

Abstract
The most important prognostic factor for atherosclerotic cardiovascular disease (ASCVD) is age, independent of all other recognized risk factors. Recently, exome sequence analyses showed that somatic mutations in blood cells, a process termed clonal hematopoiesis, is common and increases in prevalence with age, with at least 1 in 10 adults older than 70 years affected. Carriers of clonal hematopoiesis have been shown to be not only at heightened risk for hematologic malignancy but also at increased risk for ASCVD. Here, we review the prior literature of clonal selection and expansion of hematopoietic stem cells and the evidence supporting its causal association with ASCVD.
Keywords: coronary artery disease; genetics, human; genetics, rat/mouse; blood cell; clonal hematopoiesis
Journal Subject Terms: Inflammation, Aging, Cardiovascular Disease, Genetics, Atherosclerosis
INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) remains a leading cause of death in the United States and worldwide.1, 2 In fact, in a recent clinical trial where historically low low-density lipoprotein cholesterol (LDL-C) concentrations were achieved, 10% still had recurrent ASCVD events over the subsequent two years.3 As such, the identification of new risk pathways orthogonal to traditional risk factors continues to be an important research goal.
Age is the dominant risk factor for ASCVD but the mechanistic bases for why age predisposes to ASCVD is not completely understood.4, 5 While age is associated with the acquisition of and cumulative exposure of conventional risk factors, such factors do not appear to fully explain the association of age and ASCVD.5–8 Previously described plausible mechanisms accompanying aging include flow-mediated epigenetic alterations of endothelial cells9, diminished regenerative capabilities of bone marrow-derived vascular progenitor cells10, 11, alteration of vascular smooth muscle cell function12 and proliferation from epigenetic changes13, plaque instability from senescent foam macrophage cells14, with additional contributions from telomere ablation15 and oxidative stress16. Key challenges are disentangling whether proposed factors are causal for atherosclerosis or simply correlated with age.
The effect of hematopoietic cells on atherosclerosis pathogenesis has long been recognized.17–22 The acquisition of genetic mutations in cells, including hematopoietic stem cells, is a feature of aging.23 We recently observed such somatic mutations leading to clonal expansion in the absence of other hematologic abnormalities, or “clonal hematopoiesis of indeterminate potential” (CHIP), not only predisposed to risk for hematologic malignancy but, surprisingly, predisposed to risk for ASCVD.24–31 Here, we provide an overview of clonal hematopoiesis and the evidence linking it with ASCVD.
THE AGING HEMATOPOETIC SYSTEM
The hematopoietic system relies on balancing regeneration, differentiation, and senescence. Notably, much of what is known about this balance is derived from murine studies and much less is known about human aging. In adult mammals, the former two processes occur within the bone marrow, with hematopoietic stem cells differentiating into lymphoid, myeloid, erythroid, and platelet cells. A combination of intrinsic and extrinsic cellular factors influencing cytokines and related receptors determine hematopoietic lineage commitment and potential amplification. Aging can influence multiple factors influencing lineage (Table 1).32
Table 1.
Distinctive features of aging hematopoietic stem cells
| Features |
|---|
| Increased total count |
| Reduced adhesion to bone marrow stroma |
| Lineage skew favoring myeloid progenitors |
| Polarity shift |
| mTOR pathway perturbation |
| Altered DNA damage response |
| Increased reactive oxygen species |
| Global epigenetic shift |
The accumulation of genomic DNA damage is a well-known feature of cellular aging.23 Like other stem cells, hematopoietic stem cells acquire mutations during their lifetimes but most have no significant consequences or result in cell death.33 Most hematopoietic stem cells are maintained in a quiescent state to minimize the stresses of cellular metabolism and DNA replication. However, quiescence also promotes nonhomologous end joining-mediated DNA repair further contributing to mutagenesis.34 Old hematopoietic stem cells in mice show greater signs of stress from cellular replication, which is tied to a decreased expression of all subunits of the mini-chromosome maintenance helicase.35 Acquired mutations, age-related changes in DNA repair, and reduced regenerative potential with aging36, provide an ideal opportunity for clonal selection and expansion.
INITIAL EVIDENCE FOR AGE-RELATED CLONAL HEMATOPOEISIS IN ASYMPTOMATIC INDIVIDUALS
Hematopoiesis is considered to be polyclonal with largely equipotent hematopoietic stem cells. Age-related changes in hematopoietic stem cell clonal contribution was first described approximately 20 years ago through analyses of chromosome X-inactivation patterns derived from peripheral leukocytes in women.37–39 It was observed that age strongly correlated with nonrandom patterns of chromosome X-inactivation indicating allelic skewing. These data demonstrated that clonality was present in blood cells and that age was a determinant.37–39
Most mutations observed in hematopoietic stem cells are either benign or deleterious to the cell; it estimated that hematopoietic stem cells acquire approximately one exonic mutation per decade.23 More recently, sequencing of peripheral leukocytes and buccal epithelial cells of older women with chromosome X-inactivation skewing identified recurrent somatic mutations in TET2 (encoding tet methylcytosine dioxygenase 2) in leukocytes but not buccal cells in 10 of 182 individuals.40 Additionally, among non-leukemic hematopoietic stem cells from individuals with acute myeloid leukemia, a high frequency of DNMT3A (encoding DNA (cytosine-5-)-methyltransferase 3α) and TET2 mutations were observed.41, 42 These data indicate the presence of “driver” mutations shared by clonal hematopoiesis and hematologic malignancy, in support of a proposed sequential model of leukemogenesis (Figure 1).43
Figure 1. Sequential model of clonal hematopoiesis.
A sequential model of progression from normal hematopoiesis to clonal hematopoiesis.
CLONAL HEMATOPOEISIS AND RISK OF HEMATOLOGICAL MALIGNANCY
Longitudinal analyses in large numbers of apparently healthy individuals are required to better understand the prevalence and clinical consequences of clonal hematopoiesis. Such analyses became feasible over the last decade through whole exome sequencing.44 To perform exome sequencing for germline variant discovery, DNA is typically extracted from blood peripheral leukocytes. Discovery of somatic mutations in these leukocytes can be performed using separate bioinformatics approaches. Raw sequence data can be processed with conventional germline tools with increased sensitivity parameters, or dedicated somatic mutation detection software, while filtering likely germline calls.45–47 Next, rare disruptive mutations in hematologic malignancy-predisposing genes and mutations recurrently observed in hematological malignancy48 may be annotated. In 2014, three studies ascertained data from thousands of blood DNA exomes to characterize prevalence and malignancy potential (Figure 2).24, 25, 49
Figure 2. Prevalence of clonal hematopoiesis of indeterminate potential by age.
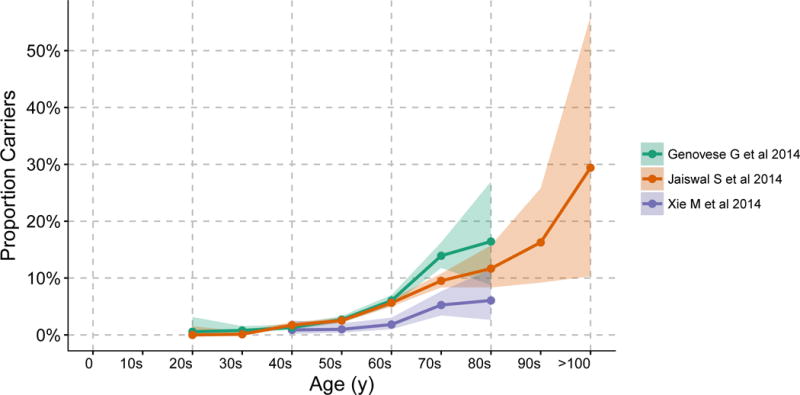
Clonal hematopoiesis of indeterminate potential prevalence by age detected by exome sequencing of blood-derived DNA in three studies of individuals without hematologic malignancy.24, 25, 49 Shaded areas represent 95% confidence intervals. Adapted from Jan M et al.43
Xie M et al analyzed blood-derived exomes from 2,278 participants of The Cancer Genome Atlas (TCGA) with one of eleven non-hematological malignancies and no prior chemotherapy or radiation from age 10 to 90 years.49 Given the unique presence of matched blood DNA and non-blood (i.e., tumor) DNA within the same individual, investigators were able to reliably discriminate acquired mutations to demonstrate hematopoietic clonal mosaicism. 77 blood-specific mutations in 58 individuals were identified. >80% of observed mutations were in 19 genes previously linked to hematologic malignancy, including: DNMT3A, TET2, ASXL1, TP53, SF3B1, BCORL1, ASXL2, and SH2B3. The prevalence of these mutations increased with age: 0.9% for cases in their 40s to 6.1% for cases in their 80s. As studied participants already had cancer, an initial concern was whether these estimates of prevalence were generalizable.
Genovese G et al analyzed blood-derived exomes from 12,380 Swedish individuals, comprising of 4,970 with schizophrenia, 1,165 with bipolar disorder, and 6,245 controls, from age 19 to 93 years.24 Health records were ascertained with 2 to 7 years of follow up data available for 11,164 participants. Genes linked to hematologic malignancy accounted for most of the observed somatic mutations at appreciable allele fractions, particularly DNMT3A, ASXL1, TET2, JAK2, and PPM1D. Observed mutations in these genes were also more likely to be protein-disruptive. Additionally, the pattern of mutations (largely C-to-T) were similar to those observed across diverse cancer types.50 1 in ~150 individuals younger than 50 years and 1 in ~17 older than 65 years had detectable clonal hematopoiesis. Individuals with clonal hematopoiesis had a nearly 13-fold increased risk of hematological malignancy and 1.4-fold increased risk of death.
We analyzed blood-derived exomes from 17,182 individuals from 22 population-based cohorts, which were largely case-control studies for type 2 diabetes (T2D), from age 19 to 108 years.25 805 candidate somatic mutations in 73 genes in 746 individuals were identified with similar increasing prevalence with age, with 1 in 10 older than 70 years having clonal hematopoiesis. The most commonly mutated genes were previously linked to hematologic malignancy, including DNMT3A, TET2, ASXL1, TP53, JAK2, and SF3B1. 93% of individuals with mutations in a hematologic malignancy-associated gene had only one such mutation consistent with the hypothesis of clonal hematopoiesis representing an initiating state. Across a median follow-up of 7.9 years among 3,342 individuals, clonal hematopoiesis was associated with a 11-fold increased risk of hematologic malignancy. A higher variant allele fraction (>0.10) was linked to a much higher risk of hematologic malignancy – nearly 50-fold. Analyses of conventional complete blood count indices showed that clonal hematopoiesis was not associated with cytopenias. Carrier status was also linked to a 1.4-fold increased risk of death among 5,132 individuals with median follow-up of 96 months.
Cancer genes are classically binned as oncogenes and tumor suppressors. The two most frequently mutated genes linked to clonal hematopoiesis, TET2, and DNMT3A, regulate DNA methylation, an epigenetic mark that is thought to influence transcription. The mutations seen in TET2 and DNMT3A are loss-of-function, consistent with these being tumor suppressor genes. The third most commonly mutated gene is ASXL1, which is involved in regulating polycomb mediated transcriptional repression, hence it is also an epigenetic regulator. However, the mutations seen in ASXL1, while truncating, are exclusively in exons 11 and 12, suggesting that they might lead to gain-of-function, or altered function (Table 2).
Table 2.
Genes with most frequent recurrent somatic mutations in blood cells.
| Gene | Function of Gene Product | References |
|---|---|---|
| DNMT3A | DNA methyltransferase performing de novo genome-wide methylation | 116–118 |
| TET2 | Methylcytosine dioxygenase catalyzing conversion of methylcytosine to 5-hydroxymethylcytosine, including downstream DNA demethylation | 26, 51, 69, 119, 120 |
| ASXL1 | Chromatin-binding protein regulating transcription through nuclear hormone receptors, such as retinoic acid receptors and peroxisome proliferator-activated receptor gamma | 121–123 |
| TP53 | Transcription factor regulating cell cycle arrest, apoptosis, senescence, DNA repair, and metabolism changes in response to diverse cellular stresses. | 124–126 |
| JAK2 | Protein tyrosine kinase associated with the prolactin receptor, thrombopoietic receptor, and interferon gamma, involved in cell growth, development, differentiation, and histone modification. | 127–130 |
| SF3B1 | Component of U2 snRNP which binds to pre-mRNA and is involved in RNA splicing regulation. | 131, 132 |
| GNB1 | A G protein beta subunit involved in transmembrane signal transduction and modulation. | 133, 134 |
| CBL | E3 ubiquitin-protein ligase promoting proteosomal degradation. | 135–137 |
| SRSF2 | Interacts with pre-mRNA and spliceosomal components to promote RNA splicing. | 138, 139 |
| GNAS | Stimulatory G-protein alpha subunit involved in transmembrane signal transduction and modulation. | 140 |
| PPM1D | PP2C family member induced by p53 and negatively regulates p38 MAPK. | 141 |
| BCORL1 | Transcriptional corepressor interacting with histone deacetylases to repress transcription. | 142 |
While competitive advantage in stem cells has been observed in Dnmt3a and Tet2 loss-of-function mice, the mechanisms predisposing to this clonal advantage are not fully understood.51, 52 Large-scale changes in epigenetic regulation may enhance self-renewal but may also limit differentiation capabilities given complex coordination required for fate determination.43 Nevertheless, despite similarities with acute myelogenous leukemia, the rate of leukemic transformation remains relatively low. Whether this reflects stochastic variation in epigenetic heterogeneity from initiating mutations or the acquisition of additional cooperating events is not understood.
Additionally, McKerrell et al resequenced 15 mutation hotspots in blood DNA from 4,219 individuals observing a largely linear association of DNMT3A mutations with age but an exponential increase in SF3B1 and SRSF2 mutations only after the age of 70 years.53 These data, suggest that spliceosome defects may preferentially select and expand clones within the aging hematopoietic stem cell niche. Splicing mutations may also yield analogous transcriptional heterogeneity, like broad epigenetic changes from DNMT3A and TET2 mutations, which can promote selection or deficiencies in differentiation.54
CLONAL HEMATOPOESIS OF INDETERMINATE POTENTIAL VERSUS MYELODYSPLASTIC SYNDROME
Clonal hematopoiesis broadly refers to the selective expansion of hematopoietic stem cells with somatic mutations. The term “clonal hematopoiesis of indeterminate potential (CHIP)” has been proposed to define individuals carrying clonal somatic mutations in genes linked to hematologic malignancy with a VAF of at least 2%, but without a known hematologic malignancy or other clonal disorder (Table 3).27, 29 Of note, sensitive deep sequencing strategies increase the apparent prevalence of clonal hematopoiesis with improved detection of somatic mutations in leukemogenic genes at lower VAF thresholds.55–57 Furthermore, using ultra-sensitive methods and a VAF threshold of at least 0.03%, almost all (95%) of healthy 50-60 year-olds are identified as having clonal hematopoiesis.27, 58 Since all hematologic malignancies in our prior study occurred among individuals with VAF > 10%, the presence of clonal hematopoiesis at such very low VAFs are unlikely to carry significant clinical relevance.
Table 3.
Diagnostic features of clonal hematopoiesis of indeterminate potential
| Features |
|---|
| Absence of definitive morphological evidence of a hematological neoplasm |
| Absence of cytopenia or dysplasia |
| Does not meet diagnostic criteria for PNH, MGUS, or MBL |
| Presence of a somatic mutation associated with hematological neoplasia at a variant allele frequency ≥ 2% in peripheral blood |
Of note, myelodysplastic syndrome (MDS) is a well-recognized low-grade clonal neoplasm with an increased risk of leukemic transformation. CHIP largely presents without cytopenia or dysplasia25 which are defining features of MDS. In addition, CHIP often has only 1 driver mutation with a low VAF, as opposed to MDS which may have several driver events at high VAF. As a result, CHIP carries a more favorable prognosis than MDS. The rate of progression to hematologic malignancy is 0.5-1% per year for CHIP24, 25, similar to rates observed for other pre-malignant clonal conditions, such as monoclonal gammopathy of undetermined significance (precursor for multiple myeloma) and monoclonal B-cell lymphocytosis (precursor for chronic lymphocytic leukemia and other B-cell lymphomas).29
CLONAL HEMATOPOESIS AND RISK OF ATHEROSCLEROTIC CARDIOVASCULAR DISEASE
To better understand the observed association of clonal hematopoiesis with all-cause mortality, we performed exploratory analyses testing CHIP status with cause-specific mortality.25 Only one individual with clonal hematopoiesis had died from hematologic malignancy. Cause-specific analyses of 5,132 individuals confirmed a lack of association with cancer death but there was an association with cardiovascular death, particularly for individuals with a variant allele fraction of at least 10% (HR 1.9). Secondary analyses suggested a potential association with coronary heart disease (CHD) and ischemic stroke despite adjusting for traditional cardiovascular risk factors, including age. A prior study showed that clonal large chromosomal rearrangements was associated with micro- and macrovascular complications among those with T2D (carriers 19/26 vs non-carriers 810/2,182).59 It is unclear whether age was accounted for in this secondary analysis and whether there is a true association for atherosclerotic cardiovascular disease (i.e., macrovascular complications). Clonal mosaicism for large chromosomal rearrangements associates with age and increased risk for hematologic malignancy, but more commonly chronic lymphocytic and chronic myeloid leukemias.60, 61 DNMT3A and TET2 mutations have not be typically observed with such large chromosomal rearrangements. Therefore, clonal large chromosomal rearrangements and CHIP may have distinct clinical consequences.
We subsequently tested the hypothesis that CHIP contributes to CHD risk.26 We analyzed blood-derived exome data from four case-control studies together comprising 4,726 with CHD and 3,529 controls. We used a nested case-control approach from two prospective cohort studies: BioImage62, 63, which was enriched for older individuals at higher cardiovascular disease risk, and Malmö Diet and Cancer (MDC)64, which had a prolonged follow-up period. We selected individuals sustaining a first CHD event and controls matched by age, sex, T2D status, and smoking history. We also used data from two retrospective case-control studies: the Atherosclerosis, Thrombosis, and Vascular Biology (ATVB) Italian Study Group65 and Pakistani Risk of Myocardial Infarction Study (PROMIS)66, 67, with cases being individuals with early-onset (<50 years) myocardial infarction.
Similar to prior studies, somatic mutations were more commonly observed in DNMT3A, TET2, ASXL1, and JAK2. Clonal hematopoiesis was associated with a 1.9-fold increased risk of CHD in both BioImage (median age: 70 years, CHIP: 17% cases vs 10% controls) and MDC (median age: 60 years, CHIP: 7% cases vs 4% controls). Surprisingly, an even stronger association for early-onset myocardial infarction was observed, carrying a 4-fold increased risk, across both ATVB (median age: 41 years cases and 40 years controls, CHIP: 2.1% cases vs 0.4% controls) and PROMIS (median age: 45 years cases and 49 years controls, CHIP: 2.0% cases vs 0.9% controls).
Somatic mutations in blood cells might influence risk of atherosclerotic cardiovascular disease in two ways—by increasing the likelihood of thrombosis, or by increasing underlying atherosclerosis. We hypothesized that individuals with clonal hematopoiesis also harbored a greater burden of subclinical coronary atherosclerosis prior to clinical events. We examined the relationship between clonal hematopoiesis and subclinical coronary atherosclerosis in BioImage through measures of coronary artery calcification (CAC) measured by cardiac computed tomography.26 Among controls, clonal hematopoiesis status was associated a 3.0-fold greater likelihood of having a CAC score greater than 615 Agatston units, a value described to indicate high CHD risk among older adults.26, 68 Similar to our observation that larger clones (variant allele fraction ≥ 10%) were more strongly associated with hematologic malignancy25, carrying a larger clone was associated with a 12-fold greater likelihood of having a CAC score greater than 615 Agatston units. Concordantly, our data suggested that the presence of a larger clone was associated with a 2.2-fold greater risk of CHD compared to 1.4-fold greater risk for carriers of smaller clones.
BY WHAT MECHANISMS MIGHT CLONAL HEMATOPOIESIS LEAD TO ATHEROSCLEROSIS?
Despite independent associations of clonal hematopoiesis with clinical and subclinical atherosclerosis in statistical models accounting for age, confounding remains a potential concern.17 Causal inference from biomarker-disease correlation in observational studies is challenging.
In murine models, two groups have tested if and how clonal hematopoiesis leads to atherosclerosis (Figure 3).26, 69 Tet2 is the second most commonly mutated gene observed in human clonal hematopoiesis and Tet2 ablation in murine hematopoietic cells led to clonal selection and expansion.70 We generatedTet2−/− mice and their bone marrow (vs control bone marrow) was engrafted into irradiated, atherosclerosis-prone Ldlr−/− mice. Compared with mice transplanted with wild-type bone marrow, chimeric mice with Tet2 deficient bone marrow had nearly 3-fold larger descending aorta atherosclerotic plaques at 17 weeks despite similar blood counts and similar serum cholesterol concentrations. The effect size was nearly identical for Ldlr−/−for bone marrow from heterozygous Tet2-deficient (Tet2+/−) mice.
Figure 3. Inflammatory mediators of clonal hematopoiesis influencing atherosclerosis.
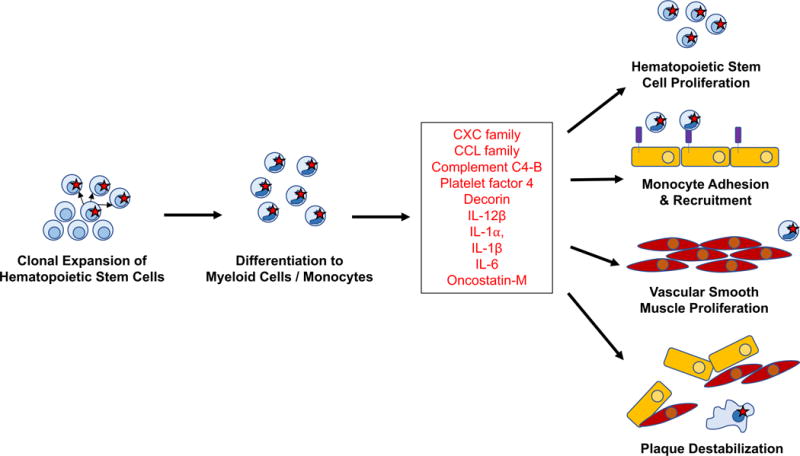
Summary of inflammatory factors observed to be upregulated in Jaiswal et al26 and Fuster et al69 and proposed influences on atherosclerosis plaque formation and destabilization.
Fuster et al also evaluated Tet2 deficiency in hematopoietic cells in mice and its effect on atherosclerosis.69 Fuster et al created Ldlr−/− mice engrafted with 10% Tet2−/− bone marrow (tagged by CD45.2) and 90% Tet2+/+ bone marrow (tagged by CD45.1) to mimic observed circulating variant allele fractions in humans; control bone marrow consisted of Tet2+/+ labeled 10% CD45.2 and 90% CD45.1. At 13 weeks after engraftment, despite having a similar total number of hematopoietic stem cells, 69% of hematopoietic stem cells in the bone marrow were CD45.2 cells in those receiving 10% Tet2−/− bone marrow. A slight predisposition toward myeloid differentiation was observed. Despite similar weight, insulin sensitivity, and plasma cholesterol levels, Ldlr−/− mice engrafted with 10% Tet2−/−/90% Tet2+/+ bone marrow had larger aortic root plaques with a proportional increase in macrophage content when compared with Ldlr−/− mice with 100% Tet2+/+ bone marrow. Furthermore, engraftment 10% Tet2−/+/90% Tet2+/+ bone marrow to Ldlr−/− mice similarly promoted atherosclerosis compared to Ldlr−/− mice with 100% Tet2+/+ bone marrow. These data demonstrate independent consistent observations between the two groups.
We tested the hypothesis that Tet2 disruption was particularly influencing myeloid cells. Ldlr−/− mice were engrafted with bone marrow from mice where Tet2 had been selectively disrupted in myeloid cells; compared with mice transplanted with wild-type bone marrow, these mice had larger aortic root atheroma at 10 weeks. Fuster et al observed that CD45.2 cells preferentially differentiated to macrophages within atherosclerotic vascular walls. The authors similarly hypothesized a central role of clonally expanded macrophages in atherosclerosis. They also generated Ldlr−/− mice selectively deficient of Tet2 in myeloid cells and reported similar results as us.
Notably, current murine bone marrow transplant models of CHIP may not faithfully replicate the kinetics clonal expansion in human CHIP. Of note, we observed among 13 individuals (17 mutations in total) with CHIP at baseline and blood-cell DNA collection 4-8 years later, 10 VAFs did not increase and 7 did increase and none developed hematologic cancer over this time frame.25 Additionally, Young et al observed a similar pattern of VAF stability in 20 individuals over ~10 years.58
In our study, Tet2−/− bone marrow-derived macrophages in vitro were exposed to native low-density lipoprotein (LDL), resulting in increased expression of several cytokines and chemokines, including Cxcl1, Cxcl2, Cxcl3, Pf4, Il1b, and Il6, compared to control macrophages. This was consistent with prior observations linking Tet2 deficient bone marrow-derived macrophages with increased inflammatory gene expression.71, 72 Notably, CXC chemokines were also elevated in serum from Ldlr−/− mice transplated with Tet2-deficient bone marrow. In human plasma, IL8, a CXC chemokine analogue that mice lack, was approximately double among those with clonal hematopoiesis compared to those without.
In complementary studies, Fuster et al exposed Tet2−/− peritoneal macrophages to lipopolysaccharide and interferon-γ in vitro and showed higher gene expression for several cytokines and chemokines, including Ccl3, Ccl4, Cxcl1, Cxcl2, Cxcl3, Cxcl5, Cxcl13, C4b, Dcn, Il12b, Il1a, Il1b, Il6, and Osm. Elevated IL-6 levels were also detected in culture supernatant. Il1b upregulation was particularly notable when exposed to a combination of oxidized LDL, tumor necrosis factor, and interferon-γ. Concordantly, unlike other chemokines, Il1b gene expression and IL-1β concentration was increased in the aortic arches of Ldlr−/− mice engrafted with 10% Tet2−/−/90% Tet2+/+ bone marrow when compared with Ldlr−/− mice with 100% Tet2+/+ bone marrow. Further, stimulation of Tet2−/− macrophages in vitro led to increased IL-1β which was mitigated by a NLRP3 inhibitor indicating that Tet2 deficiency promotes NLRP3 inflammasome-mediated IL-1β secretion. Similarly, aortic plaque size in chimeric Ldlr−/− mice with 10% Tet2−/− bone marrow treated with a NLRP3 inflammasome inhibitor was smaller compared to Ldlr−/− mice with 10% Tet2+/+ bone marrow.
Both studies strikingly converge on the role of specific chemokines, particularly CXC family chemokines, IL-6, and IL-1β, in promoting atherosclerosis in the setting of clonal hematopoiesis. CXC chemokines, such as IL-8, and their receptors have previously been shown to promote the adhesion of monocytes to vascular endothelium.73–75 Furthermore, macrophages in atheroma produce >8-fold greater IL-8 compared to circulating monocytes, with even greater production when exposed to oxidized LDL.76, 77 In the setting of oxidized LDL, IL-8 inhibits the production of specific tissue inhibitors of matrix metalloproteinases thereby contributing to local plaque instability. Additionally, CXC chemokines can recruit neutrophils, which can attach to atherosclerotic plaques via neutrophil extracellular traps (NETs) and separately promote atherothrombosis.78–83
In an exome-wide association analysis across >300,000 individuals, JAK2 p.V617F carriage detected by exome genotyping array was associated with lower (-0.32 total cholesterol and -0.30 LDL cholesterol).84 Furthermore, Ldlr−/− mice engrafted with Jak2 p.V617F bone marrow had lower total cholesterol compared to Ldlr−/− mice engrafted with wild-type bone marrow.84 The apparent discordance between low LDL cholesterol and elevated CHD risk conferred by JAK2 p.V617F requires further study. Notably, across CHIP broadly (which more frequently involves mutations in DNMT3A and TET2), there is no significant association with blood lipids.26
GERMLINE GENETIC PREDISPOSITION TO CLONAL HEMATOPOIESIS
Prior efforts have evaluated the germline genetic predisposition to the occurrence of JAK2 p.V617F at relatively small scale.85–90 JAK2 p.V617F is the most frequently observed acquired mutation recurrently identified in myeloproliferative neoplasms (MPN)91, and is observed in CHIP. Kilpivaara O et al showed that, of 321 MPN cases compared to 3,000 convenience controls, germline rs10974944-G (MAF 25%) in a JAK2 intron tagging the JAK2 46/1 haplotype was strongly associated with JAK2V617F-positive MPN (OR = 4.0, p-value = 7.7×10−22) and not JAK2V617F-negative MPN (OR = 1.6, p-value = 0.06).88 The JAK2 46/1 haplotype is estimated to account for 28% of the population attributable risk of MPN among Europeans.92
More recently, Hinds D et al performed a genome-wide association analysis for both JAK2V617F-positive MPN and JAK2V617F-positive clonal hematopoiesis among 252,637 individuals ascertained from 23andme and 726 individuals with myeloproliferative neoplasms.93 This specific mutation was chosen as it is a somatic mutation known to cause clonal hematopoiesis, occurs at appreciable frequency, and is present on most contemporary genome-wide genotyping arrays. Significant associations were observed at loci near the genes JAK2 (including variants tagging the 46/1 haplotype), TERT, SH2B3, TET2, CHEK2, ATM/PDGFD, PINT, and GFI1B. The TERT association appears to be a more general effect on all MPN, regardless of JAK2 p.V617F mutation status.86, 94
Notably, the SH2B3 and ATM/PDGFD loci were previously significantly associated with CHD in genome-wide association studies.95 SH2B3, also known as LNK, is expressed in hematopoietic cells and is a negative regulator of cellular proliferation, including negatively regulating JAK2 in stem cells.96 Rare disruptive mutations in SH2B3 predispose to MPN in humans, and targeted deletion in mice promotes multilineage expansion of hematopoietic stem cells.96–98 Wang W et al showed that both Lnk−/− mice and Ldlr−/− mice engrafted with Lnk−/− bone marrow showed myelopoiesis, this was more prominent in the latter model.98 They further observe a synergestic activation of platelets from Lnk deficiency and cholesterol loading.
While JAK2 p.V617F is observed in the setting of CHIP, mutations in other genes (i.e., DNMT3A, TET2, ASXL1, et al) are more frequently observed in CHIP. Using targeted deep sequencing (>4,000-fold mean coverage), Buscarlet M et al observed leukemogenic mutations (93% in DNMT3A and TET2) in 13.7% of 2,530 apparently healthy adults.56 Of 391 sibships within this cohort, recurrence risk of TET2 mutations between siblings was ~2.5-fold and there was no recurrence risk observed for DNMT3A mutations between siblings. Additional work is required to understand the heritable basis of such mutations more frequently observed in CHIP.
CLONAL HEMATOPOIESIS WITH UNKNOWN DRIVER MUTATIONS
A hallmark of CHIP is the presence of initiating driver mutations.29 However, Genovese et al observed the phenomenon of clonal hematopoiesis (detected by at least three putative somatic mutations at appreciable allele frequency) with unknown drivers (indicated by the lack of an initiating driver mutation in a previously implicated gene).24 Prevalence also increases with age, with 0.9% carriers among individuals younger than 50 years and 10.4% in individuals older than 65 years. Among the 439 individuals with clonal hematopoiesis, 170 (39%) did not have a known driver mutation. The risk of hematologic malignancy was similar among individuals with clonal hematopoiesis with known drivers (HR 13.73) vs unknown drivers (HR 12.89).
Detection of somatic mutations outside of known driver genes may be enhanced via deep-coverage whole genome sequencing of blood-derived DNA given ability to detect mutations across the genome. This was recently assessed in 11,262 Icelanders participating in deCODE Genetics undergoing whole genome sequencing to average genome-wide coverage 36X.99 Here, clonal hematopoiesis was defined as the presence of more than 20 singleton somatic mutations with variant allele fractions ranging 0.10-0.20 (99.5th percentile for subjects younger than 35 years). While 0.5% of participants younger than 35 years had clonal hematopoiesis, more than 50% older than 85 years had clonal hematopoiesis, approximately twice the prior yield from exome sequence analysis.24, 25 Additionally, only 1 in 8 individuals (177/1,403) with clonal hematopoiesis had a candidate driver mutation in at least 1 of 18 previously implicated genes. Deep resequencing at 54 myeloid malignancy-associated genes of a subset to variant allele fraction >0.01, identified clonal driver mutations in 40.7% still demonstrating that the majority have no detectable candidate driver mutation. However, the clinical relevance of clones with driver mutations at lower variant allele fractions (<0.10) appear to be less significant.25 As before, risk of hematologic malignancy was similar for clonal hematopoiesis with or without a candidate driver mutation.
FUTURE RESEARCH DIRECTIONS
Given that the association of clonal hematopoiesis with ASCVD has just recently been discovered, many outstanding questions exist. Some of these research questions are discussed below.
First, is the relationship between clonal hematopoiesis and atherosclerosis homogenous across implicated genes? Most known driver mutations occur in DNMT3A and TET2. In current sample sizes, effect estimates appear different for early-onset myocardial infarction – 1.4 increased odds for DNMT3A mutations and 8.3 for TET2 mutations.26 Nevertheless, risk for CHD appears similar for older individuals is similar – 1.7-fold for DNMT3A mutations and 1.9-fold for TET2 mutations. To better understand risks conferred from individual genes, larger sample sizes are required.
Second, why do some individuals develop clonal hematopoiesis without known driver mutations and do they also have elevated risk of ASCVD? Recent whole genome sequence analyses suggest clonal hematopoiesis without known driver mutations is highly prevalent in older adults.99 However, whether this phenomenon also increases risk for ASCVD is currently unknown.
Third, what are the determinants of clinical consequences among individuals with clonal hematopoiesis? Most individuals with clonal hematopoiesis followed in recent studies do not develop hematologic malignancy or atherosclerotic cardiovascular disease.24–26 Some differences may be explained by different genes. However, disruption of many such genes likely result in broad, varied epigenetic changes so specific epigenetic disruption patterns may yield differential clinical consequences. Transcriptional and epigenetic profiling in humans and model systems may provide new insights. Furthermore, smoking and diabetes have been associated with clonal hematopoiesis; the presence of such risk factors and others may influence risk of ASCVD in additive or multiplicative manners. Since CHIP promotes atherosclerosis in humans and hypercholesterolemia-prone mice, alteration of cholesterol-related pathways within hematopoietic stem cells related to dyslipidemia may differentially influence ASCVD in the setting of CHIP.
Fourth, can interventions reduce the risk for ASCVD associated with CHIP? Induction of the NLRP3 inflammasome is required for proteolytic cleavage to activate IL-1β.100 Individuals with rare Mendelian autoinflammatory conditions due to mutations activating the NLRP3 inflammasome suffer from symptoms related to overproduction of IL-1β. Cholesterol crystals can stimulate the NLRP3 inflammasome within atheromata yielding IL-1β production.101, 102 IL-1β has been shown to promote hematopoietic stem cell proliferation as well as induce endothelial cell expression of monocyte adhesion and recruitment factors.103–105 IL-1 also promotes proliferation of vascular smooth muscle cells.106, 107 IL-1 in turn further promotes the secretion of IL-6 from both endothelial cells and vascular smooth muscle cells.108, 109 Mendelian randomization studies have causally associated the IL-6 receptor with CHD.110, 111 IL-6 stimulates the production of C-reactive protein, a circulating biomarker associated with incident CHD risk.112
Recently, the Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) trial tested the efficacy and safety of IL-1β inhibition with canakinumab to prevent recurrent atherosclerotic cardiovascular disease events among individuals with elevated high-sensitivity C-reactive protein (hsCRP) levels (> 2 mg/L).113 Individuals receiving canakinumab 150 mg every three months had a 15% reduction in recurrent events at 4 years. Individuals who attained hsCRP < 2 mg/L on treatment appeared to have a greater efficacy (HR 0.75) versus those with persistent hsCRP > 2 mg/L (HR 0.90).114 A surprising secondary observation was that individuals treated with canakinumab had a lower risk of developing lung cancer (HR 0.61).115 Given that loss of Tet2 leads to increased Il1b expression in mice and that atherosclerosis can be reduced in these mice with a NLRP3 inflammasome inhibitor,69 it is possible that downstream blockade of IL-1β will reduce the risk of atherosclerotic events among humans with TET2-mutated clonal hematopoiesis. Additionally, current laboratory testing does not capture individuals with clonal hematopoiesis well. Analyses of complete blood counts, suggests that red cell distribution width (RDW) is associated but this is not adequately sensitive or specific.25 It is not known whether clonal hematopoiesis is associated with high sensitivity C-reactive protein.
CONCLUSIONS
Clonal hematopoiesis represents a new risk factor for ASCVD with an effect size estimate at least as large as conventional risk factors. Efforts to better characterize fundamental pathophysiology, identify determinants of initiation and progression, further improve diagnostic yield, and develop novel preventive and therapeutic strategies may have significant public health benefit.
Supplementary Material
Footnotes
Disclosure: None
References
- 1.Benjamin EJ, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collaborators GBDCoD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151–1210. doi: 10.1016/S0140-6736(17)32152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabatine MS, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. The New England journal of medicine. 2017 doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 4.Karmali KN, et al. A systematic examination of the 2013 ACC/AHA pooled cohort risk assessment tool for atherosclerotic cardiovascular disease. Journal of the American College of Cardiology. 2014;64:959–968. doi: 10.1016/j.jacc.2014.06.1186. [DOI] [PubMed] [Google Scholar]
- 5.Wang JC, et al. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 6.Jousilahti P, et al. Sex, age, cardiovascular risk factors, and coronary heart disease: a prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation. 1999;99:1165–1172. doi: 10.1161/01.cir.99.9.1165. [DOI] [PubMed] [Google Scholar]
- 7.Castelli WP. Epidemiology of coronary heart disease: the Framingham study. Am J Med. 1984;76:4–12. doi: 10.1016/0002-9343(84)90952-5. [DOI] [PubMed] [Google Scholar]
- 8.Tunstall-Pedoe H, et al. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90:583–612. doi: 10.1161/01.cir.90.1.583. [DOI] [PubMed] [Google Scholar]
- 9.Dunn J, et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. The Journal of clinical investigation. 2014;124:3187–3199. doi: 10.1172/JCI74792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauscher FM, et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108:457–463. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- 11.Karra R, et al. Molecular evidence for arterial repair in atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16789–16794. doi: 10.1073/pnas.0507718102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olive M, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ying AK, et al. Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc Res. 2000;46:172–179. doi: 10.1016/s0008-6363(00)00004-3. [DOI] [PubMed] [Google Scholar]
- 14.Childs BG, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (New York, NY) 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edo MD, et al. Aging, telomeres, and atherosclerosis. Cardiovasc Res. 2005;66:213–221. doi: 10.1016/j.cardiores.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 16.Finkel T, et al. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 17.Libby P, et al. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 18.Swirski FK, et al. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (New York, NY) 2013;339:161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woollard KJ, et al. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerrity RG. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol. 1981;103:181–190. [PMC free article] [PubMed] [Google Scholar]
- 21.Gerrity RG. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am J Pathol. 1981;103:191–200. [PMC free article] [PubMed] [Google Scholar]
- 22.Gerrity RG, et al. Ultrastructural identification of monocyte-derived foam cells in fatty streak lesions. Artery. 1980;8:208–214. [PubMed] [Google Scholar]
- 23.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England journal of medicine. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaiswal S, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. The New England journal of medicine. 2017 doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowman RL, et al. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell. 2018;22:157–170. doi: 10.1016/j.stem.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuster JJ, et al. Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circ Res. 2018;122:523–532. doi: 10.1161/CIRCRESAHA.117.312115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steensma DP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tall AR, et al. Cardiovascular disease: Commonality with cancer. Nature. 2017;543:45–47. doi: 10.1038/nature21505. [DOI] [PubMed] [Google Scholar]
- 31.Keaney JF., Jr CHIP-ping Away at Atherosclerosis. The New England journal of medicine. 2017;377:184–185. doi: 10.1056/NEJMe1706173. [DOI] [PubMed] [Google Scholar]
- 32.Elias HK, et al. Molecular mechanisms underlying lineage bias in aging hematopoiesis. Semin Hematol. 2017;54:4–11. doi: 10.1053/j.seminhematol.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohrin M, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flach J, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512:198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner W, et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PloS one. 2009;4:e5846. doi: 10.1371/journal.pone.0005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busque L, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 38.Gale RE, et al. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. Br J Haematol. 1997;98:512–519. doi: 10.1046/j.1365-2141.1997.2573078.x. [DOI] [PubMed] [Google Scholar]
- 39.Fey MF, et al. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood. 1994;83:931–938. [PubMed] [Google Scholar]
- 40.Busque L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jan M, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science translational medicine. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shlush LI, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jan M, et al. Clonal hematopoiesis. Semin Hematol. 2017;54:43–50. doi: 10.1053/j.seminhematol.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11 10 11–11 10 33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costello M, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic acids research. 2013;41:e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forbes SA, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic acids research. 2017;45:D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Challen GA, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKerrell T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10:1239–1245. doi: 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landau DA, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26:813–825. doi: 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arends CM, et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia. 2018 doi: 10.1038/s41375-018-0047-7. [DOI] [PubMed] [Google Scholar]
- 56.Buscarlet M, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130:753–762. doi: 10.1182/blood-2017-04-777029. [DOI] [PubMed] [Google Scholar]
- 57.Acuna-Hidalgo R, et al. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. American journal of human genetics. 2017;101:50–64. doi: 10.1016/j.ajhg.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young AL, et al. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nature communications. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonnefond A, et al. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat Genet. 2013;45:1040–1043. doi: 10.1038/ng.2700. [DOI] [PubMed] [Google Scholar]
- 60.Jacobs KB, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laurie CC, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baber U, et al. Prevalence, Impact, and Predictive Value of Detecting Subclinical Coronary and Carotid Atherosclerosis in Asymptomatic Adults: The BioImage Study. Journal of the American College of Cardiology. 2015;65:1065–1074. doi: 10.1016/j.jacc.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 63.Muntendam P, et al. The BioImage Study: novel approaches to risk assessment in the primary prevention of atherosclerotic cardiovascular disease–study design and objectives. American heart journal. 2010;160:49–57 e41. doi: 10.1016/j.ahj.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 64.Berglund G, et al. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. doi: 10.1111/j.1365-2796.1993.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 65.Atherosclerosis T, et al. No evidence of association between prothrombotic gene polymorphisms and the development of acute myocardial infarction at a young age. Circulation. 2003;107:1117–1122. doi: 10.1161/01.cir.0000051465.94572.d0. [DOI] [PubMed] [Google Scholar]
- 66.Saleheen D, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–239. doi: 10.1038/nature22034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saleheen D, et al. The Pakistan Risk of Myocardial Infarction Study: a resource for the study of genetic, lifestyle and other determinants of myocardial infarction in South Asia. European journal of epidemiology. 2009;24:329–338. doi: 10.1007/s10654-009-9334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elias-Smale SE, et al. Coronary calcium score improves classification of coronary heart disease risk in the elderly: the Rotterdam study. Journal of the American College of Cardiology. 2010;56:1407–1414. doi: 10.1016/j.jacc.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 69.Fuster JJ, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, NY) 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cull AH, et al. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70 e13. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boisvert WA, et al. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. The Journal of clinical investigation. 1998;101:353–363. doi: 10.1172/JCI1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gerszten RE, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 75.Schwartz D, et al. Role of the GRO family of chemokines in monocyte adhesion to MM-LDL-stimulated endothelium. The Journal of clinical investigation. 1994;94:1968–1973. doi: 10.1172/JCI117548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Apostolopoulos J, et al. Interleukin-8 production by macrophages from atheromatous plaques. Arteriosclerosis, thrombosis, and vascular biology. 1996;16:1007–1012. doi: 10.1161/01.atv.16.8.1007. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y, et al. Macrophages isolated from human atherosclerotic plaques produce IL-8, and oxysterols may have a regulatory function for IL-8 production. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:317–323. doi: 10.1161/01.atv.17.2.317. [DOI] [PubMed] [Google Scholar]
- 78.Warnatsch A, et al. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (New York, NY) 2015;349:316–320. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eash KJ, et al. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. The Journal of clinical investigation. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suratt BT, et al. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. 2004;104:565–571. doi: 10.1182/blood-2003-10-3638. [DOI] [PubMed] [Google Scholar]
- 81.Doring Y, et al. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ Res. 2012;110:1052–1056. doi: 10.1161/CIRCRESAHA.112.265868. [DOI] [PubMed] [Google Scholar]
- 82.Doring Y, et al. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res. 2017;120:736–743. doi: 10.1161/CIRCRESAHA.116.309692. [DOI] [PubMed] [Google Scholar]
- 83.Wantha S, et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res. 2013;112:792–801. doi: 10.1161/CIRCRESAHA.112.300666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu DJ, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. 2017;49:1758–1766. doi: 10.1038/ng.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tapper W, et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nature communications. 2015;6:6691. doi: 10.1038/ncomms7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trifa AP, et al. TERT rs2736100 A>C SNP and JAK2 46/1 haplotype significantly contribute to the occurrence of JAK2 V617F and CALR mutated myeloproliferative neoplasms - a multicentric study on 529 patients. Br J Haematol. 2016;174:218–226. doi: 10.1111/bjh.14041. [DOI] [PubMed] [Google Scholar]
- 87.Trifa AP, et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am J Hematol. 2018;93:100–106. doi: 10.1002/ajh.24946. [DOI] [PubMed] [Google Scholar]
- 88.Kilpivaara O, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41:455–459. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koh SP, et al. Genetic association between germline JAK2 polymorphisms and myeloproliferative neoplasms in Hong Kong Chinese population: a case-control study. BMC Genet. 2014;15:147. doi: 10.1186/s12863-014-0147-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pardanani A, et al. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood. 2008;111:2785–2789. doi: 10.1182/blood-2007-06-095703. [DOI] [PubMed] [Google Scholar]
- 91.Levine RL, et al. X-inactivation-based clonality analysis and quantitative JAK2V617F assessment reveal a strong association between clonality and JAK2V617F in PV but not ET/MMM, and identifies a subset of JAK2V617F-negative ET and MMM patients with clonal hematopoiesis. Blood. 2006;107:4139–4141. doi: 10.1182/blood-2005-09-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jones AV, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41:446–449. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hinds DA, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. 2016;128:1121–1128. doi: 10.1182/blood-2015-06-652941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oddsson A, et al. The germline sequence variant rs2736100_C in TERT associates with myeloproliferative neoplasms. Leukemia. 2014;28:1371–1374. doi: 10.1038/leu.2014.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.CARDIoGRAMplusC4D. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bersenev A, et al. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. The Journal of clinical investigation. 2008;118:2832–2844. doi: 10.1172/JCI35808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McMullin MF, et al. A nonsynonymous LNK polymorphism associated with idiopathic erythrocytosis. Am J Hematol. 2011;86:962–964. doi: 10.1002/ajh.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang W, et al. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ Res. 2016;119:e91–e103. doi: 10.1161/CIRCRESAHA.116.308955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zink F, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–752. doi: 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Latz E, et al. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Paramel Varghese G, et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sager HB, et al. Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation. 2015;132:1880–1890. doi: 10.1161/CIRCULATIONAHA.115.016160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shemesh S, et al. Interleukin-1 receptor type-1 in non-hematopoietic cells is the target for the pro-atherogenic effects of interleukin-1 in apoE-deficient mice. Atherosclerosis. 2012;222:329–336. doi: 10.1016/j.atherosclerosis.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 105.Kirii H, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 106.Libby P, et al. Interleukin 1: a mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. The Journal of clinical investigation. 1988;81:487–498. doi: 10.1172/JCI113346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alexander MR, et al. Interleukin-1beta modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-kappaB-dependent mechanisms. Physiol Genomics. 2012;44:417–429. doi: 10.1152/physiolgenomics.00160.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Loppnow H, et al. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. The Journal of clinical investigation. 1990;85:731–738. doi: 10.1172/JCI114498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sironi M, et al. IL-1 stimulates IL-6 production in endothelial cells. J Immunol. 1989;142:549–553. [PubMed] [Google Scholar]
- 110.Interleukin-6 Receptor Mendelian Randomisation Analysis C et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379:1214–1224. doi: 10.1016/S0140-6736(12)60110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Collaboration IRGCERF et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379:1205–1213. doi: 10.1016/S0140-6736(11)61931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ridker PM. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res. 2016;118:145–156. doi: 10.1161/CIRCRESAHA.115.306656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ridker PM, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 114.Ridker PM, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2017 doi: 10.1016/S0140-6736(17)32814-3. [DOI] [PubMed] [Google Scholar]
- 115.Ridker PM, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 116.Kaneda M, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 117.Okano M, et al. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 118.Yang L, et al. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152–165. doi: 10.1038/nrc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Delhommeau F, et al. Mutation in TET2 in myeloid cancers. The New England journal of medicine. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 120.Langemeijer SM, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 121.Boultwood J, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24:1062–1065. doi: 10.1038/leu.2010.20. [DOI] [PubMed] [Google Scholar]
- 122.Cho YS, et al. Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. The Journal of biological chemistry. 2006;281:17588–17598. doi: 10.1074/jbc.M512616200. [DOI] [PubMed] [Google Scholar]
- 123.Fisher CL, et al. A human homolog of Additional sex combs, ADDITIONAL SEX COMBS-LIKE 1, maps to chromosome 20q11. Gene. 2003;306:115–126. doi: 10.1016/s0378-1119(03)00430-x. [DOI] [PubMed] [Google Scholar]
- 124.Kern SE, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science (New York, NY) 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 125.Matlashewski G, et al. Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene. EMBO J. 1984;3:3257–3262. doi: 10.1002/j.1460-2075.1984.tb02287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McBride OW, et al. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13) Proceedings of the National Academy of Sciences of the United States of America. 1986;83:130–134. doi: 10.1073/pnas.83.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dawson MA, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sakatsume M, et al. The Jak kinases differentially associate with the alpha and beta (accessory factor) chains of the interferon gamma receptor to form a functional receptor unit capable of activating STAT transcription factors. The Journal of biological chemistry. 1995;270:17528–17534. doi: 10.1074/jbc.270.29.17528. [DOI] [PubMed] [Google Scholar]
- 129.Saltzman A, et al. Cloning and characterization of human Jak-2 kinase: high mRNA expression in immune cells and muscle tissue. Biochemical and biophysical research communications. 1998;246:627–633. doi: 10.1006/bbrc.1998.8685. [DOI] [PubMed] [Google Scholar]
- 130.Jakel H, et al. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene. 2011;30:3502–3512. doi: 10.1038/onc.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Golas MM, et al. Molecular architecture of the multiprotein splicing factor SF3b. Science (New York, NY) 2003;300:980–984. doi: 10.1126/science.1084155. [DOI] [PubMed] [Google Scholar]
- 132.Tanuma N, et al. Nuclear inhibitor of protein phosphatase-1 (NIPP1) directs protein phosphatase-1 (PP1) to dephosphorylate the U2 small nuclear ribonucleoprotein particle (snRNP) component, spliceosome-associated protein 155 (Sap155) The Journal of biological chemistry. 2008;283:35805–35814. doi: 10.1074/jbc.M805468200. [DOI] [PubMed] [Google Scholar]
- 133.Ray K, et al. Isolation of cDNA clones encoding eight different human G protein gamma subunits, including three novel forms designated the gamma 4, gamma 10, and gamma 11 subunits. The Journal of biological chemistry. 1995;270:21765–21771. doi: 10.1074/jbc.270.37.21765. [DOI] [PubMed] [Google Scholar]
- 134.Yan K, et al. Differential ability to form the G protein betagamma complex among members of the beta and gamma subunit families. The Journal of biological chemistry. 1996;271:7141–7146. doi: 10.1074/jbc.271.12.7141. [DOI] [PubMed] [Google Scholar]
- 135.Kirsch KH, et al. The adapter type protein CMS/CD2AP binds to the proto-oncogenic protein c-Cbl through a tyrosine phosphorylation-regulated Src homology 3 domain interaction. The Journal of biological chemistry. 2001;276:4957–4963. doi: 10.1074/jbc.M005784200. [DOI] [PubMed] [Google Scholar]
- 136.Petrelli A, et al. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. 2002;416:187–190. doi: 10.1038/416187a. [DOI] [PubMed] [Google Scholar]
- 137.Schmidt MH, et al. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005;6:907–918. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 138.Wu JY, et al. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 139.Zahler AM, et al. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 140.Abramowitz J, et al. XLalphas, the extra-long form of the alpha-subunit of the Gs G protein, is significantly longer than suspected, and so is its companion Alex. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8366–8371. doi: 10.1073/pnas.0308758101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fiscella M, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6048–6053. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pagan JK, et al. A novel corepressor, BCoR-L1, represses transcription through an interaction with CtBP. The Journal of biological chemistry. 2007;282:15248–15257. doi: 10.1074/jbc.M700246200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.