

 Isatinyl and indanyl nitrones (INs) were synthesized and evaluated for their antiproliferative activity and antioxidant properties.
Isatinyl and indanyl nitrones (INs) were synthesized and evaluated for their antiproliferative activity and antioxidant properties.
Abstract
A series of ketonitrones derived from isatin and indanone (INs) were synthesized and evaluated for their antiproliferative activities against several human cancer cell lines. Then, the antioxidant properties of these substrates were measured by the DPPH test to report their biological activity in terms of their spin trapping action. In particular, one substrate has showed very high biological and scavenging activity, probably due to the strong correlation between its spin trapping activity and structure.
Introduction
Small organic molecules have proven to be invaluable tools for investigating biological systems, but there is still much to learn from their use.1–3
Nitrones are small molecules that have the general chemical formula X–CH NO–Y. Their structural nature endows them their “spin-trap” ability for trapping free radical intermediates (R˙), forming stable radical adducts (X–CHR NO˙–Y).4 In fact, studies of their spin trapping activity involved the reaction of nitrones with reactive free radicals such as hydroxyl (˙OH), lipid alkoxy (˙OL) or lipid hydroxyperoxyl (˙OOL) radicals, observing the formation of more stable radicals that can be classified and quantified by electron spin resonance (ESR) or electron paramagnetic resonance (EPR) spectroscopy.5–7
Considering a) that biological systems may actively produce reactive oxygen species (ROS) and reactive nitrogen species (RNS), b) that specific oxidation products are produced from reactions between biological molecules and ROS or RNS, c) that ROS and RNS play an important role in many pathological diseases, nitrones and, in particular, PBN-nitrones, where X is a phenyl group and Y is a tert-butyl group, were recently considered as therapeutics due to their widespread anti-cancer activity.8,9 The mechanistic basis of their anti-cancer action is not known. It is probable that their ability to scavenge radical intermediates that are produced during disease processes, including cancer, is the basis for their anti-cancer activity.4 Moreover, their action on important membrane enzymes and as anti-inflammatory agents seems to contribute to the enhancement of their antioxidant activity.
Isatin and oxindole derivatives have found wide application in medicinal chemistry, for example as potential anticonvulsants,10 cyclin-dependent kinase 2-inhibitors11 or inhibitors of poxvirus,12 ectomelia,13 rhinovirus,14 HIV-115 and corona-virus,16 the latter responsible for severe acute respiratory syndrome (SARS). Moreover, the insertion of an oxindole nucleus in spiro compounds has recently attracted attention because of their prevalence in various natural products and biologically active molecules.17 In fact, the key to their activity seems to be the combination of a spiro carbon and a variously substituted oxindole core. Therefore, the oxindole moiety could be considered as a useful tool in drug discovery.
It is generally recognized that incorporating different bioactive scaffolds into one molecule is a powerful strategy to construct substrates with structural novelty and biological potential.18 In an attempt to generate novel molecular entities with amplified spin trapping and antiproliferative activity on cancer cells, we synthesized some isatin and indanone ketonitrones, combining the simultaneous presence of an oxindole-like ring and a nitrone portion.
Results and discussion
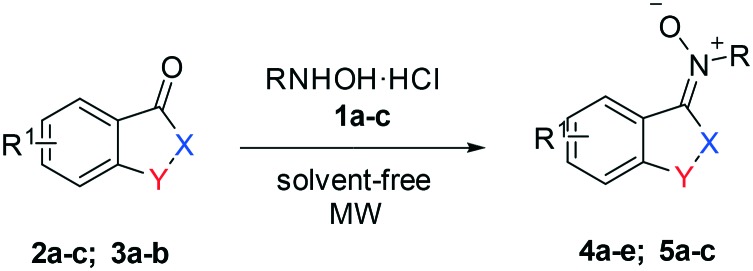
In the literature, a few examples of synthetic procedures for nitrones of oxindole derivatives have been reported thus far.19–22 Recently, we have realized an environmentally-friendly approach to synthesize aldo- and ketonitrones by solvent-free condensation of alkyl- or arylhydroxylamine hydrochlorides with aromatic aldehydes under microwave irradiation.23
Herein, we report the practical and stereoselective synthesis of isatinyl and indanyl nitrones (INs) obtained from substituted hydroxylamines 1a–c and isatin derivatives 2a–c or indanones 3a–b, as shown in Table 1. Then, we investigated the antiproliferative activity of these substrates on MG63 and TE85 human osteosarcoma cell lines and the K562 human erythroleukemic cell line; next we sought to correlate it with their antioxidant properties measured by the DPPH assay.
Table 1. Synthesis of isatinyl and indanyl nitrones.
| ||||||||
| Entry | R | R1 | Y | X | MW (W) | Time (min) | Product | Yield (%) |
| 1 | Me | 5-NO2 | NH | CO | 600 | 10 | 4a | 95 |
| 2 a | Ph | H | NH | CO | 600 | 10 | 4b | 92 |
| 3 | Bn | 5-NO2 | NH | CO | 600 | 13 | 4c | 92 |
| 4 | Bn | H | NH | CO | 600 | 12 | 4d | 95 |
| 5 | Bn | H | NMe | CO | 600 | 12 | 4e | 92 |
| 6 | Me | H | CH2 | CH2 | 400 | 30 | 5a | 82 |
| 7 | Me | 5-F | CH2 | CH2 | 400 | 28 | 5b | 83 |
| 8 | Bn | H | CH2 | CH2 | 400 | 30 | 5c | 85 |
aA ketone/alkylhydroxylamine ratio of 1 : 2 for all substrates except for entry 2 with a ratio of 1 : 3.
The target compounds 4a–e and 5a–c were prepared by following our previously reported methodology23 which consists of co-grinding the ketone and hydroxylamine in a mortar, followed by transferring the mixture without the use of solvent in an appropriate vessel and further mixing of the solids in a vortex mixer. Finally, the mixture is placed in a microwave oven without the use of solvent and is irradiated at 600 W or 400 W for variable time periods in the order of minutes.
The variously substituted products 4a–e and 5a–c were obtained in high yields (83–95%). The low reactivity of the indanone ring prompted us to reduce the MW power and to increase the reaction time. In all cases, nitrones were obtained as single isomers, (E) 4a–e and (Z) 5a–c, respectively, as confirmed by NOESY experiments. In particular, for nitrone 4b, the formation of only a single product, the E-isomer, is observed with respect to Z/E mixtures obtained by other synthetic methodologies.24
In an attempt to understand the molecular characteristics and the effects produced by the modifications introduced into our nitrones on tumour cell proliferation, the nitrones prepared according to this procedure were evaluated for their antiproliferative activity against human osteosarcoma (MG63 and TE85)25 and chronic myeloid leukemia (K562)26 cell lines. Cells were cultured for three days in the absence or in the presence of increasing concentration of nitrones and then their viability was evaluated by analyzing the activity of the oxidative metabolism by the 3-(4,5- dimethylthiazolyil-2)-2,5-diphenyltetrazolium bromide (MTT) assay.27 In Fig. 1, the proliferation of the treated cells was expressed as percentage compared to untreated control cells.
Fig. 1. Effect of nitrones 4a–e and 5a–c on cell proliferation. MG63 (A), TE85 (B) and K562 (C) cells were incubated for three days in the presence of the indicated compounds. Viable cells were measured by the MTT test and reported as % relative to untreated control. The arithmetic mean value ± standard deviation of three experiments performed in triplicate is shown.
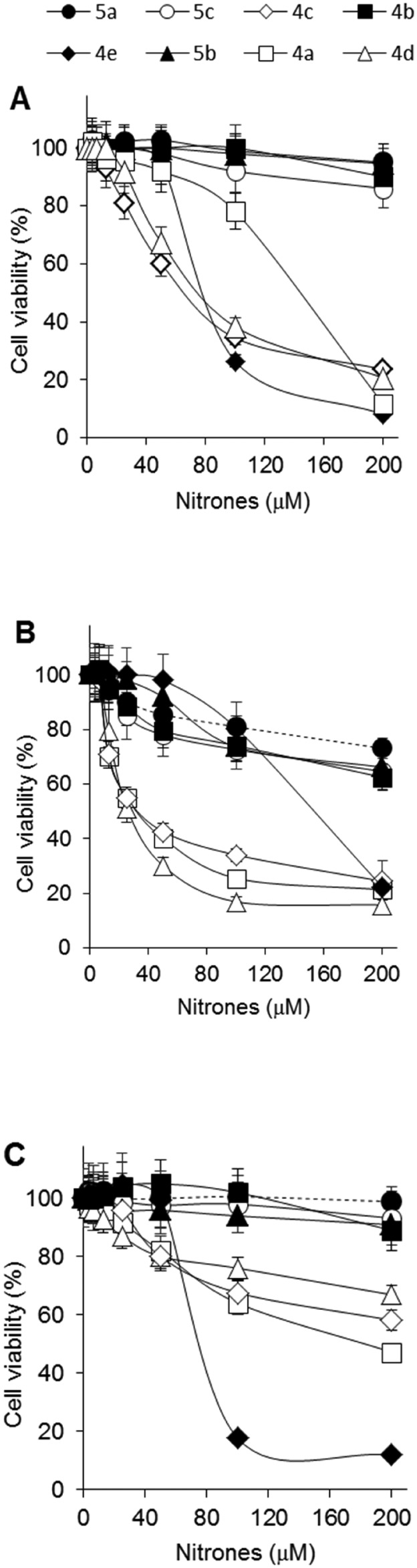
The data indicated that not all the nitrones had antiproliferative activity, since compounds 5a–c and 4b were completely inactive. Compounds 4c–e inhibited the proliferation of osteosarcoma MG63 cells compared to untreated cells (Fig. 1A, IC50 about 77 μM).
In Fig. 1B, the antiproliferative activities of nitrones 4a and 4c–d on TE85 cells (IC50 about 32 μM) are shown. The active compounds are less effective in inhibiting the proliferation of K562 cells, with the exception of nitrone 4e with an IC50 of about 78 μM (Fig. 1C). Together, these results have shown that the indanyl derivatives do not possess antiproliferative activity on the tumour cells analyzed, as well as the derivative 4b with the phenyl substituent. By contrast, isatinyl nitrones containing methyl or benzyl groups possess antiproliferative activity.
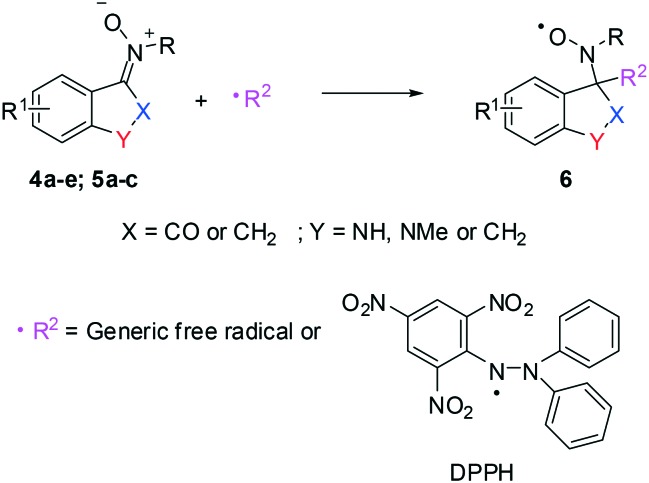
The in vitro antioxidant activity of these ketooxindole nitrones (4a–e and 5a–c) was evaluated by using the stable organic free radical DPPH (1,1-diphenyl-2-picrylhydrazyl) radical scavenging activity assay.28–31
In Scheme 1, the reaction mechanism for the spin-trapping action of nitrones 4a–e and 5a–c with a generic free radical (including the DPPH radical) is illustrated, according to the literature.32 It can be noted from the scheme below that the reaction produces the highly stable nitroxyl radical 6.33,34
Scheme 1. Reaction mechanism of nitrones 4a–e and 5a–c with free radicals.
When DPPH reacts with a radical scavenger, its maximum absorbance decreases. A freshly prepared DPPH solution displayed a deep purple colour with the absorption maximum at 517 nm. The ethanolic solution of DPPH was added to the solution of the synthesized compounds in ethanol. After 10 min of incubation in the dark, the absorbance was measured by using absolute ethanol as blank. BHT (butylated hydroxytoluene) was used as reference. In the presence of an antioxidant, the DPPH absorbance decreases. The antioxidant activity was calculated as radical scavenging activity (RSA%) as expressed in eqn (1):
| RSA% = [(A0 – Ai)/A0] × 100 | 1 |
where A0 and Ai are the DPPH absorbance at 517 nm in the absence and presence of the synthesized compounds, respectively. The RSA% for ketonitrones 4a–e and 5a–c at five different concentrations (i.e., 1.15, 2.45, 4.00, 6.25 and 9.10 × 10–6 mol L–1) of the tested compounds with DPPH at 517 nm is reported in Table 2.
Table 2. Percentage of in vitro radical scavenging activity of INs.
| Compounds | Concentrations (10–6 mol L–1) |
EC50 | ||||
| 1.15 | 2.45 | 4.00 | 6.25 | 9.10 | ||
| 4a | — | — | 1.1 ± 0.5 | 72 ± 2 | 79 ± 2 | 1.2 ± 0.2 |
| 4b | 1.1 ± 0.5 | 1.2 ± 0.5 | 1.2 ± 0.5 | 69 ± 2 | 80 ± 2 | 1.3 ± 0.2 |
| 4c | — | — | 60 ± 1 | 67 ± 2 | 80 ± 2 | 1.0 ± 0.2 |
| 4d | 1.1 ± 0.5 | 1.1 ± 0.5 | 1.2 ± 0.5 | 1.2 ± 0.5 | 78 ± 2 | 1.8 ± 0.1 |
| 4e | 1.2 ± 0.5 | 60 ± 1 | 68 ± 2 | 70 ± 2 | 79 ± 2 | 0.6 ± 0.3 |
| 5a | — | — | 1.1 ± 0.5 | 60 ± 1 | 78 ± 2 | 1.3 ± 0.2 |
| 5b | — | — | 1.1 ± 0.5 | 1.2 ± 0.5 | 79 ± 2 | 1.8 ± 0.1 |
| 5c | 1.1 ± 0.5 | 1.2 ± 0.5 | 1.3 ± 0.5 | 70 ± 2 | 79 ± 2 | 1.2 ± 0.2 |
| BHT | — | 4.6 ± 0.5 | 11.7 ± 0.5 | 23.0 ± 0.5 | 27.0 ± 0.5 | 3.8 ± 0.5 |
As can be seen in Table 2, all the compounds exhibited antioxidant activity. The substances can be divided into three main groups. The first group is composed of two substances (4c and 4e) with higher values of antioxidant activity than others. In particular, the highest values were observed for 4e, for which the RSA% is equal to 60% at a concentration of 2.45 × 10–6 mol L–1.
Four substances compose the second group with intermediate antioxidant activity (4a–b, 5a and 5c). For all of these compounds, the RSA% was higher than 60% at a concentration equal to 6.25 × 10–6 mol L–1. In the third group, only two compounds with lower antioxidant activity were included: 4d and 5b. For these last two compounds, the RSA% was 80% at a concentration equal to 9.10 × 10–6 mol L–1. As can be seen in Table 2 and Fig. 2, all nitrones 4a–e and 5a–c exhibited a radical scavenging activity significantly higher than that of the reference (BHT). We have not investigated concentrations higher than 9.10 × 10–6 mol L–1 as the RSA% trend reaches a plateau.
Fig. 2. Graphical presentation of in vitro DPPH radical scavenging activity of compounds relative to the standard antioxidant BHT.
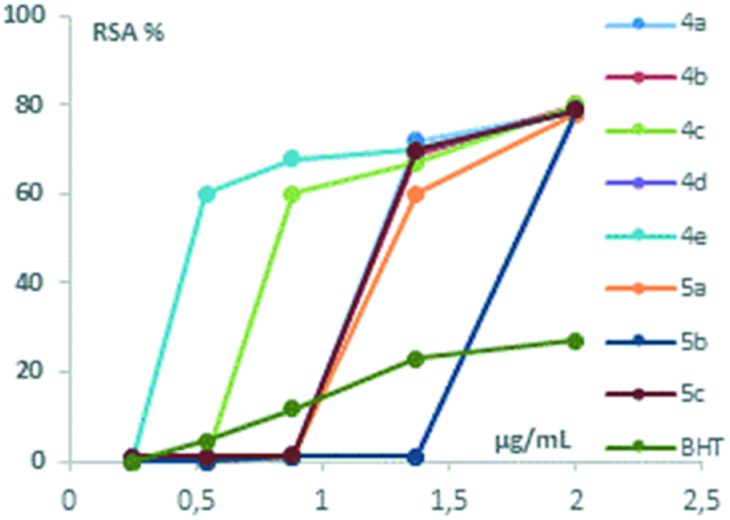
The RSA% of nitrones 4a–e and 5a–c may be compared with the value of archetypal (Z) α-phenyl-N-tert-butyl nitrone (PBN) reported in the literature.35 Its RSA% is 1.4 ± 0.9 at a concentration of 0.5 × 10–3 mol L–1 calculated through the DPPH test, demonstrating antioxidant properties significantly lower with respect to those of our nitrones.
For all the tested compounds, the EC50 values were also calculated by using the GraphPad Prism 5.01 program, as reported in the literature (Table 2).36 The EC50 is the antioxidant concentration required to obtain 50% radical inhibition. As can be seen in Table 2 the EC50 values of all eight synthesized nitrones are higher than that of the reference substance (BHT).
The antioxidant properties of INs seem to agree quite well with the biological results, confirming the importance of the isatinyl ring. In particular, the presence of both aromatic systems and electron-donating groups (e.g. methyl group) improves the performance, as can particularly be seen for substrate 4e.
Experimental
Commercial starting materials were used without further purification. Reactions were monitored by TLC using 60-F264 silica plates, commercially available from Merck. 1H and 13C NMR spectra were recorded at 300 MHz and 75 MHz, respectively, in CDCl3 and DMSO-d6 using tetramethylsilane (TMS) as an internal standard (Bruker ACP 300 MHz). Chemical shifts are given in parts per million and coupling constants in Hertz. The stereochemistry was established by NOESY experiments. Purity was verified by NMR and HPLC.
LC-MS analysis was carried out using an Agilent 6540 UHD Accurate – Mass Q-TOF LC-MS (Agilent, Santa Clara, CA) equipped with an electrospray ionisation source (Dual AJS ESI) operating in positive ion mode. Chromatographic separation was achieved using a C18 RP analytical column (Poroshell 120, SB-C18, 50 × 2.1 mm, 2.7 μm) at 30 °C with an elution gradient from 5% to 95% of B over 13 min., A being H2O (0.1% FA) and B CH3CN (0.1% FA). The flow rate was 0.4 ml min–1.
MW-assisted reactions were performed on a Synthos 3000 instrument from Anton Paar, equipped with a 4x24MG5 rotor and an IR probe as an external control of the temperature. 0.3–3 mL glass vials sealed with a dedicated PEEK screw-cup together with a reliable PTFE seal were used for all reactions. In the synthesis of all the derivatives, the temperature was always maintained at 180 °C in each experiment, except for 5a–c where the temperature was always maintained at 125 °C.
General procedure for synthesis of nitrones 4a–e and 5a–c
The selected ketone (0.50 g) and appropriate hydroxylamine derivative (2 eq. or 3 eq. for N-phenylhydroxylamine) were ground in a mortar, placed in an appropriate vessel and mixed using a vortex mixer. The mixture was transferred to a microwave oven and irradiated with suitable power. After the appropriate time, the crude oil was recrystallized with ethyl acetate (4a–e) or cyclohexane (5a–c).
(E)-N-(5-Nitro-2-oxoindolin-3-ylidene)methanamine oxide 4a
Yellow solid, 95% yield, (0.52 g). 1H NMR (300 MHz, DMSO-d6): 4.28 (s, 3H, CH3), 7.02 (d, J = 8.73 Hz, 1H, Ar), 8.23 (d, J = 8.73 Hz, 1H, Ar), 8.75 (s, 1H, Ar), 11.53 (s, 1H, NH), 13C NMR (75 MHz, DMSO-d6): δ 51.8, 110.2, 118.7, 118.7, 128.0, 133.3, 142.4, 145.2, 161.9. ESI (+)-MS: m/z [M + H] calcd for C9H8N3O4 222.0515, found: 222.0503.
(E)-N-(2-Oxoindolin-3-ylidene)aniline oxide 4b
Orange red solid, 92% yield, (0.74 g). 1H NMR (300 MHz, DMSO-d6): 6.85 (d, J = 7.80 Hz, 1H, Ar), 7.15 (t, J = 7.65 Hz, 1H, Ar), 7.40 (t, J = 7.80 Hz, 1H, Ar), 7.45–7.65 (m, 5H, Ar), 8.27 (d, J = 7.65 Hz, 1H, Ar), 10.78 (s, 1H, NH), 13C NMR (75 MHz, DMSO-d6): δ 109.7, 118.4, 121.7, 123.9, 124.1, 128.7, 130.1, 132.2, 134.4, 140.9, 146.3, 159.9. ESI (+)-MS: m/z [M + H] calcd for C14H11N2O2 239.0821, found: 239.0817.
(E)-N-(5-Nitro-2-oxoindolin-3-ylidene)-1-phenylmethanamine oxide 4c
Yellow solid, 92% yield (0.64 g). 1H NMR (300 MHz, DMSO-d6): 5.91 (s, 2H, CH2), 7.07 (d, J = 8.73 Hz, 1H, Ar), 7.31–7.58 (m, 5H, Ar), 8.28 (dd, J = 2.43 Hz, 8.73 Hz, 1H, Ar), 8.84 (d, J = 2.43 Hz, 1H, Ar), 11.70 (s, 1H, NH) 13C NMR (75 MHz, DMSO-d6): δ 65.6, 110.4, 118.8, 119.0, 128.3, 129.1, 129.1, 129.6, 133.2, 134.1, 142.6, 145.4, 161.9. ESI (+)-MS: m/z [M + H] calcd for C15H12N3O4 298.0828, found: 298.0816.
(E)-N-(2-Oxoindolin-3-ylidene)-1-phenylmethanamine oxide 4d
Orange solid, 95% yield (0.80 g). 1H NMR (300 MHz, DMSO-d6): δ 5.89 (s, 2H, CH2Bn), 6.91 (d, J = 7.78 Hz, 1H, Ar), 7.10 (dt, J = 0.91, 7.65 Hz, 1H, Ar), 7.25–7.54 (m, 6H, Ar), 8.12 (d, J = 7.65 Hz, 1H, Ar), 11.01 (s, 1H, NH), 13C NMR (75 MHz, DMSO-d6): δ 64.3, 109.8, 118.1, 122.04, 123.9, 128.5, 129.6, 129.1, 131.8, 133.4, 134.2, 139.6, 161.2. ESI (+)-MS: m/z [M + H] calcd for C15H13N2O2 253.0977, found: 253.0977.
(E)-N-(1-Methyl-2-oxoindolin-3-ylidene)-1-phenylmethanamine oxide 4e
Yellow solid, 92% yield (0.76 g). 1H NMR (300 MHz, DMSO-d6): 3.25 (s, 3H, CH3), 5.92 (s, 2H, CH2), 7.08 (t, J = 8.26 Hz, 2H, Ar), 7.37–7.50 (m, 6H, Ar), 8.15 (d, J = 7.34 Hz, 1H, Ar), 13C NMR (75 MHz DMSO-d6): δ 26.6, 65.2, 109.2, 117.8, 123.1, 124.0, 129.0, 129.0, 129.59, 132.2, 134.6, 141.5, 160.4. ESI (+)-MS: m/z [M + H] calcd for C16H15N2O2 267.1134, found: 267.1126.
(Z)-N-(2,3-Dihydro-1H-inden-1-ylidene)methanamine oxide 5a
White solid, 82% yield (0.50 g). 1H NMR (300 MHz, CDCl3): δ 2.90–3.05 (m, 2H, CH2), 3.11–3.22 (m, 2H, CH2), 3.81 (s, 3H, CH3), 7.29–7.44 (m, 3H, Ar), 8.84 (d, J = 7.81 Hz, 1H, Ar), 13C NMR (75 MHz, CDCl3): δ 28.7, 29.4, 49.6, 124.5, 127.0, 131.0, 134.4, 147.8, 149.7. ESI (+)-MS: m/z [M + H] calcd for C10H12NO 162.0919, found: 162.0912.
(Z)-N-(5-Fluoro-2,3-dihydro-1H-inden-1-ylidene)methanamine oxide 5b
White solid, 83% yield (0.48 g). 1H NMR (300 MHz, CDCl3): δ 3.01 (dd, J = 5.43 Hz, 11.73 Hz, 2H, CH2), 3.14 (dd, J = 5.55 Hz, 11.97 Hz, 2H, CH2), 6.80–7.12 (m, 2H, Ar), 8.86 (dd, J = 5.79 Hz, 8.64 Hz, 1H, Ar), 13C NMR (75 MHz, CDCl3): δ 28.8 (d, JCF = 2.28), 29.9, 49.6, 111.9 (d, J2CF = 23.02), 114.3 (d, J2CF = 22.86), 128.8 (d, J3CF = 9.03), 130.9 (d, J4CF = 2.02), 148.1, 150.5 (d, J3CF = 8.95), 164.43 (d, J1CF = 250.96). ESI (+)-MS: m/z [M + H] calcd for C10H11FNO 180.0825, found: 180.0818.
(Z)-N-(2,3-Dihydro-1H-inden-1-ylidene)-1-phenylmethanamine oxide 5c
White solid, 85% yield (0.76 g). 1H NMR (300 MHz, CDCl3): δ 2.91–3.09 (m, 2H, CH2), 3.10–3.23 (m, 2H, CH2), 5.11 (s, 2H, CH2Bn), 7.21–7.45 (m, 7H, Ar), 7.51 (d, J = 7.16 Hz, 1H, Ar), 8.92 (d, J = 7.63 Hz, 1H, Ar), 13C NMR (75 MHz, CDCl3): δ 29.0, 29.2, 66.6, 124.5, 127.1, 127.2, 128.2, 128.3, 128.9, 131.1, 133.4, 134.8, 147.7, 149.1. ESI (+)-MS: m/z [M + H] calcd for C16H16NO 238.1232, found: 238.1230.
Antiproliferative activity evaluation
Human cell lines were obtained from ATCC (Manassas, VA). MG63 (CRL-1427) and HOS TE85 (CRL-1543) osteosarcoma cells were maintained in DMEM, while K562 (CCL-243) chronic myeloid leukemia cells in RPMI 1640. The culture medium was supplemented with 10% fetal bovine serum, penicillin (100 U mL–1), streptomycin (100 U mL–1) and glutamine (2 mM); incubation was conducted at 37 °C in a 5% CO2 atmosphere. Compounds were solubilized in 40 mM DMSO, kept at –80 °C in the dark and diluted in complete medium immediately before use. The osteosarcoma cells were seeded at 4000 cells per well in a 96-well plate 6 hours before the treatment. The leukemia cells were seeded at 20 000 cells per mL. Cells were treated with the test compounds at concentrations ranging from 3 to 200 μM. Untreated cells were placed in every plate as negative control. After 3 days of culture, the inhibitory effect on cell proliferation was analysed by addition of 25 μL MTT (thiazolyl blue) staining solution, in which metabolically active cells convert the yellow tetrazolium salt to purple formazan crystals providing a quantitative determination of viable cells. Two hours later, formazan crystals were solubilized in 100 μL of lysing buffer (50% DMF + 20% SDS, pH 4.7) for 16 hours, whereupon the spectrophotometric absorbance at 570 nm was measured. The half maximal inhibitory concentration (IC50) was calculated using Scientist software. Three independent experiments were performed in triplicate.
Antioxidant evaluation
The free radical scavenging activity against DPPH was determined at five different concentrations according to a previous work.28 Thus, EtOH solutions containing known amounts of the compounds (4a–e and 5a–c) and of DPPH were prepared in a range of concentrations from 1.15 × 10–6 to 9.10 × 10–6 mol L–1 (Table 2). A decrease in the absorbance of DPPH was measured at 517 nm, against ethanol as blank, by using a UV-vis spectrophotometer (Varian Cary 50 Scan), after a period of 10 min since preparation. Experiments were carried out in triplicate and BHT (butylated hydroxytoluene) was used, in the same concentration range of the compounds, as the reference antioxidant.
Conclusions
In summary, a series of isatinyl/indanyl nitrones (INs) were synthesized and their antiproliferative activity on MG63, TE85 and K562 cells was determined. Derivatives with the isatin ring showed the highest activity, while the indanone nitrones were practically inactive. Then the antioxidant activities were evaluated through the DPPH test. The results indicated that all compounds are markedly more active than the reference compound BHT. Among them, nitrone 4e exhibits the most potent antioxidant activity at low concentration and its spin-trap action matches perfectly with its antiproliferative effect on cancer cells. Collectively, the current study may provide a new insight into the treatment of degenerative diseases such as cancer.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank the Italian Ministry of University and Scientific Research (MIUR) for a doctoral grant and the University of Calabria for financial support.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00537g
References
- Stockwell B. R. Nature. 2004;432(7019):846. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furia E., Porto R. J. Chem. Eng. Data. 2008;53(12):2739. [Google Scholar]
- Furia E., Napoli A., Tagarelli A., Sindona G. J. Chem. Eng. Data. 2013;58(5):1349–1353. [Google Scholar]
- Floyd R. A., Chandru H. K., He T., Towner R. Anti-Cancer Agents Med. Chem. 2011;11(4):373–379. doi: 10.2174/187152011795677517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen G. M., Cohen M. S., Britigan B. E., Pou S. Free Radical Res. Commun. 1990;9:187–195. doi: 10.3109/10715769009145676. [DOI] [PubMed] [Google Scholar]
- Tomasi A. and Iannone A. in ESR spin trapping artifacts in biological model systems, EMR of Paramagnetic Molecules, ed. L. J. Berliner and J. Reuben, Plenum Press, New York, 1993, pp. 353–354. [Google Scholar]
- Rhodes C. J., in Toxicology of the Human Environment, The Critical Role of Free Radicals, Taylor & Francis, New York, 2000. [Google Scholar]
- Floyd R. A., Kopke R. D., Choi C.-H., Foster S. B., Doblas S., Tower R. A., Free Radical Biol. Med., 2008, 4510 , 1361 –1374 , , 15 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd R. A., Towner R. A., Wu D., Abbott A., Cranford R., Branch D., Guo W.-X., Foster S. B., Jones I., Alam R., Moore D., Allen T., Huycke M. Free Radical Res. 2010;44(1):108–117. doi: 10.3109/10715760903321796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M., Pandeya S. N., Singh K. N., Stables J. P. Acta Pharm. 2004;54:49. [PubMed] [Google Scholar]
- Bramson H. N., Corona J., Davis S. T., Dickerson S. H., Edelstein M., Frye S. V., Gampe Jr. R. T., Harris P. A., Hassell A., Holmes W. D., Hunter R. N., Lackey K. E., Lovejoy B., Luzzio M. J., Montana V., Rocque W. J., Rusnak D., Shewchuk L., Veal J. M., Walker D. H., Kuyper L. F. J. Med. Chem. 2001;44:4339. doi: 10.1021/jm010117d. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C., Pansare S. V., Sarma K. D., Keith K. A., Kern E. R. J. Med. Chem. 2005;48:3045. doi: 10.1021/jm049147h. [DOI] [PubMed] [Google Scholar]
- Bauer D. J., Sadler P. W. Nature. 1961;190:1167. doi: 10.1038/1901167a0. [DOI] [PubMed] [Google Scholar]
- Gladych J. M. Z., Hunt J. H., Jack D., Haff R. F., Boyle J. J., Stewart R. C., Ferlauto R. J. Nat. 1969;221:286. doi: 10.1038/221286b0. [DOI] [PubMed] [Google Scholar]
- Sriram D., Bal T. R., Yogeeswari P. J. Pharm. Pharm. Sci. 2005;8:565. [PubMed] [Google Scholar]
- Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L. J. Med. Chem. 2006;49:3440. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- Yu B., Yu D.-Q., Liu H.-M. Eur. J. Med. Chem. 2015;97:673–698. doi: 10.1016/j.ejmech.2014.06.056. [DOI] [PubMed] [Google Scholar]
- Maiuolo L., Bortolini O., De Nino A., Russo B., Gavioli R., Sforza F. Aust. J. Chem. 2014;67:670. doi: 10.1016/j.bmc.2010.08.024. [DOI] [PubMed] [Google Scholar]
- Yang H.-B., Min S. Org. Biomol. Chem. 2012;10:8236. doi: 10.1039/c2ob26413g. [DOI] [PubMed] [Google Scholar]
- Aurich H. G., Weiss W. Tetrahedron. 1976;32:159. [Google Scholar]
- Wu S. Y., Ma X.-P., Liang C., Mo D.-L. J. Org. Chem. 2017;82:3232–3238. doi: 10.1021/acs.joc.6b02774. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-H., Wu M.-Y., Huang W.-C. RSC Adv. 2015;5:105825. [Google Scholar]
- Maiuolo L., De Nino A., Merino P., Russo B., Stabile G., Nardi M., D'Agostino N., Bernardi T. Arabian J. Chem. 2016;9:25. [Google Scholar]
- Wu S.-Y., Ma X.-P., Liang C., Mo D.-L. J. Org. Chem. 2017;82(6):3232–3238. doi: 10.1021/acs.joc.6b02774. [DOI] [PubMed] [Google Scholar]
- Fuchs B., Mahlum E., Halder C., Maran A., Yaszemski M., Bode B., Bolander M., Sarkar G. Gene. 2007;399:137. doi: 10.1016/j.gene.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feriotto G., Calza R., Bergamini C. M., Griffin M., Wang Z., Beninati S., Ferretti V., Marzola E., Guerrini R., Pagnoni A., Cavazzini A., Casciano F., Mischiati C. Amino Acids. 2017;49:551. doi: 10.1007/s00726-016-2339-4. [DOI] [PubMed] [Google Scholar]
- Bortolini O., De Nino A., Eliseo T., Gavioli R., Maiuolo L., Russo B., Sforza F. Bioorg. Med. Chem. 2010;18:6970. doi: 10.1016/j.bmc.2010.08.024. [DOI] [PubMed] [Google Scholar]
- Kaur M., Singh B., Singh B., Arjuna A. J. Heterocycl. Chem. 2017;54:1348. [Google Scholar]
- Fazio A., Caroleo M. C., Cione E., Plastina P. Food Packaging and Shelf Life. 2017;11:84. [Google Scholar]
- Fazio A., Terenzio D., Piccinelli A., Rastrelli L. Pharmacologyonline. 2014;2:1. [Google Scholar]
- Petkes H., Gal E., Bischin C., Lupan I., Majdik C., Cristea C., Silaghi-Dumitrescu L., Rev. Roum. Chim., 2015, 60 , 7 –8 , , 659 . [Google Scholar]
- Janzen E. G. Acc. Chem. Res. 1971;4(1):31. [Google Scholar]
- Zhdanov R. I., in Bioactive Spin Labels, Springer-Verlag, Berlin, Heidelberg, 1992. [Google Scholar]
- Hawkins C. L., Davies M. J. Biochim. Biophys. Acta. 2014;1840:708. doi: 10.1016/j.bbagen.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Samadi A., Soriano E., Revuelta J., Valderas C., Chioua M., Garrido I., Bartolomé B., Tomassolli I., Ismaili L., González-Lafuente L., Villarroya M., García A. G., Oset-Gasque M. J., Marco-Contelles J. Bioorg. Med. Chem. 2011;19:951. doi: 10.1016/j.bmc.2010.11.053. [DOI] [PubMed] [Google Scholar]
- Chen Z., Bertin R., Froldi G. Food Chem. 2013;138:414. doi: 10.1016/j.foodchem.2012.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.