

Abstract
The thermostable Geobacillus stearothermophilus GsI-IIC intron is among the few bacterial group II introns found to proliferate to high copy number in its host genome. Here, we developed a bacterial genetic assay for retrohoming and biochemical assays for protein-dependent and self-splicing of GsI-IIC. We found that GsI-IIC, like other group IIC introns, retrohomes into sites having a 5′-exon DNA hairpin, typically from a bacterial transcription terminator, followed by short intron-binding sequences (IBSs) recognized by base pairing of exon-binding sequences (EBSs) in the intron RNA. Intron RNA insertion occurs preferentially but not exclusively into the parental lagging strand at DNA replication forks, using a nascent lagging strand DNA as a primer for reverse transcription. In vivo mobility assays, selections, and mutagenesis indicated that a variety of GC-rich DNA hairpins of 7–19 bp with continuous base pairs or internal elbow regions support efficient intron mobility and identified a critically recognized nucleotide (T-5) between the hairpin and IBS1, a feature not reported previously for group IIC introns. Neither the hairpin nor T-5 is required for intron-excision or lariat formation during RNA splicing, but the 5′-exon sequence can affect the efficiency of exon ligation. Structural modeling suggests that the 5′-exon DNA hairpin and T-5 bind to the thumb and DNA-binding domains of GsI-IIC reverse transcriptase. This mode of DNA target site recognition enables the intron to proliferate to high copy number by recognizing numerous transcription terminators and then finding the best match for the EBS/IBS interactions within a short distance downstream.
Keywords: DNA-protein interactions, reverse transcriptase, retrohoming, ribozyme, SHAPE
Graphical abstract
Introduction
Mobile group II introns are bacterial and organellar retrotransposons comprised of an autocata-lytic intron RNA (a “ribozyme”) and an intron-encoded protein (IEP), which is a reverse tran-scriptase (RT) [1]. They have been of interest because of their ribozyme-based RNA splicing and mobility mechanisms; their use as bacterial gene targeting vectors (“targetrons”); as a source of novel RTs, including thermostable group II intron RTs (TGIRTs), for RNA-seq and other bio-technological applications; and as evolutionary predecessors of spliceosomal introns, the spliceo-some, non-LTR-retrotransposons, telomerase, and retroviruses in eukaryotes [1–3]. Mobile group II introns are hypothesized to have evolved in bacteria, entered ancestral eukaryotes with bacterial endosymbionts that gave rise to mitochondria, proliferated to high copy number in what became the nuclear genome, and then evolved into spliceosomal introns, with dissociated group II intron domains evolving into snRNAs that reconstitute to form the catalytic core of the spliceo-some [2–4]. While recent structural studies have strongly supported this hypothesis [5–10], the nature of the last common ancestor of group II and spliceosomal introns and how it proliferated to high copy numbers in the genomes of early eukaryotes have remained unclear.
Mobile group II introns propagate in genomes by a ribozyme-based DNA integration mechanism called “retrohoming” in which the excised intron RNA resulting from RNA splicing inserts directly into a DNA target site and is reverse transcribed by the intron-encoded RT [11–14]. First, the intron-encoded RT assists splicing by binding to the intron RNA and promoting formation of the catalytically active RNA structure. The RT then remains bound to excised in-tron lariat RNA in an RNP (ribonucleoprotein) that initiates retrohoming by using both the protein and base pairing of the intron RNA to recognize a DNA target site [15,16]. After DNA target site recognition, the excised intron RNA in the RNPs uses its ribozyme activity to integrate by reverse splicing directly into the DNA strand to which the intron RNA is base paired, leading to the insertion of the intron RNA between the two DNA exons. The integrated intron RNA is then reverse transcribed by the intron-encoded RT, yielding an intron cDNA that is fully integrated into the recipient DNA by host cell DNA recombination or repair mechanisms [17–20]. Over time, mobile group II introns retrohome to ectopic sites [21,22], and this is thought to be the major mechanism by which group II introns or their close relatives proliferated in nuclear genomes of ancestral eukaryotes before evolving into spliceosomal introns [23]. However, most bacterial group II introns are present in only one or a few copies per genome, and only a few have been found to proliferate to high copy numbers [24].
Present-day mobile group II introns have evolved into three major structural classes denoted IIA, IIB, and IIC, which differ in features of their RNA splicing and mobility mechanisms [1]. Group IIA and IIB introns are larger than group IIC introns and typically encode RTs with a C-terminal DNA endonuclease domain (EN), which nicks the target DNA to generate the primer for reverse transcription of the intron RNA [12,15,25]. By contrast, the smaller group IIC introns encode RTs lacking an EN domain and use nascent strands at DNA replication forks to prime reverse transcription [23,26]. Additionally, group IIA and IIB introns utilize three sets of base-pairing interactions to recognize exon sequences for RNA splicing and DNA integration (EBS1-IBS1 and EBS2-IBS2 in the 5′ exon, and δ-δ′ (IIA) or EBS3-IBS3 (IIB) in the 3′ exon). In contrast, group IIC introns utilize only two sets of base-pairing interactions (EBS1/IBS1 and EBS3/IBS3), and IBS2 is replaced by a 5′-exon DNA hairpin, typically from a bacterial transcription terminator or integron-insertion site, which plays a major role in DNA target site recognition [26]. Group IIC introns are thought to insert preferentially into single-stranded regions of the parental lagging strand at DNA replication forks, where formation of the 5′-exon DNA hairpin is facilitated and the intron can use a nascent lagging DNA strand as primer for reverse transcription [26–29]. Whether the 5′-exon hairpin is recognized similarly for RNA splicing and whether recognition of the hairpin involves the intron-encoded protein or intron RNA have also remained unclear.
Here we focused on the thermostable Geobacillus stearothermophilus GsI-IIC intron, which belongs to a family of group II introns found to proliferate to high copy number in a variety of mesophilic and thermophilic bacteria [30]. This intron is the source of the thermostable group II intron RT, GsI-IIC RT (sold commercially as TGIRT-III), for which we recently determined an X-ray crystal structure of the full-length protein in a catalytic conformation bound to template-primer substrate and incoming dNTP [31]. We developed an Escherichia coli genetic assay for studying the retrohoming of GsI-IIC in vivo along with biochemical assays for self-splicing and protein-dependent splicing to complement the structural analysis of GsI-IIC RT. Our studies reveal new features of group IIC intron DNA target site recognition and RNA splicing and establish an experimental system that will enable comprehensive analysis of group IIC intron splicing, mobility, and reverse transcription mechanisms.
Results
G. stearothermophilus group IIC introns
A previous study identified 17 copies of the GsI-IIC intron in what was then a partial genome sequence of G. stearothermophilus strain 10 [32]. The completed genome revealed 45 copies of the intron, denoted here GsI-IIC1 to 45, which have ≥95% sequence identity to each other and comprise ~2.3% of the genome (Table 1; Fig. S1). Forty-four of these introns are intact and range in size from 1,881 to 1,894-nt, while the remaining intron, GsI-IIC41, has a transposon inserted within the intron ORF. The GsI-IIC intron is closely related to both the Bacillus halodurans intron B.h.I1, whose splicing and mobility mechanisms have been studied previously [26,29], and to the Oceanobacillus iheyensis group II intron, whose X-ray crystal structure has been determined [5] (~60% and ~50% identity to GsI-IIC over 480-nt of the ribozyme core, respectively).
Table 1.
GsI-IIC introns in G. stearothermophilus strain 10 (accession number: CP008934). In-trons are numbered 1 to 45 in order of their genomic location. GsI-IIC3, which has the most common intron RNA sequence, was set as a standard for comparison of other introns. Length indicates the length of the intron in nt, with type A and B referring to introns containing the longer (type B) or shorter (type A) form of the DIIb stem-loop (see Fig. 1). Mutations indicate changes in the intron relative to GsI-IIC3, including base substitutions, insertions (+), and deletions (Δ). Accession numbers are listed for each RT, along with any changes compared to the RT encoded by GsI-IIC3. GsI-IIC41 has a 3,130-nt transposon inserted after amino acid residue 133 of the intron ORF. The final columns indicate the protein-coding gene upstream of the intron and the distance between its termination codon and the intron insertion site. Introns that are inserted within genes are indicated as being “Inside” with * indicating insertion in the sense (spliceable) orientation, and ** indicating insertion in the antisense (non-spliceable) orientation.
| Intron | Genomic location | Length (nt) |
Type | Mutations | RT Accession | RT Mutation (aa) |
Upstream gene | Distance (nt) |
|
|---|---|---|---|---|---|---|---|---|---|
| 5′ end | 3′ end | ||||||||
|
| |||||||||
| 1 | 90362 | 88479 | 1884 | A | C347A | WP_053413520, ALA68829 | 105 | DUF1320 domain-containing protein | 72 |
| 2 | 113253 | 111370 | 1884 | A | WP_053413546, ALA68851 | Acetoacetyl-CoA ligase | 64 | ||
| 3 | 130439 | 128546 | 1894 | B | WP_053413546, ALA68868 | Nucleotidyltransferase | In-side* | ||
| 4 | 310616 | 312509 | 1894 | B | G33A | WP_053413546, ALA69020 | Bifunctional diguanylate cyclase/phosphodiesterase | 41 | |
| 5 | 316517 | 318410 | 1894 | B | WP_053413546, ALA69026 | Cold-shock protein CspB | 73 | ||
| 6 | 362250 | 360367 | 1884 | A | G213A | WP_053413546, ALA69069 | RNA-binding protein Hfq | 62 | |
| 7 | 386691 | 384808 | 1884 | A | WP_053413546, ALA69087 | Recombinase RecA | 42 | ||
| 8 | 569299 | 567406 | 1894 | B | WP_053413546, ALA69261 | RNA polymerase sporulation sigma factor SigE | 34 | ||
| 9 | 667580 | 665697 | 1884 | A | WP_053413768, ALA69363 | 49,105,66 | YqzE family protein | 36 | |
| 10 | 834441 | 832548 | 1894 | B | WP_053413546, ALA69535 | Prephenate dehydratase | 45 | ||
| 11 | 882716 | 880833 | 1884 | A | WP_053413546, ALA69582 | YihA family ribosome biogenesis GTP-binding protein EngB | 37 | ||
| 12 | 1035372 | 1033488 | 1885 | A | +1 nt after 257 | WP_053413546, ALA69722 | Thiol peroxidase | 33 | |
| 13 | 1041637 | 1039754 | 1884 | A | A165G, U288C | WP_053413910, ALA69729 | 137 | Acyl-CoA ligase | 98 |
| 14 | 1098849 | 1096956 | 1894 | B | WP_053413546, ALA69777 | Rhodanese-like domain-containing protein | 250 | ||
| 15 | 1107091 | 1108974 | 1884 | A | A165G | WP_013522881, ALA69786 | 49,105 | Gamma carbonic anhydrase family protein | 21 |
| 16 | 1216594 | 1214701 | 1894 | B | WP_053413546, ALA69883 | Hypothetical protein | 75 | ||
| 17 | 1225036 | 1223153 | 1884 | A | WP_053413546, ALA69891 | TIGR01457 family HAD-type hydrolase | 38 | ||
| 18 | 1250285 | 1248392 | 1894 | B | WP_053413546, ALA69919 | Fe-S cluster assembly protein SufB | 201 | ||
| 19 | 1332211 | 1334094 | 1884 | A | WP_053413546, ALA69995 | ATP-dependent Clp protease proteolytic subunit | 94 | ||
| 20 | 1562077 | 1560188 | 1890 | A | +6 nt after 552 | WP_053413768, ALA70188 | 49,105,66 | DUF1861 domain-containing protein | 146 |
| 21 | 1591714 | 1589834 | 1881 | A | C430U, A1887G, Δ366-368 | WP_053414153, ALA70217 | 49,105,40,41,379 | Hypothetical protein | 32 |
| 22 | 1644550 | 1642667 | 1884 | A | WP_053413546, ALA70276 | Fructose-1,6-bisphosphate aldolase, class II | 40 | ||
| 23 | 1722290 | 1720407 | 1884 | A | WP_053413546, ALA70344 | RNA methyltransferase | Inside* | ||
| 24 | 1784959 | 1786842 | 1884 | A | WP_053413546, ALA70396 | Seryl-tRNA synthetase | 219 | ||
| 25 | 1892784 | 1894677 | 1894 | B | WP_053413546, ALA70492 | Elongation factor Tu | 48 | ||
| 26 | 1979108 | 1981001 | 1894 | B | WP_053413546, ALA70581 | Spore coat protein | 41 | ||
| 27 | 1992453 | 1994346 | 1894 | B | WP_053413546, ALA70591 | ATP-dependent helicase srmB | 13 | ||
| 28 | 2012352 | 2014245 | 1894 | B | WP_013522881, ALA70605 | 49,105 | tRNA (A(37)-N6)-threonylcarbamoyltransferase subunit TsaD | 65 | |
| 29 | 2027255 | 2029148 | 1894 | B | WP_013522881, ALA70620 | 49,105 | Class II fumarate hydratase | 41 | |
| 30 | 2142821 | 2144714 | 1894 | B | WP_053413546, ALA70707 | MFS transporter | 223 | ||
| 31 | 2230026 | 2231909 | 1884 | A | WP_053413546, ALA70779 | L-lactate permease | 182 | ||
| 32 | 2313797 | 2315690 | 1894 | B | WP_053413546, ALA70861 | Fur transcriptional repressor | 36 | ||
| 33 | 2375555 | 2377438 | 1884 | A | WP_053413546, ALA70914 | HlyC/CorC family transporter | 56 | ||
| 34 | 2532418 | 2530525 | 1894 | B | WP_053413768, ALA71042 | 49,105,66 | Metal-sulfur cluster assembly factor PaaD | 60 | |
| 35 | 2702663 | 2700780 | 1884 | A | WP_053413546, ALA71201 | RNA polymerase subunit sigma peudogene | 24 | ||
| 36 | 2735871 | 2737764 | 1894 | B | WP_013522881, ALA71232 | 49,105 | Phosphate propanoyltransferase | 60 | |
| 37 | 2752726 | 2754609 | 1884 | A | WP_053413546, ALA71246 | Hypothetical protein | 33 | ||
| 38 | 2789544 | 2791427 | 1884 | A | WP_013522881, ALA71278 | 49,105 | Hypothetical protein | 24 | |
| 39 | 2848575 | 2850468 | 1894 | B | WP_053413546, ALA71335 | Two-component sensor histidine kinase | Inside* | ||
| 40 | 2989610 | 2991503 | 1894 | B | WP_053413546, ALA71452 | HAMP domain-containing protein | 45 | ||
| 41 | 3399882 | 3404880 | 4999 | A | WP_053414828, WP_053414829 | Hypothetical protein | 20 | ||
| 42 | 3458333 | 3456444 | 1890 | A | +6 nt after 552 | WP_053413768, ALA71876 | 49,105,66 | Nicotinate phosphoribosyltransferase | 145 |
| 43 | 3492554 | 3490671 | 1884 | A | WP_013522881, ALA71896 | 49,105 | Nitrate reductase subunit beta | In-side** | |
| 44 | 3553833 | 3551940 | 1894 | B | WP_053413546, ALA71946 | Methylmalonate-semialdehyde dehydrogen-ase (CoA acylating) | 49 | ||
| 45 | 3600422 | 3598529 | 1894 | B | WP_053413546, ALA71984 | Alanyl-tRNA editing protein | In-side** | ||
Fig. 1a shows the predicted secondary structure of the GsI-IIC3 intron, with sequence differences between different copies of the intron highlighted in red. The structure is typical of group IIC introns with six conserved group II intron secondary structure domains (DI-DVI), which interact via tertiary structure contacts (Greek letters). DI contains the short EBS1 and EBS3 motifs that base pair with the complementary IBS1 and IBS3 motifs in the 5′ and 3′ exons, respectively. Comparison of the different copies of GsI-IIC in the G. stearothermophilus strain 10 genome revealed that they fall into two secondary structure classes (denoted A and B), which differ in the length of DIIb (Fig. 1a, red inset near DIIb), with 21 introns having the longer and 24 introns having the shorter DII, likely reflecting two distinct lineages of actively mobile in-trons (Table 1). Most of the other sequence differences between different introns are either single nucleotide changes or small indels in or adjacent to loops or bulges and are not expected to affect the intron’s ribozyme function. Exceptions are found in GsI-IIC2, which has a potentially disruptive single-nucleotide change at the base of DIII, and GsI-IIC21, which has the branch-point A residue in DVI changed to G and a 3-nt deletion in the loop of DIII (Fig. 1a; Table 1).
Fig. 1.

The GsI-IIC intron RNA and reverse transcriptase. (a) Predicted secondary structure of GsI-IIC3. The intron RNA is comprised of six secondary structure domains (DI-VI), which interact via tertiary contacts (Greek letters). The location of the ORF encoding the RT in DIV is indicated by a loop. Sequence variations between different copies of the intron in the G. stea-rothermophilus strain 10 genome are indicated by red letters. Red circles indicate mutations that affect base pairing or introduce deletions, green circles indicate compensatory changes in base pairs, and black circles indicate mutations in or near loops or bulged nucleotides in stems. (b) Predicted secondary structures of DIV of GsI-IIC3 and the closely related B. brevis and O. ihey-ensis group IIC introns. Green and red highlights indicate the start and stop codons of the RT ORF, respectively. (c) Schematic of the GsI-IIC RT. Conserved sequence blocks found in all RTs are denoted RT1-7 and indicated by black boxes [61]. RT0, 2a, and 3a (red) indicate additional regions conserved in group II intron and non-LTR-retrotransposon RTs [31,61,62]. Amino acid sequence variations in the different copies of the GsI-IIC RT in the genome are indicated above with the number of occurrences in parenthesis. Conserved sequence motifs that are found in group II intron RTs and known to be important for RT activity are shown below the schematic.
DIV, which encompasses the intron ORF, was partially folded with Mfold [33] using the sequences of closely related group IIC introns found in Brevibacillus brevis (strain NBRC100599) and O. iheyensis (strain HTE831) to constrain the results by assuming that their DIVs fold into similar structures (Fig. 1b). In all three introns, the ATG initiation codon of the RT ORF (green) is within the “loop” of DIVb, 9 to 16 nt from the end of the DIVb stem, and the stop codon (red) is in the stem at the base of DIV (Fig. 1b). The location of the initiation codon in these group IIC introns differs from that in the previously studied Lactococcus lactis Ll.LtrB group IIA intron where the ATG is in DIVa and overlaps a high-affinity binding site for the IEP, enabling protein-binding to autoregulate translation of the intron ORF [34,35]. The conservation of the DIVa secondary structure in this family of group IIC introns and the finding below that DIVa could not be shortened appreciably without inhibiting intron mobility are consistent with a function as a high-affinity binding site for the IEP, but perhaps no longer coupled to translational regulation at the more distant translation initiation codon.
SHAPE analysis
We investigated the structure of GsI-IIC by SHAPE analysis of a 656-nt GsI-IIC-ΔORF in-tron with a deletion of the branch-point A-residue to prevent splicing during the incubations (denoted GsI-IIC-ΔORF+ΔA; Fig. 2a). SHAPE modification was done under single-hit conditions in reaction medium containing 5 mM Mg2+, where most of the secondary structure should be present but not the tertiary structure dependent upon the IEP [36]. By using a highly processive TGIRT enzyme for mapping SHAPE modifications, we were able to map the secondary structure of the entire 656-nt intron from a single primer annealed to the 3′ exon with high signal to noise ratio (Fig. 2b). The high signal to noise ratio avoids miscalls due to RT stops by the retroviral RT SuperScript III (SSIII), which is commonly used for mapping SHAPE modifications (see example in Fig. 2, inset top). The SHAPE reactivities confirmed most of the predicted secondary structure of the intron, including that of DIV, with SHAPE accessible nucleotides confined to single-stranded loops, bulges, or the ends of stems. An exception was the DIc stem, which showed moderately reactive nucleotides on one side of the helix, possibly reflecting that this region is strained, flexible, or mispaired under the reaction conditions. Notably, some tertiary structure elements remain accessible to the SHAPE reagent, including α′, θ, ς, ς′, and EBS3, suggesting a role for the IEP in stabilizing these interactions. In the Ll.LtrB group IIA intron, ς-ς′ and a subset of other tertiary interactions were likewise found to be stabilized by the IEP at low Mg2+ concentrations [36,37].
Fig. 2.

SHAPE analysis of GsI-IIC. SHAPE was done on an in vitro transcript containing the GsI-IIC-ΔORF+ΔA intron and short flanking exons with isatoic anhydride in reaction medium containing 5 mM Mg2+, as described in Materials and Methods. Modification sites were analyzed by primer extension with TeI4c-MRF RT [37] at 60 °C using a fluorescently labeled primer an-nealed near the 3′ end of the RNA, followed by capillary electrophoresis of the resulting cDNAs. (a) Predicted secondary structure of the GsI-IIC-ΔORF+ΔA intron showing SHAPE reactivities. Nucleotides are numbered from the 5′ end of the RNA used in the experiment. Nucleotide sequences involved in long-range tertiary interactions are boxed, circled, or indicated by arrows and are named with Greek letters. Colors indicate levels of SHAPE reactivities according to the scale in panel b below. The insert at the top compares cDNA raw traces produced by TeI4c-MRF and SuperScript III (SSIII) RTs. Peaks in the raw trace from capillary electrophoresis represent reverse transcription stops at single nucleotide resolution. Premature stops during primer extensions with SSIII RT created false-positive SHAPE reactivities, whereas TeI4c-MRF produced results expected for a group IIC intron. (b) Plot of SHAPE reactivity across the GsI-IIC intron. SHAPE reactivities were calculated for each nucleotide by using QuSHAPE [55]. SHAPE reactivities: red, high; orange, medium; black, none.
GsI-IIC-encoded RTs
All 45 copies of GsI-IIC encode an RT of 420 amino acids with >99% identity to each other (Fig. 1c). The GsI-IIC34 protein has been shown to have high RT activity and a version of this RT with a proprietary solubility tag is sold commercially as TGIRT-III [37]. All of the intron-encoded RTs, except for the one with a transposon inserted in the IEP ORF, are full-length and have the conserved YADD motif at the RT active site along with other conserved RT motifs (Fig. 1c, bottom). Amino acid substitutions in the RTs encoded by different copies of the intron are present at only seven positions (Y40D, I41V, H49R, E66G, S105P, M137T, N379K; Fig. 1b). The GsI-IIC21 protein, which is encoded in the intron with a mutation in the branch-point A residue and a deletion in the loop of DIII, has the most changes in the RT (Y40D, I41V, R49H, S105P, N379K), including three changes (Y40D, I41V, N379K) that are not found in other copies of the intron, possibly reflecting that the intron and/or its encoded RT are no longer functional (Fig. 1c; Table 1).
Genomic insertion sites of the GsI-IIC intron
All 45 copies of the GsI-IIC intron are found downstream of predicted hairpin structures of 7–19 bp, and introns inserted in the top and bottom strand are largely but not completely segregated on opposite sides of the G. stearothermophilus genome (Figs. 3 and S1; Table 1). These features are as expected for a group IIC intron that inserts preferentially downstream of DNA hairpins in the parental lagging strand at DNA replication forks of a genome undergoing bidirectional replication [1,26], but also suggest some ability to insert downstream of DNA hairpins on the opposite strand, as borne out by intron mobility assays below. In 40 cases, the hairpin corresponds to a putative transcription terminator, but surprisingly, in the remaining five cases (GsI-IIC3, GsI-IIC23, GsI-IIC39, GsI-IIC43 and GsI-IIC45), the hairpin and inserted intron are located within a gene (Table 1). In two of these cases (GsI-IIC43 and GsI-IIC45), the inserted introns are in the antisense orientation and thus could not be spliced from their respective mRNAs.
Fig. 3.

GsI-IIC insertion sites in the G. stearothemophilus strain 10 gene. (a) Sequence alignments comparing the 45 GsI-IIC insertion sites. Red letters indicate nucleotides that potentially base pair to form a 5′-exon DNA hairpin structure upstream of the intron-insertion site. Green highlighting indicates nucleotides in IBS1 than can base pair with EBS1 in the intron RNA, and yellow highlighting indicates the C or T residues at the IBS3 position that can base pair with the G at EBS3 in the intron RNA. A red highlight indicates mutation of the branch-point A residue of GsI-IIC21. The short vertical lines in the alignment indicate the exon-intron boundary. The intron EBS1 and EBS3 motifs are shown at the bottom in blue and the two nucleotides downstream of EBS1, which are conserved in all 45 copies of the intron, are shown in black. A black square indicates the center of the hairpin’s loop; for loops with odd numbers of nucleotides the 5′ half of the loop contains the additional nucleotide. (b) WebLogo showing sequence conservation in the 5′ and 3′ exons generated using WebLogo3 with standard parameters [63]. (c) Distance of the intron-insertion site from the top of the 5′-exon DNA hairpin. The variable distance between the top of hairpin loop (black) and intron-insertion site can also be seen in (a).
A web logo based on the 45 intron-insertion sites shows conservation of the IBS1 and IBS3 motifs (positions −4 to +1) recognized by base pairing of the intron RNA, but surprisingly also shows strong conservation of the T residue at position −5 immediately upstream of IBS1 (42 of 45 target sites; Fig. 3b). At many sites, T-5 is part of a run of T-residues that follows a putative bacterial transcription terminator hairpin and is either in a predicted single-stranded spacer region or part of an extended hairpin that could form by base pairing with runs of A residues upstream of the terminator hairpin (Fig. 3).
The intron-insertion sites are located within a window of 16–25 nt measured from the center of the loop of the 5′-exon hairpin, with a peak from 18–22 nt (Fig. 3c). Although most of the hairpins are GC-rich (31 of 45 have >50% G-C pairs), they otherwise have relatively little sequence conservation. Most (37 out of 45) have at least one A-T or T-G pair, a bulge, or a mismatch in the middle of the hairpin separating two GC-rich stem regions. All 45 copies of GsI-IIC have the same 4-nt EBS1 (5′-UGGA) and 1-nt EBS3 (G), but IBS1 and IBS3 at the intron-insertion sites differ, with some IBS1s (16 of 45) containing as many as 2 mismatches out of the 4 possible base pairs with EBS1. IBS3 corresponds to a single nucleotide that can base pair with the G residue at EBS3 (T in 36 introns and C in 8 introns), with one site (GsI-IIC11) having a non-complementary A at the IBS3 position (Figs. 1 and 3). The very short EBS1/IBS1 and EBS3/IBS3 interactions and the relatively frequent occurrence of mispairings suggest that DNA target site recognition by GsI-IIC intron is dictated largely by the 5′-exon DNA hairpin and possibly the conserved residue T-5.
A genetic assay for GsI-IIC retrohoming and the effect of deletions in DIV
To study the GsI-IIC retrohoming mechanism, we adapted an E. coli plasmid-based mobility assay used previously for other group II introns (Fig. 4a) [25,38,39]. In this assay, an intron-donor plasmid (pADC2X-GsI-IIC-ΔORF+T7 with a chloramphenicol resistance (capR) marker) uses a T7lac promoter (PT7lac) to express a precursor RNA containing a GsI-IIC-ΔORF intron with a phage T7 promoter (PT7) inserted near its 3′ end. The RT needed to splice the intron and promote intron mobility is expressed in tandem from a position downstream of the 3′ exon (E2). The recipient plasmid (pBRR3-GeoTS, with an ampicillin resistance (ampR) marker) contains a DNA target site (TS; the ligated E1-E2 sequence) cloned upstream of a promoterless tetracycline resistance (tetR) gene, so that retrohoming of the intron carrying the T7 promoter into the target site activates that gene and confers tetracycline resistance. We made two versions of the recipient plasmid (denoted LEAD and LAG), which differ in the orientation of the replication origin relative to the target site and tetR gene and require the use of nascent leading or lagging strands, respectively, as primers for reverse transcription of the inserted intron RNA [40]. The assays were done in E. coli HMS174 (DE3), which contains an IPTG-inducible T7 RNA polymerase, with intron expression induced with IPTG for 1 h at 48 °C, the highest temperature that could be used without affecting cell viability [25]. Mobility efficiencies were determined in plating assays from the ratio of (TetR+AmpR)/AmpR colonies.
Fig. 4.

In vivo intron mobility assay and requirement of the 5′-exon hairpin structure for retrohoming. (a) Schematic of the assay showing donor and recipient plasmids and the retrohoming product. Two different versions of the recipient plasmid with the replication origin in either orientation (indicated by a bidirectional arrow) relative to the GsI-IIC target site (TS; ligated E1-E2 sequence) were used in the experiments. The two orientations are denoted LEAD or LAG depending on whether nascent leading or lagging strand DNAs could used as primers for reverse transcription of GsI-IIC. PT7, phage T7 RNA polymerase promoter; T1 and T2, E. coli rrnB T1 and T2 transcription terminators; TΦ, phage T7 Φ terminator. (b) Mobility assays with recipient plasmids containing target sites derived from three different GsI-IIC insertion sites in the G. stearothermophilus genome (TS7, TS22, and TS34 5′ exons with TS12 3′ exons, denoted TS7, TS22, and TS34, respectively). Nucleotide residues that can base pair to form the 5′-exon hairpin are boxed and highlighted by red shading, and the IBS1 sequence is boxed. The complementary EBS1 sequence in the intron is shown in blue. Base pairs are indicated by dashes. rG•dT and rU•dG are considered valid base pairs in the EBS/IBS pairings, and in some sequence contexts dG•dT pairs can form weak wobble pairs [41,64]. The bar graphs below show the mobility efficiency of the three target sites cloned in either the LEAD (blue) or LAG (orange) recipient plasmids. (c) Mobility assays of the wild-type and mutant TS34 target sites in which the 5′-exon hairpin was deleted, disrupted, or replaced. Nucleotides that differ from the TS34 target site are shown in green. Other features of the target sites are depicted as in panel (b). The bar graph below shows the mobility efficiencies (measured by the ratio of (TetR + AmpR)/AmpR) colonies) of the wild-type TS34 and mutant target sites cloned in the LAG recipient plasmid and assayed in parallel. The inset in the plot at the bottom right shows an expanded scale for mutants that have very low mobility efficiencies. In both (b) an (c), the bar shows the mean for three experiments with the error bar indicating the standard deviation.
Attempts to further streamline the GsI-IIC-ΔORF intron in the donor plasmid by deleting non-essential regions of DIV showed that deletion of DIVa and the remaining region of DIVb decreased mobility efficiency by six orders of magnitude (Fig. S2). Smaller deletions were possible, but combining the two least severe deletions (DIVa5, 31 nt, and DIVb2, 18 nt) decreased the mobility efficiency 3-fold. In view of the marginal advantage of deleting small regions of DIV, the parental GsI-IIC-ΔORF construct was used for all remaining experiments.
The 5′-exon DNA hairpin is required for GsI-IIC mobility
Comparing the 45 different genomic insertion sites of the GsI-IIC intron suggested that DNA target specificity is dictated largely by sequence elements in the 5′ exon with only the single IBS3 nucleotide in the 3′ exon contributing to DNA target site recognition, as has been found for other group IIC introns (Fig. 3; [26]). Supporting this inference, we found no significant difference in intron mobility efficiency for DNA target sites containing the TS34 5′ exon in combination with TS34, TS23, or TS12 3′ exons, all of which contain a canonical T residue at the IBS3 position, but have no other common features (Fig. S3). We therefore focused on recognition elements in the 5′ exon.
To examine the contribution of the 5′-exon hairpin to DNA target site recognition, we selected 5′-exon sequences from three different target sites (TS7, TS22, and TS34) and tested each in combination with the 3′-exon sequence from a fourth target site (TS12), thereby equalizing any contribution from the 3′ exon (Fig. 4b). The TS7, TS22, and TS34 sites differ in the length of the hairpin (10 to 14 continuous base pairs including internal T-G wobble pairs), the presence of bulged nucleotides in the hairpin, and the number of potential base pairs in the IBS1/EBS1 interactions (2 or 4). All three DNA target sites contain the conserved T-5 residue upstream of IBS1 (Fig. 3, 4).
For all three target sites, the insertion frequency into the LAG recipient plasmid was about 10-fold higher (9.4 to 11.5-fold) than in the corresponding LEAD recipient, indicating preferential use of nascent lagging strands as primers for reverse transcription (Fig. 4b). Colony PCR and sequencing of homing products from both the LEAD and LAG target sites showed correct insertion in all cases. These findings confirm that GsI-IIC preferentially uses nascent lagging strand DNAs as primers for reverse transcription of the intron RNA, but also indicate appreciable ability to integrate into the opposite strand where use of a nascent leading strand or some other type of primer would be required (see Discussion). Comparing the three target sites, mobility efficiencies were highest for TS34 and lowest for TS22, which has the fewest potential EBS1/IBS1 base pairs, as well as a bulged nucleotide within the hairpin (Fig. 4b).
Deleting the hairpin from the TS34 target site (ΔHP), changing one side of the hairpin to its complement (HP disrupt), or replacing the hairpin with an unrelated sequence of equal length (HP replace) decreased the ratio of (TetR+AmpR)/AmpR colonies in the LAG orientation by ~250-fold (Fig. 4c). Further, colony PCR and sequencing showed that this residual mobility did not reflect insertions into the mutated target site, but rather ectopic insertions into an E. coli rRNA gene T2 transcription terminator, which is present in the recipient plasmid to suppress read-through by E. coli RNA polymerase into the tetR gene (Fig. 4a; [38]). Thus, a 5′-hairpin structure is essential for DNA target site recognition by the GsI-IIC intron.
Mobility assays comparing the TS34 5′-exon hairpin with 5′-exon hairpins having continuous Watson-Crick base pairs
TS34, the most efficient of the three target sites tested above, forms a predicted DNA hairpin of 10 bp including a T-G pair (a weak context-dependent wobble pair in DNA; [41]) in the middle of the hairpin. Of the 45 5′-exon DNA hairpins at GsI-IIC insertion sites in the strain 10 genome, 19 contain one or more internal T-G pairs, 7 contain mismatches, and 8 contain bulges, leaving only 18 hairpins with just Watson-Crick base pairs (Fig. 3). T-G pairs and bulges within the 5′-exon DNA hairpin are also common in target sites for other group IIC introns, including the Bacillus halodurans intron B.h.I1 and Symbiobacterium thermophilum intron S.th.I1 [26].
To further assess critical features of the 5′-exon DNA hairpins, we carried out additional mobility assays comparing the TS34 target site with 4 additional target sites having different length DNA hairpins with continuous Watson-Crick base pairs, along with fully paired EBS/IBS interactions, and the conserved T-5 residue (Fig. 5a). Despite the difference in hairpin structure, TS34 and two other target sites, TS4 and TS31 (with 16 and 11 continuous Watson-Crick base pairs, respectively) supported similarly high mobility efficiencies (41–44%, as measured by the ratio of (TetR + AmpR)/AmpR colonies). The TS31 target site (11 bp) had a somewhat lower mobility efficiency (32%), while the TS8 target site with the shortest hairpin (7 bp) had the lowest mobility efficiency (9%), although still relatively high by absolute standards (Fig. 5b). These findings indicate that GsI-IIC can recognize and insert efficiently downstream of a variety of DNA hairpin structures.
Fig. 5.

Intron mobility assays comparing the TS34 hairpin with 5′-exon hairpins having continuous Watson-Crick base pairs. (a) Predicted secondary structures of the TS34 hairpin and a subset of 5′-exon hairpins comprised of continuous Watson-Crick base pairs from other GsI-IIC target sites in the G. stearothermophilus strain 10 genome. (b) Mobility efficiencies of the target sites containing the 5′-exon hairpins shown in (a). Mobility assays were performed with recipient plasmids containing the 5′-exon from the indicated target site combined with the same TS23 3′ exon. The target sites were cloned in the LAG recipient plasmid, and mobility efficiencies were determined by the ratio of (TetR+ AmpR)/(AmpR) colonies, as described in Fig. 4. The bar graphs show the mean and standard deviation for 3 independent experiments.
Identification of features of the TS34 5′-exon hairpin and neighboring regions required for DNA target site recognition by in vivo selection and mutagenesis
To further characterize features in the 5′-exon hairpin region that are important for intron mobility, we carried out an in vivo selection experiment using the two-plasmid mobility assay with a TS34 target site recipient plasmid in which the 5′-exon nucleotides upstream of IBS1 (positions −5 to −37) were partially randomized (“doped”) at 70% of the wild-type nucleotide and 10% of each of the other three nucleotides. After induction of donor plasmid expression with IPTG, tetracycline-resistant bacteria were selected by growing in liquid medium containing tet-racycline and homing products were amplified by PCR using a 5′ primer just upstream of the hairpin and a 3′ primer within the intron. The resulting DNAs were sequenced on an Illumina HiSeq4000 instrument to obtain 6,597,196 150-nt paired-end reads. To exclude PCR duplicates, two sets of 8-nt barcodes were included in the primers used for DNA-seq library construction, and the raw sequences were filtered so that only sequences with unique barcodes and the length expected for insertion at the homing site were analyzed (total of 5,749,781 sequences)
The sequencing data showed that the selected hairpins consist of upper and lower stem regions (6 and 4 bp, respectively), which contain strongly selected G-C base pairs, separated by the T-G ‘elbow’ region at which Watson-Crick base pairing was counterselected (Fig. 6a and b). Other strongly conserved nucleotides include T-5, which lies downstream of the hairpin and immediately upstream of IBS1 (Fig. 6a), consistent with its presence in 42 of the 45 naturally occurring GsI-IIC target sites (see Fig. 3), and four nearly invariant nucleotides within the hairpin on both sides of the elbow (G-10, G-16, C-32, and C-33).
Fig. 6.

In vivo selection of 5′-exon sequences required for GsI-IIC retrohoming. (a) Sequence and predicted secondary structure of the TS34 5′ exon and degree of selection at different nucleotide positions. The 5′-exon structure is shown to the left, with Watson-Crick base-paired region of the hairpin highlighted in a red box and the region partially randomized for the selection enclosed in a green box. The degree of selection at each nucleotide is shown via color code on the hairpin to the right: +++, nucleotides present in >99% of selected sequences; ++, remaining nucleotides present at >15% higher frequency in selected than in the unselected sequences; +, remaining nucleotides present at >5–14% higher frequency in the selected than in unselected sequences; ±, nucleotide present at similar frequencies (±4%) in selected and unselected sequences; -, nucleotide present at 5–14% lower frequency in selected than in unselected sequences; --, nucleotide present at >15% lower frequency in selected than unselected sequences. Arrows pointing to circled letters indicate nucleotides that are found >2-fold more frequently after selection. (b) Selection for or against base pairing within the hairpin and flanking regions. The horizontal bar graphs (right) show the frequency of Watson-Crick base pairs at each position in the 5′-exon hairpin in the selected and unselected sequences (blue and red, respectively). Brackets on the far right delineate the upper and lower stems in which Watson-Crick base pairing is selected for in active target sites, and the TG elbow in which Watson-Crick base pairing is selected against. (c) Intron mobility assays with mutant DNA target sites. The top shows 5′-exon sequences of wild-type TS34 and mutant target sites depicted schematically as in panel (a). Mutations are shown as green nucleotides within red boxes or circles. The bar graphs below show mobility efficiencies for wild-type and mutant target sites in the LAG recipient plasmid, with the inset showing an expanded y-axis for the T-5 mutants, which have very low mobility efficiencies. The data are the mean for three independent experiments with the error bars indicating the standard deviation.
Intron-mobility assays with mutant DNA target sites showed that replacement of the T-G elbow and adjacent A-T base pair with two G-C base pairs to make a stable continuous helix decreased the mobility efficiency by ~5-fold (Elbow 1 and 2 mutants; Fig. 6c). Thus, flexibility at these internal positions may be important for optimal recognition of the TS34 hairpin. Mutation of T-5, the conserved nucleotide that lies in the linker region between the hairpin and IBS1, to any other nucleotide residue decreased the mobility efficiency by >250-fold, whereas mutating the adjacent nucleotide G-6 to a T residue had less effect on intron mobility (4-fold decrease). Although T-5 is immediately upstream of IBS1, it does not appear to be recognized by an extended EBS1/IBS1 base-pairing interaction, as the corresponding position in the EBS1 loop is a highly conserved C residue, which cannot form a canonical base pair with T-5, and the T-5G mutation in the 5′ exon, which could potentially extend the EBS1/IBS interaction to include the -5 position, strongly decreased intron mobility (see Figs. 1a, 3, and 6c). These considerations suggest that T-5 in the DNA target site is most likely recognized by the IEP (see Discussion).
Protein-dependent and self-splicing of GsI-IIC
To investigate the splicing activity of the GsI-IIC RT, we developed an in vitro assay in which the purified protein was incubated with a 32P-labeled precursor RNA containing the 656-nt GsI-IIC-ΔORF intron with short flanking exons. As in the intron mobility assays, we started by testing splicing with precursor RNAs containing 5′-exon sequences corresponding to those at the TS7, TS22, and TS34 insertion sites. Fig. 7a shows that in reaction medium containing 5 mM Mg2+ at 50 °C, the GsI-IIC RT spliced all three constructs to produce ligated exons (confirmed by sequencing) and excised intron lariat RNA, whereas no splicing was observed under these conditions in the absence of the protein. In reaction medium containing a higher Mg2+ concentration (100 mM), all three constructs self-spliced hydrolytically (i.e., without branching) to produce linear intron RNA and ligated exons (Fig. 7a), as observed previously for other group IIC introns [26,29,42]. As was the case for intron mobility, both protein-dependent and self-splicing were most efficient with the TS34 construct and least efficient for the TS22 construct, which has two mismatches in the EBS1/IBS1 interaction (see Fig. 4b). Consequently, the TS34 construct was used for all subsequent experiments.
Fig. 7.

Protein-dependent and self-splicing of GsI-IIC. (a) Protein-dependent and self-splicing reactions of GsI-IIC from precursor RNAs with different 5′ exons. 32P-labeled precursor RNAs (10 nM) were incubated with GsI-IIC RT (20 nM) for 10 min at 50 °C in reaction medium containing 5 mM Mg2+ (protein-dependent splicing conditons), without protein in reaction medium containing 100 mM Mg2+ (self-splicing conditions), no Mg2+ (non-splicing control), or 5 mM Mg2+ (5 mM; control for self-splicing under protein-dependent splicing conditions). Bands are identified to the right of the gel. The gel is split to show differently exposed top and bottom portions. (b) Time courses of the protein-dependent splicing reaction. 40 nM GsI-IIC RNA was incubated with various amounts of purified GsI-IIC RT (20 to 200 nM, color coded as indicated to the right). Samples were taken at different times, and the products were analyzed in a denaturing 4% polyacrylamide gel, which was dried and scanned with a Phosphorimager. The sets of curves at the top and bottom show disappearence of precursor RNA and appearance of intron lariat RNA, respectively. k1 and k2 indicate the fast and slow rate constants obtained from fitting the data to an equation with two exponentials using Prism6 (GraphPad Software).
Experiments examining the splicing of the TS34 construct as a function of temperature showed that the protein-dependent and self-splicing reactions have temperature optima of 50 and 60 °C, respectively. Protein-dependent splicing was more efficient than self-splicing at temperatures below 40 °C and at 75 °C, the highest temperature tested (Fig. S4).
Time course and apparent stoichiometry of the protein-dependent splicing reaction
Time-course experiments with GsI-IIC/TS34 precursor RNA (40 nM) and different concentrations of GsI-IIC RT (20 to 200 nM) in reaction medium containing 5 mM Mg2+ at 50 °C showed that protein-dependent splicing occurred at a rate of ~6 min-1 at saturating protein concentrations with the reaction complete after ~5 min and ~73% of the precursor RNA spliced at completion (Figs. 7b and S5). The remaining precursor RNA was not spliced even at 5-fold molar excess of protein and is presumably in an inactive conformation.
Because group II intron RTs remain tightly bound to the excised intron RNA after splicing, splicing reactions are largely limited to a single turnover, and it is possible to calculate an apparent stoichiometry for the number of protein molecules required to splice a single RNA molecule [43]. In the case of GsI-IIC, the calculation is complicated by the fraction of unreactive precursor RNA, which may or may not be bound by GsI-IIC RT. For the splicing reaction at 40 nM RNA and protein, the amount of spliced RNA at completion (10 min time point) was 15.4 nM, corresponding to an apparent stoichiometry of 2.6 (assuming unreactive precursor is not bound to protein) or 1.9 (assuming unreactive precursor is bound to protein, Fig. 7b; see Materials and Methods for details of the calculation). These findings are consistent with those for the Ll.LtrB group IIA intron in which the stoichiometry determined similarly by RNA splicing or in binding assays was ~2:1 [43,44]. Although this stoichiometry has been taken to suggest that group II intron RTs function in splicing as a dimer, such measurements have limitations and other explanations are also possible (see Discussion).
Effect of 5′-exon mutations on RNA splicing
Next, we tested the effect of mutations in the 5′ exon on protein-dependent and self-splicing (Figs. 8 and 9). Because the presence of the 5′-exon hairpin causes RNAs to run anomalously in gels, even under denaturing conditions, the splicing reactions were carried out with both internally labeled and 5′-end labeled precursor RNAs to help identify splicing products (Figs. 8a and 9a, respectively). Additionally, ligated exons and free 5′ and 3′ exons resulting from the reactions were analyzed by high-throughput sequencing using a method (TGIRT-seq) that enables precise mapping of both the 5′ and 3′ ends of small RNAs to confirm the identity of products (Fig. 9b and c).
Fig. 8.

Effect of 5′-exon mutations on protein-dependent and self-splicing of GsI-IIC. Splicing reactions for wild-type and the indicated mutant introns were carried out for 10 min at 50° C using 40 nM RNA and 80 nM protein, as described in Fig. 6. The top panel shows reactions run on a denaturing 6% polyacrylamide gel, and the bottom panel shows the same samples run on a denaturing 10% polyacrylamide gel for higher resolution of small RNAs. Splicing products are identified to the right of the gel. Lane numbers are on the bottom. The positions of RNA size markers (M, Decade RNA ladder; Thermo Fisher) run in parallel lanes of the 10% polyacryla-mide gel are indicated on the left. Ligated exons, 5′ exon (E1), and 3′ exon (E2) are indicated by black, blue, and green arrows, respectively. * indicates cleaved or mis-spliced RNAs. High-throughput sequencing data show that the doublet bands for ligated exons and free 3′ exons reflect an extra, non-templated G residue at their 3′ ends in about 50% of the reads resulting from in vitro transcription.
Fig. 9.

Splicing of wild-type and 5′-exon mutant GsI-IIC and high-throughput sequencing of reaction products. (a) Splicing reactions were done as described in Fig. 7 using 5′ 32P end-labeled precursor RNAs, and the reaction products were analyzed in a denaturing 12% polyacrylamide gel, which was dried and scanned with a Phosphorimager. The positions of RNA size markers (M, Decade RNA ladder; Thermo Fisher) are shown on the left, and major splicing products are labeled on the right. Ligated exons and 5′ exon (E1) are indicated by black and blue arrows, respectively. * indicates cleaved or mis-spliced RNAs. (b) Read spans of RNA splicing products determined from concordant paired-end sequences that mapped to 5′ exon, 3′ exon, or ligated exons (including alternative ligated exons). The plots show the percent (%) of the normalized reads of different read spans that mapped to exons for wild-type and each mutant. The alternatively spliced ligated exons and free 5′ exon (lane 20 in panel a) have a good match to IBS1/3 around the cleavage site (5′-tCCT|T, where “|” indicates the intron insertion site and upper case letters indicate base pairing to EBS1/3). The 5′-truncated E2 is not a prominent band in the gels and has poor matches to IBS1/3 at the cleavage sites (5′-tgCc|T or 5′tTTa|T). (c) Bar graph showing the percentage of 5′ exon, 3′ exon, or ligated exons (including alternative ligated exons) in mapped paired-end reads for wild-type and each mutant. The 7-nt E1 from the ΔHP construct is too short for the sequence to be mapped unequivocally and is not included in the analysis shown in panels (b) and (c).
In the protein-dependent splicing reactions, neither deletion of the 5′-exon hairpin leaving only a short (7-nt) 5′ exon containing the IBS1 sequence (ΔHP) nor replacement of the hairpin region with an equal length of vector sequence (HP replace) decreased the production of excised intron lariat RNA (Fig. 8, lanes 5 and 13). However, the two mutants differed in the efficiency of exon ligation. The ΔHP mutant containing only a very short 5′ exon without the hairpin region produced only small amounts of correctly ligated exons (confirmed by sequencing) together with large amounts of free 5′-exon (Fig. 9a, lane 7), indicating substantial inhibition of exon ligation, whereas the HP replace mutant in which the 5′-exon hairpin region was replaced with vector sequence produced near wild-type levels of correctly ligated exons (Fig. 8, lane 13; Fig. 9a, lane 19; note ligated exons for the HP replace mutant, which lack a long 5′-exon hairpin, run behind wild-type ligated exons in Fig. 8, but were confirmed by sequencing in Fig. 9). The HP disrupt mutant (5′ part of the hairpin changed to its complement) showed no detectable protein-dependent splicing, reflecting either defective RNA folding or impaired binding of GsI-IIC RT under the protein-dependent splicing conditions (Fig. 8, lane 9, and Fig. 9a, lane 15). Mutation of T-5, which is required for efficient intron mobility (see above), to any other nucleotide or the G-6T mutation had no negative effect on the protein-dependent splicing reaction (Fig. 8, lanes 17–32). We conclude that neither a long 5′-exon hairpin nor T-5 is required for any step in protein-dependent splicing, but the results for the ΔHP and HP replace mutants indicate that the 5′-exon sequence can affect the efficiency of exon ligation.
In the self-splicing reaction, all of the above mutations produced substantial amounts of excised linear intron, which thus appears to depend primarily on the EBS/IBS interactions rather than recognition of the 5′-exon hairpin, in agreement with previous results for the B.h.I1 intron [29]. The ΔHP mutation resulted in higher levels of free 5′ and 3′ exons, and the HP replace mutant resulted in the use of an alternative 5′-splice site and an increased proportion of free 5′ exons (Figs. 8 and 9; note the free 7-nt 5′ exon for the ΔHP mutant is too small to be detected in Fig. 8 and is detectable but too small to be sequenced in Fig. 9). These aberrant products were not observed for protein-dependent splicing of the same mutants and presumably reflect construct-specific RNA structures that form in the mutant RNAs at high Mg2+ concentration in the absence of protein. Surprisingly, the HP disrupt mutation, which abolished protein-dependent splicing (Fig. 8, lane 9), had a much smaller effect on self-splicing, with the mutant producing substantial amounts of excised intron and correctly ligated exons (confirmed by sequencing), despite its inability to form a long 5′-exon hairpin (Fig. 8, lane 10 and Fig. 9). We conclude that although some 5′-exon mutations can affect the efficiency or accuracy of exon ligation, a 5′-exon hairpin structure is not absolutely required for any step in either protein-dependent or self-splicing. This conclusion differs from that for self-splicing of the B.h.I1 intron, where several lines of evidence indicated that recognition of a 5′-exon hairpin is required for exon ligation [29].
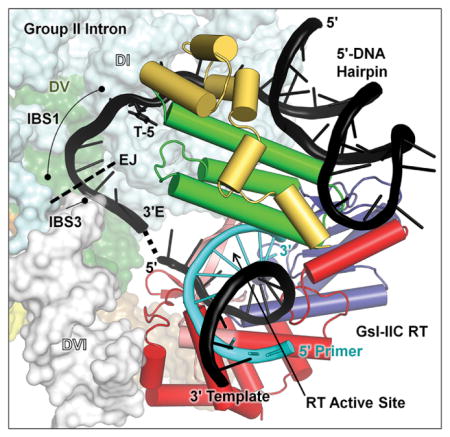
Structural modeling of DNA target site recognition by GsI-IIC RNPs
We recently determined a 3.0-Å crystal structure of full-length GsI-IIC RT bound to template/primer substrate and used it to construct a model of a GsI-IIC RNP bound to a DNA target site just prior to the reverse splicing step of retrohoming [31]. Based on the preceding results, we modified the model to incorporate features of the efficient TS34 DNA hairpin including the 5′-exon hairpin and T-5. The model shows that the 5′-exon hairpin fits in a basic cleft formed by the interface of the thumb (green) and DNA-binding domains (yellow). T-5 lies near the top of the extended helical structure formed by the thumb and D domains, in position to interact with amino acid residues in one or both of these domains. The putative hairpin-binding site in the RT may be optimized to accommodate B-form DNA helices, but able to bind A-form RNA helices and other sufficiently long 5′-exon RNAs lacking hairpins with lower affinity during RNA splicing, accounting for the stringent requirement for 5′-exon DNA hairpins for intron mobility and the variable effect of different 5′ exon mutations on the efficiency of exon-ligation. As noted previously, the model indicates that after reverse splicing of the intron RNA into the DNA strand, the RT active site is positioned to initiate reverse transcription just downstream of the integrated intron RNA, enabling seamless coupling of reverse splicing and reverse transcription for group IIC introns [31]. In the updated model, assuming no further structural rearrangements, the GsI-IIC RT is positioned to initiate reverse transcription from a nascent lagging strand primer at position 7 of the 3′ exon, raising the question of how the intervening DNA sequence is copied prior to reverse transcription of the intron RNA (see Discussion).
Discussion
Here, we studied G. stearothermophilus GsI-IIC, a group IIC intron that has proliferated to a high copy number within its host genome. We found that GsI-IIC, like other group IIC introns, inserts downstream of DNA hairpins between the IBS1 and IBS3 motifs recognized by base pairing of EBS1 and EBS3 motifs in the intron RNA. Both the distribution of genomic insertions sites (Fig. S1) and in vivo mobility assays (Fig. 4) indicated that intron insertion occurs preferentially into the strand used as the template for lagging strand DNA synthesis (LAG orientation), but that insertion can also occur in the LEAD orientation at appreciable frequency (~10% that in the LAG orientation in the mobility assays). This insertion preference presumably reflects the greater accessibility of single-stranded regions of the lagging-strand template at DNA replication forks, which facilitates the formation of the 5′-exon DNA hairpin and enables the direct use of nascent lagging strand DNAs as primers for reverse transcription. The smaller number of insertions in the LEAD orientation presumably occurred by reverse splicing of the intron into double-strand DNA prior to passage of a replication fork, possibly at transcription bubbles or DNA regions that become transiently single stranded at elevated temperature to enable formation of the 5′-exon hairpin [26]. Such insertions could have used nascent leading strands or possibly 3′ ends generated at DNA nicks as primers for reverse transcription of the intron RNA.
The 5′-exon DNA hairpin is the most critical structural feature recognized by GsI-IIC for intron mobility. All genomic insertion sites of GsI-IIC have an upstream hairpin structure, and in vivo mobility assays showed that deletion or replacement of the hairpin abolishes GsI-IIC retrohoming into the target site (Fig. 4). Mobility assays showed that DNA hairpins of 10–16 bp with either continuous Watson-Crick base pairs or an internal T-G wobble pair supported similarly high mobility efficiencies, while a shorter (7 bp) hairpin decreased mobility efficiency by about 4-fold (Fig. 5). In vivo selection and mutagenesis of an efficient 5′-exon DNA hairpin with an internal T-G pair showed that optimally recognized variants consist of two stable stem regions (4 and 5 bp) separated by a T-G elbow region at which Watson-Crick base pairing is counterselected (Fig. 6). Further, replacement of the T-G elbow and adjoining A-T base pair with two G-C pairs to make a stable continuous stem decreased mobility efficiency by five-fold (Fig. 7). Thus, a variety of GC-rich DNA hairpins of different lengths and structures can support efficient mobility, and in some cases, the hairpin benefits from a flexible elbow region that may enable it to better fit into a shared hairpin-binding site in the GsI-IIC RT (see below).
Surprisingly, in addition to the 5′-exon hairpin, we found that T-5, located in the spacer region between the hairpin and IBS1, contributes strongly to DNA target site recognition. T-5 is conserved in 42 of the 45 GsI-IIC insertion sites in the G. stearothermophilus genome (Fig. 3), and mutation of T-5 to any other residue deceases retrohoming efficiencies by >250-fold (Fig. 6c). Base recognition by a group IIC intron RT has not been reported previously and could be a secondary adaptation of GsI-IIC, perhaps related to the need for tighter binding at high temperatures. Alternatively, bacterial transcription terminator hairpins, which comprise most group IIC intron target sites, are typically flanked by runs of upstream A residues and downstream T residues, which could have obscured base recognition in the spacer region by other group IIC introns [45]. In the case of the B.h.I1 intron, deletion of one or more of the four T residues in the spacer strongly inhibited intron integration in an in vitro assay, while replacement of the four Ts with four C residues appeared to moderately decrease intron integration efficiency in the gel shown [26]. In addition, of the 21 copies of a group IIC intron found in Symbiobacterium thermophilum, 19 have T-5 [26,46].
In contrast to intron mobility, our results indicate that neither a long 5′-exon hairpin nor T-5 is essential for RNA splicing, although the length and sequence of the 5′ exon can affect the efficiency of exon ligation. Thus, constructs in which the 5′-exon hairpin was deleted leaving only a 7-nt 5′-exon (ΔHP mutant) or replaced with an equal length of unrelated vector sequence (HP replace mutant) produced substantial amounts of correctly excised intron RNA in both protein-dependent and self-splicing reactions (Figs. 8 and 9). In protein-dependent splicing reactions, the ΔHP mutant showed strongly decreased exon ligation with a corresponding increase in free 5′ exon, whereas the HP replace mutant produced wild-type levels of correctly ligated exons. In self-splicing reactions, the ΔHP and HP replace mutants used cryptic alternative cleavage and splice sites, whereas the HP disrupt mutant, which the 5′ side of the hairpin was changed to its complement, produced substantial amounts of correctly excised intron and ligated exons (Figs. 8 and 9). Previous studies of self-splicing of the B.h.I1 group IIC intron likewise indicated the EBS1/IBS1 interaction is by itself sufficient for exon-definition and precise intron excision during RNA splicing and that a 5′-exon hairpin might contribute to the efficiency of exon ligation [29]. Our findings extend these studies by showing that sufficiently long 5′ exons that are not predicted to form a long 5′-exon hairpin can support relatively efficient exon ligation in either protein-dependent or self-splicing.
We found that protein-dependent splicing of the GsI-IIC intron in vitro occurs at an apparent stoichiometry of 1.9 to 2.6 molecules GsI-IIC RT to one molecule intron RNA (Fig. 7), consistent with previous findings for the Ll.LtrB group IIA intron, which showed that the Ll.LtrB RT (LtrA protein) is monomeric in solution, but binds and splices the intron RNA at a stoichiometry of ~2:1 [43,44]. A 2:1 stoichiometry is consistent with the possibility that group II intron RTs function in splicing as a dimer, but could also reflect that splicing requires two monomers bound independently to the intron RNA, as well as limitations of the approach, in particular the difficulty of determining the concentration of active protein. Structural evidence for a group II intron RT dimer is lacking. Both the cryo-EM structure of the Ll.LtrB RT bound to intron RNA lariat and the X-ray crystal structure of GsI-IIC RT bound to template/primer showed only a single bound monomer [7,31]. A dimer interface seen in the crystal structure of a group II intron RT fragment [47] is likely non-physiological as it is not compatible with the location of the thumb in the full-length group II intron RTs in the preceding structures. Further experiments with a number of different group II introns will be required to address these issues.
Our analysis of the DNA target site of the GsI-IIC RT combined with the recent crystal structure of the full-length GsI-IIC RT primed for reverse transcription [31] enabled us to construct a structural model of a GsI-IIC intron lariat RNP bound to the DNA target site (Fig. 10). The model suggests that the 5′-exon hairpin binds in a basic cleft formed by the thumb and DNA-binding domains of the GsI-IIC RT. The binding of the hairpin in the basic cleft via non-sequence-specific electrostatic interactions could enable a variety of hairpins having different sequences and lengths to be accommodated, thereby allowing group IIC introns to recognize multiple transcription terminators for intron insertion. Further, our in vitro selections and muta-genesis (Fig. 6) suggest that some hairpins require a flexible elbow region to fit optimally into the hairpin-binding site. The recognition of the hairpin in the basic cleft between thumb and D domains of the IEP is likely to be generally relevant for group IIC introns, all of which insert downstream of hairpin structures. Sequence alignments of RTs closely related to the GsI-IIC RT show that some of the basic residues in the cleft are strongly conserved.
Fig. 10.

Model of the interaction of a GsI-IIC RNP with a DNA target site containing a 5′-exon hairpin just prior to the reverse splicing step of retrohoming. The complete model of the intron RNP is shown to the left, with the GsI-IIC RT binding region magnified in the box to the right. The position of T-5 is indicated. The model was derived from that in Stamos et al. [31] with the 5′-exon sequence of GsI-IIC34 replacing a generic hairpin. The intron model was constructed from the O. iheyensis intron lariat structure (pdb 5J02), with ligated exon DNA modeled based on an RNA bound to EBS1 and EBS3 in pdb 3IGI. The GsI-IIC RT containing a template-primer duplex (pdb 6AR1) was docked onto the intron RNA using positioning information from both the Ll.LtrB group IIA intron RNP (pdb 5G2X) and spliceosomal Prp8 (pdb 5GAN) cryo-EM structures. The hairpin was docked into the GsI-IIC RT structure using the 5′ exon from the Ll.LtrB RNP cryo-EM structure as a guide. EJ indicates the 5′-exon/3′-exon junction. Black cartoon: model DNA target site with 5′-exon DNA hairpin with the dashed line indicating the gap between the ligated exon DNA model and the RT-bound template strand; T-5 shown in stick figure. Cyan cartoon: primer; space-filling model: O. iheyensis intron; cylindrical cartoon: GsI-IIC RT, red: RT group II intron-specific inserts; salmon: RT fingers; blue: RT palm; green: RT thumb; yellow: RT D domain. The schematic at the bottom left shows the intron configuration before and after the reverse splicing step of retrohoming
Our model indicates that T-5 could be recognized by GsI-IIC RT residues near the top of the extended helical structure formed by the thumb and D domains. In the O. iheyensis intron, upon which our model is based, the EBS1/IBS1 interaction is 6 bp instead of 4 bp as in the GsI-IIC intron and a G nucleotide at position -5 is base paired with a C residue at the corresponding position of EBS1 [6]. In the GsI-IIC intron, the corresponding position in the EBS1 loop is also a C residue, but the EBS1/IBS1 interaction is not extendable for intron mobility (as shown by the negative effect of the 5′ exon T-5G mutation), and no other intron RNA nucleotide is in close enough proximity to base pair to T-5. Although these considerations favor protein recognition of T-5, we cannot exclude the possibility that T-5 is recognized by an as yet unidentified interaction with intron RNA.
A model in which a basic cleft in the IEP is capable of binding diverse 5′-exon hairpins or other 5′-exon sequences largely via non-sequence-specific electrostatic interactions could also explain the different requirements for a long 5′-exon hairpin for intron mobility and RNA splicing. Thus, the hairpin-binding cleft in the RT may bind B-form DNA helices more tightly than A-form RNA helices, enabling ligated exons produced during RNA splicing to dissociate or be displaced by 5′-exon DNA hairpins for intron mobility. At the same time, the binding of the free 5′-exon RNA to the RNP after the first step of splicing may be required for efficient exon-ligation. Such a contribution would account for our finding that a very short 5′ exon with the hairpin deleted did not support efficient exon-ligation, whereas a longer 5′-exon in which the hairpin region was replaced with vector sequence gave near wild-type levels of correctly ligated exons (Figs. 7 and 8).
According to the model, the active site of GsI-IIC is positioned to initiate reverse transcription at position 7 in the 3′ exon. The use of nascent lagging strand DNA as primer requires dissociation of the replicative polymerase or DNA primase, which presumably occurs upon encountering the group II intron RNA stably integrated in the DNA target site. Although GsI-IIC has robust DNA polymerase activity required for copying the short DNA segment [48], it initiates inefficiently from a DNA primer annealed to a DNA template, and we cannot exclude more complex scenarios in which a DNA repair polymerase or DNA primase copies all or part of the short DNA segment prior to initiation of reverse transcription of the intron RNA. Previous studies with the Ll.LtrB group IIA intron raised the possibility that such enzymes might be involved in copying the short 5′ DNA left by asymmetric cleavage of the DNA target site prior to initiating target DNA-primer reverse transcription of the intron RNA [20].
In contrast to group IIA and IIB introns, where recognition of the DNA target site requires extended base pairing interactions with the intron RNA [16,49], our results suggest that DNA target site recognition by GsI-IIC RNPs is dictated primarily by the IEP, with a only a small albeit essential contribution from the short EBS/IBS pairings. Thus, all of the genomic insertion sites have an upstream hairpin, but some can form only 2 of 4 EBS1/IBS1 base pairs and have mismatches at IBS3, and our mobility assays showed directly that GsI-IIC could still integrate correctly into the TS22 target site with 2 mismatches in the 4 base pair EBS1/IBS1 interaction. This decreased dependence on intron RNA/DNA target site base pairing could reflect in part that retrohoming in G. stearothermophilus ordinarily occurs at high temperatures, which favor DNA melting, facilitating the formation of hairpins on the separated strands, while decreasing the energetic contribution of the base-pairing interactions. The variable distance between the DNA hairpin and intron-insertion sites suggests a scenario in which group IIC intron RNAs bind first to the DNA hairpin and then sample neighboring downstream sequences for EBS/IBS pairings adequate to permit intron insertion.
Bacterial group IIA and IIB introns that have inserted outside of essential genes or into non-essential genes are frequently degenerate, presumably reflecting that intron mobility is deleterious to the host, so that strains carrying active introns are lost by purifying selection [50]. Surprisingly, despite being inserted outside of genes, nearly all the GsI-IIC introns in the G. stea-rothermophilus genome are potentially active, as judged by retention of conserved structural features of both the intron RNA and intron-encoded RT (Fig. 1). The finding that most copies of the GsI-IIC intron may be functional despite being inserted downstream of transcription terminators could reflect either recent insertion or purifying selection against inactive copies of the intron. The latter could in turn reflect that these downstream, non-coding regions are ordinarily transcribed at a low level and have important functions, which require removal of the intron by RNA splicing.
Finally, the ability of GsI-IIC to proliferate to relatively high copy number in a bacterial genome is relatively rare for group II introns, which are typically found at one or two copies per genome [24]. The high copy number of GsI-IIC could be due to the large number of transcriptional terminator hairpin sequences that provide suitable target sites [26], combined with mobility at high temperatures, which makes the intron less dependent on base-pairing interactions that would provide more specificity for DNA insertion. High temperatures were found previously to favor mobility of the Thermosynechococcus elongatus TeI4c group IIB intron by decreasing dependence on protein recognition of specific bases for local DNA melting needed for intron RNA base pairing [25]. In previously studied cases in which bacterial group II introns have proliferated to high copy number, a significant proportion of the copies are present as twintrons in which one copy of the intron has inserted into another [25,51]. GsI-IIC and presumably other group IIC introns differ in being unable to form twintrons because there are no suitable hairpin target sites located within the intron, forcing this intron to colonize new genomic sites. The combination of colonization of new sites with minimal sequence requirements, a mobility mechanism that precludes formation of twintrons, and elevated temperatures, which are thought to have prevailed on Earth during the evolution of eukaryotes [52], could have contributed to intron proliferation in the nuclear genomes of ancestral eukaryotes.
Materials and Methods
Recombinant plasmids
The GsI-IIC RT used for RNA splicing assays was expressed from plasmid pMRF-GsI-IIC and purified as described [37]. The construct expresses the GsI-IIC RT with maltose-binding protein (MBP) fused to its N-terminus via a non-cleavable rigid linker in order to maintain solubility of the protein when removed from bound nucleic acids. Protein concentrations were determined using a Qubit 2.0 fluorometer and protein concentration kit (Invitrogen) according the manufacturer’s instructions.
Intron mobility assays used intron-donor plasmid pADC2X-GsI-IIC-ΔORF+T7 and recipient plasmids pBRR-GeoTS34-LEAD and pBRR-GeoTS34-LAG or derivatives thereof. pADC2X-GsI-IIC-ΔORF+T7 is a pACYC184-based plasmid [53] that uses a T7lac promoter to express a cassette consisting of a GsI-IIC-ΔORF intron and short flanking exons with a phage T7 promoter inserted in DIVb of the intron and the ORF encoding the IEP cloned downstream of the 3′ exon. It was constructed in two steps. First, the ORF encoding the IEP was amplified from pETGsI-IIC DNA [25] using Phusion PCR mix (New England Biolabs) with 5′ primer GeoI2ORF5+SDPst, which appends PstI and NdeI sites and a Shine-Dalgarno sequence from pET3, and 3′ primer GeoI2ORF3Xho, which appends a XhoI site (Table S1). The PCR product was digested with PstI and XhoI, gel purified, and swapped for the Ll.LtrB group II intron RT (LtrA protein) in pACD2X [54] cut with the same enzymes, thereby producing intermediate construct pADC2XgeoRT. The GsI-IIC intron was assembled from two PCR products that separately amplify 5′ and 3′ segments of the intron, while replacing a ~1.4 kb segment with a phage T7 promoter. These PCRs used outside primers (5′GsI2-3 and 3′GsI2-5, respectively), which append short flanking exons (57-nt 5′ exon from TS34 and 32-nt 3′ exon from TS23) and unique cloning sites (XbaI 5′ and BamHI 3′), in combination with overlapping internal primers (DIVt3 and DIVb3), which replace the intron ORF segment in DIVb with a T7 promoter sequence and an MluI site (Table S1). The assembled PCR product was then cloned between XbaI and BamHI sites of pADC2XgeoRT (see above) resulting in pADC2X-GsI-IIC-ΔORF+T7.
Intron-recipient plasmids pBRR-GeoTS34-LEAD and pBRR-GeoTS34-LAG contain GsI-IIC intron insertion sites (positions -40 to +20) cloned into previously described Ll.LtrB intron recipient plasmids pBRR3A and pBRR3B [40], which differ in the orientation of the replication origin relative to the target site and tetR reporter gene. They were constructed by replacing the Ll.LtrB target site in plasmids pBRR3A-ltrB (LEAD) and pBRR3B-ltrB (LAG) with annealed top and bottom strand oligonucleotides that contain the desired GsI-IIC insertion sites with AatII and EcoRI site overhangs (Table S1) for direct ligation into the gel-purified pBRR3A or pBRR3B backbone digested with the same enzymes. Recipient plasmids for other hairpins or mutated TS34 sites were constructed the same way.
Recombinant plasmids used for in vitro transcription of GsI-IIC intron RNAs for SHAPE and RNA splicing were derivatives of pGsI2C-35/32. This parent plasmid contains a 656-nt GsI-IIC-ΔORF intron (nucleotides 551-1791 replaced by CGC) flanked by short 5′ and 3′ exons (35 and 32 nt, respectively) cloned downstream of a phage T3 promoter between the HindIII and BamHI sites of pUC19 (New England BioLabs). The 5′- and 3′-exon sequences are those from GsI-IIC insertion site 34 and 23, respectively, in the G. stearothermophilus strain 10 genome. GsI-IIC-ΔORF+ΔA, the construct used for SHAPE, has a further deletion of the branch-point A-residue to inhibit RNA splicing during the incubations. Derivatives of pGsI2C-35/32 with different 5′ exons were constructed by PCR stitching of two overlapping PCR products, using outside primers Stch_HindIII_T3_For and Stch_BamHI, which appended HindIII and BamHI sites, respectively (Table S1). The segment containing the 5′ exon was produced by hybridizing top- and bottom-strand primers containing the desired sequence, while the common 3′ segment containing the intron and 3′ exon of TS23 was created via PCR from pGsI2C-35/32 with primers GsI2Cintron_F and Stch_BamHI, which contains the full intron and 3′ exon sequence (Table S1). The hairpin deletion was made using primers Stch_HindIII_T3_For(DEL) and Stch_BamHI to amplify from GsI2C-35/32 (Table S1). The 5′ exons for the T-5G, T-5A, and G-6T constructs were amplified from homing products using primers -5and-6Universal and HomingProd-uct_TTCA_G (Table S1). The PCR amplicons were purified by using a Wizard PCR Clean-Up System (Promega), digested with HindIII and BamHI, and swapped for the corresponding segment of pGsI2C-35/32.
Newly constructed plasmids were sequenced prior to use.
SHAPE analysis using TGIRT enzyme for read out of SHAPE modifications
SHAPE was done using a 722-nt RNA containing the GsI-IIC-ΔORF+ΔA intron with short flanking exons produced by in vitro transcription of gel-purified pGsI-IIC-ΔORF+ΔA DNA digested with HindIII. For SHAPE, the RNA (200 nM) was incubated in 50 μl of splicing reaction medium containing 450 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl, pH 7.5 at 60 °C for 30 min, and a 9 μl-portion was added to 1 μl of freshly prepared isatoic anhydride (50 mM in DMSO), while a second 9 μl portion was added to 1 μl 100% DMSO as a negative control. After incubating the solutions at 37 °C for 36 min (~5 half-lives of isatoic acid), the RNA was ethanol precipitated (3 volumes of ethanol, one-tenth volume of 3 M sodium acetate pH 5, and 1 μl of 20 mg/ml glycogen as carrier). Primer extension of 2 pmol SHAPE-modified or control RNAs was done using fluorescently labeled primer A (Table S1) annealed to the 3′ exon. The annealed template-primer substrate was preincubated with TGIRT TeI4c-MRF (2 μM; [37]) at room tem-perature for 30 min in a reaction medium containing 450 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl pH 7.5, and 5 mM DTT. Then cDNA synthesis was initiated by adding 1.5 mM dNTPs (an equimolar mix of 1.5 mM dATP, dCTP, dGTP, and dTTP) followed by incubation at 60 °C for 1 h. Reverse transcription using SuperScript III (Invitrogen) was done in parallel according to the manufacturer’s protocol. Reactions were stopped by adding NaOH to a final concentration of 0.1 M, incubating at 95 °C for 3 min, and neutralizing with HCl. As calibration for the capillary electrophoresis, sequencing reactions were performed using TGIRT TeI4c-MRF and SuperScript III, as described above, except that unmodified RNA was used as a template and a Cy3-labeled primer B (Table S1) of identical sequence to primer A and 1.5 mM ddCTP were added to the reaction. Cy5-labeled cDNAs synthesized from SHAPE-modified RNA or control RNA were mixed with Cy3-labeled cDNAs from sequencing reactions and analyzed by electrophoresis in a single capillary of a GenomeLab™ GeXP Genetic Analysis System (Beckman Coulter). For electrophoresis, samples were denatured at 90 °C for 180 sec, injected onto the capillary array at 2.0 kV for 30 sec, and separated at 4.8 kV for 80 min. The temperature of the capillary array was maintained at 60 °C throughout the separation. The raw trace was analyzed by automated QuSHAPE software [55]. SHAPE reactivities were then overlaid onto the predicted secondary structure of the GsI-IIC intron.
Intron mobility assays
Intron mobility assays were done in E. coli HMS174(DE3) (Millipore Sigma) grown in LB medium with antibiotics added as required at the following concentrations: ampicillin (Amp), 100 mg/ml; chloramphenicol (Cap), 25 mg/ml; tetracycline (Tet), 25 mg/ml. Cells that had been co-transformed with the CapR donor (pADC2X-GsI-IIC-ΔORF+T7) and AmpR recipient plas-mids (pBRR-GeoTS-LEAD or pBRR-GeoTS-LAG or derivatives thereof) were inoculated into 5 ml of LB medium containing chloramphenicol and ampicillin and grown with shaking (200 rpm) overnight at 37 °C. A small portion (50 μl) of the overnight culture was inoculated into 5 ml of fresh LB medium containing the same antibiotics and grown for 1 h as above. The cells were then induced by adding 1 ml of fresh LB medium containing the same antibiotics and 3 mM IPTG (isopropyl β-D-1-thiogalactopyranoside, 500 μM final) and incubating for 1 h at 48 °C. After induction, the cultures were placed on ice, diluted with ice-cold LB, and plated at different dilutions onto LB agar containing ampicillin or ampicillin plus tetracycline. The plates were incubated overnight at 37 °C, and mobility efficiencies were determined by the ratio of (TetR+AmpR)/AmpR colonies.
In vitro selection of 5′-exon features required for GsI-IIC retrohoming
For selection experiments, the target site in pBRR-TS34-LAG was replaced with one in which 5′-exon positions −5 to −37 from the intron-insertion site were doped with 70% of the wild-type nucleotide and 10% of each mutant nucleotide. To construct this plasmid, a top strand oligonucleotide (Geo34TOP) was synthesized with the doped nucleotide region (IDT), and a complementary strand was made by annealing a short primer (Geo34Bot; Table S1) to the fixed 3′ end of the doped oligonucleotide and filling in the bottom strand across the doped region using the DNA polymerase activity of GsI-IIC RT (reaction conditions: 98 °C for 2 min, 50 °C for 2 min, and 72 °C 5 min in a PCR machine). GsI-IIC RT was used because it was able to efficiently synthesize the complementary strand through the DNA hairpin region, while other DNA polymerases, including Phusion, Klenow, and Taq (New England Biolabs) could not. After the reaction, the product was cleaned with a MinElute PCR Purification Kit (Qiagen), digested with AatII and EcoRI-HF (New England Biolabs), and cloned between the corresponding sites of pBRR3ltrbLAG.
For the selection, the recipient plasmid containing the doped insert was electroporated into E. coli HMS174 (DE3) and transformants were selected in 50 ml of LB medium at 37 °C over night with selection for the AmpR marker on the plasmid. 10 ml of the overnight culture (the remainder was used to isolate plasmid DNA which represents the unselected library) were then grown in 1l SOB at 37°C and made electrocompetent for introduction of the donor plasmid pACD2-GsI-IIC-ΔORF+T7. HMS174 (DE3) cells carrying both the donor and recipient plas-mids were then grown in LB medium and induced with IPTG, as described above for intron mobility assays. Instead of plating, the induced cells were grown in 200 ml LB+Tet overnight and the plasmid DNA was isolated. Illumina adapters (NG_GsI-IIC_HomingProd_3_For and NG_GsI-IIC_HomingProd_3_Rev (each containing an 8-nt bar code; Table S1) were appended to the tetracycline-selected homing product through PCR. The PCR product was then purified with Agencourt AMpure XP beads and standard Illumina tails were added by another round of PCR (6 cycles) [56]. The final libraries were cleaned up with Agencourt AMpure XP beads and sequenced at the University of Texas at Austin Genomic Sequencing and Analysis Facility on an Illumina HiSeq 4000 to obtain 150-nt paired-end reads. The raw reads (6,597,196 for the selected library and 26,745,370 for the unselected library) were filtered by their 16-nt barcode so that the filtered sequences contain only unique barcodes. Ambiguous sequences with Ns in the doped region and/or barcode were removed, as were sequences in which the doped region was shorter or longer than the expected length of 33 nt (<1% of the reads from the selected library and ~1% of the reads from the unselected library), leaving 5,749,781 unique sequences for final analysis. Galaxy was used to convert the raw NextGen sequencing data to FASTA format and for further trimming of the sequences [57]. Nucleotide and base-pair frequencies were calculated using Py-thon scripts and plotted using Excel (Microsoft).
RNA splicing assays
Splicing assays were performed by using either internally or 5′ 32P-labeled precursor RNAs containing the 656-nt GsI-IIC-ΔORF intron flanked by a 35-nt 5′ exon (positions −1 to −35 of TS34) and 32-nt 3′ exon (position +1 to +32 of TS23). Precursor RNAs were transcribed from an amplicon generated by PCR of pGsI2C-35/32 or derivatives thereof (see above) with primers Stch_HindIII_T3_For and GsI2c35_3-EX (Table S1). In vitro transcription was done by using phage T3 polymerase (60 units per 100 μl reaction; Thermo Fisher Scientific) with 2 mM of each NTP and 124 nM [α-32P] UTP (3,000 Ci/mmol; Perkin-Elmer) for 2.5 h at 37 °C. 2 mM dTTP was included in the reaction to sequester excess free Mg2+ ions, which increases hydrolytic splicing during transcription. The transcription reaction (200 μl) was treated with 8-μl RNase-free DNase I (Thermo Scientific) for 15 min at 37 °C and cleaned up with a MEGAclear Transcrip-tion Clean-Up Kit (Thermo Scientific). Unlabeled RNA was made the same way by omitting the [α-32P] UTP. 5′-end labeling of in vitro transcripts was done with [γ-32P] ATP (3,000 Ci/mmol, Perkin Elmer) and T4 polynucleotide kinase (New England Biolabs) according to the manufacturer’s protocol. Intron RNAs were re-folded prior to use by heating to 85 °C for 2 min in distilled water and then incubating at 50 °C for 2 min in 450 mM KCl, 5 mM MgCl2, and 20 mM Tris-HCl, pH 7.5.
Splicing reactions were carried out by incubating the 32P-labeled precursor RNA (10 to 40 nM) with GsI-IIC-MRF RT [37] at concentrations specified for individual experiments in 20 μl of reaction medium containing 450 mM KCl, 5 mM MgCl2, and 20 mM Tris-HCl, pH 7.5. The reactions were initiated by adding protein, which had been pre-warmed to 50 °C for 30 sec (confirmed to result in no loss of activity), incubated at 50 °C for times specified in Figure Legends for individual experiments, and terminated by adding 30-μl ice-cold phenol-chloroform-isoamyl alcohol (25:24:1; phenol-CIA) and 0.5 μl 500 mM EDTA. Splicing products were analyzed in denaturing 4% to 12% polyacrylamide gels, which were dried and scanned with a Phosphorim-ager (Typhoon FLA 9500; GE Healthcare Life Sciences). Band intensities were quantified by using ImageQuant TL (GE Healthcare Life Sciences). Data were normalized by subracting the background and fitted to a two exponential function (Y = plateau + a*e-K1t + b*e-K2t) with Prism6 (GraphPad Software).
The apparent stoichiometry of protein to RNA for the splicing reaction was calculated from the data in Fig. 7b assuming that all of the protein was active and correcting for the 26.8% (10.7 nM) of precursor RNA that remains unreactive at a saturating protein concentrations (≥80 nM). In the simplest model, this inactive precursor RNA is unable to bind protein, and we calculated the amount of protein required for splicing simply by dividing the protein input (40 nM) by the amplitude of the splicing reaction (15.4 nM, based on disappearance of precursor obtained from fitting the data to a single exponential function). This yields an apparent stoichiometry of 2.6 protein molecules per spliced RNA. In the second model, we assume that the inactive precursor RNA binds the protein in the same way as the splicing-competent precursor RNA. At 40 nM protein, 15.4 nM precursor RNA was spliced and 5.6 nM was unspliced but now assumed to be bound to protein, yielding a total of 21 nM RNA bound to protein for an apparent stoichiometry of 1.9 protein molecules bound per RNA molecule.
High-throughput TGIRT sequencing of splicing products
For analysis of ligated, free, and alternatively spliced exons, splicing reactions were done as described above with 80 nM protein and 40 nM RNA for 15 min at 50 °C, and small RNAs (< 200 nt) were isolated with an RNA Clean & Concentrator Kit (Zymo Research). After assessment and quantification on a 2100 Bioanalyzer (Agilent) using a Small RNA chip, small RNAs were analyzed by TGIRT-seq, as described [56]. Briefly, reactions were assembled by mixing RNA sample (10–12 ng), template-primer (100 nM, 34 nt RNA oligonucleotide (R2 RNA, Table S1) annealed to a 35 nt complementary DNA primer (R2R DNA; Table S1), and TGIRT enzyme (500 nM GsI-IIC RT; InGex) in 450 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl, pH 7.5, 1 mM DTT in 19.2 μl total volume. After pre-incubating at room temperature for 30 min, reactions were initiated by adding 0.8 μl 25 mM dNTPs (final concentration 1 mM dATP, dCTP, dGTP, and dTTP) and incubated for 15 min at 60 °C. The reverse transcription reaction was stopped by adding 1 μl of 5 N NaOH, and incubated at 95 °C for 3 min to degrade RNA. After cooling to room temperature, the mixtures were neutralized by adding 1 μl of 5 N HCl, and cDNAs were purified twice with a MinElute Reaction Cleanup Kit (QIAGEN). cDNAs were then ligated to a 5′-adenylated/3′-blocked (C3 spacer, 3SpC3; IDT) adapter (R1R; Table S1) by using thermostable 5′ AppDNA/RNA Ligase (New England Biolabs), and the ligated cDNAs were re-purified with a MinElute Reaction Cleanup Kit (QIAGEN) and amplified with Phusion High-Fidelity DNA polymerase (Thermo Fisher) with 200 nM of Illumina multiplex and 200 nM of barcode primers (Table S1). PCR was done at 98 °C for 5 sec pre-denaturation followed by 12 cycles of 98 °C for 5 sec, 60 °C for 10 sec, and 72 °C for 10 sec. The libraries were purified with a 1.3X volume of Agencourt AMPure XP beads (Beckman Coulter) to remove adapter dimers and sequenced on an Illumina miSeq V2 PE2x250 to obtain approximately 1 million 75-nt paired-end reads for each sample. After trimming of Illumina TruSeq adapters and PCR primer sequences with cutadapt [58] (sequencing quality score cut-off at 20; p-value < 0.01) and discarding reads of ≤7 nt, the reads were mapped to the precursor and expected ligated exons with Bowtie2 v2.2.6 [59] with local alignment, and custom setting of “ --no-mixed --norc --no-discordant --very-sensitive-local -k 2 --score-min L,-6,1.98 --mp 2” s. Unmapped reads were then remapped to precursor sequences using HISAT2 v2.0.2 [60] with default settings to capture any reads from alternative splicing.
Supplementary Material
Highlights are required for this journal. Specifications: include 3 to 5 bullet points (max. 85 characters per bullet point including spaces); only the core results of the paper should be covered. The first bullet point should state the background or context of the question. One to three bullet points should describe the principal results. The last bullet point should conclude on a clear description of the conceptual advance and significance of the work. Highlights should be submitted as a separate file in EES by selecting ‘Highlights’ from the drop-down list when uploading files. Highlights will be displayed in online search result lists, the contents List and in the online article, but will not appear in the article PDF file or print.
A thermophilic group IIC intron can retrohome to high copy number in bacteria
Retrohoming sites recognized by both intron reverse transcriptase and intron RNA
Reverse transcriptase recognizes a 5′-exon DNA hairpin and specific base
RNA splicing requires only short intron-exon base-pairing interactions
Protein recognition of multiple transcription terminators favors proliferation
Acknowledgments
This work was supported by National Institutes of Health grants GM37949 and GM37951 and Welch Foundation Grant F-1607. Jacob Grohman was supported in part by an ACS post-doctoral fellowship. We thank Dr. Eman Ghanem for construction of plasmid pGsI2C-35/32 and Laura Markham for purifying the GsI-IIC-RT protein. The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper http://www.tacc.utexas.edu/
Abbreviations
- DI to DVI
group II intron secondary structure domains I to VI
- E1
5′ exon
- E2
3′ exon
- EN
DNA endonuclease domain
- EBS
exon-binding sequence
- IBS
intron-binding sequence
- IEP
intron-encoded protein
- LAG/LEAD
lagging/leading strand primer for cDNA synthesis
- nt
nucleotide
- ORF
open reading frame
- RNP
ribonucleoprotein
- RT
reverse transcrip-tase
- SHAPE
selective 2′-hydroxyl acylation analyzed by primer extension
- TGIRT
thermostable group II intron reverse transcriptase
- TS
target site
Footnotes
Accession numbers
High through-put sequencing data shown in Fig. 9 have been deposited and can be accessed at https://www.ncbi.nlm.nih.gov/sra/SRP143565
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lambowitz AM, Zimmerly S. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. 2011;3:a003616. doi: 10.1101/cshperspect.a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cavalier-Smith T. Intron phylogeny: a new hypothesis. Trends Genet. 1991;7:145–148. doi: 10.1016/0168-9525(91)90377-3. [DOI] [PubMed] [Google Scholar]
- 3.Martin W, Koonin EV. Introns and the origin of nucleus-cytosol compartmentalization. Nature. 2006;440:41–45. doi: 10.1038/nature04531. [DOI] [PubMed] [Google Scholar]
- 4.Koonin EV. Intron-dominated genomes of early ancestors of eukaryotes. J Hered. 2009;100:618–623. doi: 10.1093/jhered/esp056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa M, Walbott H, Monachello D, Westhof E, Michel F. Crystal structures of a group II intron lariat primed for reverse splicing. Science. 2016;354:aaf9258–aaf9258. doi: 10.1126/science.aaf9258. [DOI] [PubMed] [Google Scholar]
- 7.Qu G, Kaushal PS, Wang J, Shigematsu H, Piazza CL, Agrawal RK, et al. Structure of a group II intron in complex with its reverse transcriptase. Nat Struct Mol Biol. 2016 doi: 10.1038/nsmb.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fica SM, Nagai K. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat Struct Mol Biol. 2017;24:791–799. doi: 10.1038/nsmb.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, et al. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature. 2017;542:318–323. doi: 10.1038/nature21079. [DOI] [PubMed] [Google Scholar]
- 10.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–643. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmerly S, Guo H, Eskes R, Yang J, Perlman PS, Lambowitz AM. A group II in-tron RNA is a catalytic component of a DNA endonuclease involved in intron mobility. Cell. 1995;83:529–538. doi: 10.1016/0092-8674(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 12.Zimmerly S, Guo H, Perlman PS, Lambowitz AM. Group II intron mobility occurs by target DNA-primed reverse transcription. Cell. 1995;82:545–554. doi: 10.1016/0092-8674(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Zimmerly S, Perlman PS, Lambowitz AM. Efficient integration of an intron RNA into double-stranded DNA by reverse splicing. Nature. 1996;381:332–335. doi: 10.1038/381332a0. [DOI] [PubMed] [Google Scholar]
- 14.Cousineau B, Smith D, Lawrence-Cavanagh S, Mueller JE, Yang J, Mills DA, et al. Retrohoming of a bacterial group II intron: mobility via complete reverse splicing, independent of homologous DNA recombination. Cell. 1998;94:451–462. doi: 10.1016/S0092-8674(00)81586-X. [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Zimmerly S, Perlman PS, Lambowitz AM. Group II intron endonucleases use both RNA and protein subunits for recognition of specific sequences in double-stranded DNA. EMBO J. 1997;16:6835–6848. doi: 10.1093/emboj/16.22.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh NN, Lambowitz AM. Interaction of a group II intron ribonucleoprotein endo-nuclease with its DNA target site investigated by DNA footprinting and modification interference. J Mol Biol. 2001;309:361–386. doi: 10.1006/jmbi.2001.4658. [DOI] [PubMed] [Google Scholar]
- 17.Moran JV, Zimmerly S, Eskes R, Kennell JC, Lambowitz AM, Butow RA, et al. Mobile group II introns of yeast mitochondrial DNA are novel site-specific retroele-ments. Mol Cell Biol. 1995;15:2828–2838. doi: 10.1128/MCB.15.5.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eskes R, Liu L, Ma H, Chao MY, Dickson L, Lambowitz AM, et al. Multiple homing pathways used by yeast mitochondrial group II introns. Mol Cell Biol. 2000;20:8432–8446. doi: 10.1128/MCB.20.22.8432-8446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith D, Zhong J, Matsuura M, Lambowitz AM, Belfort M. Recruitment of host functions suggests a repair pathway for late steps in group II intron retrohoming. Genes Dev. 2005;19:2477–2487. doi: 10.1101/gad.1345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao J, Truong DM, Lambowitz AM. Genetic and biochemical assays reveal a key role for replication restart proteins in group II intron retrohoming. PLoS Genet. 2013;9:e1003469. doi: 10.1371/journal.pgen.1003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickson L, Huang HR, Liu L, Matsuura M, Lambowitz AM, Perlman PS. Re-trotransposition of a yeast group II intron occurs by reverse splicing directly into ectopic DNA sites. Proc Natl Acad Sci USA. 2001;98:13207–13212. doi: 10.1073/pnas.231494498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichiyanagi K, Beauregard A, Lawrence S, Smith D, Cousineau B, Belfort M. Re-trotransposition of the Ll.LtrB group II intron proceeds predominantly via reverse splicing into DNA targets. Mol Microbiol. 2002;46:1259–1272. doi: 10.1046/j.1365-2958.2002.03226.x. [DOI] [PubMed] [Google Scholar]
- 23.Lambowitz AM, Belfort M. Mobile Bacterial Group II Introns at the Crux of Eukary-otic Evolution. Microbiol Spectr. 2015;3:MDNA3–0050–2014. doi: 10.1128/microbiolspec.MDNA3-0050-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai LX, Zimmerly S. Compilation and analysis of group II intron insertions in bacterial genomes: evidence for retroelement behavior. Nucleic Acids Res. 2002;30:1091–1102. doi: 10.1093/nar/30.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohr G, Ghanem E, Lambowitz AM. Mechanisms used for genomic proliferation by thermophilic group II introns. PLoS Biol. 2010;8:e1000391. doi: 10.1371/journal.pbio.1000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robart AR, Seo W, Zimmerly S. Insertion of group II intron retroelements after intrinsic transcriptional terminators. Proc Natl Acad Sci USA. 2007;104:6620–6625. doi: 10.1073/pnas.0700561104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Granlund M, Michel F, Norgren M. Mutually exclusive distribution of IS1548 and GBSi1, an active group II intron identified in human isolates of group B streptococci. J Bacteriol. 2001;183:2560–2569. doi: 10.1128/JB.183.8.2560-2569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeo CC, Yiin S, Tan BH, Poh CL. Isolation and characterization of group II introns from Pseudomonas alcaligenes and Pseudomonas putida. Plasmid. 2001;45:233–239. doi: 10.1006/plas.2001.1518. [DOI] [PubMed] [Google Scholar]
- 29.Toor N, Robart AR, Christianson J, Zimmerly S. Self-splicing of a group IIC intron: 5′ exon recognition and alternative 5′ splicing events implicate the stem-loop motif of a transcriptional terminator. Nucleic Acids Res. 2006;34:6461–6471. doi: 10.1093/nar/gkl820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tourasse NJ, Kolstø AB. Survey of group I and group II introns in 29 sequenced ge-nomes of the Bacillus cereus group: insights into their spread and evolution. Nucleic Acids Res. 2008;36:4529–4548. doi: 10.1093/nar/gkn372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stamos JL, Lentzsch AM, Lambowitz AM. Structure of a thermostable group II in-tron reverse transcriptase with template-primer and its functional and evolutionary implications. Mol Cell. 2017;68:926–939.e4. doi: 10.1016/j.molcel.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moretz SE, Lampson BC. A group IIC-type intron interrupts the rRNA methylase gene of Geobacillus stearothermophilus strain 10. J Bacteriol. 2010;192:5245–5248. doi: 10.1128/JB.00633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh RN, Saldanha RJ, D’Souza LM, Lambowitz AM. Binding of a group II in-tron-encoded reverse transcriptase/maturase to its high affinity intron RNA binding site involves sequence-specific recognition and autoregulates translation. J Mol Biol. 2002;318:287–303. doi: 10.1016/S0022-2836(02)00054-2. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe K, Lambowitz AM. High-affinity binding site for a group II intron-encoded reverse transcriptase/maturase within a stem-loop structure in the intron RNA. RNA. 2004;10:1433–1443. doi: 10.1261/rna.7730104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noah JW, Lambowitz AM. Effects of maturase binding and Mg2+ concentration on group II intron RNA folding investigated by UV cross-linking. Biochemistry. 2003;42:12466–12480. doi: 10.1021/bi035339n. [DOI] [PubMed] [Google Scholar]
- 37.Mohr S, Ghanem E, Smith W, Sheeter D, Qin Y, King O, et al. Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA. 2013;19:958–970. doi: 10.1261/rna.039743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo H, Karberg M, Long M, Jones JP, III, Sullenger B, Lambowitz AM. Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science. 2000;289:452–457. doi: 10.1126/science.289.5478.452. [DOI] [PubMed] [Google Scholar]
- 39.Zhuang F, Karberg M, Perutka J, Lambowitz AM. EcI5, a group IIB intron with high retrohoming frequency: DNA target site recognition and use in gene targeting. RNA. 2009;15:432–449. doi: 10.1261/rna.1378909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong J, Lambowitz AM. Group II intron mobility using nascent strands at DNA replication forks to prime reverse transcription. EMBO J. 2003;22:4555–4565. doi: 10.1093/emboj/cdg433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allawi HT, SantaLucia J. Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 42.Monachello D, Michel F, Costa M. Activating the branch-forming splicing pathway by reengineering the ribozyme component of a natural group II intron. RNA. 2016 doi: 10.1261/rna.054643.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saldanha RJ, Chen B, Wank H, Matsuura M, Edwards J, Lambowitz AM. RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry. 1999;38:9069–9083. doi: 10.1021/bi982799l. [DOI] [PubMed] [Google Scholar]
- 44.Rambo RP, Doudna JA. Assembly of an active group II intron-maturase complex by protein dimerization. Biochemistry. 2004;43:6486–6497. doi: 10.1021/bi049912u. [DOI] [PubMed] [Google Scholar]
- 45.Jeng ST, Gardner JF, Gumport RI. Transcription termination in vitro by bacteriophage T7 RNA polymerase. The role of sequence elements within and surrounding a rho-independent transcription terminator. J Biol Chem. 1992;267:19306–19312. [PubMed] [Google Scholar]
- 46.Ueda K, Yamashita A, Ishikawa J, Shimada M, Watsuji T-O, Morimura K, et al. Genome sequence of Symbiobacterium thermophilum, an uncultivable bacterium that depends on microbial commensalism. Nucleic Acids Res. 2004;32:4937–4944. doi: 10.1093/nar/gkh830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao C, Pyle AM. Crystal structures of a group II intron maturase reveal a missing link in spliceosome evolution. Nat Struct Mol Biol. 2016;23:558–565. doi: 10.1038/nsmb.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu DC, Lambowitz AM. Facile single-stranded DNA sequencing of human plasma DNA via thermostable group II intron reverse transcriptase template switching. Sci Rep. 2017;7:8421. doi: 10.1038/s41598-017-09064-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perutka J, Wang W, Goerlitz D, Lambowitz AM. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J Mol Biol. 2004;336:421–439. doi: 10.1016/j.jmb.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Leclercq S, Cordaux R. Selection-driven extinction dynamics for group II introns in Enterobacteriales. PLoS ONE. 2012;7:e52268. doi: 10.1371/journal.pone.0052268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Copertino DW, Hallick RB. Group II twintron - an intron within an intron in a chloroplast cytochrome b-559 gene. EMBO J. 1991;10:433–442. doi: 10.1002/j.1460-2075.1991.tb07965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaucher EA, Govindarajan S, Ganesh OK. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature. 2008;451:704–707. doi: 10.1038/nature06510. [DOI] [PubMed] [Google Scholar]
- 53.Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.San Filippo J, Lambowitz AM. Characterization of the C-terminal DNA-binding/DNA endonuclease region of a group II intron-encoded protein. J Mol Biol. 2002;324:933–951. doi: 10.1016/S0022-2836(02)01147-6. [DOI] [PubMed] [Google Scholar]
- 55.Karabiber F, McGinnis JL, Favorov OV, Weeks KM. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA. 2013;19:63–73. doi: 10.1261/rna.036327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nottingham RM, Wu DC, Qin Y, Yao J, Hunicke-Smith S, Lambowitz AM. RNA-seq of human reference RNA samples using a thermostable group II intron reverse tran-scriptase. RNA. 2016;22:597–613. doi: 10.1261/rna.055558.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 59.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse-transcriptase sequences. EMBO J. 1990;9:3353–3362. doi: 10.1002/j.1460-2075.1990.tb07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blocker FJH, Mohr G, Conlan LH, Qi L, Belfort M, Lambowitz AM. Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA. 2005;11:14–28. doi: 10.1261/rna.7181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sugimoto N, Nakano M, Nakano S. Thermodynamics-structure relationship of single mismatches in RNA/DNA duplexes. Biochemistry. 2000;39:11270–11281. doi: 10.1021/bi000819p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.