

Abstract
Impaired bioenergetics and oxidative damage in the mitochondria are implicated in the etiology of temporal lobe epilepsy, and hyperacetylation of mitochondrial proteins has recently emerged as a critical negative regulator of mitochondrial functions. However, the roles of mitochondrial acetylation and activity of the primary mitochondrial deacetylase, SIRT3, have not been explored in acquired epilepsy. We investigated changes in mitochondrial acetylation and SIRT3 activity in the development of chronic epilepsy in the kainic acid rat model of TLE. Hippocampal measurements were made at 48 hours, 1 week and 12 weeks corresponding to the acute, latent and chronic stages of epileptogenesis. Assessment of hippocampal bioenergetics demonstrated a ≥ 27% decrease in the ATP/ADP ratio at all phases of epileptogenesis (p < 0.05), whereas cellular NAD+ levels were decreased by ≥ 41% in the acute and latent time points (p < 0.05), but not in chronically epileptic rats. In spontaneously epileptic rats, we found decreased protein expression of SIRT3 and a 60% increase in global mitochondrial acetylation, as well as enhanced acetylation of the known SIRT3 substrates MnSOD, Ndufa9 of Complex I and IDH2 (all p < 0.05), suggesting SIRT3 dysfunction in chronic epilepsy. Mass spectrometry-based acetylomics investigation of hippocampal mitochondria demonstrated a 79% increase in unique acetylated proteins from rats in the chronic phase vs. controls. Pathway analysis identified numerous mitochondrial bioenergetic pathways affected by mitochondrial acetylation. These results suggest SIRT3 dysfunction and aberrant protein acetylation may contribute to mitochondrial dysfunction in chronic epilepsy.
Keywords: mitochondria, acetylation, SIRT3, epilepsy, mass spectrometry, proteomics
Graphical abstract
Introduction
Epilepsy is one of the most common neurological disorders with a lifetime occurrence of about 3% in the general population (Hildebrand et al., 2013). Temporal lobe epilepsy (TLE), the most common acquired epilepsy, is typically preceded by an inciting injury resulting in the transformation of a normal brain circuit to a hyperexcitable one by a process known as epileptogenesis. Metabolic and bioenergetic dysfunction, particularly in the mitochondria, is known to occur with the initial injury and subsequent epileptogenesis, and may contribute to the development of spontaneous seizures (Kovac et al., 2013; Rowley and Patel, 2013).
Impairments in mitochondrial function, such as decreased activities of TCA cycle and electron transport chain (ETC) enzymes, have been found in hippocampi of epileptic patients (Kunz et al., 2000; Vielhaber et al., 2008), and we recently demonstrated reductions in the mitochondrial respiratory and reserve capacities and inhibition of Complex I of the electron transport chain (ETC) in our rat kainic acid (KA) model of chemoconvulsant-induced epilepsy (Rowley et al., 2015; Ryan et al., 2012). Biomarkers of oxidative damage have been found in brain and plasma from patients with epilepsy (Mueller et al., 2001; Sudha et al., 2001), and our lab has demonstrated increased steady-state levels of mitochondrial ROS (Jarrett et al., 2008; Liang et al., 2000) and markers of ROS-mediated damage (Liang and Patel, 2006; Ryan et al., 2012; Ryan et al., 2014) in rodent hippocampi in our rat KA model. However, an underlying mechanism mediating these mitochondrial alterations in bioenergetics and ROS production with epilepsy are unknown.
Acetylation of lysine residues of proteins has recently emerged as a highly conserved post-translational modification (PTM) that regulates the activities of numerous metabolic enzymes within the mitochondria, and therefore may control bioenergetic homeostasis in this organelle (Duan, 2013; Weir et al., 2013). Mitochondrial acetylation is generally considered an inhibitory PTM and acetylation of specific lysine(s) is known to reduce activities of multiple enzymes in the ETC, TCA cycle, fatty acid oxidation (FAO), and ketogenesis, as well as those involved in antioxidant defenses (Ahn et al., 2008; Hebert et al., 2013; Hirschey et al., 2010; Qiu et al., 2010; Shimazu et al., 2010; Someya et al., 2010; Wagner and Payne, 2011; Yu et al., 2012). Accordingly, elevations in mitochondrial acetylation have been found in a variety of tissues in diverse disease states associated with mitochondrial dysfunction (Paulin et al., 2014; Salvatori et al., 2017; Vazquez et al., 2015; Wagner et al., 2012; Zhang et al., 2014). Unlike in the nucleus where acetylation of histones is regulated by the opposing activities of lysine deacetylases (KDACs) and lysine acetyltransferases, mitochondrial acetylation is thought to occur non-enzymatically (Wagner and Payne, 2013) and therefore, may reflect activities of mitochondrial deacetylases (Menzies et al., 2016; Wagner and Hirschey, 2014).
The sirtuins are a highly conserved protein family of Class III KDACs implicated in the maintenance of cellular homeostasis through regulation of energy metabolism and antioxidant defenses (Houtkooper et al., 2012). SIRT1-7 are nicotinamide adenine dinucleotide (NAD+)-dependent lysine deacylases and are located throughout varied cellular compartments (Canto et al., 2015; Herskovits and Guarente, 2014; Houtkooper et al., 2012). Changes in NAD+ levels have been shown to regulate sirtuin activity, possibly via altered expression and activity of the rate-limiting NAD+ biosynthetic enzyme Nampt (Imai and Guarente, 2014, 2016). SIRT1 is a nuclear-cytosolic deacylase known to control the activity of numerous enzymes and transcription factors in these cellular compartments (Boutant and Canto, 2014; Herskovits and Guarente, 2014). Mitochondrial SIRT3 has been shown to control global levels of mitochondrial acetylation and is also thought to regulate mitochondrial energy metabolism and antioxidant activities by direct deacetylation of substrate enzymes in these pathways (Kincaid and Bossy-Wetzel, 2013; Lombard et al., 2007). Many neuroprotective effects of SIRT3 have been found (Ansari et al., 2017), and it was recently shown that overexpression of SIRT3 in vitro reduces neuronal death in response to excitotoxic cell dealth (Kim et al., 2011). The role of SIRT3 in epilepsy has not yet been explored, although our laboratory has recently implicated reduced activities of manganese superoxide dismutase (MnSOD) and Complex I, two known SIRT3 substrates, during epileptogenesis (Liang et al., 2012; Ryan et al., 2012). This suggests that SIRT3 dysfunction and the resulting increase in mitochondrial acetylation may underlie the deficits in mitochondrial function and inhibition of bioenergetic enzymes known to occur with the development of chronic epilepsy.
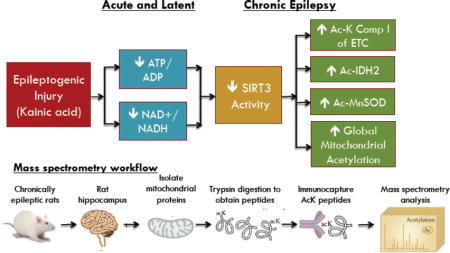
In the present study, we hypothesized that impairments in hippocampal bioenergetics are associated with reductions in SIRT3 activity and consequent increases in mitochondrial acetylation during epileptogenesis in the rat KA TLE model. Further, we sought to determine if aberrant acetylation of SIRT3 bioenergetic and metabolic substrates are associated with spontaneous seizure activity. We also aimed to characterize levels of hippocampal mitochondrial protein acetylation in hippocampi of rats with chronic epilepsy in our KA chemoconvulsant model of epilepsy using an innovative, mass spectrometry-based acetylomics approach. Additionally, we aimed to delineate affected mitochondrial biological processes by analyzing the resulting profile of acetylated proteins via gene ontology and pathway analysis.
Methods
Kainic acid-induced epileptogenesis
Animal studies were carried out in accordance to the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, revised 1996). All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Colorado, Anschutz Medical Campus. Adult, male Sprague Dawley rats (~300-350 g) received a subcutaneous injection of saline or kainic acid (KA; 11 mg/kg; dissolved in sterile saline, pH 7.4; AG Scientific, Inc. #K-1013) to induce status epilepticus (SE), a period of constant seizure activity known to incite brain injury in chronic epilepsy models. Rats were anesthetized using carbon dioxide and immediately euthanized by decapitation at 48 hours, 1 week and 12 weeks after KA administration to encompass the acute, latent and chronic periods of epileptogenesis. Hippocampi were dissected and immediately frozen in liquid nitrogen or crude mitochondria were isolated from hippocampal tissues (Liang and Patel, 2006). All tissues were stored at −80°C.
Monitoring of convulsive seizures
Rats were observed for acute seizure activity during SE and behavioral seizures were scored using a modified Racine scale (Liang et al., 2000) with only Class III, IV and V motor seizures considered. Briefly, motor seizures were scored on the following scale: Class III – unilateral forelimb clonus, Class IV – bilateral forelimb clonus with rearing, and Class V – bilateral forelimb clonus with rearing, falling, and/or hind limb clonus. SE was defined as at least five Class III or higher seizures followed by a period of continuous seizure activity. Animals that received an injection of KA but did not progress into SE were deemed “non-responders”. To confirm the development of spontaneous seizures in the chronic time point, rats were monitored with 24 hour video recording for 72 hours in the University of Colorado In Vivo Neurophysiology Core for seizure number, duration and severity at the end of the 12 week time point. Rats observed to have spontaneous, behavioral seizures during the video monitoring period were considered chronically epileptic.
Mitochondrial and cytosol isolations
Crude hippocampal mitochondria were isolated according to our previously published methodology (Liang and Patel, 2006). Briefly, hippocampal homogenates were centrifuged at 800g at 4°C for 10 min and the resulting supernatant was then centrifuged at 16,000g at 4°C for 10 min. Following the second centrifugation step, the supernatant was isolated as the cytosolic fraction and the pellet was resuspended to obtain the mitochondrial fraction. One hippocampus was used for western blot analyses from mitochondrial and cytosolic fractions and the mitochondrial acetyl-protein immunoprecipitations. For mass spectrometry-based mitochondrial acetyl-lysine immunoprecipitations, both hippocampi were used for mitochondrial isolations.
Western blot analysis
Hippocampal mitochondrial and cytosolic fractions were prepared as described above and used for protein expression analysis. Protein expression of SIRT3 (Cell Signaling 5490), NADH Dehydrogenase (Ubiquinone) 1 Alpha Subcomplex 9 (Ndufa9) (Abcam ab14713), acetyl-isocitrate dehydrogenase 2 (ac-IDH2) (Gene Tel #AC0004), SIRT1 (Millipore 08-131), Nampt (Bethyl A300-372A), and acetyl-MnSOD K122 (ac-MnSOD) (Tao et al., 2010) were determined via standard Western blot techniques, quantified with Image J software (NIH) or Image Lab 5.1 (Bio-Rad) and normalized to GAPDH (Cell Signaling 2118) or β-actin (Sigma) for cytosolic loading controls, VDAC (Abcam ab15895) for mitochondrial loading control, or total protein load for gels run using TGX Stain-free gels (Bio-Rad) (Carney et al., 2014; Tramutola et al., 2015).
HPLC analysis of ATP and ADP
ATP and ADP were measured in hippocampi according to previously published methods (Botker et al., 1994; Sellevold et al., 1986). The tissues were quickly dissected, frozen with liquid nitrogen and tissue weight obtained. Hippocampi were sonicated in 10% w/v 0.42 N perchloric acid, centrifuged at 16000g at 4°C for 15 min. 200 uL of the supernatant was neutralized with 20 uL 4 N KOH, and left at −20°C for at least 10 min. The neutralized supernatant was centrifuged at 16000g at 4°C for 15 min to ensure removal of perchlorate, and 100 uL of this supernatant was mixed with 100 uL 100 mM K2HPO4, pH 7.0. A 20 uL aliquot of this mixture was injected onto the HPLC system. ATP and ADP were analyzed using a HPLC equipped with a spectrophotometric detector (Elite LaChrom System; Hitachi, Japan) set at 258 nm and a YMC-Pack ODS-A column (4.6 × 150 mm, 3 μm, 120 Å; Waters, Milford, MA, USA). The mobile phase was composed of 100 mM K2HPO4, 12% ACN, 4 mM tetrabutylammonium bisulfate and adjusted to pH 5.0 with 4N KOH and the flow rate was set at 0.8 ml/min. The retention time of ADP was 5.78 min and that of ATP was 7.82 min.
HPLC analysis of NAD+
NAD+ was measured in hippocampi according to previously published methods (Botker et al., 1994; Revollo et al., 2004; Sellevold et al., 1986). The tissues were quickly dissected, frozen with liquid nitrogen and tissue weight obtained. Hippocampi were sonicated in 10% w/v 0.42 N perchloric acid, centrifuged at 16,000g at 4°C for 15 min. 200 uL of the supernatant was neutralized with 20 uL 4 N KOH, and left at −20°C for at least 10 min. The neutralized supernatant was centrifuged at 16000g at 4°C for 10 min, and 160 uL of this supernatant was mixed with 40 uL 100 mM K2HPO4, pH 7.0. A 20 uL aliquot of this mixture was injected onto the HPLC system. NAD+ was analyzed using a HPLC equipped with a spectrophotometric detector (Elite LaChrom System; Hitachi, Japan) set at 261 nm and a 3 uM, 15 cm × 4.6 cm SUPELCOSIL™ LC-18-T HPLC column (Sigma Aldrich). The mobile phase was composed of 100 mM K2HPO4, 5% methanol and adjusted to pH 5.0 with 4N KOH and the flow rate was set at 0.8 ml/min. The retention time of NAD+ at 25.50 min.
Hippocampal mitochondrial acetyl-lysine immunoprecipitation and protein expression
300 ug of hippocampal mitochondrial lysates were incubated with acetyl-lysine primary antibodies (Cell Signaling #9814 and #9441) overnight following 1 hr pre-clearing with Protein A/G beads (Santa Cruz sc-2003) at 4°C. Protein A/G beads were then added to the supernatant and lysates were rotated overnight at 4°C. Beads were washed with PBS-T and eluted with Laemmli Buffer at 100°C. Expression of global mitochondrial acetylated proteins (Abcam ab22550) and acetyl-Ndufa9 (ac-Ndufa9; Abcam ab14713) were determined via standard Western blot techniques and quantified with Image J software (NIH).
Acetylomics Analysis
Acetylomic analysis of mitochondrial proteins was performed according to the protocol provided by Cell Signaling Technology (Svinkina et al., 2015). Acetylated BSA was used as an internal standard for quality control. Briefly, mitochondrial proteins (2 mg) were trypsinized and enriched utilizing a mixture of monoclonal acetyl-Lys antibodies conjugated to beads (Cell Signaling #13416). Purified acetyl-peptide analysis was performed using nano-UPLC–MS/MS (Bruker Daltonics, Inc., Billerica, MA) at a flow rate of 800 nL/min with a gradient of 5–50% 0.1% formic acid in acetonitrile (ACN) over 60 min on C18 trapping (20 × 0.1 mm2) and analytical columns (150 × 0.10 mm2). The nano-UPLC was coupled to a nano-ESI source and Impact HD Q-TOF mass spectrometer (Bruker Daltonics, Inc., Billerica, MA). The instrument was operated using intensity dependent CID MS/MS. MS/MS data analysis was performed using Mascot (version 2.2.04, Matrixscience) by Proteinscape (Bruker Daltonics, Inc., Billerica, MA). Peptides were searched in Mascot using SwissProt 2016_05, allowing up to 4 missed tryptic cleavages with variable carbamidomethyl (C), deamidated (NQ), oxidation (M), and acetyl (K) modifications. Peptides with a minimum ion score of 20 were be accepted. The final protein list included only protein IDs with a probability of 99% or greater and peptide IDs required a 95% cutoff. The resulting gene list of acetylated proteins was analyzed by the Gene Ontology (GO) Consortium web toolkit to determine pathway enrichment by entering the protein accession list into the GO PANTHER website (Ashburner et al., 2000; Gene Ontology, 2015). Analysis for gene ontology used the Bonferroni correction for multiple testing with statistical significance determined by p < 0.05.
Statistical Analyses
Results are presented as means ± SEM. For all biochemical analyses, GraphPad Prism 6.0 was used to perform Student’s t-test or one-way ANOVA analysis. Tukey’s multiple comparisons post-hoc tests were used where appropriate. Significance was determined using P < 0.05.
Results
Hippocampal bioenergetics are impaired the KA model of epilepsy
We have previously shown mitochondrial respiratory deficits and alterations in the glycolytic capacity in the rat KA TLE model (Rowley et al., 2015). Since this suggests impairment in bioenergetics, we measured the ratio of ATP/ADP at three time points 48 hours, 1 week and 12 weeks after KA treatment, which correlate with the acute, latent and chronic periods of epilepsy development. The ATP/ADP ratio was decreased in the hippocampus of KA-treated rats versus controls by 28% in the acute, 35% during the latent, and by 27% in the chronic phase of KA-induced epilepsy (all p < 0.05; Fig 1A), indicating a deficit in cellular bioenergetics. Previous work from our laboratory has also shown alterations in the cellular and mitochondrial redox state, as indicated by decreases in the ratios of reduced to oxidized glutathione (GSH/GSSG) and Coenzyme A (CoASH/CoASSG), respectively, during epileptogenesis in the rat KA TLE model (Ryan et al., 2014). The cellular and mitochondrial redox states are strongly influenced by the NAD+/NADH ratios in these compartments, as is the production of ATP as these metabolic co-factors are utilized during energy production in glycolysis, the TCA cycle and during oxidative phosphorylation (Wagner and Payne, 2011). Measurement of NAD+ in the hippocampus revealed decreases of 41% and 42% at the acute and latent time points, respectively (both p < 0.05), but no change in spontaneously epileptic rats in the chronic time point relative to controls (Fig 1B). These data demonstrate impairment of hippocampal bioenergetics and NAD+ levels in the KA rat TLE model.
Figure 1.

Impaired hippocampal bioenergetics during epileptogenesis. (A) ATP/ADP ratio and (B) nicotinamide adenine dinucleotide (NAD+) in the hippocampus during epileptogenesis. Values are mean ± SEM (n = 3-18 per group). *, p < 0.05 vs Control.
Enzymes regulating bioenergetics
We next investigated the role of SIRT3 in these bioenergetics deficits and previously observed indices of mitochondrial dysfunction and oxidative stress in this model (Jarrett et al., 2008; Liang et al., 2000; Liang and Patel, 2006; Rowley et al., 2015; Ryan et al., 2012; Ryan et al., 2014) by measuring its protein expression and acetylation status of select substrates. Protein expression of SIRT3 was unchanged at the acute time point following KA-induced SE (p > 0.05), modestly elevated by 14% during the latent period and decreased by 14% in the chronic time point (both p < 0.05; Fig 2A). As SIRT3 regulates levels of mitochondrial acetylation, and increases in mitochondrial acetylation are associated with decreases in SIRT3 activity, global mitochondrial acetylation was assessed as a marker of SIRT3 activity. Hippocampal mitochondrial acetylation was unchanged during the acute and latent time points, but was elevated by 60% in spontaneously epileptic rats, indicating a reduction in SIRT3 activity in chronic epilepsy (Fig 2B).
Figure 2.
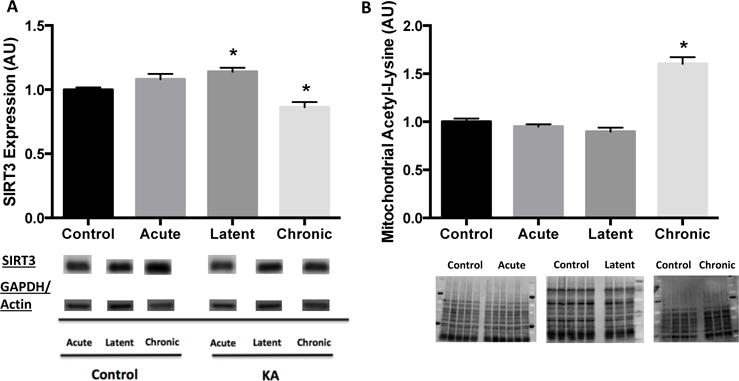
Reduced SIRT3 protein expression and activity, as assessed by global levels of mitochondrial acetylation, in hippocampus at chronic time point of epileptogenesis. Hippocampal (A) SIRT3 protein expression and (B) global levels of acetylated proteins in the mitochondria during epileptogenesis. SIRT3 expression normalized to same loading control for both Control and Kainic acid-treated at each time point (acute: GAPDH, latent: GAPDH, chronic: actin). Data are normalized to Control mean value. Representative blot images below. Values are mean ± SEM (n = 3-14 per group). *, p < 0.05 vs Control.
Given the changes in SIRT3 activity and NAD+ levels, we then assessed levels of Nampt, the rate-limiting enzyme in the production of NAD+. Hippocampal protein expression of Nampt was decreased by 26% and 30% versus controls at the acute and chronic time points, respectively (both p < 0.05), but not in the latent period (p > 0.05; Fig 3A). Protein levels of SIRT1, a nuclear-cytosolic deacetylase implicated in mitochondrial function through deacetylation of transcription factors regulating mitochondrial biogenesis and function, were also assessed. SIRT1 expression was unchanged at all time points in KA-treated versus control animals (all p > 0.05; Fig 3B), prompting us to focus our investigation into a mechanism mediating the impairments in mitochondrial function and bioenergetics during epileptogenesis on the role of SIRT3 and aberrant mitochondrial acetylation. Similarly, since we primarily found changes during the chronic time point, we decided to focus the remaining efforts on this phase of epilepsy.
Figure 3.
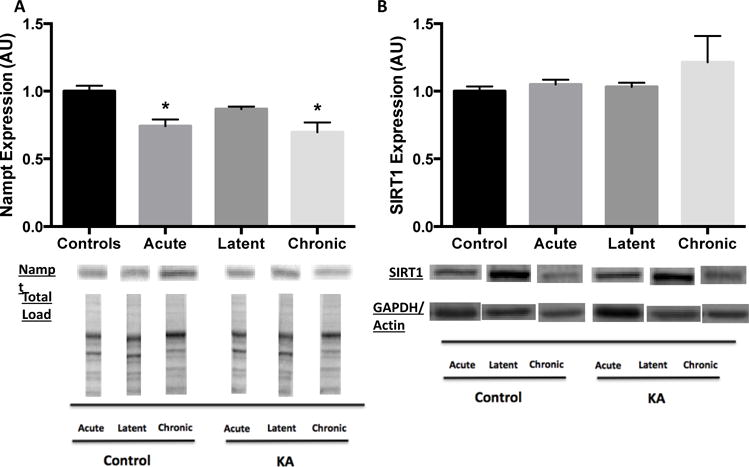
Decreased Nampt protein expression during acute and chronic phases and no change in SIRT1 protein expression at all time points of epileptogenesis in hippocampus. Hippocampal nuclear-cytoplasmic (A) Nampt and (B) SIRT1 protein expression during epileptogenesis. Nampt expression normalized to total protein load for both Control and Kainic acid (KA)-treated at all time points. SIRT1 expression normalized to same loading control for both Control and KA-treated at each time point (acute: GAPDH, latent: GAPDH, chronic: actin). Data are normalized to Control mean value. Representative blot images below. Values are mean ± SEM (n = 3-14 per group). *, p < 0.05 vs Control.
Increased acetylation of SIRT3 metabolic substrates
Since SIRT3 is known to regulate levels of mitochondrial ROS, ATP production and the mitochondrial redox state, we investigated changes in the acetylation state of SIRT3 substrates known to regulate these biological processes. SIRT3 has been shown to deacetylate, and therefore regulate the activity of, the mitochondrial antioxidant MnSOD to control levels of mitochondrial ROS (Tao et al., 2010). We have previously shown that partial deficiency of MnSOD lowers seizure threshold and complete deficiency causes epilepsy (Liang and Patel, 2004; Liang et al., 2012), raising the possibility that a decrease in MnSOD activity secondary to decreased SIRT3 activity may be associated with epileptogenesis. Indeed, acetylation of lysine 122 (K122) of MnSOD, a known SIRT3 target, was increased by 20% in spontaneously epileptic rats versus controls (p < 0.05; Fig 4A).
Figure 4.
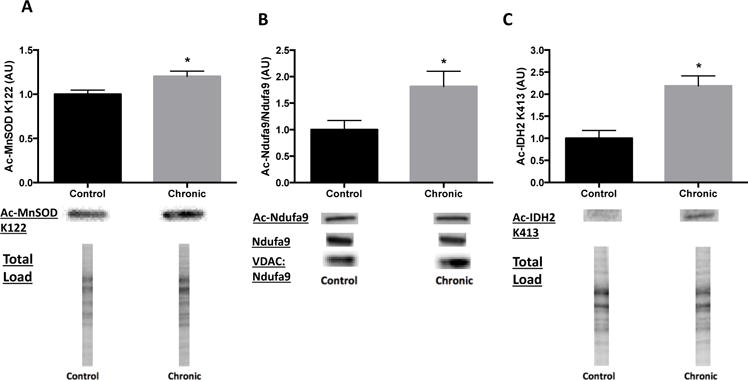
Increased hippocampal acetylation of SIRT3 protein substrates in chronic time point. Enhanced levels of (A) acetylated manganese superoxide dismutase (Ac-MnSOD) lysine 122 (K122), (B) acetylated NADH Dehydrogenase (Ubiquinone) 1 Alpha Subcomplex 9 (Ac-Ndufa9) to total Ndufa9 and (C) acetylated isocitrate dehydrogenase 2 (Ac-IDH2) in hippocampus of spontaneously epileptic rats at chronic time point. Ac-MnSOD and ac-IDH2 expression normalized to total protein load for both Control and Kainic acid (KA)-treated. Ndufa9 expression normalized to VDAC for both Control and KA-treated. Data are normalized to Control mean value. Representative blot images below. Values are mean ± SEM (n = 3-11 per group). *, p < 0.05 vs Control.
We have previously shown decreased activity of Complex I of the ETC at the acute and chronic time points in the KA model (Ryan et al., 2012), which may reduce ATP production through this mitochondrial pathway. SIRT3 has been shown to deacetylate the Ndufa9 subunit of Complex I, resulting in enhanced Complex I enzymatic activity and increases in ATP production (Ahn et al., 2008). As our current data demonstrated an impaired ATP/ADP ratio, we assessed acetylation of Ndufa9 and found an 81% increase in the acetyl-Ndufa9/Ndufa9 ratio versus controls (p < 0.05; Fig 4B).
Previous studies from our laboratory have also shown impairments in the mitochondrial redox state, as demonstrated by a decrease in the CoASH/CoASSG ratio, indicating a more oxidized mitochondrial environment, during epileptogenesis (Ryan et al., 2014). SIRT3 has been shown to regulate the mitochondrial redox state by enhancing activity of the TCA cycle enzyme, IDH2, through deacetylation of lysine 413 (K413) (Someya et al., 2010; Yu et al., 2012). IDH2 converts isocitrate to α-ketoglutarate in a reaction that converts NADP+ to NADPH, which can result in a more reduced mitochondrial redox state due to the subsequent regeneration of GSH and CoASH from NADPH (Jo et al., 2001; Someya et al., 2010). Therefore, we measured the acetylation state of IDH2 and found a 119% increase in acetyl-IDH2 K122 in spontaneously epileptic rats versus control animals (p < 0.05; Fig 4C).
Acetylomics analysis of mitochondrial protein
In order to follow up on the acetylation of specific protein targets described above, we applied an acetylomics approach to examine overall changes in mitochondrial protein acetylation in the chronic phase of epilepsy in this KA rat TLE model (Fig. 5). Here, we identified a total of 333 proteins from our enriched mitochondrial fractions from rat hippocampus, with an overlap of 227 proteins from both KA-treated and control animals. Of these mitochondrial proteins, 38 were unique to control samples alone, whereas 68 were identified only in hippocampal mitochondria from chronically epileptic rats (Supplemental File 1). These results represent an approximate 79% increase in unique acetylated proteins found in spontaneously epileptic rats versus controls. Additionally, we identified 489 and 543 acetyl-peptides in control and chronic samples, respectively, compared to 1178 and 1270 total peptides found. Our acetylomics approach provided an approximate 42% enrichment of acetyl peptides (Supplemental File 2).
Figure 5.
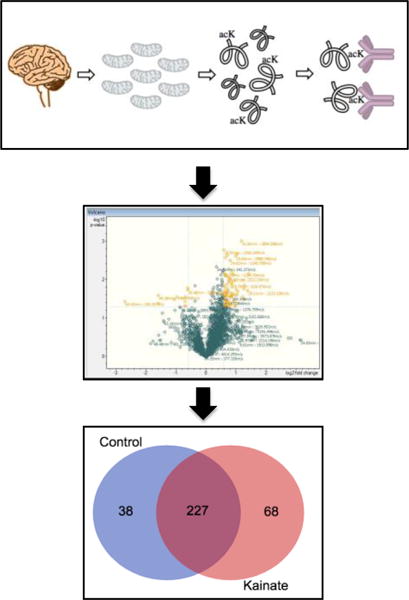
Mitochondrial acetylomics analysis in a model of KA induced epileptogenesis. Enriched mitochondrial samples were subjected to acetyl-immunopurification followed by nHPLC-MS/MS analysis. A significant number of unique features were identified in the KA samples. In total, 333 proteins were identified and include 227 which overlap between the samples, while 38 were unique to control samples alone and 68 were unique to KA treatment. This analysis identified 490 and 543 acetyl peptides in control and KA samples out of 1179 and 1270 total peptides, respectively.
The resulting protein lists were examined for biological pathway enrichment using the Gene Ontology Consortium toolkit. This search identified numerous mitochondrial pathways with significant enrichment; including, isocitrate metabolic processes, TCA cycle, energy coupled proton transport, NADH metabolic processes, aerobic respiration, ATP biosynthesis, and succinate metabolic processes, among many others (Supplemental File 3). These results confirm previously reported pathways impacted via protein lysine acetylation within the mitochondria and support a potential mechanism for acetylation-induced mitochondrial dysregulation.
Discussion
The key novel findings from the present study are that SIRT3 expression is reduced and global mitochondrial acetylation is increased, suggesting SIRT3 activity is impaired, in the hippocampus of chronically epileptic rats and this is associated with impaired bioenergetics and a decrease in protein expression of Nampt, the rate-limiting enzyme in NAD+ biosynthesis. Moreover, rats with chronic spontaneous seizures also demonstrate increased acetylation of several SIRT3 bioenergetic and metabolic substrates, including the mitochondrial antioxidant MnSOD, Ndufa9 of ETC Complex I and the TCA cycle enzyme IDH2. Mass spectrometry-based acetylomics data revealed a greater abundance of acetylated proteins and peptides in hippocampal mitochondria from chronically epileptic rats versus controls and identified multiple mitochondrial pathways affected by protein lysine acetylation. To our knowledge, this is the first study to show a reduction in the expression of SIRT3 and increased acetylation of mitochondrial enzymes with chronic epilepsy. Further, our current results also provide the first mass spectrometry acetylomics data in rat hippocampal mitochondria. Together, these results suggest a potential role for aberrant mitochondrial function due to protein acetylation via SIRT3 in chronic acquired epilepsy.
Impaired bioenergetics during epileptogenesis in rat TLE model
Our data demonstrate that epileptogenic injury impairs hippocampal bioenergetics, as assessed by a decrease in the ATP/ADP ratio. The decrease in the acute and latent time points could reflect the large metabolic demand of seizure activity following SE, which is known to deplete energy reserves and decrease cellular ATP levels (Kovac et al., 2012; Wasterlain et al., 2010). We have also shown a decrease in the maximal respiratory and reserve capacities in hippocampal synaptosomes in the acute and chronic time points in rat TLE models (Rowley et al., 2015), which could reflect a decline in the ability to produce ATP on demand during these phases of epilepsy development. Hence, the decrease in the ATP/ADP ratio during the acute and latent time points could reflect an increase in the utilization of ATP to maintain synaptic activity following SE, while the continued impairment during the chronic phase could be due to an inability to produce ATP, possibly as a result of mitochondrial dysfunction.
To our knowledge, this is the first study to show a decrease in NAD+ levels in the hippocampus of rats following epileptogenic injury. Previous studies have shown changes in NAD(P)H fluorescence in epileptic patients and rats, indicating an alteration in the total NADPH and NADH pools (Kann et al., 2005), as well as an increase in the NADH/NAD+ ratio, as assessed by the lactate dehydrogenase ratio, following seizure activity in rats (Folbergrova et al., 1985). Further, others have shown a decrease in NAD+ levels in vitro following excitotoxic injury (Liu et al., 2009; Liu et al., 2008). We detected an early decline in hippocampal NAD+ levels at 24 hours (data not shown) and 48 hours after KA-induced SE, which is similar to decreases in brain NAD+ found shortly after injury in rodent models of cerebral ischemia, axon degeneration and retinal degeneration, which are all associated with SIRT3 dysfunction (Dai et al., 2017; Gerdts et al., 2015; Lin et al., 2016; Magnifico et al., 2013; Yang et al., 2002).
Technical limitations currently prevent us from accurately measuring the mitochondrial pool of NAD+, so it is possible that the lack of change found in total NAD+ levels in the chronic period reflects maintenance of cytoplasmic NAD+ levels, which would sustain glycolysis to produce ATP. This is possible, as we have shown no change in the glycolytic rate during the chronic time point of epileptogenesis (Rowley et al., 2015). However, our finding of increased mitochondrial protein acetylation during the chronic time point is reflective of a decrease in mitochondrial NAD+ levels, as manipulation of the activity of Complex I of the ETC to decrease the mitochondrial NAD+/NADH ratio can increase mitochondrial acetylation, whereas supplementation of the NAD+ precursor nicotinamide mononucleotide to boost NAD+ levels has been shown to reduce global levels of mitochondrial acetylation (Karamanlidis et al., 2013).
Reduced SIRT3 activity and Nampt expression and increased mitochondrial acetylation
Our observation of an increase in SIRT3 expression in the latent time point following KA-induced SE, but a decrease during the chronic period, is similar to in vitro findings from others. It was recently shown that low dose of administration of glutamate and the glutamate receptor agonists NMDA and KA increased SIRT3 protein expression in mouse primary cortical neurons, however high dose exposure decreased SIRT3 expression (Cheng et al., 2016). This may suggest an initial compensatory increase in SIRT3 expression to maintain mitochondrial function upon glutamate receptor activation following seizure activity, as has also been shown acutely in vitro in mouse primary cortical neurons with NMDA administration (Kim et al., 2011).
The increase in mitochondrial acetylation during the chronic time point suggests decreased activity of SIRT3. SIRT3 has been shown to be the major mitochondrial deacetylase as knockout of SIRT3, but not deficiency of mitochondrial SIRT4 or SIRT5, increases mitochondrial acetylation levels in mice (Lombard et al., 2007). This decline in SIRT3 activity is also consistent with the known impairments in mitochondrial function found in chronic epilepsy (Kunz et al., 2000; Rowley et al., 2015; Rowley and Patel, 2013; Vielhaber et al., 2008), as SIRT3 has been shown to positively regulate mitochondrial function by enhancing the activity of multiple enzymes in all energy producing pathways in the mitochondria via direct deacetylation (Ahn et al., 2008; Hirschey et al., 2010; Houtkooper et al., 2012; Someya et al., 2010; Yu et al., 2012). Similarly, a loss of SIRT3 activity could also contribute to the decrease in the ATP/ADP ratio found in chronically epileptic rats, which is supported by a recent study demonstrating decreased ATP levels in the cerebral cortex and hippocampus of mice deficient in SIRT3 (Cheng et al., 2016). Decreases in SIRT3 activity and consequent increases in mitochondrial acetylation have been shown in other pathological states known to involve mitochondrial dysfunction and metabolic impairments (Hirschey et al., 2011; Salvatori et al., 2017; Wagner et al., 2012; Zhang et al., 2014). However, to our knowledge this is the first study to demonstrate dysregulation of SIRT3 and associated increases in mitochondrial acetylation in chronic epilepsy.
The reduction in Nampt protein expression during the acute and chronic time points may contribute to the decrease in NAD+ levels we found during the acute and latent time points, as well as the bioenergetics impairments during these phases of epilepsy. As the rate-limiting enzyme in the NAD+ salvage pathway, Nampt converts nicotinamide to nicotinamide mononucleotide, which is then further converted to NAD+ by nicotinamide mononucleotide adenylyl transferase (NMNAT) 1-3, which reside in different cellular compartments (Imai and Yoshino, 2013; Verdin, 2015). This salvage pathway is the primary method to regenerate NAD+ from the nicotinamide produced as a byproduct of the activities of NAD+-consuming enzymes, such as the sirtuins (Verdin, 2015). It is possible that a reduction in the level of this important NAD+ biosynthetic enzyme could also lead to an increase in cellular levels of nicotinamide, which has also been shown to inhibit sirtuin activity (Verdin, 2015). Further, Nampt inhibition has been shown to enhance neuronal injury in postischemic stroke (Wang et al., 2011) and methods to increase Nampt activity are neuroprotective (De Jesus-Cortes et al., 2012; Tesla et al., 2012; Wang et al., 2014; Yin et al., 2014; Zhao et al., 2015). Reduced expression of hippocampal Nampt protein has been found with aging, concurrent with decreases in NAD+ (Liu et al., 2012; Stein and Imai, 2014), but to our knowledge this is the first study to show changes in Nampt protein levels in epilepsy.
We did not find a change in the expression of the nuclear-cytoplasmic deacetylase SIRT1. Two recent studies have assessed SIRT1 in the acute time point following SE in rats. In the pilocarpine rat model of TLE, SIRT1 protein expression and activity were shown to increase in the hippocampus acutely following SE (Wang et al., 2015). Interestingly, another study found no change in total cellular or cytoplasmic SIRT1 protein in rat following KA-induced SE, but demonstrated a large increase in nuclear levels of SIRT1 protein during this acute time point (Brennan et al., 2016). However, neither of these studies examined SIRT1 in the chronic time point of epileptogenesis.
Increased acetylation of SIRT3 metabolic and antioxidant substrates
Overall, our analysis of rat hippocampal mitochondrial fractions in this model provides an in-depth look at potential mechanistic acetylation targets. Additionally, these results confirm previous analyses in whole brain fractions, which demonstrate a preponderance of protein acetylation among mitochondrial metabolic and antioxidant pathways (Dittenhafer-Reed et al., 2015). The increased acetylation state of the known SIRT3 substrates MnSOD, Ndufa9 of ETC Complex I, and IDH2 in spontaneously epileptic rats further demonstrates a decrease in SIRT3 activity in the chronic phase. As increases in lysine acetylation of these proteins have been shown to impair enzymatic activity (Ahn et al., 2008; Someya et al., 2010; Tao et al., 2010; Yu et al., 2012), these changes are not only reflective of reduced SIRT3 activity, but may also contribute to the metabolic dysfunction and impaired redox state known to occur during epileptogenesis.
An increase in acetylation of MnSOD on K122 has been shown to impair enzymatic activity and increase mitochondrial levels of ROS (Assiri et al., 2017; Tao et al., 2010). Several lines of evidence implicate elevated levels of mitochondrial ROS with epilepsy. In studies with mice partially and completely deficient for MnSOD, Sod2+/− and Sod2−/− mice, we have previously shown that loss in the activity of this critical mitochondrial antioxidant increases markers of mitochondrial ROS and results in spontaneous seizure activity (Liang and Patel, 2004; Liang et al., 2012). The increase in ROS with loss of MnSOD activity was causally implicated in seizure activity as seizure duration and seizure numbers per day were attenuated with administration of a catalytic antioxidant (Liang et al., 2012). Further, we have shown that antioxidant administration ameliorates the increased mitochondrial ROS and neuronal death in the KA rat TLE model (Liang et al., 2000). More recently, it was demonstrated that a combination of the antioxidant compounds sulforaphane, a Nrf2 inducer, and N-acetyl cysteine decreased epilepsy progression in rats (Pauletti et al., 2017). These results demonstrate an important role for the loss of MnSOD activity and elevated levels of mitochondrial ROS in the development of chronic epilepsy and our current data suggests acetylation of MnSOD could play a role.
Enhanced acetylation of the ETC Complex I subunit Ndufa9 has previously been shown to decrease Complex I activity resulting in diminished production of ATP (Ahn et al., 2008). Importantly, loss of Complex I activity has also been found in hippocampus tissue of epileptic patients (Kunz et al., 2000). Impaired Complex I activity has also been demonstrated by us in chronically epileptic rats in this same model of KA-induced epilepsy (Ryan et al., 2012), as well as in Sod2 −/− mice with spontaneous seizures (Liang et al., 2012). Thus, increased acetylation of Ndufa9 in chronic epilepsy could result in decreased Complex I activity and ATP production, which is further supported by our current results demonstrating a reduction in the ATP/ADP ratio.
Increased acetylation of the TCA cycle enzyme IDH2 on lysine 413 (K413) has been shown to reduce enzymatic activity, thereby decreasing conversion of the cofactor NADP+ to NADPH, which impairs regeneration of GSH, resulting in a more oxidized cellular environment, (Someya et al., 2010; Yu et al., 2012). We have previously demonstrated a shift in the redox state towards a more oxidized cellular and mitochondrial environment in the chronic phase of epilepsy in this rat model of TLE (Ryan et al., 2014). Further, a decrease in levels of GSH in brain tissue from epileptic patients has also been shown (Mueller et al., 2001). Loss of IDH2 activity has been shown to contribute to mitochondrial dysfunction in Parkinson disease models (Kim et al., 2016) and mutations in IDH2 are associated with seizures as the initial symptom in patients with low-grade gliomas (Stockhammer et al., 2012; Zhong et al., 2015). Additionally, TCA cycle dysfunction has been documented by our lab and others in animal models of epilepsy (Cock et al., 2002; Kovac et al., 2013; Liang et al., 2000). Specifically, we have shown inactivation of the oxidant-sensitive enzyme aconitase (Liang et al., 2000), which precedes IDH2 in the TCA cycle, and others have demonstrated a loss in the activity of alpha-ketoglutarate dehydrogenase (Cock et al., 2002), the enzyme following IDH2 in the TCA cycle. Therefore, increased acetylation, and possible subsequent decreased activity, of these TCA cycle enzymes is another possible source of the metabolic dysfunction and redox imbalance found in chronic epilepsy.
Conclusions
Our current results demonstrating increases in mitochondrial protein lysine acetylation in chronic epilepsy suggests an intriguing link to the known deficits in mitochondrial function and the redox dysfunction known to occur during epileptogenesis. Identification of additional enzymes with aberrant hyperacetylation and the associated affected pathways during epileptogenesis may provide further mechanistic insight into the development of spontaneous seizures. Further, activation of SIRT3 during the epileptogenic cascade may attenuate the resulting hyperacetylation of mitochondrial proteins and prevent the associated metabolic dysfunction. Methods to enhance cellular levels of NAD+ are known to activate SIRT3 (Brown et al., 2014; Karamanlidis et al., 2013; Lin et al., 2016; Traba et al., 2015) and compounds found to boost cellular levels of NAD+ have shown efficacy in rodent models of other neurodegenerative diseases such as Alzheimer’s disease, Parkinson disease, traumatic brain injury, amyotrophic lateral sclerosis, axon degeneration, diabetic neuropathies and retinal dysfunction (De Jesus-Cortes et al., 2012; Gerdts et al., 2015; Gong et al., 2013; Lin et al., 2016; Tesla et al., 2012; Trammell et al., 2016; Wang et al., 2014; Yin et al., 2014). Therefore, methods to increase neuronal NAD+ to activate SIRT3 may be effective therapies to prevent mitochondrial dysfunction during epileptogenesis and attenuate the development of chronic epilepsy.
Supplementary Material
Acknowledgments
We thank the University of Colorado Anschutz Medical Campus In Vivo Neurophysiology Core for providing the facilities to acquire video data and the School of Pharmacy Mass Spectrometry Facility Core for their technical assistance. This work was supported by American Epilepsy Society Postdoctoral Fellowship (LBG) and NIH grants RO1NS039587 (MP), F32NS090808 (LBG), RO1CA182506 (AV), RO1AA022146 (KSF), and UL1TR001082 (NR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell. 2017;16:4–16. doi: 10.1111/acel.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assiri MA, Roy SR, Harris PS, Ali H, Liang Y, Shearn CT, Orlicky DJ, Roede JR, Hirschey MD, Backos DS, et al. Chronic Ethanol Metabolism Inhibits Hepatic Mitochondrial Superoxide Dismutase via Lysine Acetylation. Alcohol Clin Exp Res. 2017;41:1705–1714. doi: 10.1111/acer.13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botker HE, Kimose HH, Helligso P, Nielsen TT. Analytical evaluation of high energy phosphate determination by high performance liquid chromatography in myocardial tissue. Journal of molecular and cellular cardiology. 1994;26:41–48. doi: 10.1006/jmcc.1994.1006. [DOI] [PubMed] [Google Scholar]
- Boutant M, Canto C. SIRT1 metabolic actions: Integrating recent advances from mouse models. Mol Metab. 2014;3:5–18. doi: 10.1016/j.molmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan GP, Dey D, Chen Y, Patterson KP, Magnetta EJ, Hall AM, Dube CM, Mei YT, Baram TZ. Dual and Opposing Roles of MicroRNA-124 in Epilepsy Are Mediated through Inflammatory and NRSF-Dependent Gene Networks. Cell Rep. 2016;14:2402–2412. doi: 10.1016/j.celrep.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, Jaffrey SR. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell metabolism. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell metabolism. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney KE, Milanese M, van Nierop P, Li KW, Oliet SH, Smit AB, Bonanno G, Verheijen MH. Proteomic analysis of gliosomes from mouse brain: identification and investigation of glial membrane proteins. Journal of proteome research. 2014;13:5918–5927. doi: 10.1021/pr500829z. [DOI] [PubMed] [Google Scholar]
- Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell metabolism. 2016;23:128–142. doi: 10.1016/j.cmet.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock HR, Tong X, Hargreaves IP, Heales SJ, Clark JB, Patsalos PN, Thom M, Groves M, Schapira AH, Shorvon SD, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002;48:157–168. doi: 10.1016/s0920-1211(01)00334-5. [DOI] [PubMed] [Google Scholar]
- Dai SH, Chen T, Li X, Yue KY, Luo P, Yang LK, Zhu J, Wang YH, Fei Z, Jiang XF. Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK-mTOR pathway. Free radical biology & medicine. 2017;108:345–353. doi: 10.1016/j.freeradbiomed.2017.04.005. [DOI] [PubMed] [Google Scholar]
- De Jesus-Cortes H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17010–17015. doi: 10.1073/pnas.1213956109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, Denu JM. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell metabolism. 2015;21:637–646. doi: 10.1016/j.cmet.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W. Sirtuins: from metabolic regulation to brain aging. Frontiers in aging neuroscience. 2013;5:36. doi: 10.3389/fnagi.2013.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folbergrova J, Ingvar M, Nevander G, Siesjo BK. Cerebral metabolic changes during and following fluorothyl-induced seizures in ventilated rats. Journal of neurochemistry. 1985;44:1419–1426. doi: 10.1111/j.1471-4159.1985.tb08778.x. [DOI] [PubMed] [Google Scholar]
- Gene Ontology, C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science. 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Pan Y, Vempati P, Zhao W, Knable L, Ho L, Wang J, Sastre M, Ono K, Sauve AA, et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiology of aging. 2013;34:1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Molecular cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits AZ, Guarente L. SIRT1 in neurodevelopment and brain senescence. Neuron. 2014;81:471–483. doi: 10.1016/j.neuron.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand MS, Dahl HH, Damiano JA, Smith RJ, Scheffer IE, Berkovic SF. Recent advances in the molecular genetics of epilepsy. Journal of medical genetics. 2013;50:271–279. doi: 10.1136/jmedgenet-2012-101448. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nature reviews. Molecular cell biology. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. Nature Publishing Group. 2016;2:1–6. doi: 10.1038/npjamd.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15(Suppl 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Liang LP, Hellier JL, Staley KJ, Patel M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiology of disease. 2008;30:130–138. doi: 10.1016/j.nbd.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001;276:16168–16176. doi: 10.1074/jbc.M010120200. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovacs R, Njunting M, Behrens CJ, Otahal J, Lehmann TN, Gabriel S, Heinemann U. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain. 2005;128:2396–2407. doi: 10.1093/brain/awh568. [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell metabolism. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim SH, Cha H, Kim SR, Lee JH, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and dopaminergic neurotoxicity: implications for Parkinson’s disease. Free Radic Res. 2016;50:853–860. doi: 10.1080/10715762.2016.1185519. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lu HF, Alano CC. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PloS one. 2011;6:e14731. doi: 10.1371/journal.pone.0014731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Frontiers in aging neuroscience. 2013;5:48. doi: 10.3389/fnagi.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac S, Abramov AY, Walker MC. Energy depletion in seizures: anaplerosis as a strategy for future therapies. Neuropharmacology. 2013;69:96–104. doi: 10.1016/j.neuropharm.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Kovac S, Domijan AM, Walker MC, Abramov AY. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J Cell Sci. 2012;125:1796–1806. doi: 10.1242/jcs.099176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz WS, Kudin AP, Vielhaber S, Blumcke I, Zuschratter W, Schramm J, Beck H, Elger CE. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Annals of neurology. 2000;48:766–773. [PubMed] [Google Scholar]
- Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101:563–570. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free radical biology & medicine. 2004;36:542–554. doi: 10.1016/j.freeradbiomed.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free radical biology & medicine. 2006;40:316–322. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Liang LP, Waldbaum S, Rowley S, Huang TT, Day BJ, Patel M. Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: attenuation by a lipophilic metalloporphyrin. Neurobiology of disease. 2012;45:1068–1076. doi: 10.1016/j.nbd.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, Nakamura R, Zapata N, Kubota M, Tsubota K, et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016;17:69–85. doi: 10.1016/j.celrep.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Pitta M, Mattson MP. Preventing NAD(+) depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann N Y Acad Sci. 2008;1147:275–282. doi: 10.1196/annals.1427.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LY, Wang F, Zhang XY, Huang P, Lu YB, Wei EQ, Zhang WP. Nicotinamide phosphoribosyltransferase may be involved in age-related brain diseases. PloS one. 2012;7:e44933. doi: 10.1371/journal.pone.0044933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Molecular and cellular biology. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnifico S, Saias L, Deleglise B, Duplus E, Kilinc D, Miquel MC, Viovy JL, Brugg B, Peyrin JM. NAD+ acts on mitochondrial SirT3 to prevent axonal caspase activation and axonal degeneration. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:4712–4722. doi: 10.1096/fj.13-229781. [DOI] [PubMed] [Google Scholar]
- Menzies KJ, Zhang H, Katsyuba E, Auwerx J. Protein acetylation in metabolism - metabolites and cofactors. Nat Rev Endocrinol. 2016;12:43–60. doi: 10.1038/nrendo.2015.181. [DOI] [PubMed] [Google Scholar]
- Mueller SG, Trabesinger AH, Boesiger P, Wieser HG. Brain glutathione levels in patients with epilepsy measured by in vivo (1)H-MRS. Neurology. 2001;57:1422–1427. doi: 10.1212/wnl.57.8.1422. [DOI] [PubMed] [Google Scholar]
- Pauletti A, Terrone G, Shekh-Ahmad T, Salamone A, Ravizza T, Rizzi M, Pastore A, Pascente R, Liang LP, Villa BR, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain. 2017;140:1885–1899. doi: 10.1093/brain/awx117. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell metabolism. 2014;20:827–839. doi: 10.1016/j.cmet.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell metabolism. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Rowley S, Liang LP, Fulton R, Shimizu T, Day B, Patel M. Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy. Neurobiology of disease. 2015;75:151–158. doi: 10.1016/j.nbd.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley S, Patel M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free radical biology & medicine. 2013;62:121–131. doi: 10.1016/j.freeradbiomed.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K, Backos DS, Reigan P, Patel M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:11250–11258. doi: 10.1523/JNEUROSCI.0907-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K, Liang LP, Rivard C, Patel M. Temporal and spatial increase of reactive nitrogen species in the kainate model of temporal lobe epilepsy. Neurobiology of disease. 2014;64:8–15. doi: 10.1016/j.nbd.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori I, Valle C, Ferri A, Carri MT. SIRT3 and mitochondrial metabolism in neurodegenerative diseases. Neurochem Int. 2017 doi: 10.1016/j.neuint.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Sellevold OF, Jynge P, Aarstad K. High performance liquid chromatography: a rapid isocratic method for determination of creatine compounds and adenine nucleotides in myocardial tissue. Journal of molecular and cellular cardiology. 1986;18:517–527. doi: 10.1016/s0022-2828(86)80917-8. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell metabolism. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockhammer F, Misch M, Helms HJ, Lengler U, Prall F, von Deimling A, Hartmann C. IDH1/2 mutations in WHO grade II astrocytomas associated with localization and seizure as the initial symptom. Seizure. 2012;21:194–197. doi: 10.1016/j.seizure.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Sudha K, Rao AV, Rao A. Oxidative stress and antioxidants in epilepsy. Clin Chim Acta. 2001;303:19–24. doi: 10.1016/s0009-8981(00)00337-5. [DOI] [PubMed] [Google Scholar]
- Svinkina T, Gu H, Silva JC, Mertins P, Qiao J, Fereshetian S, Jaffe JD, Kuhn E, Udeshi ND, Carr SA. Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Anti-acetyl-lysine Antibodies and an Optimized Proteomic Workflow. Mol Cell Proteomics. 2015;14:2429–2440. doi: 10.1074/mcp.O114.047555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Molecular cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17016–17021. doi: 10.1073/pnas.1213960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traba J, Kwarteng-Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, Waclawiw MA, Han K, Pelletier M, Sauve AA, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125:4592–4600. doi: 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, Brenner C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Scientific reports. 2016;6:26933. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. Journal of neurochemistry. 2015;133:739–749. doi: 10.1111/jnc.13037. [DOI] [PubMed] [Google Scholar]
- Vazquez EJ, Berthiaume JM, Kamath V, Achike O, Buchanan E, Montano MM, Chandler MP, Miyagi M, Rosca MG. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc Res. 2015;107:453–465. doi: 10.1093/cvr/cvv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Vielhaber S, Niessen HG, Debska-Vielhaber G, Kudin AP, Wellmer J, Kaufmann J, Schonfeld MA, Fendrich R, Willker W, Leibfritz D, et al. Subfield-specific loss of hippocampal N-acetyl aspartate in temporal lobe epilepsy. Epilepsia. 2008;49:40–50. doi: 10.1111/j.1528-1167.2007.01280.x. [DOI] [PubMed] [Google Scholar]
- Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Molecular cell. 2014;54:5–16. doi: 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Payne RM. Mitochondrial acetylation and diseases of aging. Journal of aging research. 2011;2011:234875. doi: 10.4061/2011/234875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Pride PM, Babbey CM, Payne RM. Friedreich’s ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Human molecular genetics. 2012;21:2688–2697. doi: 10.1093/hmg/dds095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014;158:1324–1334. doi: 10.1016/j.cell.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, Zhai QW, Su DF, Miao CY. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Annals of neurology. 2011;69:360–374. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Zhao XH, Chen W, Bo N, Wang XJ, Chi ZF, Wu W. Sirtuin 1 activation enhances the PGC-1alpha/mitochondrial antioxidant system pathway in status epilepticus. Mol Med Rep. 2015;11:521–526. doi: 10.3892/mmr.2014.2724. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Thompson KW, Suchomelova L, Niquet J. Brain energy metabolism during experimental neonatal seizures. Neurochem Res. 2010;35:2193–2198. doi: 10.1007/s11064-010-0339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir HJ, Lane JD, Balthasar N. SIRT3: A Central Regulator of Mitochondrial Adaptation in Health and Disease. Genes & cancer. 2013;4:118–124. doi: 10.1177/1947601913476949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Klaidman LK, Nalbandian A, Oliver J, Chang ML, Chan PH, Adams JD., Jr The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neuroscience letters. 2002;333:91–94. doi: 10.1016/s0304-3940(02)01005-4. [DOI] [PubMed] [Google Scholar]
- Yin TC, Britt JK, De Jesus-Cortes H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, et al. P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep. 2014;8:1731–1740. doi: 10.1016/j.celrep.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012;287:14078–14086. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhu Y, Zhan G, Fenik P, Panossian L, Wang MM, Reid S, Lai D, Davis JG, Baur JA, et al. Extended wakefulness: compromised metabolics in and degeneration of locus ceruleus neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:4418–4431. doi: 10.1523/JNEUROSCI.5025-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Guan YF, Zhou XM, Li GQ, Li ZY, Zhou CC, Wang P, Miao CY. Regenerative Neurogenesis After Ischemic Stroke Promoted by Nicotinamide Phosphoribosyltransferase-Nicotinamide Adenine Dinucleotide Cascade. Stroke. 2015;46:1966–1974. doi: 10.1161/STROKEAHA.115.009216. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Wang Z, Wang Y, You G, Jiang T. IDH1/2 mutation is associated with seizure as an initial symptom in low-grade glioma: A report of 311 Chinese adult glioma patients. Epilepsy Res. 2015;109:100–105. doi: 10.1016/j.eplepsyres.2014.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.