP3-binding domain3,33. Crystal structures of isolated IP3-binding domains in the presence and absence of IP3 reveal that IP3 binding is accompanied by a closing of the clamshell-like IP3-binding domain around IP3 (refs34–37). A cryo-electron microscopy (cryo-EM) structure of rat type 1 IP3R (rIP3R1) in a ligand-free state at a resolution of 4.7 Å revealed that this IP3-binding domain is located more than 70 Å from the pore38. However, side-chain detail is lacking for most residues in the rIP3R1 density map and the majority of the structural model was therefore built as a poly-alanine backbone. Thus, the architecture of IP3R at atomic resolution remains unknown and it is unclear how IP3 binding induces conformational changes that are propagated over such a long distance to influence the gating state of the pore. Therefore, high-resolution structural studies are required to understand the mechanisms through which Ca2+ and IP3 regulate IP3Rs. Here, we determined structures of full-length human type 3 IP3R (hIP3R3) in different states, allowing us to start to delineate the mechanistic basis for IP3R regulation by IP3 and Ca2+.
Results
Architecture of ligand-free human type 3 IP3 receptor
To resolve the atomic structure of a full-length human IP3R, we first determined the structure of hIP3R3 in a ligand-free state by single-particle cryo-EM at an overall resolution of 3.5 Å with C4 symmetry imposed (Supplementary Fig. 1 and Table 1). The map was of excellent quality, enabling us to de novo model most parts of the channel. However, some of the peripheral regions in the cytoplasmic domain and the carboxy-terminal domain were less well resolved. The density for the cytoplasmic domain was improved by employing symmetry expansion, signal subtraction and focused refinement39 (Supplementary Fig. 1 and Supplementary Table 1). Using the cytoplasmic domain focused refinement map, we were able to build and register most of the cytoplasmic domain. In contrast, focused refinement approaches were unable to sufficiently improve the density for the small carboxy-terminal domain due to its extreme conformational flexibility. Thus, while the carboxy-terminal domain is present in the density map as a four-helix bundle as has been observed for rIP3R138, it is not modeled in the final structure (Supplementary Fig. 2).
Table 1.
Cryo-EM data collection, refinement and validation statistics.
| hIP3R3 apo (EMD-7978) (PDB 6DQJ) | hIP3R3 IP3 class 1 (EMB-7981) (PDB 6DQN) | hIP3R3 IP3 class 2 (EB-7984) (PDB 6DQV) | hIP3R3 IP3 class 3 (EMB-7983) (PDB 6DQS) | hIP3R3 IP3 class 4 (EMB-7986) (PDB 6DQZ) | hIP3R3 IP3 class 5 (EMB-7987) (PDB 6DR0) | hIP3R3 Ca2+-bound (EMB-7988) (PDB 6DR2) | hIP3R3 Low IP3-Ca2+ (EMB-7991) (PDB 6DRA) | hIP3R3 High IP3-Ca2+ (EMB-7994) (PDB 6DRC) | |
|---|---|---|---|---|---|---|---|---|---|
| Data collection and processing | |||||||||
| Magnification | 105,000x | 22,500x | 22,500x | 22,500x | 22,500x | 22,500x | 22,500x | 22,500x | 22,500x |
| Voltage (kV) | 300kV | 300kV | 300kV | 300kV | 300kV | 300kV | 300kV | 300kV | 300kV |
| Electron exposure (e–/Å2) | 60 | 61 | 61 | 61 | 61 | 61 | 61 | 61 | 61 |
| Defocus range (μm) | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 | −1.0 – 2.5 |
| Pixel size (Å) | 1.096 | 1.088 | 1.088 | 1.088 | 1.088 | 1.088 | 1.088 | 1.088 | 1.088 |
| Symmetry imposed | C4 | C4 | C4 | C1 | C1 | C1 | C4 | C4 | C4 |
| Initial particle images (no.) | 52,767 | 302,966 | 302,966 | 302,966 | 302,966 | 302,966 | 49,060 | 74,277 | 170,308 |
| Final particle images (no.) | 26,325 | 38,777 | 40,531 | 37,910 | 9,535 | 27,334 | 33,807 | 49,087 | 131,437 |
| Map resolution (Å) | 3.49 | 3.33 | 3.82 | 4.12 | 6.01 | 4.47 | 4.33 | 3.96 | 3.92 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 300-3.49 | 300-3.33 | 300-3.82 | 300-4.12 | 300-6.01 | 300-4.47 | 300-4.33 | 300-3.96 | 300-3.92 |
| Refinement | |||||||||
| Initial model used (PDB code) | 3UJ4 | ||||||||
| Model resolution (Å) | 3.80/3.35 | 3.88/3.36 | 4.53/3.93 | 4.83/3.90 | 7.63/6.33 | 7.06/4.23 | 7.44/4.34 | 4.49/3.91 | 4.43/3.92 |
| FSC threshold | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 | 0.50/0.143 |
| Model resolution range (Å) | 300-3.5 | 300-3.3 | 300-3.8 | 300-4.1 | 300-6.0 | 300-4.5 | 300-4.3 | 300-4.0 | 300-4.0 |
| Map sharpening B factor (Å2) | −90 | −90 | −90 | −90 | −90 | −90 | −90 | −90 | −90 |
| Model composition | |||||||||
| Non-hydrogen | 69,220 | 69,412 | 70,144 | 69,600 | 69,828 | 69,986 | 69,3572 | 69,572 | 69,508 |
| Protein residues | 8,744 | 8,744 | 8,752 | 8,750 | 8,748 | 8,750 | 8,764 | 8,764 | 8,744 |
| Ligands | 4 | 8 | 8 | 8 | 8 | 8 | 12 | 12 | 16 |
| Mean B factors (Å2) | |||||||||
| Protein | 95.1 | 116.1 | 169.0 | 193.5 | 376.7 | 217.0 | 239.9 | 240.0 | 217.9 |
| Ligand | 53.5 | 96.95 | 133.0 | 177.4 | 362.0 | 182.9 | 174.7 | 163.6 | 263.3 |
| R.m.s. deviations | |||||||||
| Bond lengths (Å) | 0.007 | 0.003 | 0.005 | 0.003 | 0.003 | 0.003 | 0.004 | 0.003 | 0.004 |
| Bond angles (°) | 0.688 | 0.490 | 0.648 | 0.520 | 0.536 | 0.538 | 0.660 | 0.517 | 0.623 |
| Validation | |||||||||
| MolProbity score | 1.53 | 1.21 | 1.36 | 1.28 | 1.22 | 1.26 | 1.26 | 1.24 | 1.33 |
| Clashscore | 2.16 | 2.09 | 2.22 | 2.41 | 2.43 | 2.37 | 2.07 | 2.96 | 2.14 |
| Poor rotamers (%) | 1.97 | 1.06 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.42 | 0.00 |
| Ramachandran plot | |||||||||
| Favored (%) | 95.52 | 96.70 | 94.74 | 96.14 | 96.8 | 96.33 | 95.91 | 97.12 | 95.04 |
| Allowed (%) | 4.48 | 3.30 | 5.26 | 3.86 | 3.20 | 3.67 | 4.09 | 2.88 | 4.96 |
| Disallowed (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
hIP3R3 is a tetrameric channel comprised of 2671 residues per subunit with a large amino-terminal cytoplasmic domain (CD) followed by a juxtamembrane domain (JD) and a transmembrane domain (TMD), an architecture that resembles that of rIP3R138 (Fig. 1a). Starting at the amino-terminus, the large CD can be divided into two β-trefoil-containing domains (BTF1, BTF2), an armadillo repeat domain (ARM1), the first segment of the central linker domain (CLD), a second armadillo repeat domain (ARM2), the second segment of the CLD and the third armadillo repeat domain (ARM3) (Supplementary Note 1). The CD is stabilized by a number of inter-subunit interactions including a central BTF ring that is composed of the BTF1 and BTF2 domains from all four subunits (Fig. 1b). The BTF ring, which contains several residues involved in IP3 binding, is connected to the JD through the largely helical ARM1, CLD and ARM3 domains while the helical ARM2 domain is positioned at the periphery of the channel. The JD is comprised of the two segments that flank the TMD and is stabilized by a Zn2+ ion that is coordinated by Cys2538, Cys2541, His2558 and His2563. The Zn2+ binding site is also present in the structure of rIP3R1 and in ryanodine receptor structures, indicating that the Zn2+-binding site is broadly conserved38,40.
Figure 1. Structure of human type 3 IP3R in a ligand-free state.

a, Structure of hIP3R3 viewed in the plane of the membrane with the cytoplasmic domain at the top. A single subunit is colored according to domain with BTF1 in purple, BTF2 in blue, ARM1 in violet, the CLD in cyan, ARM2 in green, ARM3 in yellow, the JD in orange and the TMD in red. Gray lines represent the approximate position of the membrane. b, Structure of the cytoplasmic domain viewed from the cytoplasm. A single subunit is colored according to domain. ARM3 is removed for clarity. c, Structure of the transmembrane domain viewed from the cytoplasm. S1–S4 helices are colored red, S1′ and S1″ blue and S5, S6 and pore helix green. d–e, Structure of the ion conduction pathway viewed in the plane of the membrane and plot of pore radius. Residues comprising the luminal vestibule, the selectivity filter and S6 gate are highlighted. Front and rear subunits are removed for clarity.
While modeling the TMD, we noticed the presence of multiple hydrophobic lipid or detergent molecules forming a bilayer-like structure at the periphery of the TMD as well as two additional densities that could not be attributed to lipid molecules (Supplementary Fig. 3 and Supplementary Table 1). Inspection of these densities revealed that they correspond to two additional transmembrane helices located between the canonical S1 and S2 helices of the 6TM ion channel fold, which we will refer to as S1′ and S1″ (Fig. 1c). S1″ is positioned adjacent to S1 and S2 and is relatively well ordered in the density map, which allowed us to register its sequence. S1′ is positioned further from the S1–S4 domain on the outside of S1″ and is less well ordered in the density map. Focused classification and refinement improved the density for S1′, but the register could not be confidently determined and we thus modeled S1′ as an unregistered poly-alanine helix (Supplementary Fig. 3). Sequence conservation of the region between S1 and S2 is high among the three human IP3R types, especially within S1′. Indeed, close inspection of the rIP3R1 density map revealed densities in similar positions as S1′ and S1″, suggesting that S1′ and S1″ are conserved features of IP3Rs (Supplementary Fig. 3). S1′ and S1″ extend away from the channel in the membrane where they may potentially facilitate regulatory protein-protein interactions with other membrane-embedded proteins41.
The ion conduction pathway of hIP3R3 is formed by S5, S6 and the re-entrant pore helices of each subunit (Fig. 1d). Starting from the luminal side of the membrane, ions first pass through an electronegative vestibule formed by the extended loops between S5 and the pore helix and between the pore helix and S6. A total of 12 negatively charged residues (Glu2398, Asp2400 and Asp2478 from each subunit) are resolved in the luminal vestibule. The electronegative environment of the vestibule may serve to concentrate cations before entering the pore, potentially facilitating Ca2+ conductance. Two constrictions are present in the pore between the luminal vestibule and cytoplasmic opening. The first constriction is created by the backbone carbonyl oxygen of Asn2472, which resides on the pore helix (Fig. 1d and Fig. 1e). The constriction has a radius of about 3 Å at its narrowest point and along with neighboring residues may contribute to ion selectivity, although IP3R3 is only weakly selective for Ca2+ over other cations42. The second constriction is a hydrophobic gate formed by the side chains of Phe2513 and Ile2517 of S6, which constricts the pore to a radius of less than 1 Å, too narrow to conduct Ca2+. The ligand-free state of hIP3R3 is thus a non-conductive state, consistent with electrophysiological recordings showing that Ca2+ and IP3 are required for IP3R activation2,18,32.
IP3-bound conformations
To resolve the conformational changes associated with IP3 binding in the context of the full channel, we collected images of hIP3R3 in the presence of saturating (50 μM) IP3. Successive rounds of three-dimensional classification revealed an ensemble of distinct IP3-bound conformational states (Supplementary Fig. 4). Five of the conformational states were sufficiently well populated to generate 3D reconstructions at resolutions up to 3.3 Å, from which we could model their structures.
A reconstruction of the particles assigned to the first class, IP3 class 1, at 3.3 Å revealed an overall conformation that is similar to the apo state (Fig. 2a and Table 1). The structure of IP3 class 1 has an all-atom RSMD of approximately 1 Å compared to the apo structure. Despite this overall structural similarity, local conformational changes are present in the IP3-binding domain. The ARM1-proximal region of BTF2 moves 4 Å towards ARM1 in IP3 class 1, narrowing the interface between the two domains without disrupting the BTF ring. In the interface between BTF2 and ARM1, we identified a three-lobed density that we modeled as an IP3 molecule (Supplementary Fig. 5). The IP3 molecule is coordinated by the positively charged side chains of Arg266 and Arg270 from BTF2 and Arg503, Lys507, Arg510, Arg568 and Lys569 from ARM1 (Fig. 2b and Supplementary Fig. 5), many of which have been previously identified as residues that directly bind IP3 in crystal structures of isolated IP3-binding domains34,36,37.
Figure 2. IP3 binding site in two IP3-bound conformations.

a, d, Superposition of (a) IP3 class 1 (colored by domain) and apo (grey) or (d) IP3 class 2 (colored by domain) and apo (grey) viewed from in the plane of the membrane (left) and the cytoplasm (right). b, e, Superposition of IP3-binding domain of (b) IP3 class 1 (colored by domain) and apo (grey) or (e) IP3 class 2 (colored by domain) and apo (grey) aligned by BTF1 and BTF2. IP3 and the side chains of IP3-coordinating residues are shown as sticks. c, f, Superposition of ARM3, the JD and S6 of (c) IP3 class 1 (colored by domain) and apo (grey) or (f) IP3 class 2 (colored by domain) and apo (grey) aligned by the TMD and viewed from the lumen.
Whereas IP3 class 1 closely resembles apo hIP3R3, a reconstruction of the particles assigned to IP3 class 2 at 3.8 Å reveals significant conformational changes (Supplementary Fig. 4 and Table 1). Compared to the apo structure, the length of IP3 class 2 is 5 Å shorter along the four-fold axis and the conformation of the CD is substantially different (Fig. 2d). To better understand the origin of these conformational changes, we examined the IP3-binding site of class 2. Similar to class 1, an IP3 molecule is coordinated by the side chains of Arg266, Arg270, Arg503, Lys507, Arg510, Arg568 and Lys569 from BTF2 and ARM1 (Fig. 2e and Supplementary Fig. 5). However, the configuration of the IP3-binding domain differs in class 2. In class 2, IP3 binding is accommodated by a rigid-body rotation of ARM1 towards BTF2, which adopts an apo-like conformation (Fig. 2e). The only changes in BTF2 compared to the apo state are slight movements of the loops that directly coordinate IP3.
Because ARM1 moves relative to the fixed BTF ring structure in IP3 class 2, IP3 binding is accompanied by conformational changes throughout the channel (Fig. 2d). These conformational changes can be separated into two major movements. The first is a coupled movement of the CLD and ARM3 that results in a 5˚ rotation of the JD (Fig. 2f, Supplementary Video 1 and Supplementary Video 2). While the TMD is nearly unchanged relative to the apo state, these concerted movements provide a model for how ligand binding can be transmitted through multiple helical domains to the pore. The second conformational change is a 17 Å movement of ARM2 to position itself near the CLD of the same subunit (Fig. 2d, Supplementary Video 1 and Supplementary Video 2). Besides coming nearly into contact with the CLD, the movement of ARM2 also alters its interactions with the neighboring subunit. ARM2 no longer makes contacts with ARM1 of the neighboring subunit and its interaction with BTF2 of the neighboring subunit is shifted by 2 armadillo repeats from α63 to α68.
When examining the density maps of IP3 classes 3, 4 and 5, we realized that they are not four-fold symmetric like the maps of classes 1 and 2 (Supplementary Fig. 4). The asymmetry of these classes is most apparent in the positions of the four ARM2 domains, which can adopt either a class 1-like extended conformation or a class 2-like compact conformation (Fig. 3a). We therefore calculated reconstructions of classes 3, 4 and 5 without imposing symmetry at resolutions of 4.1, 6.0 and 4.5 Å, respectively (Table 1). Structures of classes 3, 4 and 5 were initially modeled by rigid-body docking domains of classes 1 or 2 into the density map and then comparing which of the two structures fit best into the density map. The docking demonstrated that these classes were composed of both class 1-like and class 2-like domains (Fig. 3b).
Figure 3. Ensemble of IP3-bound conformations.

a, Structures of IP3 class 1, IP3 class 32, IP3 class 4, IP3 class 5 and IP3 class 2 colored by domain. The ARM2 domain of subunits marked with an * adopt a class 2-like conformation. b, Structures of IP3 class 1, IP3 class 3, IP3 class 4, IP3 class 5 and IP3 class 2. Domains adopting class 1-like domains are colored orange and domains adopting class 2-like conformations are colored cyan.
Even at the moderate resolutions of these reconstructions, inspection of the IP3-binding domains reveals densities consistent with IP3 molecules at the BTF2-ARM1 interfaces in all three density maps (Supplementary Fig. 5). Thus, the asymmetry in these classes is not caused by variations in IP3-binding site occupancies. Rather, the asymmetry arises from variations in the conformation of the individual subunits. Notably, a single subunit can be composed of domains that adopt class 1-like and domains that adopt class 2-like conformations. For example, one of the subunits in class 5 has a class 1-like IP3-binding domain, while the rest of the subunit is class 2-like (Fig. 3b).
Thus, despite IP3R possessing a single IP3 binding site, the configuration of the IP3-binding domain can vary when IP3 is bound and these variations are coupled to large changes in global channel conformation (Fig. 2). Notably, both configurations can be present in the same channel indicating that the binding modes are in dynamic exchange and that stable intermediates exist between them (Fig. 3). Multiple crystal structures have been determined of isolated IP3-binding domains in complex with IP3 (refs34,36,37). The position of ARM1 relative to BTF2 varies among these structures, adopting a range of conformations between those present in IP3 class 1 and IP3 class 2 suggesting that the rest of the channel may influence the configuration of the IP3-binding domain (Supplementary Fig. 5).
Ca2+-bound conformations
To identify the Ca2+ binding sites and visualize the effects of Ca2+ binding on IP3R conformation, we next determined a structure of hIP3R3 at 4.3 Å resolution in the presence of approximately 2 mM Ca2+ with C4 symmetry imposed, which we will refer to as Ca2+-bound (Supplementary Fig. 6 and Table 1). Examination of the map revealed that the peripheral portions of the CD, including BTF1, BTF2 and ARM1 were poorly resolved and not strictly four-fold symmetrical. We therefore employed two different focused refinement strategies to better resolve the channel. We first calculated a four-fold symmetric reconstruction of ARM3, the JD and the TMD to a resolution of 4.2 Å (Supplementary Fig. 6 and Supplementary Table 1). Next, to improve the density of the flexibly attached CD, we employed signal subtraction, symmetry expansion and focused refinement, resulting in a 4.7 Å reconstruction in which the CD is clearly resolved (Supplementary Fig. 6 and Supplementary Table 1). The structure of Ca2+-bound hIP3R3 was modeled using both the full channel and focused refinement maps. In addition to densities corresponding to hIP3R3, both the full channel and focused refinement maps contained densities corresponding to two bound Ca2+ ions per subunit (Fig. 4a and Supplementary Fig. 7). The first Ca2+, which we will call the CD Ca2+, is located at the interface between CLD and ARM2 where it is coordinated by main-chain and side-chain oxygen atoms from both domains (Fig. 4a,b). The CLD contributes the main-chain carbonyl oxygen of Arg743, while ARM3 contributes the side chain of Glu1125 and the main-chain carbonyl oxygen of Glu1122. From the apo state, ARM2 moves 28 Å to complete the binding site (Fig. 4c). Notably, ARM2 in the Ca2+-bound state is in a similar conformation to its location in IP3 class 2. From its position in IP3 class 2, Glu1122 and Glu1125 on ARM2 must move only an additional 3 Å to complete the coordination of the CD Ca2+. The second Ca2+, which we will refer to as the JD Ca2+, occupies a cavity between the JD and ARM3 (Fig. 4a and Supplementary Fig. 7). The JD Ca2+ is coordinated by the side chains of two glutamate residues on ARM3 (Glu1882 and Glu1946) and by main-chain and side-chain carbonyl oxygens of one residue on the JD (Thr2581) (Fig. 4d). To bring the side chains of Glu1882 and Glu1946 into close proximity of Thr2581 and complete the Ca2+ coordination, ARM3 must rotate 11˚ compared to the apo state (Fig. 4e). In the Ca2+-bound conformation, the structure of the JD binding site almost perfectly matches that of a Ca2+ binding site identified in the rabbit ryanodine receptor structure suggesting that some aspects of channel regulation are conserved between IP3Rs and ryanodine receptors40 (Supplementary Fig. 7d).
Figure 4. Ca2+ binding sites in Ca2+-bound hIP3R3.

a, Monomeric structure of Ca2+-bound hIP3R3 colored by domain viewed in the plane of the membrane. Ca2+ and Zn2+ ions are shown as spheres. b, CD Ca2+-binding site at the CLD-ARM2 interface. Residues coordinating the Ca2+ are shown as sticks and the CD Ca2+ ion is shown as a magenta sphere. c, Superposition of the CLD and ARM2 of Ca2+-bound (colored by domain) and apo (grey, left) or Ca2+-bound (colored by domain) and IP3 class 2 (grey, right) aligned by the CLD. d, JD Ca2+-binding site at the ARM3-JD interface. Residues coordinating the Ca2+ are shown as sticks and the Ca2+ ion is shown as a green sphere. e, Superposition of ARM3 and the JD of Ca2+-bound (colored by domain) and apo (grey) aligned by the JD.
Figure 5 shows a comparison of the tetrameric Ca2+-bound and apo structures revealing the conformational changes induced by Ca2+ binding at the CD and JD sites. The most obvious differences between the structures are a nearly 30 Å dilation of the CD and a complete disruption of the BTF ring that anchors inter-subunit interactions (Fig. 5 and Supplementary Video 3). Indeed, close inspection reveals that Ca2+ binding eliminates all of the inter-subunit interactions present in the apo and the IP3-bound structures. Dissociation of the CDs yields a channel in which the four CDs are physically uncoupled from one another, explaining the increased mobility of the CDs compared to the apo state. The dissociation also explains the reduced retention time of Ca2+-bound hIP3R in gel filtration experiments compared to unliganded channels and is consistent with FRET measurements demonstrating global conformational changes in the presence of Ca2+ (ref31) (Supplementary Fig. 6a).
Figure 5. Ca2+ binding disrupts CD intra- and inter-subunit interactions.
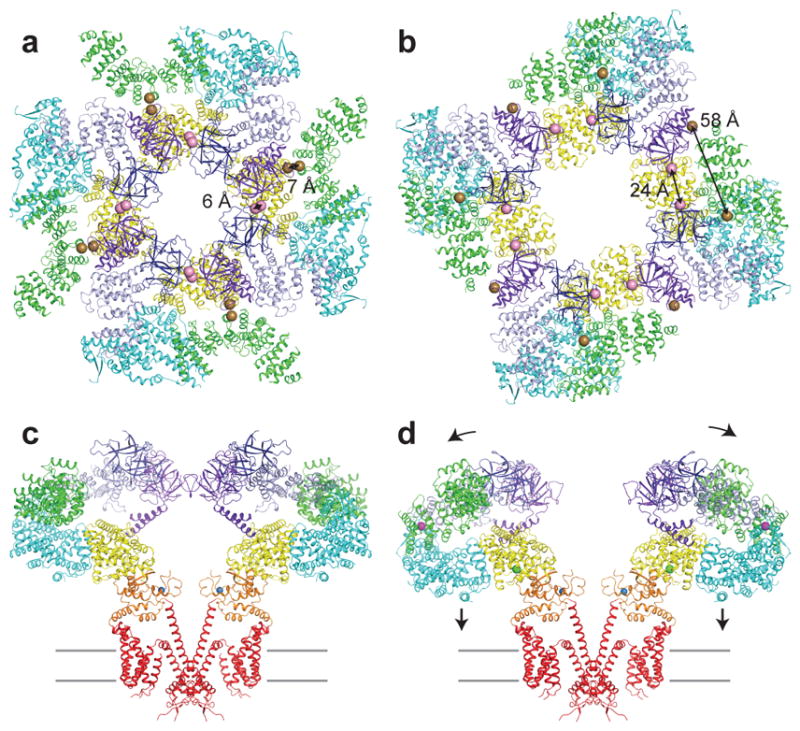
a–b, Cytoplasmic domain of (a) apo and (b) Ca2+-bound colored by domain, viewed from the cytoplasm. Spheres represent the Cα position of Trp168 of BTF1 and Lys426 of BTF2 (pink) and Pro140 of BTF1 and Ala1291 of ARM2 (brown). c–d, Structure of (c) apo and (d) Ca2+-bound colored by domain, viewed in the plane of the membrane. Front and rear subunits are removed for clarity. Ca2+ and Zn2+ ions are shown as spheres. Grey lines represent the approximate position of the membrane.
Ca2+- and IP3-bound conformations
To understand how IP3 and Ca2+ coordinate to regulate channel gating, we determined structures of hIP3R3 in the presence of approximately 2 mM Ca2+ and either 10 μM (low) or 50 μM (high) IP3 at 4.0 Å and 3.9 Å, respectively (Supplementary Fig. 8, Supplementary Fig. 9 and Table 1). Similar to the Ca2+-bound state, the CD was poorly ordered in both the low IP3-Ca2+ and high IP3-Ca2+ reconstructions. We therefore calculated two sets of focused refinement maps for each condition. The TMD focused refinement maps were resolved to 3.8 Å for the low IP3-Ca2+ state and 3.7 Å for the high IP3-Ca2+ state while the CD focused refinement maps were resolved to 4.2 Å for the low IP3-Ca2+ state and 3.8 Å for the high IP3-Ca2+ state (Supplementary Figure 8, Supplementary Figure 9, Supplementary Table 1). Inspection of the ligand-binding sites in the high IP3-Ca2+ condition revealed density consistent with the IP3-binding site and both Ca2+ sites being occupied (Supplementary Fig. 10). To bind IP3, ARM1 in the high IP3-Ca2+ condition is rotated 15˚ towards BTF2 to adopt a conformation that is similar to IP3 class 2. In the low IP3-Ca2+ condition densities are clearly present at both of the Ca2+ sites, but the IP3-binding site is unoccupied and the IP3-binding domain adopts an apo-like conformation.
Despite IP3 being bound, the overall structure of the high IP3-Ca2+ state closely resembles the Ca2+-bound structure (Fig. 6a,c and Supplementary Video 3). In both structures the BTF rings are dissociated and the four CDs are detached from one another. Indeed, the only difference in the high IP3-Ca2+ structure compared to the Ca2+-bound structure is a clamping down of the IP3-binding domain around IP3. In contrast to the similarity between the Ca2+-bound and high IP3-Ca2+ structures, the structure of IP3 class 2 demonstrates that global conformational changes can occur following IP3 binding (Fig. 6b,d). Whereas the network of inter-subunit interactions in the cytoplasmic domain enable conformational changes to be propagated throughout the channel in IP3 class 2, their disruption in the high IP3-Ca2+ structure results in four CDs that are physically uncoupled from one another and from the pore. Thus, the Ca2+ binding sites stabilize the high IP3-Ca2+ structure in an inhibited conformation. Furthermore, Ca2+ binding also stabilizes the separation of the CD domains in the low IP3-Ca2+ and Ca2+-bound structures indicating that Ca2+-dependent IP3R inhibition may be independent of IP3-binding state (Fig. 5).
Figure 6. IP3-induced conformational changes.
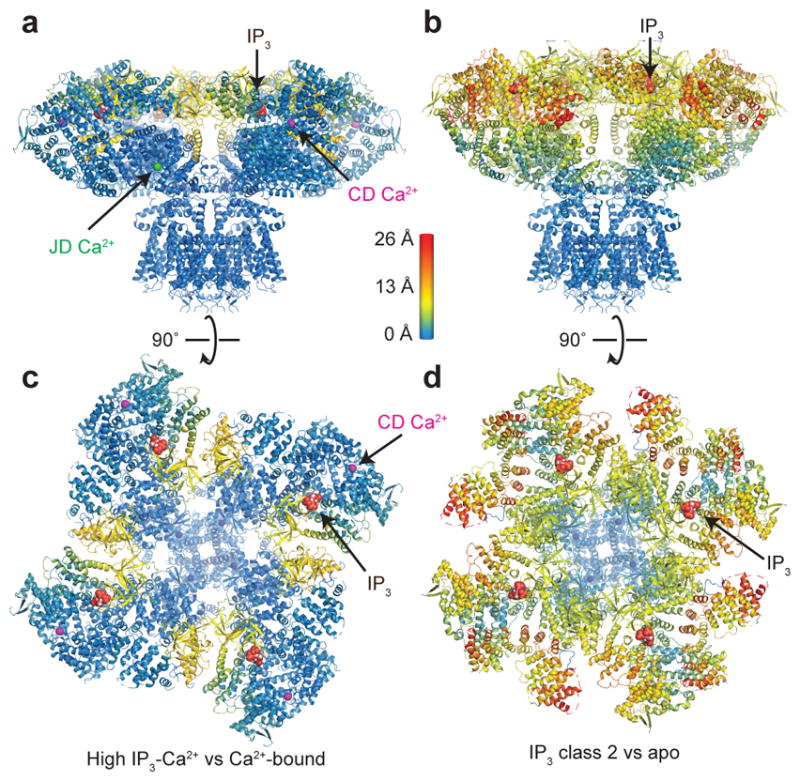
a, High IP3-Ca2+ structure colored by distance of Cα deviation from the Ca2+-bound structure viewed in the plane of the membrane. b, IP3 class 2 colored by distance of Cα deviation from the apo structure viewed in the plane of the membrane. c, Cytoplasmic domain of high IP3-Ca2+ structure colored by distance of Cα deviation from the Ca2+-bound structure viewed from the cytoplasm. d, Cytoplasmic domain of IP3 class 2 colored by distance of Cα deviation from the apo structure viewed from the cytoplasm. IP3 and Ca2+ ions are shown as spheres.
Discussion
The ensemble of structures presented here outlines a model for how IP3 and Ca2+ control the gating of IP3R. The structures support a model in which a network of inter-subunit interactions enables the propagation of conformational changes from peripheral ligand binding domains to the JD and TMD. It has long been recognized that domains such as BTF1, which do not directly contribute to ligand binding, are essential for IP3R gating42–45. The IP3-bound structures demonstrate that these domains are essential because of their incorporation into the inter-subunit interactions network. BTF1, for example, is a key component of a central ring structure that serves to anchor the inter-subunit network and forms a fulcrum against which movements of the IP3-binding domain are propagated throughout the channel. Thus, this network ensures that conformational changes in the IP3-binding domain are coupled to the gating state of the pore (Fig. 2). Ca2+ binding disrupts this network leading to an inhibited state where movements due to IP3 binding are insulated from global channel conformation (Fig. 6).
While high concentrations of Ca2+ inhibit IP3R, low concentrations of Ca2+ are required for activation. In the Ca2+-bound inhibited structures two Ca2+ ions are present: one in the periphery of the CD (CD Ca2+) and one at the CD-JD interface (JD Ca2+). The presence of two Ca2+ binding sites at distinct locations within IP3R is consistent with different apparent Ca2+ binding affinities for activation and inhibition32. While the functional role of the two sites cannot be distinguished from the structural data alone, it is possible that the two sites play distinct roles in gating, with one regulating IP3R activation and the second inhibition.
Aligning the structures into a trajectory allows us to posit a model for how IP3 and Ca2+ binding leads to activation of hIP3R3 (Fig. 7). Due to the similarity between the apo state and IP3 class 1, we hypothesize that upon IP3 binding the channel first adopts a structure similar to IP3 class 1 in which conformational changes are limited to the local environment of the IP3-binding domain. Spontaneous rearrangements of individual IP3-binding domains into class 2-like configurations may then allow the channel to adopt asymmetric intermediate conformations analogous to IP3 classes 3, 4 and 5 in which subunits within the channel can adopt different conformations. The presence of multiple stable intermediate states suggests that the conformational exchange between the two IP3-binding domain configurations occur on a per-subunit basis rather than as a concerted mechanism in which all four subunits act as one (Fig. 3). Compared to the apo state, ARM3 and the JD are rotated as more of the IP3-binding domains adopt the IP3 class 2-like configuration suggesting that IP3-bound structures are intermediates on the pathway to activation (Fig. 2 and Fig. 3). We speculate that Ca2+ binding at one of the sites may induce further rotation of the JD leading to an opening of the S6 gate and a conductive state. Between the selectivity filter and the S6 gate, a portion of S6 adopts a short five-turn or π-helical region in the apo structure. Relaxation from this high-energy π-helix into a lower energy α-helix upon JD rotation may help to stabilize the pore in a conductive state. Transitions between π-helical and α-helical conformations within the S6 helices during gating have been recently described in TRPV646. It is also possible that all of the IP3-bound states can directly access an activated state. Single-channel recordings have identified three reversible modes with distinct gating kinetics whose open channel probability vary by more than 100-fold without changing Ca2+ or IP3 concentrations47. These gating modes may result from the existence of different IP3-bound states that open with stably different probabilities. IP3 class 1, which is nearly identical to the apo state, would require large conformational changes and despite the presence of bound IP3 may be the least likely to open. In contrast, IP3 class 2, in which IP3 binding is accompanied by conformational changes that propagate towards the pore, may be more likely to open. Thus, the ensemble of structures provide a foundation for beginning to understand the complex gating of IP3Rs.
Figure 7. Model for IP3R regulation by IP3 and Ca2+.
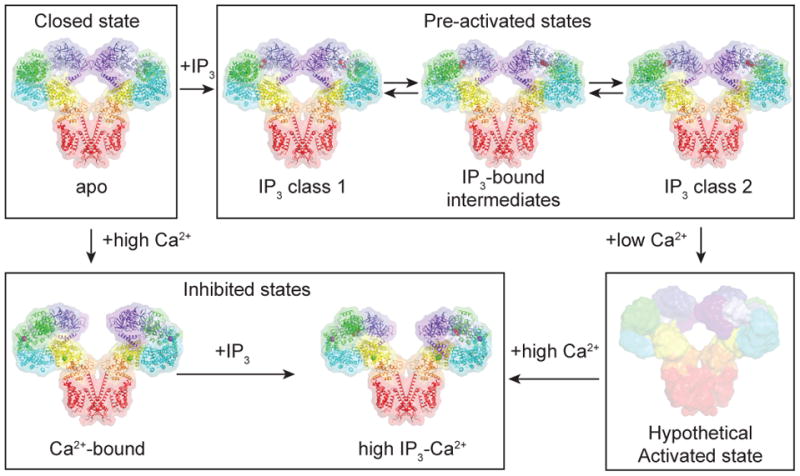
In the absence of Ca2+ and IP3, IP3R adopts a closed state. In the presence of IP3, IP3R adopts one of an ensemble of pre-activated states that are in equilibrium. In the presence of high Ca2+, the BTF ring is dissociated and the channel adopts an inhibited state. In the presence of high Ca2+ and IP3, the BTF ring is dissociated and the channel adopts an inhibited state. In the presence of low Ca2+ and IP3, the channel adopts a hypothetical activated state in which the S6 gate is opened.
Online Methods
Cell Lines
HEK293S GnTI- (human embryonic kidney, N-acetylglucosaminyltransferase I knockout) cells were used for protein expression and maintained in Freestyle 293 Expression Media (Gibco) supplemented with 2% fetal bovine serum (FBS) at 37˚ C.
Constructs
A gene for human type 3 IP3R (HsITPR3) in pENTR223 vector was obtained from HMS PlasmID (HsCD00399229). Internal restriction sites for XhoI and EcoRI were removed by site directed mutagenesis. Polymerase chain reaction (PCR) was used to amplify the gene and add restriction sites for XhoI and EcoRI for in-frame subcloning into a bacmam expression vector with an N-terminal GFP and PreScission protease cut site48,49. The final product (N-GFP-HsITPR3) was sequenced in its entirety.
hIP3R3 Expression & Purification
N-GFP-HsITPR3 was used to transform DH10Bac E. coli cells to generate bacmid DNA. Recombinant baculovirus was produced by three rounds of viral amplification in Sf9 (Spodoptera frugiperda) cells maintained in Grace’s media supplemented with 10% FBS, 1% penicillin/streptomycin, and 0.1% Pluronic F-68. Viral particles were separated from cell debris by centrifugation and filtration, and used to infect HEK293S GnTI- cells for protein production. For the first 24 hours of infection, cultures were grown at 37˚ C, after which sodium butyrate was added to a final concentration of 10 mM and cells moved to 30˚ C to grow for an additional 48–72 hours prior to harvesting. Cell pellets were washed in phosphate-buffered saline solution and flash frozen in liquid nitrogen.
Thawed cell pellets were solubilized for 1 hour in 2% lauryl maltose neopentyl glycol, 4 mM cholesterol hemisuccinate (CHS), 20 mM HEPES pH 7.5, 150 mM NaCl with protease inhibitor cocktail (1 mM PMSF, 2.5 ug/mL aprotinin, 2.5 ug/mL leupeptin, 1 ug/mL pepstatin A, 0.5 mM AEBSF, 1 mM benzamidine), and DNAse. Whole cell lysates were centrifuged at 75,000xg for 40 minutes, and the soluble fraction was bound to GFP-nanobody beads for 1–3 hours on a rotator50. Beads were collected on a column by gravity and then washed with 50 mL of wash buffer (150 mM NaCl, 50 mM Tris pH 8.0, 2 mM DTT, 0.06% digitonin). The protein was digested with PreScission protease diluted in wash buffer on the column for 3 hours to separate the channel from the GFP and then eluted with 12 mL of wash buffer.
Concentrated protein was further purified by size-exclusion chromatography on a Superose 6 Increase (GE Life Sciences) column in gel filtration buffer (150 mM NaCl, 50 mM Tris pH 8.0, 2 mM DTT, 0.06% digitonin) with either addition of 100 μM Ca2+, 1 mM Ca2+, 5 mM EGTA or 10 mM HEDTA, based on the conditions required for vitrification. Peak fractions were pooled and concentrated to ~8 mg/mL for cryo-EM analysis. For cryo-EM samples containing IP3 (d-myo-Inositol 1,4,5-Trisphosphate), a stock solution of 1 mM IP3 (Calbiochem, SR5440-1102-669) in ddH2O was added to bring the solution to the appropriate concentration approximately 30 minutes before vitrification.
Electron Microscopy Sample Preparation and Imaging
3.5–5 μl of purified channel at a concentration of ~8 mg/mL was pipetted onto glow-discharged 400 mesh gold Quantifoil R1.2/1.3 holey carbon grids (Quantifoil, Q25869) approximately 5–20 seconds before blotting (blot force = 1) for two seconds and plunging into liquid nitrogen-cooled liquid ethane using an FEI Vitrobot Mark IV (FEI ThermoFisher). Precise control over free Ca2+ concentrations in the vitrified samples is complicated by the presence of high concentrations of Ca2+ in the filter paper used for blotting excess solution away from the grid. Prior to our data collection, we performed initial screening experiments using various Ca2+ and EGTA concentrations, which suggested that the final concentration of free Ca2+ may be as much as 2 mM higher than those used during sample preparation. This estimate was based upon the concentrations of Ca2+ and EGTA needed to completely eliminate the inhibited conformation. Efforts to more precisely estimate the free Ca2+ following blotting were complicated by the extremely small quantity of solution remaining on the grid following blotting (less than 0.01 μl). The Ca2+ concentrations presented here are thus best estimates and may not reflect the true concentration of free Ca2+ to which the samples are exposed. Additional conditions were tested, but the presence of channels in Ca2+-inhibited states indicated that the free Ca2+ was not well controlled. For the ligand-free condition, we added no free Ca2+ and either 5 mM EGTA or 10 mM HEDTA and predict a free Ca2+ below 100 nM. For the IP3-bound condition, we added no free Ca2+ and 5 mM EGTA and predict a free Ca2+ below 100 nM. For the Ca2+-bound condition, we added 1 mM Ca2+ and no EGTA and predict a free Ca2+ above 1 mM. For the low IP3-Ca2+ condition, we added 100 μM Ca2+ and no EGTA and predict a free Ca2+ above 1 mM. For the high IP3-Ca2+ condition, we added 100 μM Ca2+ and no EGTA and predict a free Ca2+ above 1 mM.
Grids were transferred to an FEI Titan Krios cryo-EM operating at 300 kV. Images were recorded in an automated fashion using SerialEM or Leginon with a defocus range of −1.0 to −2.5 μM over 8 seconds as 40 sub-frames using a Gatan K2 Summit direct electron detector in super-resolution mode51,52. For the apo data collection, images were recorded at a calibrated super-resolution pixel size of 0.548 Å/pixel with energy filter slit width of 20 eV. For all other data sets, images were recorded at a calibrated super-resolution pixel size of 0.544 Å/pixel. Electron dose was 9 e−/pix/s at the detector for a total accumulated dose of 60 e−/Å2 for the apo data set and 61 e−/Å2 for the other data sets. Images were two-times Fourier cropped and aligned using whole frame and local correction algorithms by Motioncor2 resulting in a final calibrated pixel size of 1.096 Å/pixel for the apo data set and 1.088 Å for the other data sets53. The effects of the contrast transfer function were estimated using CTFfind 4.1.8 (ref54). A total of 1801, 4795, 944, 1409, and 4076 images were recorded for the apo, IP3-bound, Ca2+-bound, low IP3-Ca2+ and high IP3-Ca2+ data sets, respectively. After removing images with excessive drift, bad ice or poor contrast, 1767, 4519, 926, 1409, and 3974 images were kept for processing for the apo, IP3-bound, Ca2+-bound, low IP3-Ca2+ and high IP3-Ca2+ data sets, respectively.
Cryo-EM analysis
Approximately 1000 particles from each data set were manually selected to generate initial templates for autopicking using Relion39. Several rounds of 2D classification and autopicking were used to generate improved templates for autopicking. An initial model of the apo condition was generated from the autopicked particles using CryoSPARC55 and used as the starting model for all data sets. Two-dimensional and three-dimensional classification in Relion were used to eliminate false-positives or damaged particles resulting in 53k, 303k, 49k, 74k, and 170k images for the apo, IP3-bound, Ca2+-bound, low IP3-Ca2+ and high IP3-Ca2+ data sets, respectively. The particle images were polished in Relion to realign particles on a per-particle basis and correct for dose-dependent beam-induced specimen damage.
For the apo condition, the polished particle stack was subjected to one round of three-dimensional classification resulting in a particle stack of 26k homogenous particles. For refinement, the particles were analyzed by cisTEM with C4 symmetry imposed56. For the initial global search in cisTEM, the reference map generated in Relion was filtered to 20 Å. For the final iterations, the references were filtered to 5 Å. Using the 0.143 FSC criterion, the resolution was estimated to be 3.49 Å57.
To improve the features of the CLD and ARM2 in the apo reconstruction, the stack of 53k particles was subjected to focused three-dimensional classification, resulting in a stack of 30k particles that was symmetry expanded to 120k particles in C1 and the signal was subtracted for the CD and JD of three of the subunits, and the entire TMD. Focused refinement in Relion using a soft mask that included only the CD and JD of one subunit resulted in a reconstruction whose resolution was estimated to be 3.76 Å by the 0.143 FSC criterion.
To improve the features of S1′ and S1″ in the apo reconstruction, the stack of 30k particles was symmetry expanded to 120k particles in C1 and the signal was subtracted for the CD of three of the subunits. Focused refinement in Relion using a soft mask that included the TMD and JD of all four subunits and the CD of one subunit followed by focused classification using a mask that just included the S1–S4 domain of one subunit yielded one major class (81k particles) in which S1′ and S1″ were improved. Focused refinement in Relion of the 81k particles using a soft mask that just included the TMD and JD and all four subunits and the CD of one subunit resulted in a reconstruction whose resolution was estimated to be 3.69 Å by the 0.143 FSC criterion.
For the IP3 condition, the stack of 303k polished particles was subjected to one round of three-dimensional classification specifying 7 classes without imposing symmetry using a reference in which the four ARM2 domains had been removed. After the first round, one class was poorly resolved (40k particles) and excluded from later processing steps. The remaining particles were merged into 3 groups based upon the positions of the ARM2 domain and reclassified using a reference lacking the ARM2 domains into 7 classes without imposing symmetry. After the second round, the classes were merged into 5 groups based upon the positions of the ARM2 domain. The particles were then reclassified and merged two additional times. The merged particle stacks were then refined without symmetry and classified without angular sampling to remove any misclassified particles. The final cleaned particle stacks were then refined in cisTEM. Classes 1 (39k particles) and 2 (41k particles) were refined in cisTEM with C4 symmetry. For the initial global search in the refinement of classes 1 and 2 in cisTEM, the reference maps generated in Relion were filtered to 20 Å. For the final iterations, the references were filtered to 4.8 Å for class 1 and 6 Å for class 2. Using the 0.143 FSC criterion, the resolutions were estimated to be 3.33 Å for class 1 and 3.82 for class 2. Classes 3, 4 and 5 were refined in cisTEM without symmetry. For the initial global search of classes 3 (38k particles), 4 (10k particles) and 5 (27k particles) in cisTEM, the reference map generated in Relion was filtered to 20 Å. For the final iterations, the references were filtered to 6, 10 and 7 Å, for classes 3, 4 and 5 respectively. Using 0.143 FSC criterion, the resolutions for classes 3, 4 and 5 were estimated to 4.12, 6.01 and 4.47 Å, respectively.
For the Ca2+-bound condition, the polished particle stack was subjected to one round of focused three-dimensional classification using a soft mask including ARM3, the JD and the TMD with C4 symmetry imposed resulting in a final particle stack of 34k particles. The particle stack was analyzed in cisTEM with C4 symmetry imposed. For the initial global search in cisTEM, the reference map generated in Relion was filtered to 20 Å. For the final iterations, the references were filtered to 7 Å. Using the 0.143 FSC criterion, the resolution was estimated to be 4.33 Å. To improve the alignment of the more rigid ARM3, JD and TMD, which includes the JD Ca2+ binding site, a soft mask was applied to those domains during refinement in Relion, yielding a reconstruction at 4.2 Å. To improve the features of the CD, the stack of 34k particles was symmetry expanded to 135k particles in C1 and the signal was subtracted for the CD of three of the subunits. Focused refinement in Relion using a soft mask including the TMD and JD of all four subunits and the CD of one subunit resulted in a reconstruction whose resolution was estimated to be 4.69 Å by the 0.143 FSC criterion.
For the low IP3-Ca2+ condition, the polished particle stack of 74k particles was classified into 6 classes without imposing symmetry. Four of the six classes, totaling 49k particles, were selected for refinement in cisTEM with C4 symmetry imposed. For the initial global search in cisTEM, the reference map generated in Relion was filtered to 20 Å. For the final iterations, the references were filtered to 7 Å. Using the 0.143 FSC criterion, the resolution was estimated to be 3.96 Å. To improve the alignment of the more rigid ARM3, JD and TMD, which includes the JD Ca2+ binding site, a soft mask was applied to those domains during refinement in Relion, yielding a reconstruction at 3.80 Å. To improve the features of the CD, the stack of 196k particles was symmetry expanded to C1 and the signal was subtracted for the JD and CD of three of the subunits, and the entire TMD. Focused classification in Relion using a soft mask that included the JD and CD of one subunit yielded a single major class of 158k particles. Refinement of the 158k particles in Relion using a soft mask that included the JD and CD of one subunit resulted in a reconstruction whose resolution was estimated to be 4.22 Å by the 0.143 FSC criterion.
For the high IP3-Ca2+ condition, the polished particle stack of 170k particles was analyzed in cisTEM with C4 symmetry imposed. One round of three-dimensional classification into seven classes identified six similar classes containing a total of 131k particles. Refinement of these 131k particles in cisTEM resulting in a reconstruction with an estimated resolution of 3.92 Å using reference filtered to 7 Å. To improve the alignment of the more rigid ARM3, JD and TMD, which includes the JD Ca2+ binding site, a soft mask was applied to those domains during classification and refinement in Relion, yielding a reconstruction at 3.66 Å. To improve the features of the CD, the stack of 170k particles was symmetry expanded to C1 and the signal was subtracted for the CD of three of the subunits. Focused classification identified a population of 350k particles that when refined in cisTEM using a soft mask that included the JD and CD of one subunit resulted in a reconstruction whose resolution was estimated to be 3.78 Å by the 0.143 FSC criterion.
Model building and coordinate refinement
The structure of ligand-free IP3-binding domain of rIP3R1 (PDB: 3UJ4)37 was docked into the full channel apo state density map using UCSF Chimera58 and then manually rebuilt according to the density in Coot59. The remainder of the protein was built de novo into the apo state full channel, apo state CD focused refinement and S1–S4 focused refinement density maps. The refined apo state model contains residues 5-80, 85-321, 350-527, 534-674, 690-893, 961-1003, 1024-1036, 1043-1129, 1167-1432, 1587-1687, 1717-1804, 1863-2038, 2043-2074, 2111-2226, 2260-2403 and 2449-2611. Unregistered poly-alanine helices were modeled for S1′ and α71-α75 at the C-terminal end of ARM2 using Jpred460. The coordinates were refined against one of the full-channel half-maps (work) using Rosetta 3.761 and phenix.real_space_refine62 with secondary structure and Ramachandran restraints maintained throughout. To monitor the effects of over fitting during coordinate refinement, Fourier shell correlations were compared against the half-map excluded from refinement (free). Map to model correlations were determined using phenix.mtriage (https://www.phenix-online.org/newsletter/CCN_2017_07.pdf).
For IP3 classes 1 and 2 and the Ca2+-bound structures, the final refined model of apo hIP3R was docked into the density map using UCSF Chimera and then manually rebuilt according to the density in Coot. The coordinates were refined against one of the full-channel half-maps (work) using phenix.real_space_refine with secondary structure and Ramachandran restraints maintained throughout. To monitor the effects of over fitting during coordinate refinement, Fourier shell correlations were compared against the half-map excluded from coordinate refinement (free). Map to model correlations were determined using phenix.mtriage.
For IP3 classes 3, 4 and 5, the refined models of IP3 classes 1 and 2 were docked into the density on a subunit-by-subunit basis by rigid-body fitting in coot. Once the overall fit of the models were completed, the models of IP3 classes 1 and 2 were split into domain fragments and docked into the density map by rigid-body fitting. The rigid-body fit model was refined by real-space refinement for classes 3 and 5 in phenix.real_space_refine against one of the half-maps and rigid-body refinement for class 4 in phenix.real_space_refine against the full reconstruction.
Software was compiled by SBGrid63. Local resolution estimates were performed using ResMap64 and structure figures were prepared with UCSF Chimera and PyMol (PyMol version 1.8.0 Schrodinger LLC).
Data availability
Cryo-EM maps have been deposited in the EMDB under accession codes EMD-7978 for the full channel apo map, EMD-7979 for the apo CD focus refinement map, EMD-7980 for the apo S1–S4 focus refinement map, EMD-7981 for the IP3 class 1 map, EMD-7982 for the IP3 class 1 CD focus refinement map, EMD-7984 for the IP3 class 2 map, EMD-7982 for the IP3 class 2 CD focus refinement map, EMD-7985 for the IP3 class 3 map, EMD-7986 for the IP3 class 4 map, EMD-7987 for the IP3 class 5 map, EMD-7888 for the full channel Ca2+-bound map, EMD-7990 for the Ca2+-bound TMD focus refinement map, EMD-7989 for the Ca2+-bound CD focus refinement map, EMD-7991 for the full channel low IP3-Ca2+ map, EMD-7993 for the low IP3-Ca2+ TMD focus refinement map, EMD-7992 for the low IP3-Ca2+ CD focus refinement map, EMD-7994 for the high IP3-Ca2+ map, EMD-7995 for the high IP3-Ca2+ TMD focus refinement map and EMD-7996 for the high IP3-Ca2+ CD focus refinement map. Atomic coordinates are available from the RCSB Protein Data Bank under accession codes 6DQJ for the apo structure, 6DQN for IP3 class 1, 6DQV for IP3 class 2, 6DQS for IP3 class 3, 6DQZ for IP3 class 4, 6DR0 for IP3 class 5, 6DR2 for the Ca2+-bound structure, 6DRA for the low IP3-Ca2+ structure and 6DBC for the high IP3-Ca2+ structure. All other source data are available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank M. de le Cruz at the Memorial Sloan Kettering Cancer Center Cryo-EM facility and staff at the New York Structural Biology Simons Electron Microscopy Center for help with data collection and S.B. Long and T. Walz for comments on the manuscript. This work was supported in part by National Institutes of Health grant CA008748, the Josie Robertson Investigators Program and the Searle Scholars Program.
Footnotes
Accession Codes
Cryo-EM maps have been deposited in the EMDB under accession codes EMD-7978 for the full channel apo map, EMD-7979 for the apo CD focus refinement map, EMD-7980 for the apo S1–S4 focus refinement map, EMD-7981 for the IP3 class 1 map, EMD-7982 for the IP3 class 1 CD focus refinement map, EMD-7984 for the IP3 class 2 map, EMD-7982 for the IP3 class 2 CD focus refinement map, EMD-7985 for the IP3 class 3 map, EMD-7986 for the IP3 class 4 map, EMD-7987 for the IP3 class 5 map, EMD-7888 for the full channel Ca2+-bound map, EMD-7990 for the Ca2+-bound TMD focus refinement map, EMD-7989 for the Ca2+-bound CD focus refinement map, EMD-7991 for the full channel low IP3-Ca2+ map, EMD-7993 for the low IP3-Ca2+ TMD focus refinement map, EMD-7992 for the low IP3-Ca2+ CD focus refinement map, EMD-7994 for the high IP3-Ca2+ map, EMD-7995 for the high IP3-Ca2+ TMD focus refinement map and EMD-7996 for the high IP3-Ca2+ CD focus refinement map. Atomic coordinates are available from the RCSB Protein Data Bank under accession codes 6DQJ for the apo structure, 6DQN for IP3 class 1, 6DQV for IP3 class 2, 6DQS for IP3 class 3, 6DQZ for IP3 class 4, 6DR0 for IP3 class 5, 6DR2 for the Ca2+-bound structure, 6DRA for the low IP3-Ca2+ structure and 6DBC for the high IP3-Ca2+ structure.
Author contributions
N.P. and R.K.H. designed, performed and analyzed the experiments. N.P. and R.K.H. prepared the manuscript.
Competing financial interest statement
The authors declare no competing interests.
References
- 1.Burgess GM, Mckinney JS, Fabiatoq A, Leslie BA, Putney JW. Calcium Pools in Saponin-permeabilized Guinea Pig Hepatocytes. J Biol Chem. 1983;258:35336–153345. [PubMed] [Google Scholar]
- 2.Ehrlich BE, Watras J. Inositol 1,4,5-trisphosphate activates a channel from smooth muscle sarcoplasmic reticulum. Nature. 1988;336:583–586. doi: 10.1038/336583a0. [DOI] [PubMed] [Google Scholar]
- 3.Furuichi T, et al. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–38. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- 4.Mignery GA, Südhof TC, Takei K, De Camilli P. Putative receptor for inositol 1,4,5-trisphosphate similar to ryanodine receptor. Nature. 1989;342:192–5. doi: 10.1038/342192a0. [DOI] [PubMed] [Google Scholar]
- 5.Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:67–69. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- 6.Foskett JK, White C, Cheung KH, Mak DOD. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vervloessem T, Yule DI, Bultynck G, Parys JB. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochimica et biophysica acta. 2015;1853:1992–2005. doi: 10.1016/j.bbamcr.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferris CD, Huganir RL, Supattapone S, Snyder SH, Solomon H. Purified inositol 1,4,5-trisphosphate receptor mediates calcium flux in reconstituted lipid vesicles. Nature. 1989;342:87–9. doi: 10.1038/342087a0. [DOI] [PubMed] [Google Scholar]
- 9.Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 10.Jean T, Klee CB. Calcium modulation of inositol 1,4,5-trisphosphate-induced calcium release from neuroblastoma x glioma hybrid (NG108-15) microsomes. J Biol Chem. 1986;261:16414–16420. [PubMed] [Google Scholar]
- 11.Suematsu E, Hirata M, Hashimoto T, Kuriyama H. Inositol 1,4,5-trisphosphate releases Ca2+from intracellular store sites in skinned single cells of porcine coronary artery. Biochem Biophys Res Commun. 1984;120:481–485. doi: 10.1016/0006-291x(84)91279-8. [DOI] [PubMed] [Google Scholar]
- 12.Armant DR. Intracellular Ca2+ signaling and preimplantation development. Adv Exp Med Biol. 2015;843:151–171. doi: 10.1007/978-1-4939-2480-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berridge MJ. Calcium signalling remodelling and disease. Biochem Soc Trans. 2012;40:297–309. doi: 10.1042/BST20110766. [DOI] [PubMed] [Google Scholar]
- 14.Kania E, Roest G, Vervliet T, Parys JB, Bultynck G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front Oncol. 2017;7:140. doi: 10.3389/fonc.2017.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ando H, Kawaai K, Bonneau B, Mikoshiba K. Remodeling of Ca2+signaling in cancer: Regulation of inositol 1,4,5-trisphosphate receptors through oncogenes and tumor suppressors. Advances in Biological Regulation. 2017 doi: 10.1016/j.jbior.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Berridge MJ. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol Rev. 2016;96:1261–1296. doi: 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- 17.Egorova PA, Bezprozvanny IB. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 2017 doi: 10.1111/febs.14366. [DOI] [PubMed] [Google Scholar]
- 18.Bezprozvanny I, et al. Bell-shaped calcium-response curves of lns(l,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 19.Mikoshiba K. Role of IP3 receptor signaling in cell functions and diseases. Adv Biol Regul. 2015;57:217–227. doi: 10.1016/j.jbior.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Gerasimenko OV, Gerasimenko JV, Belan PV, Petersen OH. Inositol trisphosphate and cyclic ADP-ribose-mediated release of Ca2+from single isolated pancreatic zymogen granules. Cell. 1996;84:473–480. doi: 10.1016/s0092-8674(00)81292-1. [DOI] [PubMed] [Google Scholar]
- 21.Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J Gen Physiol. 1990;95:1103–22. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stehno-Bittel L, Lückhoff A, Clapham DE. Calcium release from the nucleus by InsP3 receptor channels. Neuron. 1995;14:163–167. doi: 10.1016/0896-6273(95)90250-3. [DOI] [PubMed] [Google Scholar]
- 23.Bootman MD, Berridge MJ, Lipp P. Cooking with calcium: The recipes for composing global signals from elementary events. Cell. 1997;91:367–373. doi: 10.1016/s0092-8674(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 24.Marchant JS, Parker I. Role of elementary Ca2+ puffs in generating repetitive Ca2+ oscillations. EMBO J. 2001;20:65–76. doi: 10.1093/emboj/20.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker I, Choi J, Yao Y. Elementary events of InsP3-induced Ca2+liberation in Xenopus oocytes: Hot spots, puffs and blips. Cell Calcium. 1996;20:105–121. doi: 10.1016/s0143-4160(96)90100-1. [DOI] [PubMed] [Google Scholar]
- 26.Hajnóczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO J. 1997;16:3533–3543. doi: 10.1093/emboj/16.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshall IC, Taylor CW. Two calcium-binding sites mediate the interconversion of liver inositol 1,4,5-trisphosphate receptors between three conformational states. Biochem J. 1994;301(Pt 2):591–598. doi: 10.1042/bj3010591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sienaert I, et al. Molecular and functional evidence for multiple Ca2+-binding domains in the type 1 inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1997;272:25899–906. doi: 10.1074/jbc.272.41.25899. [DOI] [PubMed] [Google Scholar]
- 29.Striggow F, Ehrlich BE. The inositol 1,4,5-trisphosphate receptor of cerebellum. Mn2+ permeability and regulation by cytosolic Mn2+ J Gen Physiol. 1996;108:115–124. doi: 10.1085/jgp.108.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamada K, Terauchi A, Mikoshiba K. Three-dimensional Rearrangements within Inositol 1,4,5-Trisphosphate Receptor by Calcium. J Biol Chem. 2003;278:52881–52889. doi: 10.1074/jbc.M309743200. [DOI] [PubMed] [Google Scholar]
- 31.Shinohara T, et al. Mechanistic basis of bell-shaped dependence of inositol 1,4,5-trisphosphate receptor gating on cytosolic calcium. Proc Natl Acad Sci U S A. 2011;108:15486–15491. doi: 10.1073/pnas.1101677108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mak DOD, McBride S, Foskett JK. Inositol 1,4,5-tris-phosphate activation of inositol tris-phosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci. 1998;95:15821–15825. doi: 10.1073/pnas.95.26.15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maranto AR. Primary structure, ligand binding, and localization of the human type 3 inositol 1,4,5-trisphosphate receptor expressed in intestinal epithelium. J Biol Chem. 1994;269:1222–1230. [PubMed] [Google Scholar]
- 34.Bosanac I, et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 35.Bosanac I, et al. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol Cell. 2005;17:193–203. doi: 10.1016/j.molcel.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 36.Lin CC, Baek K, Lu Z. Apo and InsP3-bound crystal structures of the ligand-binding domain of an InsP3 receptor. Nat Struct Mol Biol. 2011;18:1172–1174. doi: 10.1038/nsmb.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo MD, et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature. 2012;483:108–112. doi: 10.1038/nature10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan G, et al. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature. 2015;527:336–341. doi: 10.1038/nature15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scheres SHW. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods in Enzymology. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 40.des Georges A, et al. Structural Basis for Gating and Activation of RyR1. Cell. 2016;167:145–157.e17. doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prole DL, Taylor CW. Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. Journal of Physiology. 2016;594:2849–2866. doi: 10.1113/JP271139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boehning D, Joseph SK. Direct association of ligand-binding and pore domains in homo- and heterotetrameric inositol 1,4,5-trisphosphate receptors. EMBO J. 2000;19:5450–5459. doi: 10.1093/emboj/19.20.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan J, et al. Structural Studies of Inositol 1,4,5-Trisphosphate Receptor. J Biol Chem. 2010;285:36092–36099. doi: 10.1074/jbc.M110.140160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchida K, Miyauchi H, Furuichi T, Michikawa T, Mikoshiba K. Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2003;278:16551–16560. doi: 10.1074/jbc.M300646200. [DOI] [PubMed] [Google Scholar]
- 45.Yamazaki H, Chan J, Ikura M, Michikawa T, Mikoshiba K. Tyr-167/Trp-168 in type 1/3 inositol 1,4,5-trisphosphate receptor mediates functional coupling between ligand binding and channel opening. J Biol Chem. 2010;285:36081–36091. doi: 10.1074/jbc.M110.140129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGoldrick LL, et al. Opening of the human epithelial calcium channel TRPV6. Nature. 2018;553:233–237. doi: 10.1038/nature25182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ionescu L, et al. Mode Switching Is the Major Mechanism of Ligand Regulation of InsP 3 Receptor Calcium Release Channels. J Gen Physiol. 2007;130:631–645. doi: 10.1085/jgp.200709859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goehring A, et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc. 2014;9:2574–85. doi: 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whicher JR, MacKinnon R. Structure of the voltage-gated K+channel Eag1 reveals an alternative voltage sensing mechanism. Science (80-) 2016;353:664–669. doi: 10.1126/science.aaf8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirchhofer A, et al. Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol. 2010;17:133–139. doi: 10.1038/nsmb.1727. [DOI] [PubMed] [Google Scholar]
- 51.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 52.Suloway C, et al. Automated molecular microscopy: The new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Punjani A, Rubinstein JL, Fleet DJ, Brubaker M. A cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 56.Grigorieff N, Grant T, Rohou A. cisTEM: User-friendly software for single-particle image processing. 2018:257618. doi: 10.1101/257618. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 58.Pettersen EF, et al. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 59.Emsley P, Cowtan K IUCr. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 60.Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang RYR, et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife. 2016;5 doi: 10.7554/eLife.17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morin A, et al. Collaboration gets the most out of software. Elife. 2013;2013:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps have been deposited in the EMDB under accession codes EMD-7978 for the full channel apo map, EMD-7979 for the apo CD focus refinement map, EMD-7980 for the apo S1–S4 focus refinement map, EMD-7981 for the IP3 class 1 map, EMD-7982 for the IP3 class 1 CD focus refinement map, EMD-7984 for the IP3 class 2 map, EMD-7982 for the IP3 class 2 CD focus refinement map, EMD-7985 for the IP3 class 3 map, EMD-7986 for the IP3 class 4 map, EMD-7987 for the IP3 class 5 map, EMD-7888 for the full channel Ca2+-bound map, EMD-7990 for the Ca2+-bound TMD focus refinement map, EMD-7989 for the Ca2+-bound CD focus refinement map, EMD-7991 for the full channel low IP3-Ca2+ map, EMD-7993 for the low IP3-Ca2+ TMD focus refinement map, EMD-7992 for the low IP3-Ca2+ CD focus refinement map, EMD-7994 for the high IP3-Ca2+ map, EMD-7995 for the high IP3-Ca2+ TMD focus refinement map and EMD-7996 for the high IP3-Ca2+ CD focus refinement map. Atomic coordinates are available from the RCSB Protein Data Bank under accession codes 6DQJ for the apo structure, 6DQN for IP3 class 1, 6DQV for IP3 class 2, 6DQS for IP3 class 3, 6DQZ for IP3 class 4, 6DR0 for IP3 class 5, 6DR2 for the Ca2+-bound structure, 6DRA for the low IP3-Ca2+ structure and 6DBC for the high IP3-Ca2+ structure. All other source data are available from the corresponding author upon request.