

TLKs promote nucleosome assembly and prevent fork collapse and DNA damage upon checkpoint or PARP inhibition.
Abstract
DNA sequence and epigenetic information embedded in chromatin must be faithfully duplicated and transmitted to daughter cells during cell division. However, how chromatin assembly and DNA replication are integrated remains unclear. We examined the contribution of the Tousled-like kinases 1 and 2 (TLK1/TLK2) to chromatin assembly and maintenance of replication fork integrity. We show that TLK activity is required for DNA replication and replication-coupled nucleosome assembly and that lack of TLK activity leads to replication fork stalling and the accumulation of single-stranded DNA, a phenotype distinct from ASF1 depletion. Consistent with these results, sustained TLK depletion gives rise to replication-dependent DNA damage and p53-dependent cell cycle arrest in G1. We find that deficient replication-coupled de novo nucleosome assembly renders replication forks unstable and highly dependent on the ATR and CHK1 checkpoint kinases, as well as poly(adenosine 5′-diphosphate–ribose) polymerase (PARP) activity, to avoid collapse. Human cancer data revealed frequent up-regulation of TLK genes and an association with poor patient outcome in multiple types of cancer, and depletion of TLK activity leads to increased replication stress and DNA damage in a panel of cancer cells. Our results reveal a critical role for TLKs in chromatin replication and suppression of replication stress and identify a synergistic lethal relationship with checkpoint signaling and PARP that could be exploited in treatment of a broad range of cancers.
INTRODUCTION
Faithful duplication of DNA and its organization into chromatin is essential to maintain genome integrity and function. During genome replication, progression of the replication machinery can be challenged by limitations in nucleotide supply and physical obstacles on the DNA template, including naturally occurring DNA lesions and difficult to replicate secondary structures. To ensure correct and complete duplication of the genome, cells have evolved a network of safeguards and repair mechanisms that protect replication forks (1). When replication forks are challenged, long stretches of single-stranded DNA (ssDNA) can accumulate because of uncoupling of the replicative helicase from stalled DNA polymerases (1). Replication protein A (RPA)–coated ssDNA, along with the 9-1-1 (RAD9-RAD1-HUS1) DNA clamp complex and TOPBP1, recruits and activates ATR, the upstream kinase in the replication checkpoint. ATR activation and subsequent activation of CHK1 act to stabilize arrested forks, suppress late origin firing, and trigger activation of DNA repair machinery to deal with lesions (1). In addition, poly(adenosine 5′-diphosphate–ribose) polymerase (PARP) activity is required for CHK1 retention at stalled forks, activation of the S-phase checkpoint, and the restart of stalled replication forks (2, 3). Prolonged fork arrest poses a risk of fork collapse and generation of DNA double-strand breaks (DSBs) potentially due to nuclease attack (1).
DNA replication and checkpoint control is tightly integrated with histone dynamics and chromatin assembly (4), but little is known about how this is linked to fork protection mechanisms. The function of the H3-H4 chaperone ASF1 and the assembly of newly synthesized DNA into chromatin are required for DNA replication in mammalian cells (5–7). The regulation of ASF1 has been linked to the Tousled-like kinases (TLKs), a relatively unexplored family of nuclear serine/threonine kinases whose activity peaks during unperturbed DNA replication and is regulated by CHK1. The TLKs could therefore act at the interface between DNA replication, chromatin assembly, and genome integrity (8–10).
Mammalian cells express two TLK isoforms, TLK1 and TLK2, which are highly active in S phase, can interact with each other, and are negatively regulated by CHK1 (8–10). Recent studies have identified TLK1 at replication sites (11) and observed an S-phase delay in TLK1-depleted cells (12). The histone H3-H4 chaperones ASF1a and ASF1b are the primary established TLK1 and TLK2 targets (13) and are required to deliver histones to CAF-1 and HIRA for replication-coupled and replication-independent chromatin assembly, respectively (14). Recently, TLK1 was reported to phosphorylate RAD9—part of the 9-1-1 proliferating cell nuclear antigen (PCNA)–like clamp—and this was linked to G2-M checkpoint release (12, 15), while other reports implicated TLK2, but not TLK1, in G2-M checkpoint recovery through an ASF1a-mediated transcriptional regulation (16). However, TLK activity peaks in S phase, and its loss during early and rapid cell divisions in Drosophila and Caenorhabditis elegans causes severe chromatin and proliferation defects and cell death (17, 18), suggesting that TLKs serve central conserved functions during DNA replication.
TLKs have been proposed to promote histone provision in S phase (11) based on the finding that phosphorylation of ASF1 stimulates histone H3-H4 binding and interaction with the downstream chaperones CAF-1 and HIRA. However, the requirement of TLK activity for DNA replication and chromatin assembly remains to be established. Understanding the precise relationship of TLK activity to these processes is particularly important because TLK1/2, as S-phase active kinases, are attractive druggable targets for cancer therapy—a notion supported by recent evidence that amplification of TLK-ASF1 signaling is indicative of poor prognosis in breast cancer (19, 20).
Here, we examine the relative impact of TLK1 and TLK2 depletion and show that these enzymes are required for DNA replication and chromatin assembly. Depletion of TLK activity led to replication stress and impaired chromatin assembly, accumulation of DNA damage, and subsequent cell cycle arrest. The deleterious effects of TLK depletion were exacerbated by treatment with replication checkpoint inhibitors and the PARP inhibitor olaparib. Inhibition of new histone biosynthesis, but not ASF1 depletion, recapitulated this synthetic lethality response, arguing that TLK activity protects replication fork stability by stimulating de novo histone deposition. Analysis of The Cancer Genome Atlas (TCGA) data sets revealed that amplifications of TLK-ASF1 pathway genes occur frequently in human cancers and the requirement for TLK activity to maintain genome integrity applies across multiple cancer types. Together, these data argue that TLK activity by promoting nucleosome assembly plays a central role in replication fork progression and that targeted inhibition of TLK1/2 could be an attractive strategy to enhance replication stress and augment the effects of ATR, CHK1, and PARP-1 inhibitors in many types of cancers.
RESULTS
TLK2 is required for efficient DNA replication
TLK activity peaks during S phase, when DNA is replicated and packaged into chromatin (9). We therefore set out to investigate the influence of TLK activity on DNA replication by measuring the incorporation of thymidine analogs. The small interfering RNA (siRNA)–mediated depletion of TLK1 in U-2-OS cells did not affect the number of 5-bromo-2′-deoxyuridine (BrdU)–positive cells (Fig. 1A), and we could readily generate stable TLK1Δ CRISPR (clustered regularly interspaced short palindromic repeats) knockouts from the MDA-MB-231 breast cancer cell line. These TLK1Δ cells exhibited similar proliferation rates and levels of BrdU incorporation as the parental cells (Fig. 1B and fig. S1, A to D). Depletion of TLK2 reduced the rate of DNA replication and the overall number of BrdU-positive or 5-ethynyl-2′-deoxyuridine (EdU)–positive cells significantly (Fig. 1, A to E, and fig. S1E). Notably, the siRNA-mediated depletion of TLK2 also often resulted in the reduction of TLK1 protein levels, an effect that was more dramatic in U-2-OS cells than in MDA-MB-231 cultures (Fig. 1C), probably reflecting the fact that heterodimerization influences TLK1 stability (10). Stable expression of siRNA-resistant wild-type (WT) TLK2 rescued the replication defect of U-2-OS cells, whereas a kinase-dead (KD) mutant (D590A) (10) of TLK2 failed to do so (Fig. 1E and fig. S1F). These demonstrate that TLK2 promotes DNA replication through its kinase activity.
Fig. 1. TLK2 is required for efficient DNA replication.
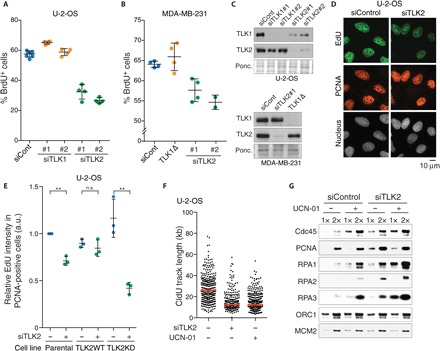
(A) U-2-OS cells were pulsed with BrdU 48 hours after transfection with siRNAs against TLK1 or TLK2. The percentage of S-phase cells was subsequently quantified by analysis of DNA content using propidium iodide (PI) and staining for BrdU. Means and SD from technical replicates performed in at least biological duplicate are shown. (B) The percentage of S-phase cells was quantified in MDA-MB-231 cells pulsed with BrdU 72 hours after transfection and analyzed as in (A). (C) Western blot analysis of TLK1 and TLK2 from whole-cell lysates of U-2-OS (top) or MDA-MB-231 cells (bottom) 48 or 72 hours after siRNA transfection, respectively. (D) Immunofluorescence analysis of EdU incorporation in U-2-OS cells pulsed with EdU 30 hours after TLK2 siRNA transfection. Representative images are shown with PCNA as a marker for S-phase cells. (E) DNA replication in complemented U-2-OS cells. U-2-OS cells stably expressing siRNA-resistant WT or KD TLK2 were analyzed as in (D). See fig. S1F for Western blots for TLK2-WT or KD. EdU average intensities relative to parental cells from n = 3 independent biological replicates are shown with means and SD. One-sample and unpaired two-tailed t tests were used for statistical analysis of parental U-2-OS cells and complemented cell lines (TLK2WT and KD), respectively. **P < 0.01; n.s., not significant; a.u., arbitrary units. (F) Analysis of replication fork speed by DNA combing analysis. Length of CldU-labeled tracks (n > 250) was measured. One representative experiment of two biological replicates is shown, and median is indicated by a red line. (G) Analysis of replication factor chromatin loading in U-2-OS cells treated with or without UCN-01 30 hours after transfection. Cells were preextracted, and the chromatin pellet was subjected to Western blotting. One representative experiment of two biological replicates is shown.
To further dissect the role of TLK2 in replication, we used DNA combing to analyze replicating DNA molecules to monitor fork elongation rates. We carried out the analysis 30 hours after siRNA treatment, the earliest time where a moderate reduction in EdU incorporation was evident (Fig. 1E and fig. S1, E and G). We incorporated 5-chloro-2′-deoxyuridine (CldU) into newly replicating DNA and then quantified elongation rates by measuring the length of CldU-labeled tracks. As a control, we included the CHK1 inhibitor UCN-01, which triggers hyperactivation of cyclin-dependent kinase 2 (CDK2) kinase activity and slowdown of fork progression (21, 22), as indicated by reduced CldU track length (Fig. 1F) (21, 22). Similarly, siRNA-mediated depletion of TLK2 reduced fork speed substantially (Fig. 1F and fig. S1H). The loading of replication fork factors CDC45 and PCNA, but not prereplication complex components ORC1 and MCM2, was increased upon TLK2 depletion (Fig. 1G), and CldU intertrack distances were reduced (fig. S1I). This mimicked the response to CHK1 inhibition and argued that, while origin licensing was not affected, origin firing was increased in cells depleted of TLK activity as expected when replication fork speed is reduced (fig. S1I) (1). Collectively, these results demonstrated that multiple cell lines are dependent on TLK activity for DNA replication and indicated that impaired replication fork progression underlies the replication defect in cells lacking TLKs.
TLK activity is required for chromatin assembly
The histone chaperone ASF1 is a well-established TLK substrate (13), and its phosphorylation by TLK is proposed to facilitate provision of canonical histones H3.1-H4 and H3.2-H4 during DNA replication (11). Given that nucleosome assembly is required for replication fork progression (5), we asked whether TLK depletion influenced the deposition of new histones using U-2-OS cells stably expressing SNAP-tagged histones (Fig. 2A and fig. S2A) (23). We included both canonical histone H3.1 and the replacement variant H3.3, as ASF1 supplies histones to both replication-dependent and replication-independent pathways (14). Using quench-chase-pulse experiments, we found that depletion of TLK activity using siRNA against TLK2 impaired de novo deposition of both histones H3.1 and H3.3 (Fig. 2, B to D, and fig. S2B). However, because H3.1-H4 incorporation is replication-dependent, the observed defect in histone H3.1 and H4 incorporation in TLK-depleted cells could potentially be secondary to impaired DNA replication or alterations in the cell cycle (Fig. 1, A to E, and fig. S2C). To resolve this point, we specifically assayed chromatin assembly on newly synthesized DNA, as previously described (5). We labeled replicating DNA with radioactive thymidine and measured the sensitivity of nascent chromatin to nuclease digestion (Fig. 2E). Consistent with previous work (5), inhibition of histone biosynthesis upon depletion of FLASH (24) strongly increased the accessibility of nascent chromatin to micrococcal nuclease (MNase) (Fig. 2F) and reduced DNA replication fork speed (fig. S2D). Following TLK depletion, nascent chromatin was also more sensitive to MNase digestion (Fig. 2F), demonstrating that TLK activity is required for efficient replication-coupled nucleosome assembly. Collectively, this shows that TLK activity promotes nucleosome assembly and maintains chromatin organization, extending previous findings that TLK phosphorylation of ASF1 stimulates histone binding and interaction with CAF-1 and HIRA (11).
Fig. 2. TLK2 is required for replication-coupled chromatin assembly.
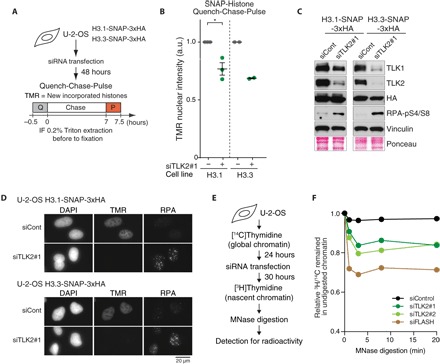
(A) Experimental design for assaying histone incorporation in cell lines stably expressing SNAP-tag histones H3.1 and H3.3. For quench (Q)–chase–pulse (P) experiments, U-2-OS cells were pulsed with SNAP-Block, chased for 7 hours, and then pulsed with TMR-Star. IF, immunofluorescence. (B) Quantification of SNAP-tag histone incorporation from n = 3 (H3.1) and n = 2 (H3.3) independent experiments as described in (A). Tetramethylrhodamine (TMR) intensity relative to mock-transfected cells is plotted. For each data point, n > 300 nuclei were analyzed. Means and SEM are indicated. For H3.1, a two-tailed t test was used for statistical analysis (*P < 0.05). (C) Western blot analysis of whole-cell lysates of U-2-OS cells stably expressing SNAP-tag histones 48 hours after siRNA transfection. (D) Representative images of TMR signal in H3.1- and H3.3-SNAP–expressing U-2-OS cell lines. 4′,6-Diamidino-2-phenylindole (DAPI) and RPA staining are also shown. (E) Experimental design of chromatin assembly assay measuring resistance to MNase digestion of nascent chromatin relative to bulk chromatin. (F) Nascent chromatin assembly analyzed as in (E) in U-2-OS cells transfected with the indicated siRNAs for 30 hours. One representative experiment of n = 3 biological replicates is shown. siRNA against FLASH that inhibits new histone biosynthesis and was included as a positive control.
Loss of TLK activity leads to DNA unwinding, checkpoint activation, and loss of viability
Replication fork stalling, origin hyperactivation, and defects in chromatin assembly have all been implicated in fork collapse and genome instability (1, 5, 25). Sustained depletion of TLK2 in MDA-MB-231 cells, on either a WT or TLK1Δ background, significantly increased the levels of chromatin-bound RPA, H2AX phosphorylation at S139 (γH2AX), and phosphorylation of RPA at S4/S8 (Fig. 3, A to C, and fig. S3, A and B). Similar results were obtained in U-2-OS cells, where TLK depletion led to accumulation of chromatin-bound RPA and increased phosphorylation of RPA at S4/S8, a marker of replication fork collapse (Fig. 3, D to F). This was indicative of increased ssDNA accumulation, checkpoint activation, and replication fork collapse (1), and as expected for a genuine replication stress response, depletion of the replication initiation factor CDC45 blocked RPA accumulation and DNA damage (Fig. 3, E and F). Consistent with these results in human cells, conditional depletion of both TLK1 and TLK2 in mouse embryonic fibroblasts resulted in reduced DNA replication and elevated replication stress signaling (fig. S3, C and D).
Fig. 3. Loss of TLK activity leads to DNA unwinding, accumulation of DNA damage, and loss of viability.
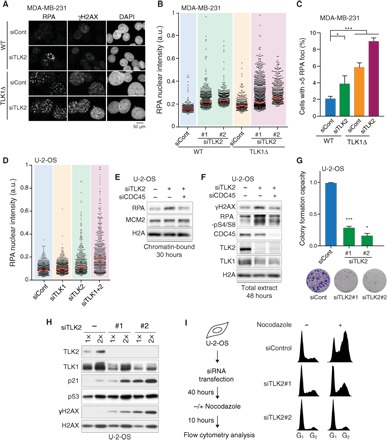
(A) Immunofluorescence analysis of MDA-MB-231 cells costained with antibodies against RPA, γH2AX, and DAPI. (B) High-throughput microscopy (HTM) analysis of chromatin-bound RPA in parental MDA-MB-231 (WT) or TLK1-null cells (TLK1Δ) 72 hours after siRNA transfection. One representative experiment of n = 3 biological replicates is shown; median is indicated in red. After normalization to siCont (WT) average, one-sample t test was used for statistical analysis of n = 3 independent experiments: significance of P = 0.1475 was observed for siTLK2#1 (WT), **P = 0.0015 for siTLK2#2 (WT), P = 0.0917 for siTLK2#1 (TLK1Δ), and **P = 0.0014 for siTLK2#2 (TLK1Δ) relative to siCont (WT). Differences between siCont (TLK1Δ) and siCont (WT) were not significant. (C) Quantification of the MDA-MB-231 cells with more than five RPA foci. At least 300 nuclei were analyzed, and the mean with SEM is shown for independent cultures (n = 10 for siCont; n = 6 for siTLK2). An unpaired two-tailed t test was used for statistical analysis. *P < 0.05, ***P < 0.001. (D) HTM analysis of the chromatin-bound RPA in U-2-OS cells 48 hours after siRNA transfection. One representative experiment of n = 6 biological replicates is shown; median is indicated in red. After normalization to siCont average, one-sample t test was used for statistical analysis of at least n = 6 independent experiments: significance of P = 0.0514 was observed for siTLK1, P = 0.0005 for siTLK2, and P = 0.0120 for siTLK1+2. (E and F) Analysis of replication stress response in U-2-OS cells transfected with TLK2 siRNAs alone or together with CDC45 siRNAs for 30 hours (E) or 48 hours (F). One representative experiment of two biological replicates is shown for (E) and (F). (G) Colony formation assay in U-2-OS cells transfected with TLK siRNAs. Data represent means and SEM of biological replicates (n = 5 for siCont, n = 4 for siTLK2#1, and n = 2 for siTLK2#2), each done at least in technical duplicates. A one-sample t test was used for statistical analysis. ***P = 0.0001 for siTLK2#1 and *P = 0.0324 for siTLK2#2. (H) Western blot analysis of U-2-OS cell lysates after 48 hours of siRNA transfection, representative of three independent experiments. (I) Cell cycle progression analyzed by flow cytometry of cells treated with nocodazole and stained with PI. The experimental design (left) and representative cell cycle profiles (right) from one of n = 3 biological replicates are shown.
TLK-depleted cells showed reduced colony-forming capacity, providing evidence of reduced viability and/or proliferative capacity (Fig. 3G). DNA damage signaling elicits a delay in cell cycle progression due to the activation of cell cycle checkpoints. Upon sustained TLK2 depletion, we observed a moderate increase in p53, a major inducer of G1-S checkpoint arrest, along with a p53-dependent induction of the CDK inhibitor p21 (Fig. 3H and fig. S3E). Consistent with this finding, the number of S-phase cells was progressively reduced from 48 to 72 hours of TLK2 depletion (fig. S1, E and G), and nocodazole trap experiments indicated that G1-S transition was impaired (Fig. 3I and fig. S3, F and G). Co-depletion of p53 was sufficient to partially rescue this block in G1-S transition (fig. S3G), arguing that, as DNA damage accumulates during prolonged TLK2 depletion, the p53-p21 checkpoint is activated and blocks S-phase entry.
TLK depletion is synthetic lethal with checkpoint inactivation and PARP inhibition
Whereas sustained TLK depletion for 48 to 72 hours generated DNA damage (Fig. 3), short-term reduction of TLK activity impaired fork progression without eliciting a strong DNA damage response (Figs. 1F and 4, A and B, and fig. S4, A and B). However, we noted that the combined inhibition of TLK2 and the replication checkpoint kinase CHK1 strongly augmented RPA loading at early time points (Fig. 1G). We reasoned that this effect might reflect the fact that destabilized replication forks in TLK-depleted cells strongly rely on the replication checkpoint to avoid collapse, and we therefore addressed the possibility of a synthetic sickness between these two pathways. As expected from previous work (22), we observed DNA damage signaling in cells treated with the checkpoint inhibitor UCN-01 (Fig. 4A and fig. S4A). Co-depletion of TLK2 substantially enhanced this response (Fig. 4A and fig. S4A), elevating the phosphorylation of ATM-S1981, CHK2-T68, p53-S15, RPA2-S4/S8, and γH2AX. Notably, these experiments were performed at 24 hours after transfection before any detectable DNA damage occurred in cells depleted for TLK2. Similar results were obtained in U-2-OS cells treated with two different CHK1 inhibitors (Gö-6976 and MK-8776; fig. S4B), in MDA-MB-231 cells treated with a CHK1 inhibitor (AZD7762; fig. S4C), and upon treatment with ATR inhibitors (AZ20 and ETP-46464; fig. S4, D and E) (26–29).
Fig. 4. TLK depletion causes genomic instability and sensitizes cells to checkpoint inactivation and PARP inhibition.
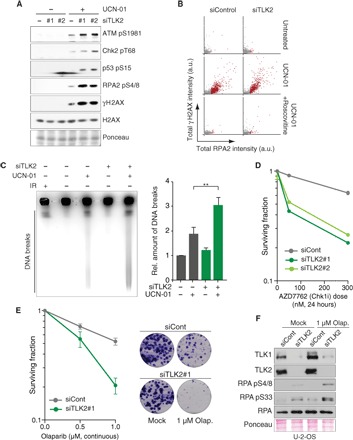
(A) Analysis of DNA damage signaling in U-2-OS cells 24 hours after siRNA transfection and treatment with or without UCN-01 for 2 hours. One representative experiment of three biological replicates is shown. (B) HTM analysis of RPA accumulation and γH2AX in TLK2-depleted U-2-OS cells treated with UCN-01 alone or together with roscovitine for 2 hours. One representative experiment of two biological replicates is shown (n > 1800). (C) PFGE analysis of DNA DSBs in U-2-OS cells transfected with siRNAs for 24 hours and treated with UCN-01 for 4 hours. Representative result (left) and quantification (right) of DNA breaks relative to untreated control from n = 3 independent experiments are shown with the means and SD. An unpaired two-tailed t test was used for statistical analysis. **P < 0.01. IR, ionizing radiation, 20 Gy. (D) Sensitivity of TLK-depleted cells to the CHK1 inhibitor AZD7762 measured by colony formation assay. Representative experiment of two biological replicates performed in technical duplicate is shown. (E) Sensitivity to PARP inhibitor olaparib measured by colony formation assay; representative images are shown. Means and range of two biological replicates performed in technical duplicate are shown. (F) Western blotting of RPA and phospho-RPA following TLK depletion and olaparib treatment in U-2-OS cells.
High-content imaging showed that DNA damage occurred in cells with hyperloading of RPA on chromatin (Fig. 4B), as previously found upon replication fork collapse in response to ATR inhibition in hydroxyurea-treated cells (30). Cotreatment with the CDK2 inhibitor roscovitine suppressed both RPA accumulation and DNA damage (Fig. 4B), suggesting that fork collapse was a result of CDK2-induced unscheduled origin firing and, possibly, titration of replication factors away from forks with chromatin assembly defects (30, 31). The application of pulsed-field gel electrophoresis (PFGE) to directly measure DSBs revealed that combined inactivation of TLK2 and inhibition of the replication checkpoint synergistically increased the numbers of DSBs observed (Fig. 4C). Consistent with this result, TLK2-depleted MDA-MB-231 cells displayed a further reduction in colony formation capacity following CHK1 inhibition by AZD7762 (Fig. 4D), and TLK2 depletion also enhanced the cytotoxicity of CHK1 inhibitors in U-2-OS cells (fig. S4F).
In addition to CHK1 and ATR inhibitors, the PARP inhibitor olaparib is of interest in current clinical studies, as it was shown to cause synthetic phenotypes with defects in homologous recombination–mediated repair (32, 33). We therefore asked whether olaparib would enhance the defects of TLK-depleted cells in a manner similar to checkpoint inhibition. We found that the addition of olaparib strongly decreased the survival of MDA-MB-231 cells depleted for TLK activity (Fig. 4E and fig. S4G) and that this was accompanied by enhanced RPA phosphorylation (Fig. 4F and fig. S4H).
De novo nucleosome assembly defects synergize with checkpoint inhibition
New histone deposition and nucleosome assembly are required for replication fork progression (5), and nucleosome assembly defects lead to a gradual decline in replication fork stability (5), as observed in cells depleted for TLK activity. However, DNA unwinding at stalled replication forks is strongly impaired in ASF1-depleted cells (6, 7), in contrast to what we observed upon TLK depletion (Fig. 3, A to D). It was thus important to address whether the synthetic lethal relationships were linked to the function of TLKs in chromatin assembly or to a distinct function that has yet to be identified. We thus compared TLK depletion to depletion of either ASF1 (a and b) or FLASH, a key regulator of histone biosynthesis (24), in a time course analysis. At 48 hours after transfection, when extensive RPA phosphorylation was evident in TLK-depleted cells, little signaling was observed in cells depleted for ASF1 (a and b) (Fig. 5A). In contrast, FLASH depletion led to increased RPA phosphorylation, albeit to lower levels than TLK depletion, indicating that replication stress was induced (Fig. 5A).
Fig. 5. Defects in new histone deposition are synthetic lethal with checkpoint inhibition.
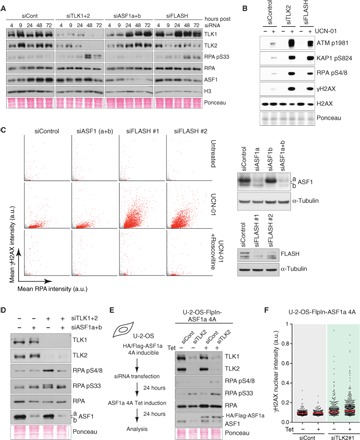
(A) Time course Western blot analysis of replication stress signaling following the siRNA depletion of TLK1/2, ASF1a/b, and FLASH in U-2-OS cells. (B) Analysis of DNA damage signaling in U-2-OS cells treated with siRNAs and UCN-01 for 24 and 2 hours, respectively. One representative experiment of two biological replicates is shown. (C) HTM analysis of RPA accumulation and γH2AX in ASF1a/b or FLASH-depleted U-2-OS cells as in Fig. 4B (left). Western blot controls for siRNA treatment (right). One representative experiment of two biological replicates is shown (n > 4800). (D) Western blot analysis of replication stress signaling following the siRNA depletion of the indicated combinations of TLK1/2 and ASF1a/b in U-2-OS cells. One representative experiment of three biological replicates is shown. (E) Schematic of the experiment (left). FlpIn-ASF1a-4A U-2-OS cells were induced with tetracycline for 24 hours following transfection with siRNA against TLK2, and indicated proteins were monitored by Western blotting (right). One representative experiment of four biological replicates is shown. (F) HTM analysis of the nuclear intensity of γH2AX was measured in ASF1 4A cells. Representative results from one representative experiment of three biological replicates performed are shown. After normalization to siCont (−Tet) average, one-sample t test was used for statistical analysis of n = 3 independent experiments: significance of P = 0.0266 was observed for siTLK2 (−Tet) and P = 0.0011 for siTLK2 (+Tet). In addition, a two-tailed unpaired t test was performed between siTLK2 (−Tet) and siTLK2 (+Tet), and P = 0.0484 was observed.
Next, we addressed whether defects in ASF1 function or in new histone biosynthesis per se were sufficient to sensitize cells to treatment with checkpoint inhibitors. Similar to TLK2 depletion, FLASH knockdown also exacerbated UCN-01–induced DNA damage, mainly in cells exhibiting an accumulation of RPA-coated ssDNA (Fig. 5, B and C). Surprisingly, this synthetic relationship was not recapitulated by depletion of ASF1 (a and b) (Fig. 5C), suggesting that fork arrest differs in TLK- and ASF1-depleted cells. As ASF1 is the most well-characterized substrate of TLKs, we analyzed the effects of co-depleting TLK1/2 and ASF1a/b. This led to the suppression of RPA phosphorylation observed following TLK depletion (Fig. 5D), further confirming that ASF1 is necessary for unwinding and indicating that its phosphorylation by TLKs is dispensable for this function. This was corroborated by the overexpression of a nonphosphorylatable ASF1a mutant (ASF1a-4A) that enhanced the levels of RPA phosphorylation observed following TLK depletion (Fig. 5E). In addition to delivering new histones to CAF-1 for de novo deposition, ASF1 has been implicated in histone recycling together with MCM2-7 (6, 34, 35). Given that inhibition of new histone biosynthesis mimics the hypersensitivity to checkpoint inhibitors in TLK-depleted cells, the most parsimonious explanation is that this phenotype is linked specifically to de novo histone deposition and not to other functions of ASF1 in histone dynamics. We cannot exclude that TLKs serve functions at the replication fork independent of histones, for example, through targets other than ASF1 (36). However, we note that the exacerbated DNA damage response in FLASH-depleted cells could be suppressed by roscovitine treatment (Fig. 5C), suggesting that it was linked to origin hyperactivation, as in the case of TLK depletion. Together, these results indicated that replication forks stalled because of de novo nucleosome assembly defects are vulnerable to collapse and that cells rely on basal levels of checkpoint signaling to prevent fork collapse, rampant genomic instability, and cell death.
TLK levels influence cancer cell proliferation
Previous work has linked high expression levels of either TLK2 or ASF1B with poor patient outcome in subsets of breast cancer patients (19, 20). As our results indicated that TLK activity was critical for nucleosome assembly during DNA synthesis and for replication fork stability, we considered that it was likely to be maintained or amplified in many cancer cell types to support proliferation. We examined the available data sets from TCGA for copy number alterations (CNAs), mutations, or relative expression of the TLK1 and TLK2 genes (table S1). As a basis of comparison, we also included ASF1A/B, ATR, and CHEK1 (encoding CHK1), as well as several genes related to proliferation (MKI67, MCM2, and E2F1) and well-characterized tumor suppressors or oncogenes (TP53, ATM, MYC, PTEN, and EGFR) to provide scale for comparison (table S2). Analyzing data from more than 7000 patient samples (n = 7343), we found that TLK1, TLK2, and ASF1B more frequently exhibited copy number increases (CNIs) rather than copy number decreases (CNDs) or mutations (Fig. 6, A and B, and fig. S5, A to C). In contrast, CNDs were the most frequent type of alteration observed for ASF1A, consistent with recent reports (Fig. 6A and fig. S5B) (37). As alterations in gene expression can also occur independently of CNAs, we examined gene expression in the data sets where it was available and asked whether there were correlations between the expression of TLK-ASF1 and ATR-CHEK1 genes, as well as the proliferation markers MKI67 and MCM2 (fig. S5D). At the expression level, TLK1 and ASF1A showed a strong correlation with each other, and neither gene was significantly correlated with the expression of proliferation markers (MKI67 or MCM2). In contrast, expression of ASF1B and CHEK1 showed a highly significant correlation with the expression of both MKI67 and MCM2 across nearly all TCGA data sets, supporting the proposed utility of ASF1B as a proliferation marker (fig. S5D and table S3) (19). TLK2 expression also correlated positively with proliferative markers across the pan-cancer data set, albeit to a much lesser degree than ASF1B, and was significantly correlated with MKI67 expression in a more limited subset of individual cancers (fig. S5D and table S3). Together, these data demonstrated that TLK1, TLK2, and ASF1B CNIs occur more frequently than other types of alterations and in diverse cancer types.
Fig. 6. TLKs are amplified and suppress replication stress in multiple cancer types.
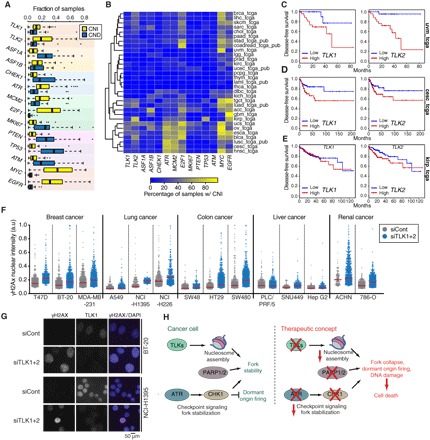
(A) Box plots of the fraction of patients with CNIs or CNDs among the TCGA cohorts (details in table S1) for the indicated genes. Box plots show first to third quartiles, with whiskers at third quartile +1.5*IQR (interquartile range) and first quartile −1.5*IQR. Analysis of mutation frequency for the same genes is provided in fig. S5A. (B) Heat map showing the percentage of samples in individual TCGA cohorts with CNIs for the indicated gene. TCGA cohort abbreviations and chromosome locations of each gene are provided in tables S1 and S2, and heat maps for CNDs and mutations are provided in fig. S5 (B and C). (C to E) Kaplan-Meier plots of multivariate disease-free survival analysis of the TCGA uveal melanoma (uvm; D), cervical squamous cell carcinoma and endocervical adenocarcinoma (cesc; E), and cervical kidney renal papillary cell carcinoma (kirp; F) cohorts based on the expression of the indicated gene. The hazard ratios (HR) and [likelihood ratio test (LRT)] P values were calculated using expression as a continuous variable: uvm (low, n = 30; high, n = 29), HR = 3.29 (1.44 to 7.53) for TLK1(LRT-pv = 0.0007744) and HR = 2.52 (1.19 to 5.35) for TLK2(LRT-pv = 0.0064077); cesc (low and high, n = 84), HR = 1.49 (1.04 to 2.12) for TLK1(LRT-pv = 0.031458) and HR = 1.45 (1.06 to 1.97) for TLK2(LRT-pv = 0.028423); kirp (low, n = 112; high, n = 110), HR = 0.80 (0.58 to 1.11) for TLK1(LRT-pv = 0.17713) and HR = 1.28 (0.99 to 1.66) for TLK2(LRT-pv = 0.075713). Additional supporting data are provided in tables S4 and S5. (F) HTM analysis of γH2AX 48 hours after siRNA treatment in the indicated cell line. One representative experiment of two biological replicates is shown. Median is indicated by a red line. (G) Representative image examples for the quantification in (F). Staining for γH2AX and TLK1 (to assess depletion levels) is shown. A minimum of 150 nuclei were evaluated for each cell line in each experiment. (H) Model for the combined influence of chromatin assembly and checkpoint signaling in the protection of stalled replication forks. Assembly of new nucleosomes on replicating DNA is required for fork progression and is dependent on ASF1-mediated histone provision. Reduced TLK activity impairs de novo nucleosome assembly, slowing down replication forks and reducing their stability. Under these circumstances, basal checkpoint and PARP activity are required to maintain the stability of stalled forks and prevent new origin firing that would accelerate fork collapse and increase levels of DNA damage.
We next wanted to examine the potential relationship between the genomic alterations or expression differences of TLK1 and TLK2 genes and patient outcome. We first carried out a univariate analysis to identify cancer types where TLK1 or TLK2 expression was significantly associated with survival or other clinical parameters (table S4). We then performed a multivariate analysis on several of these cohorts that showed high levels of CNIs (Fig. 6B) or high expression of TLK1 and TLK2 (fig. S5E). We included age, sex, MKI67 expression level (proliferation), and tumor stage as covariables to determine whether expression levels of TLK1 or TLK2 significantly correlated with patient outcomes. High expression levels of both TLK1 and TLK2 correlated with reduced disease-free survival in the uveal melanoma (uvm_tcga) and cervical squamous cell carcinoma and endocervical adenocarcinoma (cesc_tcga) cohorts (Fig. 6, C and D). Furthermore, high expression of TLK2, but not of TLK1, was associated with reduced disease-free survival in kidney renal papillary cell carcinoma (kirp_tcga), which was among the cohorts that exhibited the highest number of TLK2 CNIs among the TCGA data sets (Fig. 6E), as well as estrogen receptor–positive breast cancer, as previously reported (fig. S5F) (20). Collectively, these results show that the TLK-ASF pathway is amplified frequently in many cancer types and that, in several cases, high expression of TLK1 and TLK2 correlates with poor patient outcome, further suggesting that TLK kinase activity may be a valuable therapeutic target that can enhance the efficacy of checkpoint or PARP inhibitors in a variety of cancer types.
The analysis of human cancer data suggested that TLK activity may be higher in many cancers, potentially reflecting a dependency on TLK activity to suppress replication stress. We therefore examined the effects of total TLK depletion in a panel of human cancer cell lines from different tissues, including breast, lung, liver, colon, and kidney. Depletion of both TLK1 and TLK2 in most of the cell lines examined led to increased phosphorylation of H2AX and RPA, although the extent of the response was variable (Fig. 6F and fig. S5G). We also observed that, while all cell lines seemed to respond to TLK depletion with replication stress signaling, those with higher levels of basal replication stress tended to be more sensitive. For example, NCI-H226 lung cancer cells or SW480 colon cancer cells showed higher basal levels of γH2AX and a much stronger increase following TLK depletion (Fig. 6, F and G). This prominent DNA damage induction upon TLK depletion may indicate that those tumors that rapidly proliferate and exhibit higher levels of oncogene-induced replication stress could also be more susceptible to TLK inhibition, alone or in combination with checkpoint or PARP inhibitors (Fig. 6H).
DISCUSSION
Here, we demonstrate a key role for TLK activity in promoting chromatin assembly and maintaining replication fork stability. The depletion of TLK activity impaired nucleosome assembly and led to replication-coupled ssDNA accumulation and fork stalling. Over time, DNA damage accumulated and induced a p53-dependent G1 arrest. In cells experiencing short-term TLK depletion or histone deprivation, inactivation of the replication checkpoint led to massive accumulation of DSBs and loss of viability. This line of evidence argues that TLK activity governs replication fork integrity and chromatin restoration on newly synthesized DNA, with checkpoint kinases and PARP activity being crucial to prevent the collapse of forks arrested with chromatin assembly defects in the absence of TLK activity (Fig. 6H).
Our work shows that proper control of TLK activity is critical for chromosomal integrity and survival in cancer cells. Furthermore, it also provides an explanation as to why the loss of TLK activity during early and rapid cell divisions in Drosophila and C. elegans causes severe chromatin and proliferation defects and cell death (17, 18). Chromatin is central to all DNA-based processes and governs cell fate specification as well as genome integrity. Failure to assemble the newly synthesized DNA into chromatin, as a result of histone deprivation (5) or loss of CAF1 (38), ASF1 (6), or TLK1/2 (this work), causes severe DNA replication defects. Available evidence indicates that ASF1 is the prime TLK target in metazoans (13, 17, 18) and that it coordinates DNA synthesis with histone supply for chromatin assembly (11). However, TLK depletion does not simply recapitulate the phenotype of ASF1 loss of function. Loss of ASF1, as well as inhibition of histone biosynthesis, reduces replication fork speed and arrests cells in S phase (5, 6). In this setting, exposure of ssDNA is not evident at stalled forks (5, 6). In contrast, lack of TLK activity reduces fork speed concomitant with accumulation of ssDNA. While this might reflect that the TLKs have other targets, such as RAD9 (12, 15), our data support that ASF1 is physically required for unwinding through a function that is independent of its phosphorylation by TLKs (Fig. 5, D and F). Given that, in addition to delivery of new histones, ASF1 forms a histone-dependent complex with the MCM2-7 helicase that may control DNA unwinding and ssDNA exposure (6, 35), we favor that TLK-independent functions of ASF1 in helicase regulation explain the different phenotypes of TLK- and ASF1-depleted cells. We find that, in contrast to ASF1 depletion, inhibition of new histone biosynthesis leads to low levels of replication stress and, most remarkably, mimics the hypersensitivity of TLK-depleted cells to checkpoint inhibitors. This points toward defective new histone deposition as a major contributor to fork instability in TLK2-depleted cells.
It has been demonstrated that TLK activity is inhibited by CHK1 phosphorylation upon DNA damage (8, 9), indicating that the responses are interlinked. Nevertheless, it is remarkable that concomitant depletion of TLK and inhibition of CHK1 or ATR activity showed strong synergistic effects, leading to genome breakage and lethality. Given that new histones via H4K20me0 provide an important platform for recruitment of the TONSL-MMS22L homologous recombination complex (39, 40), we propose that impaired nucleosome assembly impairs TONSL-MMS22L recruitment in the absence of TLK activity and sensitizes replication forks for collapse upon CHK1-dependent new origin firing and RPA exhaustion (30). This model is consistent with our finding that roscovitine prevents fork collapse in this setting and with the recent finding that long-term depletion of CAF-1 and ASF1 for 72 hours impairs recruitment of TONSL-MMS22L to DNA DSBs (25).
Both TLK1 and TLK2 target ASF1 (13), but specialized functions for TLK1 and TLK2 have been proposed (12, 15, 16, 20). We could generate TLK1-deleted MDA-MB-231 cell lines, but we were unable to propagate lines that initially appeared to have lost TLK2 (fig. S1A). Thus, we cannot exclude a TLK2-specific function important for cell viability. However, our data are most compatible with redundant functions of TLK1/2 in chromatin assembly and suppression of replication fork stability, with their relative levels and, potentially, regulation varying across different cell types. This inference is consistent with previous conclusions from the study of knockout mice (36). Our analysis of genomic data from more than 7000 patients in the TCGA data sets revealed that amplifications were the most common genomic alteration observed for either TLK1 or TLK2, as well as ASF1B, while few mutations or deletions were found in either of these genes. We have extended previous observations from the analysis of breast cancer (20, 41) and showed that high expression of both TLK1 and TLK2 is associated with reduced disease-free survival in additional cancer types from multiple tissues. Given the central role of TLKs in promoting histone provision during DNA replication, we propose that TLK activity is likely to be important for genomic stability in most proliferating cells and is thus a potentially valid target in many types of highly proliferative cancers, regardless of the association between expression and patient outcome. This proposition is further supported by the observation that TLK depletion leads to increased replication stress across a wide variety of cancer types (Fig. 6F and fig. S5G).
Together, we have shown that loss of TLK activity cripples chromatin assembly and fork stability, suggesting that highly proliferative cancer cells may generally require the integrity, and in some cases the amplification, of TLK-ASF1 signaling to support elevated levels of DNA replication and reduce toxic replication stress. The development of specific TLK1 and TLK2 inhibitors may therefore provide new therapeutic opportunities in a broad range of cancers (42), particularly in conjunction with ATR, CHK1, and PARP inhibitors, as cells with reduced TLK activity become highly dependent on the ATR-CHK1–mediated replication checkpoint and PARP activity for survival.
MATERIALS AND METHODS
Study design
All experiments were carried out using a minimum of two biological replicates. No statistical method was used to predetermine sample size. No samples were excluded from the analyses. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment. The primary research objectives were to test the hypothesis that TLK activity was required for DNA replication. The resulting data led to the secondary hypotheses that TLK depletion would synergize with agents that exacerbate replication stress (namely, checkpoint and PARP inhibitors) and that TLK activity would be beneficial to many types of cancer cells. Experimental design was primarily controlled laboratory experiments using standard cell culture techniques. It also involved the retrospective statistical analysis of TCGA cancer data sets.
Cell culture, stable cell lines, viral production, and drug treatment
U-2-OS, TIG3, MDA-MB-231, T47D, A549, SW48, HT29, SW480, and PLC/PRF/5 cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen); BT-20, HepG2, and ACHN cells were grown in minimum essential medium (Invitrogen); and NCI-H1395, NCI-H226, SNU449, and 786-O cells were grown in RPMI 1640 (Invitrogen), all with 10% fetal bovine serum (HyClone or Sigma) and 1% penicillin/streptomycin. Lentivirus was produced (Applied Biological Materials Inc.) using pLenti6/UbC/V5-Dest (Invitrogen) with TLK2 WT or KD mutant (D591A) (10) resistant to siTLK2#1. U-2-OS cells were infected with the virus supernatant supplemented with polybrene (Millipore) and subjected to single-clone selection with blasticidin (5 μg/ml). SNAP-tag histone lines were generated using the retrovirus vector pBABE-Blast-H3.1, H3.3, or H4-SNAP-3XHA [gift from L. Jansen (43)]. U-2-OS cells were infected with the virus supernatant supplemented with polybrene (Millipore) and selected with blasticidin (2.5 μg/ml). U-2-OS Flp-In inducible cell lines expressing ASF1a 4A were previously described (11). For expression of HA/Flag-ASF1a 4A mutant, cells were induced with tetracycline (1 μg/ml) for 24 hours. For siRNA transfection, cells were transfected with siRNAs at a final concentration of 50 to 100 nM (Sigma) using Oligofectamine or Lipofectamine RNAiMAX (Invitrogen). Oligo sequences, generation of knockout cell lines using CRISPR/CAS9, drug treatments, and other details can be found in Supplementary Materials and Methods.
Immunocytochemistry and microscopy
Cells were preextracted with CSK-T [10 mM Pipes (pH 7), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100], fixed with 2% formaldehyde, and processed as described (7). EdU staining was performed using the Click-iT EdU Alexa Fluor 488/647 High-Throughput Imaging (HCS) Assay Kit (Invitrogen) according to the manufacturer’s instructions. In brief, cells were labeled with 40 μM EdU for 15 min, preextracted, fixed, and imaged. The treatment of cold methanol at −20°C for 15 min was used for antigen retrieval of endogenous PCNA. Images were collected using a DeltaVision system (Applied Precision) with UApo/340 40×/1.35 numerical aperture (NA) oil objective lens or ScanR system with UPlanSApo 20×/0.75 NA objective lens. All images in the individual panels were acquired under room temperature with the same settings and adjusted for brightness and contrast identically using Adobe Photoshop CS6. Analysis was done with SoftWoRx software (Applied Precision), Volocity image analysis software (PerkinElmer), or ScanR analysis software. All experiments were carried out in biological triplicates, and mean intensities are displayed in graphs unless otherwise indicated.
For HTM, cells grown in Lab-Tek II Chamber slides (Labclinics) were preextracted in 0.2% Triton X-100/phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (PFA). Fixed cells were blocked in 3% bovine serum albumin (BSA)/0.1% Tween/PBS for 1 hour and stained. Forty-eight images per well were automatically acquired with a robotized fluorescence microscopy station (ScanR, Olympus) at ×40 magnification and nonsaturating conditions. Images were segmented using the DAPI staining to generate masks matching cell nuclei from which the corresponding signals were calculated using an in-house–developed package based on CellProfiler. Antibody information and technical details can be found in Supplementary Materials and Methods.
Cell cycle and BrdU labeling analysis by flow cytometry
Fluorescence-activated cell sorting (FACS) analysis of cell cycle was described previously (5). In brief, cells were fixed overnight in ice-cold ethanol (70%), stained with PI [50 μg/ml in PBS supplemented with ribonuclease (RNase) A (0.25 mg/ml)] for 45 min at 37°C, and analyzed by a BD FACSCalibur equipped with CellQuest software. Data were analyzed and processed by FlowJo (version 8.8.4). For BrdU labeling, cells were pulsed with 10 μM BrdU for 1 hour (U-2-OS) or 4 hours (MDA-MB-231) and fixed overnight with 70% ethanol. DNA was denatured with 0.1 M HCl and incubated at 100°C, and BrdU was detected using the BD Pharmingen FITC Mouse Anti-BrdU Set Antibody (BD Biosciences). Cells were resuspended in 400 μl of PBS containing PI (25 μg/ml) and RNase A (0.1 mg/ml) and subjected to FACS analysis. The percentage of BrdU-positive cells was analyzed with FlowJo software. Results are representative of biological duplicates, at a minimum.
Clonogenic assay
siRNA-transfected U-2-OS or MDA-MB-231 cells were seeded onto six-well plates in technical triplicates or on 6-cm plates in technical duplicates. After 8 to 14 days, cells were fixed and colonies were stained with crystal violet (Sigma-Aldrich). For colony formation capacity, graphs show the average of at least two biological replicates and SEM. For survival analysis upon drug treatments, MDA-MB-231 cells were seeded according to the plating efficiency. After 24 hours of plating, cells were treated with AZD7762 for 24 hours and then washed and grown in fresh medium or were treated with olaparib and left in the culture continuously. Cells were incubated at 37°C for 10 to 14 days. Colonies were fixed and stained as described above. The colonies were counted using an in-house–built ImageJ macro using a Trainable Weka Segmentation plugin. On the basis of the colony number, plating efficiency (PE = number of colonies formed/number of cells seeded) and surviving fraction (SF = number of colonies formed after siRNA treatment/number of cells seeded × PE) were calculated and plotted.
MNase digestion assay
Chromatin was prelabeled with [14C]thymidine (0.5 pCi/ml) for 24 hours before siRNA transfection. Thirty hours after transfection, nascent chromatin was labeled with [3H]thymidine (25 nCi/ml). To compensate for lower replication rate in TLK2- and FLASH-depleted cells, labeling times were adjusted to obtain similar [3H]thymidine incorporation (10, 15, and 30 min for siRNA control, siTLK2, and siFLASH, respectively). Cells were lysed in hypotonic buffer [10 mM tris (pH 7.4), 2.5 mM MgCl2, and 0.5% NP-40, protease and phosphatase inhibitors], and nuclei were resuspended in digestion buffer [10 mM tris (pH 7.4), 10 mM NaCl, 5 mM MgCl2, and 2 mM CaCl2, protease and phosphatase inhibitors] and subjected to digestion with MNase (0.01 U/μl) (Worthington Biochemical Co.) at 37°C. Undigested chromatin was pelleted by centrifugation at 1500g for 2 min, and 14C and 3H activity in supernatant and undigested chromatin were measured with a liquid scintillation counter (LS 6500, Beckman Coulter). Readings were corrected for 14C bleed through into the 3H channel. Graphs show one representative experiment of three biological replicates.
SNAP-tag histone imaging
Cells were plated in Lab-Tek II Chamber slides (Labclinics) 24 hours after siRNA transfection. SNAP labeling was initiated 48 hours after transfection (24 hours after plating). For quench-chase-pulse experiments, cells were incubated with 5 μM SNAP-Cell Block (S9106S, New England Biolabs) for 30 min at 37°C. After two PBS washes, cells were incubated in medium for 30 min, followed by two washes. Cells were incubated in medium for the chase period (6 to 7 hours). Cells were incubated with 1 μM TMR-Star (S9105S, New England Biolabs) for 30 min, washed twice, incubated in medium for 30 min, washed twice, preextracted for 5 min in 0.2% Triton X-100/PBS on ice, and fixed for 10 min in 4% PFA.
DNA fiber assay and DNA combing
For fiber assays, 30 hours after siRNA transfection, U-2-OS cells were pulsed with 10 μM 5-iodo-2′-deoxyuridine (IdU) (Sigma-Aldrich) for 10 min, followed by 20-min labeling with 100 μM CldU (MP Biomedicals). Cells resuspended in 2 μl of ice-cold PBS were incubated with 7 μl of spreading buffer [200 mM tris-HCl (pH 7.5), 0.5% SDS, and 50 mM EDTA] for 3 min on a slide, and DNA fibers were stretched by tilting. After fixation with methanol/acetic acid (3:1), DNA was denatured with 2.5 M HCl and blocked (PBS with 1% BSA and 0.1% Triton X-100) before staining with primary antibodies. Single-molecule analysis of DNA replication by molecular combing was performed as described in protocol 36 available from the EpiGeneSys Network of Excellence website. In brief, after 30 hours of siRNA transfection, U-2-OS cells were labeled with 10 μM IdU (Sigma-Aldrich) for 10 min, followed by 20-min labeling with 100 μM CldU (MP Biomedicals). Cells were harvested immediately after the pulse and molded into low-melting agarose plugs. Plugs were treated with proteinase K buffer, melted at 67°C, and then digested by β-agarase. DNA was combed on silanized coverslips (Genomic Vision), denatured by 2.5 M HCl, and probed by the primary antibodies. Graphs show one representative experiment of two biological replicates, and lines represent medians. Images for both assays were collected using a DeltaVision system (Applied Precision) with a UApo/340 40×/1.35 NA oil objective lens, and the length of CldU-labeled tracks was measured with SoftWoRx 5.0.0 (Applied Precision).
Chromatin fractionation
U-2-OS cells were transfected with TLK2 siRNA and treated with or without UCN-01 for 2 hours. Soluble proteins were removed by preextraction with cold CSK-T buffer containing protease/phosphatase inhibitors on ice for 5 min, and proteins associated with chromatin were subjected to Western blotting. Results are representative of two biological replicates.
Pulsed-field gel electrophoresis
U-2-OS cells were transfected with siRNA for 24 hours and treated with UCN-01 for 4 hours or irradiated with 20 Gy of ionizing radiation. Cells (1 × 106) were molded into 1% low-melting agarose plugs (InCert Agarose, Lonza), followed by treatment with proteinase K buffer [10 mM tris-Cl (pH 7.5), 50 mM EDTA, 1% N-laurylsarcosyl, proteinase K (2 mg/ml)] at 50°C for 48 hours. Plugs were then subjected to PFGE (1% agarose, CHEF-DR II system; Bio-Rad Laboratories; 120° angle, 60- to 240-s switch time, and 4 V/cm) for 20 hours. DNA was visualized by ethidium bromide staining. Quantification was performed using ImageJ software.
Statistical analysis
Statistical analysis was performed using Prism 6 (GraphPad Software), and the n and tests used were stated in the figure legends. Statistical tests were performed and are only reported in figures and figure legends, where n ≥ 3. Statistical analysis of TCGA data sets is described in Supplementary Materials and Methods.
Supplementary Material
Acknowledgments
We thank P. Menard for help with cloning and C. Alabert and Z. Jasencakova for discussion and comments on the manuscript, A. Lladó and S. Tosi for ImageJ macro writing, the IRB Barcelona Advanced Digital Microscopy Facility and Y. Antoku for help with microscopy, C. Cortina for advice regarding CRISPR/CAS9 editing, and L. Jansen for SNAP-tag histone plasmids. Funding: A.G. was supported by the Danish National Research Foundation to the Center for Epigenetics (DNRF82), the European Commission Innovative Training Networks FP7 “aDDRess,” European Research Council starting grants (ERC2011StG no. 281,765 and ERC2016CoG no. 724436), the Danish Cancer Society, the Independent Research Fund Denmark, the NEYE Foundation, the Novo Nordisk Foundation, and the Lundbeck Foundation. T.H.S. was supported by a grant from the Spanish Ministry of Economy and Competitiveness (MINECO) (BFU2015-68354), Ayudas para incentivar la incorporación estable de doctores 2015, and institutional funding from MINECO through the Centres of Excellence Severo Ochoa award and from the Centres de Recerca de Catalunya Programme of the Catalan Government. S.-B.L. was supported by postdoctoral fellowships from the Lundbeck Foundation and the Danish Cancer Society and by junior researcher grants from Taipei Medical University (TMU104-AE1-B23) and the Ministry of Science and Technology (MOST 105-2311-B-038-002 and 106-2311-B-038-001). S.S.-B. was supported by a fellowship from Fundació La Caixa, and M.V.-P. was supported by a Severo Ochoa Formación de Personal Investigador fellowship (MINECO). Author contributions: S.-B.L. and S.S.-B. performed most of the experiments, designed experiments, analyzed data, prepared figures, and edited the manuscript. G.S. and M.A.M.T. helped with high-content imaging of EdU, DNA damage, and RPA. M.V.-P. performed replication stress experiments in numerous cell lines and analyzed data. T.-Y.C. performed Western blots and data analysis. C.S.-O.A. performed the TCGA data analysis. T.H.S. and A.G. supervised all experiments, analyzed data, prepared figures, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat4985/DC1
Supplementary Materials and Methods
Fig. S1. TLK2 is required for efficient DNA replication.
Fig. S2. TLK2 is required for replication-coupled chromatin assembly.
Fig. S3. Sustained depletion of TLK activity leads to DNA damage and checkpoint-induced G1 arrest.
Fig. S4. TLK2 depletion causes genomic instability that is amplified by checkpoint and PARP inactivation.
Fig. S5. Analysis of TLK status in cancer.
Table S1. TCGA cohort designations.
Table S2. Chromosome locations of genes analyzed.
Table S3. Analysis of correlated gene expression.
Table S4. Survival analysis of TCGA patient data.
Table S5. Multivariate survival analysis of TCGA patient data.
REFERENCES AND NOTES
- 1.Zeman M. K., Cimprich K. A., Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant H. E., Petermann E., Schultz N., Jemth A.-S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T., PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Min W., Bruhn C., Grigaravicius P., Zhou Z.-W., Li F., Krüger A., Siddeek B., Greulich K.-O., Popp O., Meisezahl C., Calkhoven C. F., Bürkle A., Xu X., Wang Z.-Q., Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 4, 2993 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Alabert C., Groth A., Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 13, 153–167 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Mejlvang J., Feng Y., Alabert C., Neelsen K. J., Jasencakova Z., Zhao X., Lees M., Sandelin A., Pasero P., Lopes M., Groth A., New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 204, 29–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groth A., Corpet A., Cook A. J. L., Roche D., Bartek J., Lukas J., Almouzni G., Regulation of replication fork progression through histone supply and demand. Science 318, 1928–1931 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Groth A., Ray-Gallet D., Quivy J.-P., Lukas J., Bartek J., Almouzni G., Human Asf1 regulates the flow of S phase histones during replicational stress. Mol. Cell 17, 301–311 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Krause D. R., Jonnalagadda J. C., Gatei M. H., Sillje H. H. W., Zhou B.-B., Nigg E. A., Khanna K., Suppression of Tousled-like kinase activity after DNA damage or replication block requires ATM, NBS1 and Chk1. Oncogene 22, 5927–5937 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Groth A., Lukas J., Nigg E. A., Silljé H. H. W., Wernstedt C., Bartek J., Hansen K., Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. EMBO J. 22, 1676–1687 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silljé H. H. W., Takahashi K., Tanaka K., Van Houwe G., Nigg E. A., Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 18, 5691–5702 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klimovskaia I. M., Young C., Strømme C. B., Menard P., Jasencakova Z., Mejlvang J., Ask K., Ploug M., Nielsen M. L., Jensen O. N., Groth A., Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat. Commun. 5, 3394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly R., Davey S. K., Tousled-like kinase-dependent phosphorylation of Rad9 plays a role in cell cycle progression and G2/M checkpoint exit. PLOS ONE 8, e85859 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silljé H. H. W., Nigg E. A., Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr. Biol. 11, 1068–1073 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Hammond C. M., Stromme C. B., Huang H., Patel D. J., Groth A., Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 18, 141–158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunavala-Dossabhoy G., De Benedetti A., Tousled homolog, TLK1, binds and phosphorylates Rad9; TLK1 acts as a molecular chaperone in DNA repair. DNA Repair 8, 87–102 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Bruinsma W., van den Berg J., Aprelia M., Medema R. H., Tousled-like kinase 2 regulates recovery from a DNA damage-induced G2 arrest. EMBO Rep. 17, 659–670 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han Z., Riefler G. M., Saam J. R., Mango S. E., Schumacher J. M., The C. elegans Tousled-like kinase contributes to chromosome segregation as a substrate and regulator of the Aurora B kinase. Curr. Biol. 15, 894–904 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrera P., Moshkin Y. M., Grönke S., Silljé H. H. W., Nigg E. A., Jäckle H., Karch F., Tousled-like kinase functions with the chromatin assembly pathway regulating nuclear divisions. Genes Dev. 17, 2578–2590 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corpet A., De Koning L., Toedling J., Savignoni A., Berger F., Lemaître C., O’Sullivan R. J., Karlseder J., Barillot E., Asselain B., Sastre-Garau X., Almouznia G., Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J. 30, 480–493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J. A., Tan Y., Wang X., Cao X., Veeraraghavan J., Liang Y., Edwards D. P., Huang S., Pan X., Li K., Schiff R., Wang X.-S., Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat. Commun. 7, 12991 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petermann E., Woodcock M., Helleday T., Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. U.S.A. 107, 16090–16095 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sørensen C. S., Syljuåsen R. G., Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 40, 477–486 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clément C., Vassias I., Ray-Gallet D., Almouzni G., Functional characterization of histone chaperones using SNAP-tag-based imaging to assess de novo histone deposition. Methods Enzymol. 573, 97–117 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Barcaroli D., Bongiorno-Borbone L., Terrinoni A., Hofmann T. G., Rossi M., Knight R. A., Matera A. G., Melino G., De Laurenzi V., FLASH is required for histone transcription and S-phase progression. Proc. Natl. Acad. Sci. U.S.A. 103, 14808–14812 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang T. H., Fowler F. C., Chen C.-C., Shen Z.-J., Sleckman B. P., Tyler J. K., The histone chaperones ASF1 and CAF-1 promote MMS22L-TONSL-mediated Rad51 loading onto ssDNA during homologous recombination in human cells. Mol. Cell 69, 879–892 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foote K. M., Blades K., Cronin A., Fillery S., Guichard S. S., Hassall L., Hickson I., Jacq X., Jewsbury P. J., McGuire T. M., Nissink J. W. M., Odedra R., Page K., Perkins P., Suleman A., Tam K., Thommes P., Broadhurst R., Wood C., Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-y l}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 56, 2125–2138 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Zabludoff S. D., Deng C., Grondine M. R., Sheehy A. M., Ashwell S., Caleb B. L., Green S., Haye H. R., Horn C. L., Janetka J. W., Liu D., Mouchet E., Ready S., Rosenthal J. L., Queva C., Schwartz G. K., Taylor K. J., Tse A. N., Walker G. E., White A. M., AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 7, 2955–2966 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Guzi T. J., Paruch K., Dwyer M. P., Labroli M., Shanahan F., Davis N., Taricani L., Wiswell D., Seghezzi W., Penaflor E., Bhagwat B., Wang W., Gu D., Hsieh Y., Lee S., Liu M., Parry D., Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 10, 591–602 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Kohn E. A., Yoo C. J., Eastman A., The protein kinase C inhibitor Gö6976 is a potent inhibitor of DNA damage-induced S and G2 cell cycle checkpoints. Cancer Res. 63, 31–35 (2003). [PubMed] [Google Scholar]
- 30.Toledo L. I., Altmeyer M., Rask M. B., Lukas C., Larsen D. H., Povlsen L. K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J., ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Dungrawala H., Rose K. L., Bhat K. P., Mohni K. N., Glick G. G., Couch F. B., Cortez D., The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 59, 998–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T., Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Farmer H., McCabe N., Lord C. J., Tutt A. N. J., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M. B., Jackson S. P., Smith G. C. M., Ashworth A., Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Jasencakova Z., Scharf A. N., Ask K., Corpet A., Imhof A., Almouzni G., Groth A., Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell 37, 736–743 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Huang H., Strømme C. B., Saredi G., Hödl M., Strandsby A., González-Aguilera C., Chen S., Groth A., Patel D. J., A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat. Struct. Mol. Biol. 22, 618–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segura-Bayona S., Knobel P. A., González-Burón H., Youssef S. A., Peña-Blanco A., Coyaud É., López-Rovira T., Rein K., Palenzuela L., Colombelli J., Forrow S., Raught B., Groth A., de Bruin A., Stracker T. H., Differential requirements for Tousled-like kinases 1 and 2 in mammalian development. Cell Death Differ. 24, 1872–1885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee K. Y., Im J. S., Shibata E., Dutta A., ASF1a promotes non-homologous end joining repair by facilitating phosphorylation of MDC1 by ATM at double-strand breaks. Mol. Cell 68, 61–75.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoek M., Stillman B., Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 12183–12188 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saredi G., Huang H., Hammond C. M., Alabert C., Bekker-Jensen S., Forne I., Reverón-Gómez N., Foster B. M., Mlejnkova L., Bartke T., Cejka P., Mailand N., Imhof A., Patel D. J., Groth A., H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714–718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piwko W., Buser R., Peter M., Rescuing stalled replication forks: MMS22L-TONSL, a novel complex for DNA replication fork repair in human cells. Cell Cycle 10, 1703–1705 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Kelemen L. E., Wang X., Fredericksen Z. S., Pankratz V. S., Pharoah P. D. P., Ahmed S., Dunning A. M., Easton D. F., Vierkant R. A., Cerhan J. R., Goode E. L., Olson J. E., Couch F. J., Genetic variation in the chromosome 17q23 amplicon and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 18, 1864–1868 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobbelstein M., Sørensen C. S., Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 14, 405–423 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Bodor D. L., Valente L. P., Mata J. F., Black B. E., Jansen L. E. T., Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Mol. Biol. Cell 24, 923–932 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N., The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curtis C., Shah S. P., Chin S.-F., Turashvili G., Rueda O. M., Dunning M. J., Speed D., Lynch A. G., Samarajiwa S., Yuan Y., Gräf S., Ha G., Haffari G., Bashashati A., Russell R., McKinney S.; METABRIC Group, Langerød A., Green A., Provenzano E., Wishart G., Pinder S., Watson P., Markowetz F., Murphy L., Ellis I., Purushotham A., Børresen-Dale A.-L., Brenton J. D., Tavaré S., Caldas C., Aparicio S., The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat4985/DC1
Supplementary Materials and Methods
Fig. S1. TLK2 is required for efficient DNA replication.
Fig. S2. TLK2 is required for replication-coupled chromatin assembly.
Fig. S3. Sustained depletion of TLK activity leads to DNA damage and checkpoint-induced G1 arrest.
Fig. S4. TLK2 depletion causes genomic instability that is amplified by checkpoint and PARP inactivation.
Fig. S5. Analysis of TLK status in cancer.
Table S1. TCGA cohort designations.
Table S2. Chromosome locations of genes analyzed.
Table S3. Analysis of correlated gene expression.
Table S4. Survival analysis of TCGA patient data.
Table S5. Multivariate survival analysis of TCGA patient data.