

 Cancer is a black spot on the face of humanity in this era of science and technology.
Cancer is a black spot on the face of humanity in this era of science and technology.
Abstract
Cancer is a black spot on the face of humanity in this era of science and technology. Presently, several classes of anticancer drugs are available in the market, but issues such as toxicity, low efficacy and solubility have decreased the overall therapeutic indices. Thus, the search for new promising anticancer agents continues, and the battle against cancer is far from over. Imidazole is an aromatic diazole and alkaloid with anticancer properties. There is considerable interest among scientists in developing imidazoles as safe alternatives to anticancer chemotherapy. The present article describes the structural, chemical, and biological features of imidazoles. Several classes of imidazoles as anticancer agents based on their mode of action have been critically discussed. A careful observation has been made into pharmacologically active imidazoles with better or equal therapeutic effects compared to well-known imidazole-based anticancer drugs, which are available on the market. A brief discussion of the toxicities of imidazoles has been made. Finally, the current challenges and future perspectives of imidazole based anticancer drug development are conferred.
Introduction
In the 21st century, cancer is still a problem. It is a genetic disease that progresses via development of multistep carcinogenesis with the participation of various physiological systems in the human body, such as cell signaling and apoptosis, and as a result, cancer is extremely complicated to combat.1 Generally, cancer begins as a local disease, but it metastasizes over time, making it difficult to cure. It is the leading cause of death in developed countries. The possible signs and symptoms of cancer are new lumps, abnormal bleeding, prolonged cough, unexplained weight loss, and change in bowel movements, among others.2 However, it is not presently clear how nutritional imbalance affect one's cancer risk.3 Globally, cancer is the second leading cause of death, and it is the number one cause in the US.4–6 From all over the world, cancer has drawn unusual attention, and extensive research (radiation and chemotherapy) has been devoted towards the development of effective anticancer modalities.7 As per the prediction of the World Health Organization in 2012, the number of annual cancer cases will rise from 14 million to 22 million within the next 2 decades.8 According to a 2014 UN DESA report, the world population of 7.3 billion is expected to reach 8.5 billion by 2030, 9.7 billion in 2050 and 11.2 billion in 2100. Out of these, about 1.5 billion cancer cases are expected with about 1.2 billion deaths.9 Moreover, total deaths are expected to reach 2.14 billion in the year 2030 with an expected 8.5 billion population.10,11 Breast and lung cancers are the common causes of deaths (a total of 522 000 deaths in 2012) in women and men, respectively. These two types of cancers are most frequently diagnosed in about 140 countries.12
Despite the accessibility of several anticancer drugs, common problems like multi-drug resistance, less therapeutic efficacy, solubility, adverse side effects, and/or poor bioavailability issues necessitate the development of new anticancer agents.13,14 For more than a century, heterocyclic compounds (particularly bearing nitrogen atoms) have been the major molecules in organic chemistry because of their remarkable pharmaceutical activities, especially their anticancer properties.15–17 Since the discovery of imidazole in the 1840s, the research and development of imidazole based compounds have been interesting and active fields of research.18–22 Imidazole (five membered aromatic ring) is an organic alkaloid that acts as an important pharmacophore in drug discovery. The imidazole ring is present in various natural23–25 and synthetic26–29 bioactive compounds. Natural compounds include biotin, essential amino acids, histidine, histamine, and pilocarpine alkaloids, and the synthetic compounds include cimetidine, losartan26 fungicides, herbicides,27 plant growth regulators28 and therapeutic agents.29 Imidazoles have a wide range of biological activities, such as anticancer,30 antifungal,31 antiviral,32 antibacterial,33 antitubercular,34 antiparasitic,35 antihistaminic,36 anti-inflammatory,37 antineuropathic,38 antiobesity,39 and antihypertensive,40 and other medicinal properties.41–47 Among various pharmaceutical activities, imidazoles have been used as effective anticancer agents. Presently, several imidazoles, dacarbazine, temzolomide, zoledronic acid, mercaptopurine, nilotinib, tipifarnib etc., are being used in clinics to cure various cancers (Fig. 1). Briefly, these molecules have remarkable pharmaceutical potencies to control cancer. The interesting anticancer properties of imidazoles inspired the prevalent development of their derivatives with the hope of increasing the efficiency and lowering the side effects. The experimental evidence indicated that the different types of cancers originate because of a change in the structure and function of DNA (deoxyribonucleic acid), VEGF (vascular endothelial growth factor), topoisomerase I & II, mitotic spindle microtubules, histone deacetylases, receptor tyrosine kinases, topoisomerases, CYP26A1 enzyme, etc. Among the various targets, DNA is one of the leading targets for anticancer molecules. Thus, it has become the epicenter of attraction for the scientific community to identify and characterize the interactions of small molecules with DNA to help design new and efficient therapeutic agents for controlling gene expression.48–50 Remarkably, imidazoles interact with DNA via covalent or non-covalent interactions (electrostatic, intercalation and groove binding) and halt cell division.51 In comparison to other heterocyclic molecules, imidazoles easily binds to protein molecules,52 and some imidazole drugs at high concentrations directly inhibit the synthesis of essential cell membrane components without interference from sterols and sterol esters.53
Fig. 1. Market faces and chemical structures of some imidazole-based anticancer drugs.

The literature survey revealed around 1180 research papers on imidazoles with interesting anticancer results. An analysis of the number of publications on “imidazoles as anticancer agents” from 2002 to 2016 indicated steadily growing interest in research on this topic. The period from 2002 to 2016 was divided into three intervals, viz. 2002–06, 2007–11 and 2012–16, (Fig. 2). It is clear from Fig. 2 that there were 261 publications during 2002–2006, 420 during 2007–2011 and 500 from 2012–2016. Further, the literature indicated that some review papers on related topics have appeared on imidazoles.54–57 However, none of the articles discussed the classes of imidazoles as anticancer agents based on the mode of action, and a review has not appeared since 2010. Therefore, an updated review of the recent advances in this field is missing. Thus, it was thought worthwhile to address the recent advances in the development of imidazoles as anticancer agents of clinical interest. In this review, the recent advances with a special emphasis on the structural, chemical, and biological features of imidazoles are addressed. Attempts have been made to outline the mechanism of action of imidazoles, and careful observations of pharmacologically active imidazoles with better or equal therapeutic effects to that of the standard drugs have been presented. In addition, the issues of the toxicity of imidazoles have been highlighted. Finally, market status, current challenges and future perspectives have been addressed. We hope that this paper will open new opportunities for researchers to design future generation novel and potent imidazole containing anticancer drugs.
Fig. 2. A graphic representation of the increasing interest in research on imidazoles as anticancer agents from 2002–06 through 2007–11 to 2012–16.

Imidazole: an exciting heterocyclic moiety
Imidazole is an important chemical ring that is present in many existing drugs.58 This ring exists in two equivalent tautomeric forms as the hydrogen atom is located on either of the two nitrogen atoms. In 1858, imidazole was first synthesized by Heinrich Debus59 from glyoxal and formaldehyde (Scheme 1a). The % yield in this protocol was quite low, but the protocol is still being used to create C-substituted imidazoles. Later, in 1977, Van Leusen synthesized imidazole using a three component reaction with aldimines and tosylmethyl isocyanide (Scheme 1b).60 However, the yield was low. Therefore, the thrust of its synthesis continues. Consequently, in 1996, Zhang et al.61 reported a constructive approach for imidazoles syntheses (16–56% overall yields) via a Ugi four component reaction of arylglyoxals, primary amines, carboxylic acids, and isocyanide on a Wang resin (Scheme 1c). Initially, ketoamides were formed, which were treated with 60 equivalent of NH4OAc in AcOH at 100 °C for 20 h, followed by reaction with 10% TFA-DCM at 23 °C for 20 min for cyclization of the corresponding imidazoles. The Ugi reaction was carried out in a mixture of CHCl3–MeOH–pyridine (1 : 1 : 1) at 65 °C for 3 days. With the passage of time, high yielding productive approaches were given in the mid-2000's. In the year 2003, a novel one pot synthesis of tetra substituted imidazoles under solvent-free conditions and microwave irradiation was reported by Balalaie et al., (Scheme 1d) and the yield of imidazoles was quite high (80–92%).62 Another efficient, simple, and high yielding constructive approach for synthesis of imidazoles from 1,2-diketones and aldehydes in the presence of NH4OAc was reported by Wolkenberg et al. (Scheme 1e). Under microwave irradiation, alkyl, aryl, and heteroaryl substituted imidazoles were formed in yields ranging from 80 to 99%.63 Besides, there are many other methods available for the synthesis of imidazoles with good yields.64–70
Scheme 1. Heinrich debus synthesis 1a, Van Leusen three-component reaction (vl-3CR) 1b, synthesis of imidazoles via Ugi reaction on Wang resin 1c, one pot synthesis 1d and short synthesis 1e.

At the molecular level, medicinal chemistry is concerned with the discovery, development, interpretation, and identification of the mechanism of action of biologically active compounds.71 As already noted above, this exciting heterocyclic moiety constructs several compounds that possess an extensive spectrum of pharmacological activities. This paper aims to review the anticancer activities of imidazoles during the past years.
Imidazoles as anticancer agents
Many research papers have revealed that imidazoles possess anticancer drug potentials. The success of imidazoles as anticancer agents started with dacarbazine, which triggered interest in the development of imidazole agents. Several classes of various structured heterocyclic molecules, which include imidazoles, for different types of cancer treatment have been designed and developed to cure cancer via various targets.12 Imidazoles have the potential to overcome the various drawbacks of currently available clinical drugs and to develop anticancer agents. Therefore, attempts have been made to highlight some important classes of imidazoles based on mechanisms of action via various targets like DNA, VEGF, mitotic spindle microtubules, histone deacetylases, receptor tyrosine kinases, topoisomerases, CYP26A1 enzyme, rapid accelerated fibrosarcoma (RAF) kinases, etc.
Imidazoles as antiangiogenic agents
Angiogenesis is essential to the growth of cells and tissues.72–75 Tumors cannot grow through a defined volume if they are not vascularized. The vascular endothelial growth gactor (VEGF) is an important angiogenic factors secreted by tumor cells. Most human cancer cells express elevated levels of VEGF and VEGF receptors on their surface. An antiangiogenic drug (e.g., bevacizumab) impairs the VEGF pathway and tumor vasculature by targeting VEGF76 or their receptors. In the last two decades, several small molecules have been evaluated as antiangiogenic agents, but only a few molecules have worked.77 For example vincristine, taxol and etoposide are currently being used for the treatment of acute lymphocytic leukemia, ovarian, and lung cancers, respectively.78–80 Several imidazoles have been explored as anticancer agents with antiangiogenesis as the mode of action. Hansen et al. reported a series of levamisole derivatives 1–4i (Fig. 3) for the in vitro inhibition of angiogenesis.81 The authors summarized the morphological effect of the three groups of levamisole. In the first group, the authors found p-bromolevamisole 2 was an equivalent angiogenic agent to levamisole, and N-methyllevamisoles 4a and 4b were more potent than the parent inhibitor, levamisole. In the second one, bromolevamisole was highlighted as having a higher antiangiogenic agent than levamisole. Finally, the third group comprised N-alkylated analogues, and they were considered good inhibitors of angiogenesis. Gopinath et al.77 constructed another series of imidazoles 5a–8f (Fig. 3) by the reaction of chrome-3-carboxylic acids with substituted acyl bromides in the presence of TEA followed by reflux with NH4OAc in toluene. The reported imidazoles were screened in vitro for the inhibition of KRAS/Wnt and their anti-angiogenesis properties. In addition, they carried out a molecular docking analysis and found the higher binding affinity of 8f with KRAS/Wnt and VEGF. Finally, based on the angiogenesis inhibition mechanism, it was concluded that imidazole 8f is a potent anticancer agent.
Fig. 3. Imidazoles reported as antiangiogenic agents.

The focal adhesion kinase (FAK) enzyme is a key pathogenic mediator of various diseases, including cancer. It stimulates the tumor progression and metastasis via regulation of the cell migration, invasion, extracellular matrix, and angiogenesis. Therefore, this enzyme is a promising therapeutic target, as it is highly expressed at both the transcriptional and translational levels in various cancers.82–84 In this countenance, Dao et al. highlighted the anticancer efficiencies of imidazoles (triazines incorporated with imidazole rings) and explored their antiangiogenic activities against human umbilical vein endothelial cells and anticancer efficacy against several cancer cell lines with the aid of focal adhesion kinase (FAK) inhibition.85 Besides, the authors checked the in vitro enzymatic activities of the developed imidazole based heterocyclic compounds with reference to the standard inhibitor TAE-226. It is an ATP competitive FAK inhibitor that has shown potent anti-proliferative and antitumor effects in vitro and in vivo in several malignancies, including brain tumors, esophageal cancer, breast cancer, ovarian cancer, and gastrointestinal stroma tumor. The IC50 value of the standard TAE-226 is 0.007 ± 0.002 μM. The series of compounds displayed IC50 values in the range of 10–7–10–8 M. Among the series, several inhibitors, 9a–9d (Fig. 4), showed high anticancer activities against four cancer cell lines (U87-MG, HCT-116, MDA-MB-231, and PC-3).
Fig. 4. Potent antiangiogenic imidazoles.

Imidazoles as antiproliferative agents
Among various cancer cures, the antiproliferative approach is one of the significant ways to control this deadly disease. The various heterocyclic molecules, particularly imidazoles, have great antiproliferative potential. Chen et al.86 reported seventeen molecules of imidazole and imidazoline analogues as antiproliferative agents for melanoma. In this work, the authors investigated the antiproliferative activities of these imidazole and imidazoline analogs in two melanoma cell lines (human A375 cells and mouse B16 cells) using a fibroblast cell line as a control unit. The authors studied the anticancer activities of 10a–10g imidazoles (Fig. 5) using the standard sulforhodamine B (SRB) assay and sorafenib (Bay43-9006) as a reference standard drug. As per the authors, 10 μM concentrations of some imidazoles showed good activity. Moreover, a few analogues had similar activities to the sorafenib standard drug. The IC50 values of the reported compounds are given in Fig. 5. It is clear from this figure that these values ranged from 16.1 to >100 μM.
Fig. 5. Potent antiproliferative imidazoles.

Jamalian and his colleagues87 demonstrated in their review that the potential of 1,8-acridinones as antiproliferative agents could be enhanced by incorporating imidazole moieties bearing electron withdrawing groups. In this regard, Sarkarzadeh et al.88 investigated a series of heterocyclic molecules containing imidazole moieties for their antiproliferative effects on cervical (HeLa), colon (LS180), breast (MCF-7) and Jurkat human cancer cell lines by MTT assay. Interestingly, the authors found that the compounds 11a–11c, bearing the imidazole-2-yl moiety on the C11-position of the dihydropyridine ring, exhibited superior antiproliferative activities compared to cis-platin, especially in the Jurkat cell line. The chemical structures of the potent imidazoles along with the IC16 and IC50 values are given in Fig. 5. Benzimidazoles are complex compounds with a broad range of pharmacological properties, like antitumor.89 These compounds have been studied extensively and are reported to be very potent cytotoxic agents against various cancer cell lines.90 Roopashree et al. investigated the antiproliferative effect of newly synthesized benzimidazole derivatives 12a–12d (Fig. 6) on Hela cells.91 Although the reported compounds were found to be less effective with reference to IC50 > 50, the compound 12a, with a high alkyl group, was considered more potent than the standard drug with an IC50 = 25.3 μM. In addition, the compounds 12b–12d were found to be good cytotoxic agents with IC50 = 30.2 μM, 31.9 μM, and 30 μM, respectively. Due to the close structural similarities of benzimidazoles with the purine nucleotide, these moieties are applied for multiple targeting, especially in antitumor activity, which make these heterocycles good pharmacophores. Alkahtani et al. synthesized another series of benzo[d]imidazoles and evaluated their anticancer activities.92 In this work, the authors found some imidazoles 13a–13c (Fig. 6) carrying chloro, cylopentyl, –CH3, –SOCH3 and –SO2CH3 substituents as potent antiproliferative agents in cancer cell lines.
Fig. 6. Antiproliferative benzimidazoles.

Imidazoles as CYP26A1 enzyme inhibitors
All trans retinoic acids (ATRAs), physiological metabolites of vitamin A, are useful in the treatment of cancer,93–95 skin related diseases, and neurodegenerative and autoimmune diseases, particularly in oncology against acute promyelocytic leukemia (APL). ATRAs can change the prognosis of APL from fatal to highly curable.96–99 However, the emergence of resistance has severely hampered clinical applications along with the easily metabolism of ATRAs into 4-hydroxyl-RA by the CYP26A1 enzyme. Therefore, inhibition of the CYP26A1 metabolic enzyme denotes a promising strategy for the discovery of new specific anticancer agents. Few research papers have been published wherein imidazoles have been reported as inhibitors of the CYP26A1 enzyme. Several families of retinoic acid metabolism blocking agents (RAMBAs) targeting the CYP26A1 enzyme with imidazoles have been exploited from generation to generation. The first generation imidazoles as RAMBAs were ketoconazole, miconazole and liarozole100,101 (Fig. 7), which were evaluated as potent blocking agents to inhibit CYP26A1 activities. However, the lack of CYP26 isoform specificity and the extensive structure activity relationship (SAR) studies on imidazoles led to the discovery of the second generation CYP26A1 inhibitors [OSI Pharma, naphthyl compound (Fig. 7)] and other azoles.102–105 It was found that these second generation RAMBAs exhibited higher potency and better specificity against the CYP26A1 enzyme than the first generation compounds, and some proceeded to clinical studies where they showed encouraging preclinical and clinical results with improved specific activities.106,107 Recently, Sun et al. described the synthesis and biological evaluation of fifteen imidazoles 14a–14o (Fig. 8) as retinoic acid metabolism blocking agents (RAMBAs) towards the CYP26A1 enzyme.108 It was found that all the compounds exhibited applicable activities towards the CYP26A1 enzyme with IC50 values ranging from 0.22–1.11 μM. Additionally, the authors observed that 14f and 14i were promising inhibitors with IC50 values of 0.22 and 0.46 μM towards the CYP26A1 enzyme, respectively. Furthermore, these two compounds were evaluated for CYP selectivity and the metabolic profile of ATRA and found to show higher selectivities for CYP26A1 in comparison to CYP2D6, CYP3A4. Gomaa et al. synthesized another series of imidazoles109 (Fig. 9). The imidazoles were evaluated for their ability to inhibit CYP26-mediated retinoic acid metabolism (CYP26) inhibitory activity using a cell-free microsomal assay with the standards liarozole (IC50 = 540 nM) and R116010 (IC50 = 10 nM). All the imidazoles showed poor response, but two imidazoles, namely 3-[4-(6-bromo-pyridin-3-ylamino)-phenyl]-3-imidazol-1-yl-2,2-dimethyl-propionic acid methyl ester 15a and 3-[4-(benzo[1,3]dioxol-5-ylamino)-phenyl]-3-imidazol-1-yl-2,2-dimethyl-propionic acid methyl ester 15b, exhibited potent activities against the CYP26 enzyme. The chemical structures of the potent imidazoles are shown in Fig. 9.
Fig. 7. Chemical structures of the first generation (A) and second generation (B) imidazoles as CY26A1 inhibitors.

Fig. 8. Imidazoles as CY26A1 inhibitors.

Fig. 9. Imidazoles as CY26A1 inhibitors.

Gomaa et al. synthesized 3-(1H-imidazol)-2,2-dimethyl-3-(4-(naphthalen-2-ylamino)phenyl)propyl derivatives 16a–16k (Fig. 9) and carried out a MCF-7 CYP26A1 microsomal assay for the assessment of anticancer activities.110 Almost all the compounds have shown good CYP26A1 inhibitory activities compared to the standard drugs (liarozole and R116010). Amongst all the compounds, 2-[3-imidazol-1-yl-5-(naphthalen-2-ylamino)-phenyl]-2-methyl-propionic acid methyl ester 16a had the most promising IC50 value, which was less than the standard drug.
Imidazoles as topoisomerase inhibitors
DNA topoisomerases are the enzymes that regulate DNA strand supercoiling by catalyzing winding and unwinding. These have been accepted as an important target in anticancer drug discovery.111–115 There are several clinically important classes of antitumorals drugs like anthracycline (doxorubicin and daunorubicin), epipodophyllotoxin (etoposide and teniposide), mitoxanthrone, amonafide, topotecan, irinotecan, camptothecin and amsacrine classes that are considered topoisomerase (TOP) inhibitors.116–119 About 50% of anticancer agents available target cancer via inhibition of the topoisomerase mechanism.120,121 However, high toxicity, the poor solubility and short action duration hinders their continuous use. Many efforts have been devoted to searching for safer and more effective TOP inhibitors. Due to two typical nitrogen atoms, imidazoles can form hydrogen bonds, which are favorable for the water solubility of the target compounds. For this reason, imidazole has been considered as a structural fragment and is frequently introduced into other bioactive skeletons. By multiple component reactions, Baviskar et al. constructed a series of N-fused imidazoles and evaluated them against human topoisomerase IIα (hTopoIIα) in decatenation, relaxation, cleavage complex, and DNA intercalation in vitro assays.122 The results of the study revealed that five imidazoles 17a–17e (Fig. 10) acted as potent DNA non-intercalating topoisomerase IIα catalytic inhibitors. In addition, the authors observed higher anticancer activities compared to etoposide and 5-fluorouracil in kidney and breast cancer cell lines with an apoptotic effect in the G1/S phase. Besides, these imidazoles showed low toxicity to normal cells. Recently, Negi et al. reported the microwave synthesis of ten imidazoles conjugated with imine/amides and screened them for anticancer activities with seven cancer cell lines (A-459, Hep-G2, H-460, HeLa, MCF7, PC3 and IGROV-1).123 Only three imidazoles 18a–18c (Fig. 10) showed promising in vitro anticancer activities with low micromolar IC50 values against A-459, Hep-G2 and H-460 cancer cell lines.
Fig. 10. Topoisomerase-II-α inhibitors.

Indimitecan 19a is a clinical anticancer candidate with good solubility to treat refractory solid tumors. It has improved stability without the lactone moiety in comparison to the conventional Top-I inhibitors. Moreover, it can induce long lasting DNA breaks and Top-I cleavage strands at unique genomic positions. Besides, it can bind with various targeting sites and cause cell cycle arrest in both the S and G(2)-M phases,124 which reveals its robust potency to overcome drug resistance.
Camptothecin (CPT) was the first effective TOP-I inhibitor derived from the plant alkaloid Camptotheca acuminate, but its lower water solubility largely limits drug administration.125 Introduction of ethyl and imidazolyl groups into camptothecin at the 7- and 10-positions yielded compound 19b. Due to these groups, it was observed that both the water solubility and TOP I inhibitory ability of CPT were improved. The IC50 values (0.16 and 0.06 μM) of 19b were low against breast (MCF-7) and colon cancer cell lines (HCT-8), and they were comparable to topotecan (0.15 and 0.07 μM) but inferior to camptothecin (0.02 and <0.01 μM), respectively.125 In addition, the research revealed that the E-lactone ring in camptothecin is unstable and produced toxic carboxylates in the human body. However, 19c and 19d with the imidazolyl at the 20-position were found to increase the stabilities of the lactone rings and decrease the toxicity. Besides, both exhibited stronger anticancer potencies than camptothecin towards the PC-3, MCF-7 and HCT-8 cancer cell lines.126 Cinelli et al. reported imidazole based aromathecin 19e, which had a better target TOP I inhibitory activity than aromathecin (non-CPT TOP I inhibitor).127 Indenoisoquinolines without the unstable hydroxyl lactone are also an important non-CPT TOP I inhibitors. However, the imidazole analog 19f displayed better water solubility and affinity towards DNA than the indenone analog (19g), due to the interaction of the incorporated NH moiety with the phosphates of the DNA backbone.
Podophyllotoxin derivatives have showed encouraging clinical efficacy toward several types of neoplasms by inhibiting DNA-TOP II. However, their imidazole derivative displayed more potent anticancer activities towards human cell lines, i.e., HeLa, K562, and K562/ADM, than etoposide.128 It is well established that acridine derivatives like amsacrine are a class of DNA-TOP II inhibitors as they can intercalate into the DNA base pair, leading to cell cycle arrest and apoptosis. The acridine-based imidazole 19h can satisfy the stereochemical and electronic requirements of the target receptor in a suitable way and displayed better anticancer activity towards MCF-7, HEP-2, COLO-205, HCT-15, A-549, and IMR-32 human cancer cell lines at a concentration of 10 μM in comparison to adriamycin.129 This result suggested that the introduction of an imidazole ring into the 9-amino acridine is an effective approach to exploit new anticancer drugs. Nor-indenoisoquinolines were also found to be a novel class of TOP I inhibitors with poor solubility. The imidazole derivative (19i) showed good water solubilities and improved anticancer efficacies towards MCF-7, IGROVI, HCT-116, and SN12C.130 The chemical structures of the podophyllotoxin derivatives (19j–19n) and imidazoles as topoisomerase inhibitors are shown in Fig. 11.
Fig. 11. Imidazoles as topoisomerase inhibitors.

Imidazoles as rapid accelerated fibrosarcoma (RAF) kinase inhibitors
Among important biological targets in the discovery of anticancer drugs, RAF kinases (A, B and C) are regarded as attractive targets because of their relationship with cell growth, differentiation and proliferation. Currently, sorafenib is clinically available for the treatment of renal cancer as an RAF inhibitor.131 However, its imidazole analogs 20a and 20b, possessing an amide linker and urea bridge, respectively, were found to possess remarkable CRAF inhibitory efficiency and more anticancer activities with GI50 values of 0.62 and 0.65 μM, respectively, compared to sorafenib (GI50 = 0.78 μM) towards the melanoma cancer cell line WM3629. Particularly, compound 20a showed a 99% inhibitory efficacy towards CRAF at a concentration of 10 μM with high selectivity. Astonishingly, for 20b, the substitution of the chlorine atom of the benzene ring decreased the bioactivities.132 All these findings revealed that imidazole analogs have great potential to act as RAF inhibitors for the treatment of melanoma. In another work reported by Rajitha et al., imidazole 20c was identified to be an excellent RAF inhibitor and exhibited superior anticancer activities towards the A549 and DLD-1 cell lines compared to sorafenib.133 Furthermore, notable antiproliferative activity was obtained when it was tested against acute myeloid leukemia (MV4-11) and compared to sorafenib. Moreover, modeling studies of 20c were carried out, and they showed the compound occupying the ribose position of the ATP binding pocket, which is necessary for anticancer activity. Yu et al. reported some imidazoles incorporated pyrazole moieties.134 This work revealed that the compound 20d exhibited 96% inhibitory activity towards CRAF at a concentration of 10 μM and good antiproliferative activities with GI50 values of 2.24 and 0.86 μM, respectively, in contrast to sorafenib (GI50 = 5.58 and 0.65 μM) against the human melanoma cell lines A375P and WM3629. Niculescu–Duvaz et al. reported the imidazole containing pyrazole tricyclic derivative 20e to be an active BRAF inhibitor for melanoma with good cellular activity (IC50 = 0.24 μM) and a pharmacokinetic profile.135 Furthermore, the study indicated that both the pyrazole tricyclic moiety and imidazole ring played crucial roles in exerting bioactivity. The replacement of the tricyclic pyrazole by a tricyclic triazole or six membered ring decreased the inhibitory potency. This might be attributed to the steric clash or unfavorable electrostatic interaction with the BRAF binding pocket. The structures for imidazoles as RAF inhibitors are shown in Fig. 12.
Fig. 12. Imidazoles as rapid accelerated fibrosarcoma kinase inhibitors.

Imidazoles as microtubule destabilizing anticancer agents
Microtubules, the important cytoskeletal filaments in cells, are assembled by polymerized tubulins. These filaments are proven molecular targets for anticancer drugs.136–140 The tubulin binding agents interact with the tubulins on the binding sites and cause aberrations in the tubulin assembly kinetics and, thus, alter the stabilities of the microtubule structure. The inhibition of tubulin polymerization or interfering with microtubule disassembly disturbs some cellular functions, like cell motility and mitosis. There are a number of microtubule targeted tubulin binding agents that are clinically effective drugs in cancer treatments, like vincristine, vinblastine, a third-generation vinca alkaloid (vinorelbine), combretastatin A-4 phosphate (under clinical trial), paclitaxel, docetaxel, epothilones, etc.131–136 Paclitaxel has a specific binding site on the microtubule polymer that makes it unique among chemotherapeutic agents, and the ability of paclitaxel to polymerize tubulin in the absence of cofactors like guanosine triphosphate and microtubule-associated proteins is unusual. It blocks cells in the G2/M phase of the cell cycle, and such cells are unable to form a normal mitotic apparatus. Vincristine also works via a microtubule destabilizing mechanism, via binding to the tubulin protein, and stopping the cell from separating its chromosomes during the metaphase; the cell then undergoes apoptosis.
Some papers141–146 have been published under the context of microtubule destabilizing agents in which the anticancer potencies of imidazoles are well addressed. The structures for imidazoles as microtubule destabilizing agents are shown in Fig. 13. In 2010, Li et al. reported 2-amino-1-arylidenaminoimidazoles as orally active anticancer agents via a microtubule target mechanism.147 In this work, the authors used a high throughput screening platform for the discovery of these imidazoles. The reported imidazoles were initially evaluated for cytotoxicity in the human gastric cancer NUGC-3 cells. The NUGC-3 cells were immunostained for microtubules by fluorescein isothiocyanate (FITC) and treated with one of the imidazoles at 1.0 μM for 16 h, and a DMSO vehicle control was included. Compared to the well-arranged microtubule structures in the untreated cancer cells, the disarranged microtubule structures were observed as diffused green fluorescent tubulinimmunoreactive activities. Therefore, the molecular interaction effects of 2-amino-1-arylidenaminoimidazoles on targeted microtubules were visualized by fluorescence imaging. It has been found that the substituents on the phenyl ring of arylidene 21a and 21b indicated comparable activities with IC50 values of 0.06 and 0.05 μM, respectively. However, imidazoles 21c, 21d and 21e, containing arylidene substituents of phenyl, 3-methyl-2-thiophenyl, and 4-quinolyl, respectively, exhibited cytotoxic IC50 values of 0.84, 0.91, and >10 μM against NUGC-3 cancer cells. Among these, the phenyl substitution was superior to the others. So, substitution at the 4-position of the phenyl ring played a critical role in the cytotoxicity. The authors also observed a similar phenomenon with substituents of 3,4-dichlorophenyl 21f and 2,3-dichlorophenyl 21g on arylidene and showed IC50 values of 0.74 and 5.12 μM, respectively. Furthermore, it has been observed that imidazoles 21h and 21i containing 3-thiophenyl and 2-thiophenyl at the 4-position of the imidazole ring exhibited the most potent cytotoxic activities against the NUGC-3 cancer cells, more than 21j and 21k containing 2-pyridinyl and 1,3-thiazol-2-yl, respectively. Compound 21l, with a 3-pyridyl group at the 4-position of the imidazole, revealed a similar activity to 21j, containing a 2-pyridyl group. In addition, 21m, with a substitution of the 4-fluorophenyl of arylidene and 3-pyridyl group at the 4-position of imidazole, exhibited a potent cytotoxic IC50 of 0.06 μM. Substitution on the 5th position of the imidazole caused a loss of activity, like that shown by compound 21n. In this work, the authors also carried out tubulin polymerization assays to access the anticancer activities of 2-amino-1-arylidenaminoimidazoles regarding colchicine and found imidazoles 21a and 21l were potent microtubule assembly inhibitors. In addition, in this study, an ex vivo antitumor activity assessment was performed against histocultured human gastric (MKN-45) and colorectal (SW-480) tumors with some imidazoles for 96 h and a reduction in the number of MKN-45 and SW-480 tumor cells was observed. The inhibition on the tubulin polymerization and colchicine binding kinetics provided evidence at the molecular level for interference in the microtubule assembly, which is one of the proven molecular mechanisms of anticancer action for the compounds presented in the study.
Fig. 13. Imidazoles as microtubule-destabilizing agents.

In another work, a group of imidazoles linked with a thiazole moiety were promoted as orally active microtubule destabilizing anticancer agents.148 The reported imidazoles were evaluated for cytotoxic activity against the human gastric cancer NUGC-3 cells. Among the group, compound 22 (1-(4-phenylthiazol-2-yl)-4-(thiophen-2-yl)-1H-imidazol-2-amine) with an IC50 value of 0.05 μM was found to be potent against human cancer cells. Its anticancer action was explained based on interferences in the colchicine binding sites of tubulins and the kinetics of microtubule assembly, which cause cell cycle arrest at the G2/M phase, inhibition of tumor cell proliferation, and induction of tumor cell apoptosis. Besides, 22 showed oral activity and significantly prolonged (75% longer compared to that of the vehicle control group vinblastine 83%) the lifespan of leukemia mice.
In one more study, Kamal et al. constructed chalcone conjugates featuring the imidazo[2,1-b]thiazole scaffold and evaluated their cytotoxic activities against five human cancer cell lines (MCF-7, A549, HeLa, DU-145 and HT-29) as inhibitors of microtubules.149 Promising cytotoxic activities with IC50 values ranging from 0.64 to 30.9 μm were observed. The imidazole 23 showed significant activity against the five human cancer cell lines and among the tested cancer cell lines. A549 cells were the most sensitive to 23 (IC50 = 0.64 mm). Furthermore, a flow cytometric analysis was also carried on 23, which resulted in cell cycle arrest in the G2/M phase, leading to apoptotic cell death. Besides, Hoechst staining, activation of caspase-3, DNA fragmentation analysis, Annexin V-FITC assay and molecular docking studies were carried out to support the results. Finally, it was concluded that the combo of imidazole and chalcone is a potent tubulin polymerization inhibitor (23a–23d). Recently, Stepanov et al. reported a series of benzoimidazolyl furazanamines and evaluated their microtubule destabilizing properties in the sea urchin embryo model and in cultured human cancer cell lines.150 Some imidazoles were studied in vitro using a tubulin polymerization assay and cell cycle distribution analysis. Further, these compounds were screened against a panel of human cancer cell lines to assess their cytotoxicities. The authors followed a simple protocol that includes a fertilized egg test for antimitotic activity and the compounds displayed cleavage alteration/arrest and swimming pattern observation of blastulae treated by the compounds after hatching. The lack of forward movement, settling to the bottom of the culture vessel, and rapid spinning of the embryos around the animal vegetal axis suggested the microtubule destabilizing activity caused by the molecule (video illustrations are available at ; http://www.chemblock.com). The attainment of a specific tuberculate shape of the arrested eggs (typical for microtubule destabilizing agents) was considered indirect evidence of targeting microtubules.151,152 In this work, the authors found some imidazoles 23e–23j very interesting and considered them as active microtubule destabilizing agents. In the experiment, it was found that the arrested sea urchin eggs acquired a tuberculate shape typical of microtubule destabilizers.151–153
Imidazoles as CHK-1 and CHK-2 inhibitors
The checkpoint kinases (CHK-1 and CHK-2) are serine/threonine protein kinases, which function as key regulatory kinases in cellular DNA damage response pathways limiting cell-cycle progression in the presence of DNA damage. The development of checkpoint kinase inhibitors for the treatment of cancer has been a major objective in drug discovery over the past decade, as evidenced by three checkpoint kinase inhibitors entering clinic trials since late 2005.154–158 Since cell cycle arrest is a mechanism by which tumor cells can overcome the damage induced by cytotoxic agents, abrogation of the G2/M checkpoint with novel small compounds may increase the sensitivity of p53-deficient tumors to chemotherapy. Importantly, in contrast to many current therapies for cancer, this mechanism potentially carries with it only a low risk of toxicity against nonmalignant cells, as CHK-1 inhibition is most effective in p53-defective cancer cells. The major advantage of CHK-1 inhibitors in the treatment of cancer is their selective activity in conjunction with cytotoxicity, such as DNA-damaging reagents. Until now, several CHK-1 inhibitors have been reported, but UCN-01 (7-hydroxystaurosporine) has been found to be a potent inhibitor (Ki = 5.6 nM) that has modest selectivity among other kinases of CHK-1 and is currently under clinical trials.159–165 However, the checkpoint kinase CHK-2 is activated by ATM (ataxia-telangiectasia mutated) in response to DNA damage and controls the downstream p53-dependent apoptosis machinery.166 Deletion of CHK-2 in mice blocks apoptosis after lethal radiation in several cell types and increases cell survival.167,168 Tumor tissue is, generally, more apoptosis resistant than normal cells. Generally, inhibition of CHK-2 would not protect these tissues and becomes an important target for anticancer drugs. However, imidazole analogues also showed potency as CHK inhibitors. Ni et al. constructed a series of benzimidazoles as serine/threonine checkpoint kinase (CHK-1) inhibitors with synergistic effects and DNA-damaging agents in tumor cells.169 Among the series, the unsubstituted benzimidazole linked with quinolone 24 showed moderate activity (IC50 = 0.73 l μM) against CHK-1. However, its substitution by various heterocyclic rings increased the activity potencies by more than 10 fold. Replacement of the amine hydrogen with the aza-bicyclo-octane ring 25 increased the potency (IC50 = 0.35 nM). Moreover, the imidazoles containing methyl 26 and chloro 27 groups also showed high potential against CHK-1 with IC50 = 0.50 nM and IC50 = 0.32 nM, respectively. In another work, the imidazoles 28 (IC50 = 2 nM) and 29 (IC50 = 15 nM) were identified as potent inhibitors of CHK-2.170,171 Based on molecular modeling, it was predicted that the high affinity of compound 28 was potentially due to a hydrogen bonding interaction between an active site residue (histamine 157) and the hydroxyl group of the terminal phenol. In view of the potencies of the 28 and 29 imidazoles, Neff et al. exploited a series of benzimidazoles as CHK-2 inhibitors.172 All compounds showed significantly less inhibitory activity than 28 and 29 but more overall solubility. The structures for imidazoles as CHK-1 and CHK-2 inhibitors are shown in Fig. 14.
Fig. 14. Imidazoles as CHK-1 and CHK-2 inhibitors.

Miscellaneous biological targets of imidazoles
Benzimidazole is an important scaffold in various biological active molecules. It is an isostere of a purine based nucleic acid and is extensively explored for the development of anticancer agents. Earlier benzimidazoles 30–32 as anticancer agents act via a DNA alkylation mechanism through cleavages of the G and A bases.173–175 Various benzimidazole substituted derivatives 33–35 are reported as cytotoxic against lung and breast cancers.176,177 Abdel-Mohsen et al. developed benzimidazoles conjugated with pyrimidine and explored their anticancer activities for twelve carcinoma cell lines (KB, SK OV-3, SF-268, NCI H460, RKOP27 HL60 U937 K562 G361 SK-MEL-28 GOTO NB-1).178 Compound 36 emerged as the most active against non-small lung cancer (NCI H460), ovarial carcinoma (SK OV-3) and leukemia (HL60) but was still less active than the standards. Compound 36a was observed as the most active against cervical carcinoma (KB), and the phenylhydrazino compound 36b was one of the most active compounds against melanoma (G 361) and neuroblastoma (NB-1). In addition, the 4-aminopyridino compound 36c and the 2-aminothiazolo compound 36d were found to be the most active compounds against CNS cancer (SF-268), while the 2-aminopyridino compound 36e was the most active against leukaemia (U937) and neuroblastoma (GOTO). The thiosemicarbazido compound 36f was one of the most active compounds against neuroblastoma (GOTO), while the uryl compound 36g was one of the most active compounds against colon adenocarcinoma (RKOP27). Compound 37a was the most active against melanoma (SK-MEL-28), and the N-methylpiperazino compound 37b was the most active among the series against leukemia (K562) and neuroblastoma (NB-1). The N-benzoylpiperazine compound 37c and the 4-nitrobenzoylpiperazino compound 37d were found to be the most active compounds against melanoma (G 361), and the phenylacetylpiperazino compound 37e was active against leukemia (U937). The benzenesulfonylpiperazino compound 37f was among the most active compounds against cervical carcinoma (KB) and non-small lung cancer (NCI H460), and the tosyl piperazine compound 37g was one of the most active compounds against colon adenocarcinoma RKOP27. The 2-nitrobenzenesulfonylpiperazino compound 37h was among the most active compounds against neuroblastoma (NB-1). Compound 38a was one of the most active compounds against colonadenocarcinoma (RKOP27), and 38b was one of the most active compounds against cervical carcinoma (KB). In other work, benzimidazole derivatives were synthesized, and compounds 39 (IC50 = 4.2 μM) and 40 (IC50 = 8.29 μM) were found to have high cytotoxic activity against breast cancer (MCF7).179 Compounds 41 and 42 possess significant inhibitory activity against Burkitt's lymphoma promotion,177 and 43, 44 (log GI50 = –5.61) and 45 have high cytotoxic activity against non-small lung cancer and breast cancer, 43 (log GI50 = 5.77) and 44 (log GI50 = 5.36).176,178 Development of benzimidazoles is a continuous goal of researchers, and its pyrazoline 46 analogues180 and thiazole 47 derivatives181 have been developed as potent anticancer agents for various cancer cell lines. Furthermore, another series of benzimidazoles as anti-tumour agents have been reported by Refaat,182 and some imidazoles, 4-amino-thioxothiazole 48, 4-oxothiazolidine 49, 4-fluorobenzylidene 50, and cycloalkylidene were found to be potent anti-tumour agents. Ramla et al. evaluated the antitumor activity of benzimidazoles and revealed the imidazole 51 incurs the maximum antitumor activity.179 Ng et al. explored some 2,5,6-trihalogenobenzimidazoles as androgen receptor antagonists for their use in prostate cancer and revealed that benzimidazole possessed the 1-(4-bromobenzyl) substituent (52) as the most potent androgen receptor antagonist.183 Similarly, another series of benzimidazoles containing 5-carboxylic acid have been evaluated as anti-leukemic agents by Gowda et al., and compound 53 was found to activate apoptosis.184 Hranjec et al. reported a series of benzimidazoles covalently linked to a quinolone ring and found compound 54 (IC50 of 0.8–30 μM) was the most potent on all cancer cell lines.185 This potency was predicted via the potential to insert into the space between the base pairs of DNA, resulting in DNA cleavage. Recently, more fused planar benzimidazole derivatives have been reported to exhibit potent cytotoxicity. The examplaries include 55 (pyrimido[1,2-a]benzimidazole-3(4H)-one)186 and 56 (1,3-diarylpyrazino[1,2-a]benzimidazole).187 Bis-benzimidazoles are another class of compounds exploited for the discovery of anticancer agents. Hoechst-33342 57a and Hoechst-33258 57b are the first of this series exhibiting in vitro antitumor as well as DNA top I inhibitory activities.188–192 Yang et al. synthesized novel hybrid compounds between imidazole and 2-phenylbenzofuran, and imidazole 57 with the 2-bromobenzyl group was the most active potent compound against four human tumor cell lines (SMMC-7721, SW480, MCF-7 and HL-60) and more active than cisplatin (DDP).41 Ozkay et al. constructed a series of 2-substituted-N-[4-(1-methyl-4,5-diphenyl-1H-imidazole-2-yl)phenyl] acetamide derivatives and revealed that three compounds 58a–58c were the most active anticancer agents against HT-29 and MCF-7 carcinoma cell lines amongst the series.193 Wang et al. reported another series of imidazoles covalently linked with 2-benzylbenzofurane and found two imidazoles, 59a and 59b, were the most potent against five human tumor cell lines (HL-60, A549, SW480, MCF-7 and SMMC-7721).194 In addition, these two compounds were more active than cisplatin (DDP). In another research article reported by Singh et al., novel imidazole conjugates were designed and synthesized regioselectively; wherein, compound 60 was found as the most potent cytotoxic scaffold against HCT-116, DLD-1 cell lines and relatively non-toxic to MDA-MB-231 and HUVEC cell lines.195 The structures for benzimidazoles as potent anticancer agents are shown in Fig. 15(A–C). Copp et al. reported 2-aminoimidazole alkaloid naamidine A 61, which was isolated from a Fijian Leucetta species sponge as an inhibitor of the epidermal growth factor (EGF) receptor.196 In this part of the work, the authors claimed that the compound exhibited a potent ability to inhibit the EGF signaling pathway and was more specific for the EGF-mediated mitogenic response than for the insulin-mediated mitogenic response. Furthermore, 85% growth inhibition at the maximal tolerated dose of 25 mg kg–1 was observed after the evaluation of an A431 xenograft tumor model in athymic mice. In another research article by Hassan et al., extractionof Naamine G 62 from Indonesian Sponge Leucetta chagosensis has been reported, and it revealed a mild cytotoxicity against mouse lymphoma (L5178Y) and human cervix carcinoma (HeLa) cell lines.197 Similarly, girolline 63 and preclathridine A 64, the other two important natural imidazoles as anticancer agents, have been reported.198–200 The structures of some naturally occurring 2-aminoimidazoles as anticancer agents are shown in Fig. 16.
Fig. 15. (A): Benzimidazoles as potent anticancer agents. (B): Benzimidazoles as potent anticancer agents. (C): Benzimidazoles as potent anticancer agents.

Fig. 16. Some naturally occurring 2-aminoimidazoles as anticancer agents.

Market status of Imidazole-based anticancer drugs
Imidazole-based anticancer drugs possess remarkable potential due to their ability to interfere first with DNA synthesis and then halt the cell growth and division. Until now, many imidazole-based anticancer drugs: azathioprine, misonidazole, pimonidazole, nilotinib, tipifarnib, fadrozole, indimitecan, acadesine, bleomycin, bombesin, dacarbazine, zoledronic acid, clotrimazole, mitozolomide, mercaptopurine, pilocarpine, radotinib, plinabulin, SB-431542, temozolomide, etanidazole and bendamustine, have been developed and play an important role in the clinic (Table 1). Dacarbazine is an alkylating agent that destroys cancer cells by adding an alkyl group to DNA. It has applications for metastatic malignant melanoma, hodgkin lymphoma, sarcoma and islet cell carcinoma of the pancreas.201 Azathioprine is a prodrug of mercaptopurine that has applications for the treatment of childhood acute lymphoblastic leukemia.202 Moreover, 2-nitroimidazoles (misonidazole, etanidazole and pimonidazole) act as radiosensitizers of hypoxic tumor cells.203 Pimonidazole is reduced in hypoxic environments, as in tumor cells, and it can be used as a hypoxia marker.204 Bendamustine is an alkylating agent synthesized in the 1960s by Ozegowski and Krebs in East Germany,205 which is used in the treatment of chronic lymphocytic leukemia and lymphomas.206,207 Etanidazole also possess potential applications in cancer chemotherapy by depleting the glutathione concentration, and it inhibits glutathione transferase. This enhances the cytotoxicity of ionizing radiation.208 Zoledronic acid is an inhibitor of osteoclast mediated bone resorption and is used in the management of patients with cancer. It slows down bone resorption, allowing the bone forming cells time to rebuild normal bone and bone remodeling.209 Nilotinib is a novel and selective small molecule Bcr-Abl; whereas, tipifarnib is a non-peptidomimetic quinolone analog of imidazole and a potent farnesyl transferase inhibitor developed by Johnson & Johnson Pharmaceutical for the treatment of acute myeloid leukemia.210 Fadrozole is of great interest to medicinal chemists as it has been recommended as a first-line drug in the therapy of breast cancer.211 Furthermore, indimitecan has been demonstrated to inhibit topoisomerase-I enzyme by intercalating between the DNA base pairs and stabilizing a ternary complex. Also, it produces a unique pattern of DNA cleavage sites relative to camptothecins and may target genes differently, which could result in a spectrum of anticancer activities.212 Attempts have been made to compile the imidazole drugs available in the market and clinical trials and are given in Table 1.
Table 1. The chemical structures and applications of imidazole drugs in cancer chemotherapy.
| S. no | Drugs | Structures and chemical names | Brand names | Market status | Applications |
| 1 | Acadesine |
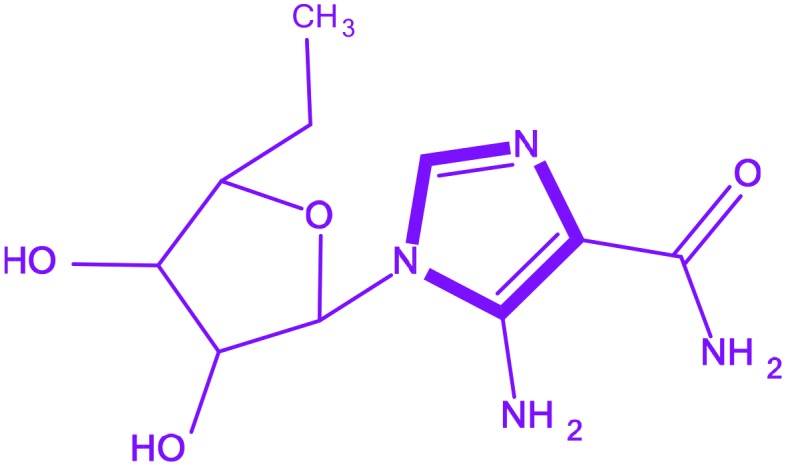 5-Amino-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-imidazole-4-carboxamide 5-Amino-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-imidazole-4-carboxamide |
— | Phase III | Acute lymphoblastic leukemia |
| 2 | Azathioprine |
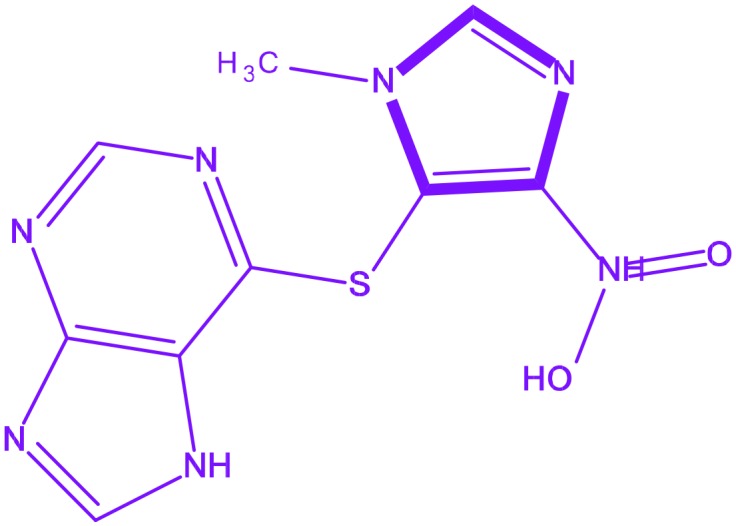 6-[(1-Methyl-4-nitro-1H-imidazol-5-yl)sulfanyl]-7H-purine 6-[(1-Methyl-4-nitro-1H-imidazol-5-yl)sulfanyl]-7H-purine |
Imuran | Phase I | Childhood acute lymphoblastic leukemia |
| 3 | Bleomycin |
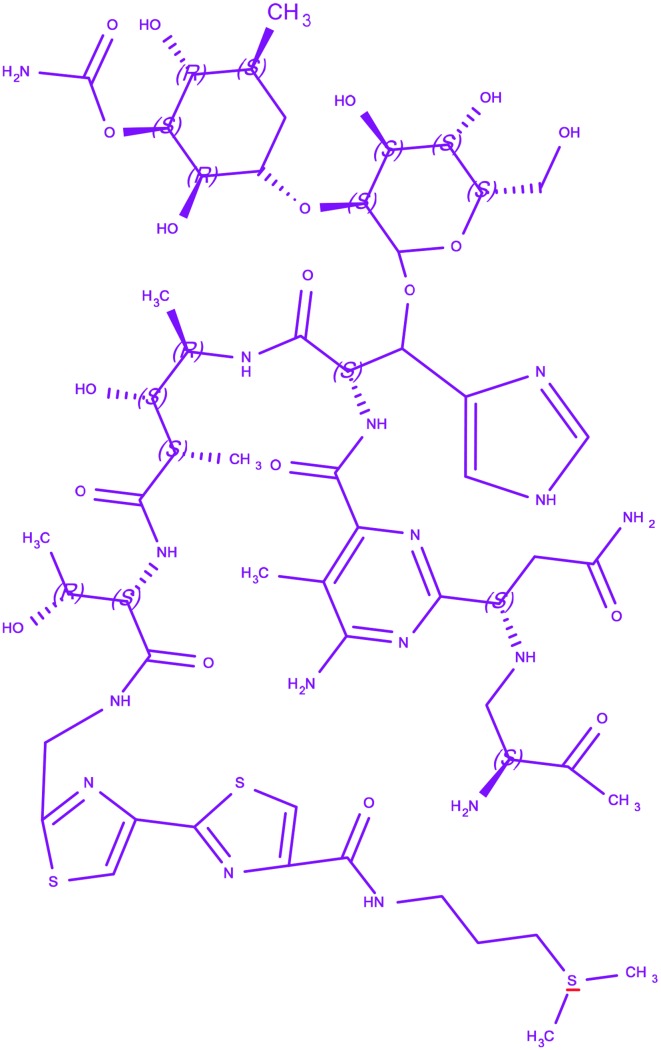 (3-{[(2′-{(5S,8S,9S,10R,13S)-15-{6-Amino-2-[(1S)-3-amino-1-{[(2S)-2,3-diamino-3-oxopropyl]amino}-3-oxo propyl]-5-methylpyrimidin-4-yl}-13-[{[(2R,3S,4S,5S,6S)-3-{[(2R,3S,4S, 5R,6R)-4-(carbamoyloxy)-3,5-dihydr oxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxy}-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxy}(1H-imidazol-5-yl)methyl]-9-hydroxy-5-[(1R)-1-hydr oxyethyl]-8,10-dimethyl-4,7,12,15-tetraoxo-3,6,11,14-tetraazapentadec-1-yl}-2,4′-bi-1,3-thiazol-4-yl)carbon yl]amino}propyl)dimethylsulfonium (3-{[(2′-{(5S,8S,9S,10R,13S)-15-{6-Amino-2-[(1S)-3-amino-1-{[(2S)-2,3-diamino-3-oxopropyl]amino}-3-oxo propyl]-5-methylpyrimidin-4-yl}-13-[{[(2R,3S,4S,5S,6S)-3-{[(2R,3S,4S, 5R,6R)-4-(carbamoyloxy)-3,5-dihydr oxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxy}-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxy}(1H-imidazol-5-yl)methyl]-9-hydroxy-5-[(1R)-1-hydr oxyethyl]-8,10-dimethyl-4,7,12,15-tetraoxo-3,6,11,14-tetraazapentadec-1-yl}-2,4′-bi-1,3-thiazol-4-yl)carbon yl]amino}propyl)dimethylsulfonium |
Blenoxane | US-FDA approval in 1973 | Hodgkin's lymphoma, non-Hodgkin's lymphoma, testicular, ovarian and cervical cancer |
| 4 | Bendamustine |
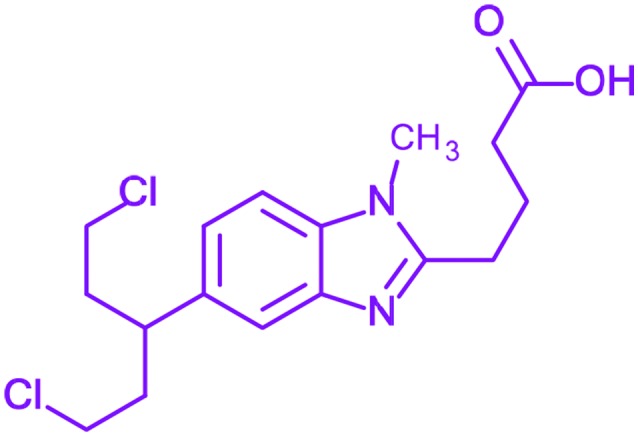 4-[5-[Bis(2-chloroethyl)amino]-1-met hylbenzimidazol-2-yl]butanoic acid 4-[5-[Bis(2-chloroethyl)amino]-1-met hylbenzimidazol-2-yl]butanoic acid |
Treanda | US-FDA approval in 2008 | Chronic lymphocytic leukemia and lymphomas |
| 5 | Bombesin |
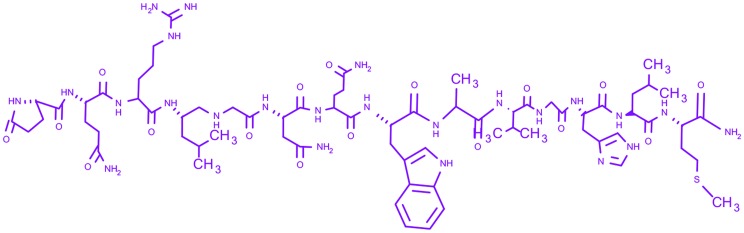 Pyr-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 Pyr-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2
|
— | Phase-III | Small cell carcinoma of lung, gastric, pancreatic and neuroblastoma |
| 6 | Dacarbazine | Fig. 1 5-(3,3-Dimethyl-1-triazenyl)imidazole-4-carboxamide | DTIC-Dome | US-FDA approval in 1975 | Malignant melanoma, Hodgkin's lymphoma, sarcoma and islet cell carcinoma of the pancreas |
| 7 | Mitozolomide |
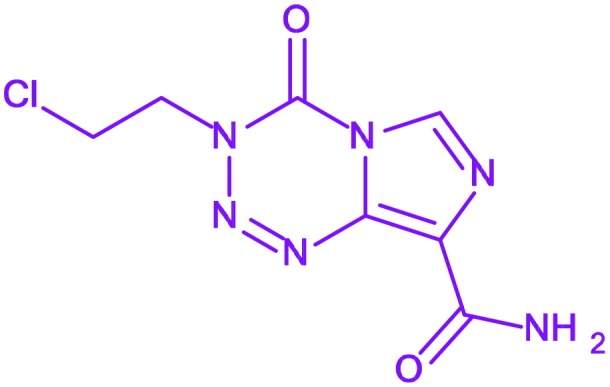 3-(2-Chloroethyl)-4-oxo-3,4-dihydr oimidazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide 3-(2-Chloroethyl)-4-oxo-3,4-dihydr oimidazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide |
— | Discontinued during phase II | — |
| 8 | Mercaptopurine | Fig. 1 3,7-Dihydropurine-6-thione | Purinethol | Phase I | Acute lymphocytic leukemia |
| 9 | Nilotinib | Fig. 1 4-Methyl-N-[3-(4-methyl-1H-imid azol-1-yl)-5-(trifluoromethyl)phenyl]-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]benzamide | Tasigna | US-FDA approval in 2009 | Philadelphia chromosome(Ph+)-positive chronic myelogenous leukaemia |
| 10 | Pilocarpine |
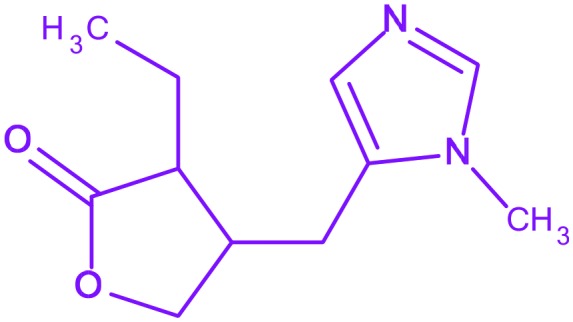 3-Ethyl-4-((1-methyl-1H-imidazol-5-yl)methyl)dihydrofuran-2(3H)-one 3-Ethyl-4-((1-methyl-1H-imidazol-5-yl)methyl)dihydrofuran-2(3H)-one |
Salagen | Under clinic | As a side effect of radiation therapy for head and neck cancer |
| 11 | Plinabulin |
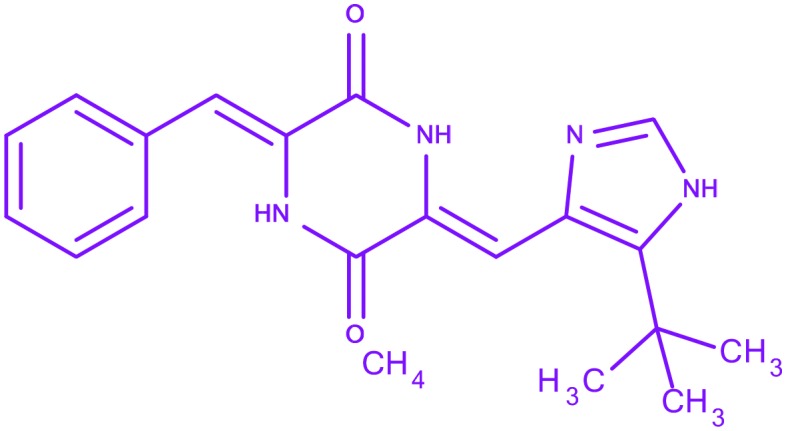 3-Benzylidene-6-{[5-(2-methyl-2-propanyl)-1H-imidazol-4-yl]methylene}-2,5-piperazinedione 3-Benzylidene-6-{[5-(2-methyl-2-propanyl)-1H-imidazol-4-yl]methylene}-2,5-piperazinedione |
Phase I and II | Non-small cell lung cancer | |
| 12 | Radotinib |
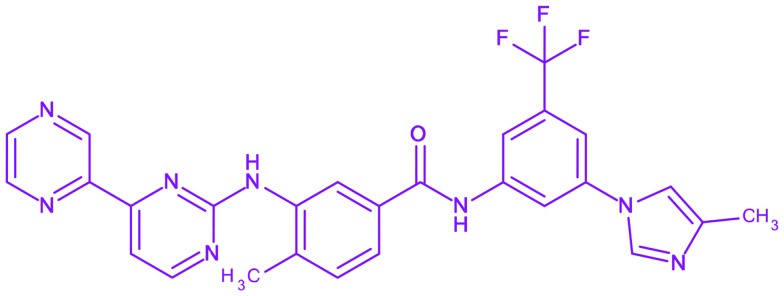 4-Methyl-N-[3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[(4-pyrazin-2-ylpyrimidin-2-yl)amino]benzamide 4-Methyl-N-[3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[(4-pyrazin-2-ylpyrimidin-2-yl)amino]benzamide |
Supect | Phase II | Chronic myeloid leukemia |
| 13 | SB-431542 |
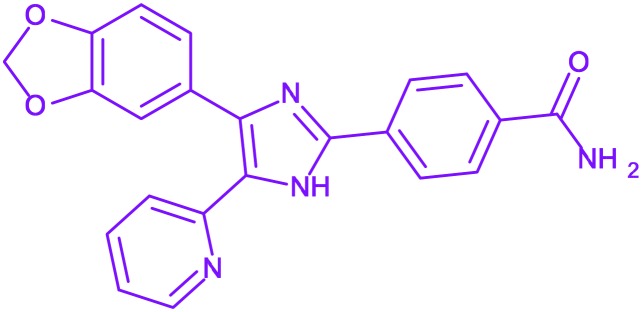 4-(4-Benzo[1,3]dioxol-5-yl-5-pyridin-2-yl-1H-imidazol-2-yl)-benzamide 4-(4-Benzo[1,3]dioxol-5-yl-5-pyridin-2-yl-1H-imidazol-2-yl)-benzamide |
— | No clinical trials | It suppress the TGF-beta-induced proliferation of osteosarcoma cells in humans |
| 14 | Tipifarnib | Fig. 1 6-[Amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlor ophenyl)-1-methylquinolin-2(1H)-one | Zarnestra | US-FDA disapproval in 2005 | Acute myeloid leukemia |
| 15 | Temozolomide | Fig. 1 4-Methyl-5-oxo-2,3,4,6,8-pentaza bicyclo[4.3.0]nona-2,7,9-triene-9-carboxamide | Temodar | Astrocytoma and glioblastoma multiforme | |
| 16 | Zoledronic acid | Fig. 1 1-Hydroxy-2-(1H-imidazol-1-yl)ethane-1,1-diyl]bis(phosphonic acid) | Zometa | Under clinic | In management of patients with multiple myeloma and prostate cancer |
| 17 | Indimitecan |
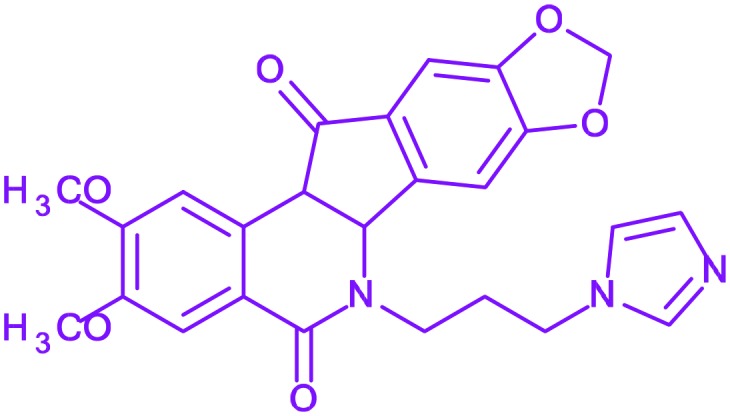
|
— | Phase I | Topoisomerase-I inhibitor |
| 18 | Fadrozole |
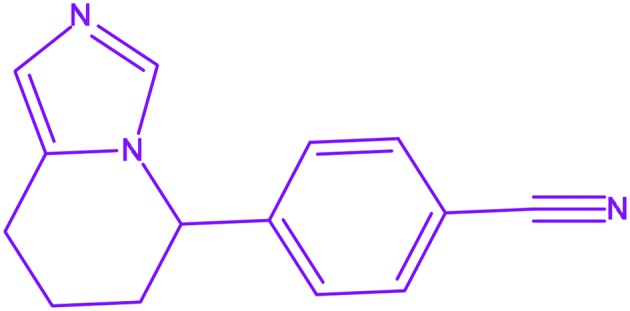 4-(5,6,7,8-Tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile 4-(5,6,7,8-Tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile |
Afema | Under clinic | Breast cancer |
Toxicity issues of imidazoles
While developing anticancer drugs, toxicity is one of the serious issues of importance. Drug toxicity is acceptable when the drugs are less harmful than the disease. The main toxicities associated with the use of imidazoles are nausea, vomiting, and hepatic dysfunction. Toxicity of imidazoles interfere with steroid biosynthesis that may result in impaired reproduction, alterations in (sexual) differentiation, growth, and development and the development of certain cancers. The toxicity of imidazole entities (1-alkyl-3-methylimidazolium) increases with the length of the side chain alkyl groups. Similar results were also found for the 1-vinylimidazole cation. At the same time, the toxicity of 1-alkyl-3-methylimidazolium and 1-vinylimidazole ionic liquids also depends on the associated anion species. The EC50 values are largest for tetrafluroborate, whereas bis(trifluromethylsulfonyl) imide results in the lowest EC50 value.213 In 1969, Nishie et al. reported the convulsant and lethal effects of imidazoles in mice. The authors found methylimidazole as the most potent convulsant. Furthermore, it was observed that the imidazoles tested at high dosage levels produced varying degrees of tremor, restlessness, running, sialorrhea, opisthotonus, straub tail and tonic extensor seizure terminating in death, whereas at lower doses, a loss of balance was the common finding. Toxicity experiments were carried out in 3 species, rabbit, mouse, and chicken, and the toxic manifestations, like abrupt paralysis of respiration, cyanosis, and gradual cardiac arrest, were observed in rabbit and mice. In addition, in rabbits, the heart rate decreased on the order of 33% below the control, while the respiratory rate increased to three times the normal values.214 Generally, the use of higher concentrations of any drug is always a problem. Norregaard et al. carried out an experiment in which the toxicity of temzolomide on U87MG glioblastoma cells was studied in order to confirm the activity of the drug, and the toxic effects of TMZ were observed at 50 and 500 μM concentrations.215 Moreover, Sing et al. reported the regioselective synthesis of aminoimidazole quinoline hybrids and cytotoxic evaluation against two cancer cells and toxicity against normal endothelial cells (HUVEC) and found some imidazoles 65a–65f had a good cytotoxicity against the cancer cell line. However, unfortunately against the HUVEC cells, the IC50 values decreased and resulted in an increment in the toxicity, which is a matter of concern.216 Furthermore, compound 60 showed selective toxicity against colon cancer cells and was relatively non-toxic to breast cancer cells. Moreover, the imidazole substituted with the theinyl moiety at position-4 reduces the anticancer activity of the imidazole derivative, and substitution of the imidazole derivative on the phenyl ring at position-4 leads to a decrease in the anticancer potential and, unfortunately, increases the toxicity to normal cells. The chemical structures of compounds 61–6f are shown in Fig. 17. Based on toxicological evaluation of imidazole and the latest update in 2006 (; www.bgchemie.de/toxicologicalevaluations), it has been found to be harmful via oral administration with signs of intoxication including salivation, ophisthotonus, decreased food consumption, increased blood pressure, disturbance of balance, vasodilatation, apathy, accelerated breathing, dyskinesia, lethargy, effects on liver enzyme, convulsion, tremors, etc. In addition, imidazole had an analgesic, anti-inflammatory, mild antipyretic, vasodilatory effect in animals and inhibition of platelet aggregation.
Fig. 17. The chemical structures of imidazoles that are less toxic to cancer cells and more toxic to normal cells.

It is clear from the discussion that some imidazole derivatives exhibit toxic effects towards normal cells. Some of the derivatives are mildly toxic, while some exhibit pronounced toxicities. However, there are limited studies available on this subject, and more investigations are needed to further comment on this issue. Besides, there is a need to carry out toxicity studies of imidazole derivatives both in vitro and in vivo to visualize the potential of such compounds in anticancer chemotherapy.
Molecular physicochemical properties and topological steric effect indexes
Molecular docking is a useful tool for selecting molecules with a certain structure. It is also necessary to consider other theoretical tools to estimate pharmacological activity. With the use of online software's important pharmacological properties, including absorption, bioavailability, permeability, blood brain barrier penetration, metabolism, and excretion, to achieve greater affinity for the biological receptor, lower toxicity and better scores can be considered. Therefore, the physicochemical properties of some of the most potent imidazole based anticancer agents discussed in this article were theoretically studied. The different properties were the atom & bond count (A&BC), dreiding & MMFF94 energy, van der Waals volume (VWV), polar surface area (PSA), number of rotatable bonds (NB), the partition coefficient (log P), distribution coefficient (log D), donor & acceptor count (D&AC) and donor & acceptor sites (D&AS). These results demonstrated that the future of imidazoles is quite bright in the field of medicinal chemistry. In addition, a drug should obey Lipinski's rule (Rule of Five), which states that most molecules with good membrane permeability have log P < 5, molecular weight <400, number of hydrogen bond acceptors <10, and number of hydrogen bond donors <5. This rule is widely used as a filter for drug-like properties. An analysis of small drug-like molecules suggests a filter of log D > 0 and <3, which enhances the probability of a compound to exhibit good intestinal permeability. A poor permeation or absorption is more likely when there are more than 5 H-bond donors and 10 H-bond acceptors. It is clear from Table 2 that imidazole based compounds have obeyed the ‘Rule of Five’ with log P values <5 and HBAs ≤ 10.
Table 2. Molecular physicochemical and topological properties of some selected potent imidazoles.
| Mol. wt | ABC, rotable bond count | PSA (2D) | log(p) | log(D) | No. of HBD's | No. of HBA's | Drieding & MMFF94 (kcal mol–1) | VWV | |
| 8f | 410 | 53, 57, 6 | 56.37 | 4.93 | 4.92 | 1 | 4 | 95.88 & 106.53 | 366.45 |
| 9d | 388 | 49, 52, 6 | 123.88 | 2.86 | 0.75 | 5 | 8 | 53.58 & –27.78 | 337.88 |
| 11c | 390 | 47, 51, 2 | 107.13 | 1.45 | 1.45 | 1 | 9 | 91.94 & 66.28 | 324.89 |
| 13b | 319 | 35, 36, 5 | 54.10 | 3.88 | 3.88 | 0 | 9 | 59.7 & 39.7 | 262.32 |
| 14a | 349 | 45, 47, 7 | 73.22 | 2.44 | 2.37 | 1 | 5 | 95.33 & 63.58 | 317.03 |
| 14f | 387 | 45, 47, 7 | 91.04 | 1.33 | 1.26 | 1 | 9 | 69.97 & 76.06 | 322.95 |
| 16a | 385 | 56, 59, 6 | 46.92 | 4.38 | 4.30 | 1 | 4 | 142.58 & 85.21 | 371.68 |
| 19a | 483 | 57, 62, 6 | 107.80 | 0.82 | 0.75 | 0 | 9 | 219.3 2 & 90.52 | 407.10 |
| 19i | 427 | 53, 58, 4 | 82.45 | 1.80 | 1.74 | 1 | 9 | 197.15 & 85.83 | 366.33 |
| 20c | 435 | 48, 52, 4 | 67.01 | 5.14 | 5.14 | 2 | 9 | 120.79 & 120.28 | 354.00 |
| 20e | 475 | 65, 71, 4 | 73.64 | 3.67 | 3.11 | 2 | 6 | 123.79 & 111.28 | 388.00 |
| 21a | 310 | 37, 39, 3 | 57.45 | 4.02 | 3.83 | 2 | 6 | 96.22 & 78.31 | 270.39 |
| 21b | 294 | 37, 39, 3 | 57.45 | 3.56 | 3.37 | 2 | 6 | 96.22 & 78.31 | 270.39 |
| 24 | 278 | 35, 38, 1 | 85.42 | 1.68 | 0.75 | 4 | 4 | 60.31 & 54.62 | 240.48 |
| 25 | 401 | 57, 62, 3 | 77.63 | 3.00 | 1.57 | 3 | 5 | 147.01 & 100.72 | 370.63 |
| 27 | 395 | 50, 54, 3 | 86.42 | 2.18 | –0.48 | 4 | 8 | 120.81 & 43.48 | 343.41 |
| 36 | 359 | 39, 42, 4 | 123.78 | 4.31 | 4.31 | 2 | 8 | 127.81 & 43.48 | 363.41 |
| 36a | 373 | 42, 45, 5 | 141.60 | 4.00 | 4.00 | 4 | 8 | 75.82 & 41.19 | 312.83 |
| 37a | 437 | 52, 66, 5 | 123.40 | 4.19 | 2.78 | 2 | 8 | 95.64 & 74.00 | 370.77 |
| 37b | 441 | 55, 59, 5 | 110.03 | 4.57 | 4.40 | 1 | 8 | 101.48 & 76.03 | 388.03 |
| 48 | 340 | 40, 43, 3 | 124.99 | 2.78 | 0.50 | 4 | 9 | 86.78 & 30.73 | 285.90 |
| 49 | 376 | 39, 42, 3 | 138.21 | 3.56 | 3.25 | 2 | 9 | 94.11 & 76.56 | 301.71 |
| 50 | 307 | 33, 55, 3 | 92.60 | 3.49 | 2.97 | 2 | 9 | 71.79 & 46.86 | 251.19 |
| 53 | 449 | 53, 56, 8 | 96.49 | 5.45 | 5.45 | 0 | 9 | 114.11 & 76.56 | 341.71 |
Current challenges and future perspectives
In the last couple of decades, there have been mammoth advances in the era of science, but, despite that, the advance in the cure for cancer remains a major challenge.217 Cancer is a leading threat to human beings and a serious challenge to scientists and society. The uses of the presently available anticancer drugs are limited due to their side effects, and other drug issues and higher numbers of deaths are reported compared to the cases of survival. Moreover, these drugs are too expensive and cannot be afforded by everyone. Until now, there have been significant advances in the understanding of the molecular etiology of cancer, but ultimate therapeutic strategies are still largely missing. Therefore, it is very crucial to develop new therapeutic agents against this vulnerable disease. Anticancer drugs with rapid action, selective nature, efficient bioavailability and reduced or no side effects (magic bullets) are the needs of the present day. So, it would be beneficial to explore imidazoles with various molecular features and topologies as anticancer drugs. Besides, targeting and activation strategies may be helpful in the development of future generations of anticancer drugs with ability to overcome the disadvantages of the already available conventional drugs. The novel imidazoles should be obtained with a broad spectrum of activity, no onset of drug resistance and fewer or no side effects. In the modern era of science, methods like density functional theory (DFT), molecular operating environment (MOE) and techniques like high resolution electrospray mass spectrometry and multinuclear polarization transfer NMR (nuclear magnetic resonance) spectroscopy are significantly helpful and can surely improve our understanding of the chemical and biochemical reactivity of drugs, which might help in establishing some meaningful structure activity relationships. Thus, the chemical studies of anticancer agents under physiologically-relevant conditions (e.g., biological screening conditions) become very important in drug development rationales. Nanotechnology has brought a grand revolution in the pharmaceutical field, especially in drug delivery, and resolves several issues of chemotherapeutic agents. The nanodrugs showed some advantages over conventional drugs, such as controlled drug release, enhanced permeability, and tumor specific targeting. Besides, nanodrugs exhibit few or no side effects to normal cells with the potential to cross the barriers of cancer cells easily.218 Hence, it would be of great interest if anticancer agents are trapped in nanocages, which might avoid their poor bioavailability and ensure the selective, specific, and fast action of the trapped drugs. Nanocapsules of imidazoles may be expected to be effective replacements of their conventional counterparts. In the present, science, software, and simulation programmes can be used to estimate a drug therapeutic efficiency even before their actual syntheses. Hence, simulated drugs with good bioavailability, maximum solubility, remarkably fewer side effects, and higher efficiency need to be designed and developed. In view of the discussion in this section and the understanding of the molecular mechanism of action in cancer by imidazoles, we foresee the design and development of novel imidazoles along with the development of nanocounterpart imidazoles. Throughout the world, scientists and researchers are currently working on the development of safe and effective anticancer agents (either in the nano regime or the normal size range), and it will be great to see how they work. Briefly, the future of imidazole-based drugs is quite bright for the treatment of cancer. A pictorial depiction of the current challenges and future perspectives of the development of imidazole-based anticancer drugs is shown in Fig. 18.
Fig. 18. A pictorial depiction of the current challenges and future perspectives of the development of imidazole based anticancer drugs.

Concluding remarks
The above critical assessment about various imidazoles is a significant class of heterocyclic compounds. It has been perceived so far that amendments on the imidazole core displayed promising anticancer activities, and it is interesting to observe that many imidazoles possess stimulating pharmacological profiles. Furthermore, it is still to be revealed the best mode of action of imidazoles, and it will be interesting to observe that in the future many new pharmacological profiles will be added as leads for future development of safer and more effective compounds. Therefore, we strongly suggest that imidazoles be used in pre-clinical and clinical studies at both the conventional and nano levels.
Conflict of interest
Authors declare that there is no conflict of interest regarding the publication of this article.
Acknowledgments
Mohammad Nadeem Lone acknowledges the UGC, New-Delhi for providing UGC-BSR Meritorious research fellowship.
Footnotes
†The authors declare no competing interests.
References
- Reichert J. M., Wenger J. B. Drug Discovery Today. 2008;13(1–2):30–37. doi: 10.1016/j.drudis.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Kumar A. and Amanulla M. K. M., A Study on Cancer Perpetuation using the Classification Algorithms, 2015.
- Society AC: Cancer facts & figures: The Society, 2008.
- Molina J. R., Yang P., Cassivi S. D., Schild S. E. and Adjei A. A., Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship, in Mayo Clinic Proceedings, Elsevier, 2008, pp. 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navada S., Lai P., Schwartz A. and Kalemkerian G., Temporal trends in small cell lung cancer: analysis of the national Surveillance, Epidemiology, and End-Results (SEER) database, in ASCO Annual Meeting Proceedings, 2006, vol. 2006, p. 7082. [Google Scholar]
- Sher T., Dy G. K. and Adjei A. A., Small cell lung cancer, in Mayo Clinic Proceedings, Elsevier, 2008, pp. 355–367. [DOI] [PubMed] [Google Scholar]
- Grasso C. S., Wu Y.-M., Robinson D. R., Cao X., Dhanasekaran S. M., Khan A. P., Quist M. J., Jing X., Lonigro R. J., Brenner J. C. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer IAfRo, World cancer report 2014, WHO, Geneva, 2014.
- Bray F., Møller B. Nat. Rev. Cancer. 2006;6(1):63–74. doi: 10.1038/nrc1781. [DOI] [PubMed] [Google Scholar]
- Ali I., Wani W. A., Saleem K. Cancer Ther. 2011;8(1):56. [Google Scholar]
- Pereira C., Leão M., Soares J., Bessa C., Saraiva L. Curr. Pharm. Des. 2012;18(27):4223–4235. doi: 10.2174/138161212802430422. [DOI] [PubMed] [Google Scholar]
- Ali I., Nadeem Lone M., Alothman Z. A., Al-Warthan A., Marsin Sanagi M. Curr. Drug Targets. 2015;16(7):711–734. doi: 10.2174/1389450116666150309115922. [DOI] [PubMed] [Google Scholar]
- Rana A., Alex J. M., Chauhan M., Joshi G., Kumar R. Med. Chem. Res. 2015;24(3):903–920. [Google Scholar]
- Saleem K., Wani W. A., Haque A., Lone M. N., Hsieh M.-F., Jairajpuri M. A., Ali I. Future Med. Chem. 2013;5(2):135–146. doi: 10.4155/fmc.12.201. [DOI] [PubMed] [Google Scholar]
- Bacolini G. Top. Heterocycl. Syst.: Synth., React. Prop. 1996;1:103. [Google Scholar]
- Besson T. and Thiery V., Microwave-assisted synthesis of sulfur and nitrogen-containing heterocycles, Microwave-Assisted Synthesis of Heterocycles, Springer, 2006, pp. 59–78. [Google Scholar]
- Alcázar J., Oehlrich D. Future Med. Chem. 2010;2(2):169–176. doi: 10.4155/fmc.09.144. [DOI] [PubMed] [Google Scholar]
- Hill R. A. Annu. Rep. Prog. Chem., Sect. B: Org. Chem. 2003;99:183–207. [Google Scholar]
- Forte B., Malgesini B., Piutti C., Quartieri F., Scolaro A., Papeo G. Mar. Drugs. 2009;7(4):705–753. doi: 10.3390/md7040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z. Nat. Prod. Rep. 2011;28(6):1143–1191. doi: 10.1039/c0np00074d. [DOI] [PubMed] [Google Scholar]
- Gao G., Xiao R., Yuan Y., Zhou C.-H., You J., Xie R.-G. J. Chem. Res. 2002;2002(6):262–263. [Google Scholar]
- Jiang H.-Y., Zhou C.-H., Luo K., Chen H., Lan J.-B., Xie R.-G. J. Mol. Catal. A: Chem. 2006;260(1):288–294. [Google Scholar]
- Jochims J., Katritzky A., Rees C. and Scriven E., Comprehensive Heterocyclic Chemistry II, Pergamon Press, Oxford, 1996, vol. 4, p. 179. [Google Scholar]
- Faulkner D. J. Nat. Prod. Rep. 2000;17(1):7–55. doi: 10.1039/a809395d. [DOI] [PubMed] [Google Scholar]
- Ho J. Z., Mohareb R. M., Ahn J. H., Sim T. B., Rapoport H. J. Org. Chem. 2003;68(1):109–114. doi: 10.1021/jo020612x. [DOI] [PubMed] [Google Scholar]
- Lombardino J. G., Wiseman E. H. J. Med. Chem. 1974;17(11):1182–1188. doi: 10.1021/jm00257a011. [DOI] [PubMed] [Google Scholar]
- Maier T., Schmierer R., Bauer K., Bieringer H., Buerstell H. and Sachser B., German Pat., 317094 (1983); in Chem. Abstr., 1984, 1984, 85699. [Google Scholar]
- Schmierer R., Mildenberger H. and Buerstell H., German Pat., 361464, 1987; in Chem. Abstr., 1988, 1988, 37838. [Google Scholar]
- Lo Y. S., Nolan J. C., Maren T. H., Welstead Jr W. J., Gripshover D. F., Shamblee D. A. J. Med. Chem. 1992;35(26):4790–4794. doi: 10.1021/jm00104a003. [DOI] [PubMed] [Google Scholar]
- Cai J., Li S., Zhou C., Wu J. Zhongguo Xinyao Zazhi. 2009;18:598–608. [Google Scholar]
- Piérard G., Vroome V., Borgers M., Cauwenbergh G., Pierard-Franchimont C. Curr. Top. Pharmacol. 2006;10(1):59–65. doi: 10.1159/000089143. [DOI] [PubMed] [Google Scholar]
- Sharma D., Narasimhan B., Kumar P., Judge V., Narang R., De Clercq E., Balzarini J. Eur. J. Med. Chem. 2009;44(6):2347–2353. doi: 10.1016/j.ejmech.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Khabnadideh S., Rezaei Z., Motazedian M., Eskandari M. Daru, J. Pharm. Sci. 2007;15(1):17–20. [Google Scholar]
- Stover C. K., Warrener P., VanDevanter D. R., Sherman D. R., Arain T. M., Langhorne M. H., Anderson S. W., Towell J. A., Yuan Y., McMurray D. N. Nature. 2000;405(6789):962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Sánchez-Moreno M., Gómez-Contreras F., Navarro P., Marín C., Ramírez-Macías I., Olmo F., Sanz A. M., Campayo L., Cano C., Yunta M. J. J. Antimicrob. Chemother. 2012;67(2):387–397. doi: 10.1093/jac/dkr480. [DOI] [PubMed] [Google Scholar]
- Łażewska D., Więcek M., Ligneau X., Kottke T., Weizel L., Seifert R., Schunack W., Stark H., Kieć-Kononowicz K. Bioorg. Med. Chem. Lett. 2009;19(23):6682–6685. doi: 10.1016/j.bmcl.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Cherkofsky S. C. and Sharpe T. R., inventors; EI Du Pont de Nemours, assignee. Anti-inflammatory 4, 5-diarly-2-(substituted-thio) imidazoles and their corresponding sulfoxides and sulfones, United States Pat., US4190666, 1980.
- Galley G., Stalder H., Goergler A., Hoener M. C., Norcross R. D. Bioorg. Med. Chem. Lett. 2012;22(16):5244–5248. doi: 10.1016/j.bmcl.2012.06.060. [DOI] [PubMed] [Google Scholar]
- Hancock A. A., Bennani Y. L., Bush E. N., Esbenshade T. A., Faghih R., Fox G. B., Jacobson P., Knourek-Segel V., Krueger K. M., Nuss M. E., Pan J. B. Eur. J. Pharmacol. 2004;487(1):183–197. doi: 10.1016/j.ejphar.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Carini D. J., Duncia J. V., Aldrich P. E., Chiu A. T., Johnson A. L., Pierce M. E., Price W. A., Santella 3rd J. B., Wells G. J., Wexler R. R. J. Med. Chem. 1991;34(8):2525–2547. doi: 10.1021/jm00112a031. [DOI] [PubMed] [Google Scholar]
- Yang W.-C., Li J., Li J., Chen Q., Yang G.-F. Bioorg. Med. Chem. Lett. 2012;22(3):1455–1458. doi: 10.1016/j.bmcl.2011.11.115. [DOI] [PubMed] [Google Scholar]
- Uçucu Ü., Karaburun N. G., Işikdağ İ. Farmaco. 2001;56(4):285–290. doi: 10.1016/s0014-827x(01)01076-x. [DOI] [PubMed] [Google Scholar]
- Lu X., Liu X., Wan B., Franzblau S. G., Chen L., Zhou C., You Q. Eur. J. Med. Chem. 2012;49:164–171. doi: 10.1016/j.ejmech.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Padmavathi V., Venkatesh B., Padmaja A. Eur. J. Med. Chem. 2011;46(11):5317–5326. doi: 10.1016/j.ejmech.2011.08.032. [DOI] [PubMed] [Google Scholar]
- Yang X.-D., Wan W.-C., Deng X.-Y., Li Y., Yang L.-J., Li L., Zhang H.-B. Bioorg. Med. Chem. Lett. 2012;22(8):2726–2729. doi: 10.1016/j.bmcl.2012.02.094. [DOI] [PubMed] [Google Scholar]
- Vijesh A., Isloor A. M., Telkar S., Peethambar S., Rai S., Isloor N. Eur. J. Med. Chem. 2011;46(8):3531–3536. doi: 10.1016/j.ejmech.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Zhan P., Liu X., Zhu J., Fang Z., Li Z., Pannecouque C., De Clercq E. Bioorg. Med. Chem. 2009;17(16):5775–5781. doi: 10.1016/j.bmc.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Li X.-L., Hu Y.-J., Wang H., Yu B.-Q., Yue H.-L. Biomacromolecules. 2012;13(3):873–880. doi: 10.1021/bm2017959. [DOI] [PubMed] [Google Scholar]
- Alniss H. Y., Anthony N. G., Khalaf A. I., Mackay S. P., Suckling C. J., Waigh R. D., Wheate N. J., Parkinson J. A. Chem. Sci. 2012;3(3):711–722. [Google Scholar]
- Ketron A. C., Denny W. A., Graves D. E., Osheroff N. Biochemistry. 2012;51(8):1730–1739. doi: 10.1021/bi201159b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Li Y., Gong S., Fu D. Spectrosc. Lett. 2002;35(6):751–756. [Google Scholar]
- Groessl M., Reisner E., Hartinger C. G., Eichinger R., Semenova O., Timerbaev A. R., Keppler B. K. J. Med. Chem. 2007;50(9):2185–2193. doi: 10.1021/jm061081y. [DOI] [PubMed] [Google Scholar]
- Shalini K., Sharma P. K., Kumar N. Chem. Sin. 2010;1:36–47. [Google Scholar]
- Baroniya S., Anwer Z., Sharma P. K., Dudhe R., Kumar N. Chem. Sin. 2010;1(3):172–182. [Google Scholar]
- Iradyan M., Iradyan N., Stepanyan G., Arsenyan F., Garibdzhanyan B. Pharm. Chem. J. 2010;44(4):175–182. [Google Scholar]
- Gaba M., Mohan C. Med. Chem. Res. 2016;25(2):173–210. [Google Scholar]
- Verma A., Joshi S., Singh D. Journal of Chemistry. 2013;2013:329412. [Google Scholar]
- Williams A. and Lemke L., Foye's Principles of Medicinal Chemistry Fifth Edition. W: Antiviral Agents and protease inhibitors, Lippincott Williams & Wilkins, Philadelphia, 2002. [Google Scholar]
- Debus H. Justus Liebigs Ann. Chem. 1858;107(2):199–208. [Google Scholar]
- Vanleusen A. M., Wildeman J., Oldenziel O. J. Org. Chem. 1977;42(7):1153–1159. [Google Scholar]
- Zhang C., Moran E. J., Woiwode T. F., Short K. M., Mjalli A. M. Tetrahedron Lett. 1996;37(6):751–754. [Google Scholar]
- Balalaie S., Hashemi M. M., Akhbari M. Tetrahedron Lett. 2003;44(8):1709–1711. [Google Scholar]
- Wolkenberg S. E., Wisnoski D. D., Leister W. H., Wang Y., Zhao Z., Lindsley C. W. Org. Lett. 2004;6(9):1453–1456. doi: 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]
- Lantos I., Zhang W. Y., Shui X., Eggleston D. S. J. Org. Chem. 1993;58(25):7092–7095. [Google Scholar]
- Bleicher K. H., Gerber F., Wüthrich Y., Alanine A., Capretta A. Tetrahedron Lett. 2002;43(43):7687–7690. [Google Scholar]
- Balalaie S., Hashemi M. M., Akhbari M. Tetrahedron Lett. 2003;44(8):1709–1711. [Google Scholar]
- Balalaie S., Arabanian A. Green Chem. 2000;2(6):274–276. [Google Scholar]
- D'Souza D. M., Mueller T. J. Chem. Soc. Rev. 2007;36(7):1095–1108. doi: 10.1039/b608235c. [DOI] [PubMed] [Google Scholar]
- Zaman S., Mitsuru K., Abell A. D. Org. Lett. 2005;7(4):609–611. doi: 10.1021/ol047628p. [DOI] [PubMed] [Google Scholar]
- Sparks R. B., Combs A. P. Org. Lett. 2004;6(14):2473–2475. doi: 10.1021/ol049124x. [DOI] [PubMed] [Google Scholar]
- Singh H. and Kapoor V., Medicinal and Pharmaceutical Chemistry, Vallabh Prakashan, 1996. [Google Scholar]
- Carmeliet P. Nat. Med. 2003;9(6):653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Nature. 2005;438(7070):932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Coultas L., Chawengsaksophak K., Rossant J. Nature. 2005;438(7070):937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- Bao P., Kodra A., Tomic-Canic M., Golinko M. S., Ehrlich H. P., Brem H. J. Surg. Res. 2009;153(2):347–358. doi: 10.1016/j.jss.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz H., Fehrenbacher L., Novotny W., Cartwright T., Hainsworth J., Heim W., Berlin J., Baron A., Griffing S., Holmgren E. N. Engl. J. Med. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Gopinath G., Sagurthi S. R., Perugu S., Achaiah G., Krupadanam G. D. RSC Adv. 2014;4(99):56489–56501. [Google Scholar]
- Sartorelli A. C., Creasey W. A. Annu. Rev. Pharmacol. 1969;9(1):51–72. doi: 10.1146/annurev.pa.09.040169.000411. [DOI] [PubMed] [Google Scholar]
- McGuire W. P., Rowinsky E. K., Rosenshein N. B., Grumbine F. C., Ettinger D. S., Armstrong D. K., Donehower R. C. Ann. Intern. Med. 1989;111(4):273–279. doi: 10.7326/0003-4819-111-4-273. [DOI] [PubMed] [Google Scholar]
- Stähelin H. F., von Wartburg A. Cancer Res. 1991;51(1):5–15. [PubMed] [Google Scholar]
- Hansen A. N., Bendiksen C. D., Sylvest L., Friis T., Staerk D., Jørgensen F. S., Olsen C. A., Houen G. PLoS One. 2012;7(9):e45405. doi: 10.1371/journal.pone.0045405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Hochwald S. N. Pharmacol. Ther. 2014;142(2):154–163. doi: 10.1016/j.pharmthera.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cance W. G., Kurenova E., Marlowe T., Golubovskaya V. Sci. Signaling. 2013;6(268):pe10. doi: 10.1126/scisignal.2004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechertier T., Hodivala-Dilke K. J. Pathol. 2012;226(2):404–412. doi: 10.1002/path.3018. [DOI] [PubMed] [Google Scholar]
- Dao P., Smith N., Tomkiewicz-Raulet C., Yen-Pon E., Camacho-Artacho M., Lietha D., Herbeuval J.-P., Coumoul X., Garbay C., Chen H. J. Med. Chem. 2014;58(1):237–251. doi: 10.1021/jm500784e. [DOI] [PubMed] [Google Scholar]
- Chen J., Wang Z., Lu Y., Dalton J. T., Miller D. D., Li W. Bioorg. Med. Chem. Lett. 2008;18(11):3183–3187. doi: 10.1016/j.bmcl.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamalian A., Shafiee A., Hemmateenejad B., Khoshneviszadeh M., Miri R., Madadkar-Sobhani A., Bathaie S., Moosavi-Movahedi A. J. Iran. Chem. Soc. 2011;8(4):1098–1112. [Google Scholar]
- Sarkarzadeh H., Miri R., Firuzi O., Amini M., Razzaghi-Asl N., Edraki N., Shafiee A. Arch. Pharmacal Res. 2013;36(4):436–447. doi: 10.1007/s12272-013-0032-7. [DOI] [PubMed] [Google Scholar]
- Oksuzoglu E., Tekiner-Gulbas B., Alper S., Temiz-Arpaci O., Ertan T., Yildiz I., Diril N., Sener-Aki E., Yalcin I. J. Enzyme Inhib. Med. Chem. 2008;23(1):37–42. doi: 10.1080/14756360701342516. [DOI] [PubMed] [Google Scholar]
- Abonia R., Cortés E., Insuasty B., Quiroga J., Nogueras M., Cobo J. Eur. J. Med. Chem. 2011;46(9):4062–4070. doi: 10.1016/j.ejmech.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Roopashree R., Mohan C. D., Swaroop T. R., Jagadish S., Rangappa K. S. Asian J. Pharm. Clin. Res. 2014;7(5):309–313. [Google Scholar]
- Alkahtani H. M., Abbas A. Y., Wang S. Bioorg. Med. Chem. Lett. 2012;22(3):1317–1321. doi: 10.1016/j.bmcl.2011.12.088. [DOI] [PubMed] [Google Scholar]
- Smith M. A., Parkinson D. R., Cheson B. D., Friedman M. A. J. Clin. Oncol. 1992;10(5):839–864. doi: 10.1200/JCO.1992.10.5.839. [DOI] [PubMed] [Google Scholar]
- Gudas L. J., Wagner J. A. J. Cell. Physiol. 2011;226(2):322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S., Newman R. A., Lippman S. M., Huber M. H., Minor T., Raber M. N., Krakoff I. H., Hong W. K. J. Clin. Oncol. 1993;11(5):959–966. doi: 10.1200/JCO.1993.11.5.959. [DOI] [PubMed] [Google Scholar]
- Armstrong J., Ruiz M., Boddy A., Redfern C., Pearson A., Veal G. Br. J. Cancer. 2005;92(4):696–704. doi: 10.1038/sj.bjc.6602398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozpolat B., Mehta K., Lopez-Berestein G. Leuk. Lymphoma. 2005;46(10):1497–1506. doi: 10.1080/10428190500174737. [DOI] [PubMed] [Google Scholar]
- McSorley L. C., Daly A. K. Biochem. Pharmacol. 2000;60(4):517–526. doi: 10.1016/s0006-2952(00)00356-7. [DOI] [PubMed] [Google Scholar]
- Thacher S. M., Vasudevan J., Tsang K.-Y., Nagpal S., Chandraratna R. A. J. Med. Chem. 2001;44(3):281–297. doi: 10.1021/jm0000214. [DOI] [PubMed] [Google Scholar]
- Ahmad M. Sci. Pharm. 2011;79(4):921. doi: 10.3797/scipharm.1106-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster R., Wouters W., Van Ginckel R., End D., Krekels M., Coene M.-C., Bowden C. J. Steroid Biochem. Mol. Biol. 1992;43(1):197–201. doi: 10.1016/0960-0760(92)90208-z. [DOI] [PubMed] [Google Scholar]
- Gomaa M. S., Bridgens C. E., Aboraia A. S., Veal G. J., Redfern C. P., Brancale A., Armstrong J. L., Simons C. J. Med. Chem. 2011;54(8):2778–2791. doi: 10.1021/jm101583w. [DOI] [PubMed] [Google Scholar]
- Stoppie P., Borgers M., Borghgraef P., Dillen L., Goossens J., Sanz G., Szel H., Van Hove C., Van Nyen G., Nobels G. J. Pharmacol. Exp. Ther. 2000;293(1):304–312. [PubMed] [Google Scholar]
- Van Ginckel R., Bruwiere H., Moelans P., Janssen B., Floren W., Van der Leede B., Van Dun J., Sanz G., Venet M., Dillen L. Br. J. Cancer. 2002;86(4):605–611. doi: 10.1038/sj.bjc.6600056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill M. J., Kan J. L., Beck P., Bittner M., Cesario C., Cooke A., Keane D. M., Nigro A. I., Nillson C., Smith V. Bioorg. Med. Chem. Lett. 2005;15(6):1669–1673. doi: 10.1016/j.bmcl.2005.01.044. [DOI] [PubMed] [Google Scholar]
- Verfaille C., Thissen C., Bovenschen H., Mertens J., Steijlen P., Van De Kerkhof P. J. Eur. Acad. Dermatol. Venereol. 2007;21(8):1038–1046. doi: 10.1111/j.1468-3083.2007.02158.x. [DOI] [PubMed] [Google Scholar]
- Geria A. N., Scheinfeld N. S. Curr. Opin. Invest. Drugs. 2008;9(11):1228–1237. [PubMed] [Google Scholar]
- Sun B., Liu K., Han J., Zhao L.-Y., Su X., Lin B., Zhao D.-M., Cheng M.-S. Bioorg. Med. Chem. 2015;23(20):6763–6773. doi: 10.1016/j.bmc.2015.08.019. [DOI] [PubMed] [Google Scholar]
- Gomaa M. S., Lim A. S., Lau S. W., Watts A.-M., Illingworth N. A., Bridgens C. E., Veal G. J., Redfern C. P., Brancale A., Armstrong J. L. Bioorg. Med. Chem. 2012;20(20):6080–6088. doi: 10.1016/j.bmc.2012.08.044. [DOI] [PubMed] [Google Scholar]
- Gomaa M. S., Bridgens C. E., Veal G. J., Redfern C. P., Brancale A., Armstrong J. L., Simons C. J. Med. Chem. 2011;54(19):6803–6811. doi: 10.1021/jm200695m. [DOI] [PubMed] [Google Scholar]
- Andoh T., DNA Topoisomerases in Cancer Therapy: Present and Future, Springer Science & Business Media, 2003. [Google Scholar]
- Nitiss J. L. Nat. Rev. Cancer. 2009;9(5):338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. K., Morris-Natschke S. L., Lee K. H. Med. Res. Rev. 1997;17(4):367–425. doi: 10.1002/(sici)1098-1128(199707)17:4<367::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Haglof K. J., Popa E., Hochster H. S. Update Cancer Ther. 2006;1(2):117–145. [Google Scholar]
- Pommier Y. Chem. Rev. 2009;109(7):2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunino F., Capranico G. Anti-Cancer Drug Des. 1990;5(4):307–317. [PubMed] [Google Scholar]
- Nitiss J. L., Liu Y.-X., Harbury P., Jannatipour M., Wasserman R., Wang J. C. Cancer Res. 1992;52(16):4467–4472. [PubMed] [Google Scholar]
- Pommier Y. Nat. Rev. Cancer. 2006;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Thomas C. J., Rahier N. J., Hecht S. M. Bioorg. Med. Chem. 2004;12(7):1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- Hande K. R. Biochim. Biophys. Acta, Gene Struct. Expression. 1998;1400(1):173–184. doi: 10.1016/s0167-4781(98)00134-1. [DOI] [PubMed] [Google Scholar]
- Hande K. Eur. J. Cancer. 1998;34(10):1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- Baviskar A. T., Madaan C., Preet R., Mohapatra P., Jain V., Agarwal A., Guchhait S. K., Kundu C. N., Banerjee U. C., Bharatam P. V. J. Med. Chem. 2011;54(14):5013–5030. doi: 10.1021/jm200235u. [DOI] [PubMed] [Google Scholar]
- Negi A., Alex J. M., Amrutkar S. M., Baviskar A. T., Joshi G., Singh S., Banerjee U. C., Kumar R. Bioorg. Med. Chem. 2015;23(17):5654–5661. doi: 10.1016/j.bmc.2015.07.020. [DOI] [PubMed] [Google Scholar]
- Holleran J. L., Parise R. A., Yellow-Duke A. E., Egorin M. J., Eiseman J. L., Covey J. M., Beumer J. H. J. Pharm. Biomed. Anal. 2010;52(5):714–720. doi: 10.1016/j.jpba.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.-Y., Deng X.-Q., Zu Y.-G., Lv H., Su L., Yao L., Zhang Y., Li L. Eur. J. Med. Chem. 2010;45(7):3200–3206. doi: 10.1016/j.ejmech.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Li Q., Lv H., Zu Y., Qu Z., Yao L., Su L., Liu C., Wang L. Bioorg. Med. Chem. Lett. 2009;19(2):513–515. doi: 10.1016/j.bmcl.2008.11.031. [DOI] [PubMed] [Google Scholar]
- Cinelli M. A., Morrell A. E., Dexheimer T. S., Agama K., Agrawal S., Pommier Y., Cushman M. Bioorg. Med. Chem. 2010;18(15):5535–5552. doi: 10.1016/j.bmc.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang H., Chen H., Zhao D., Tang X., Liu Y., Pan L., Cheng M. Arch. Pharm. 2012;345(1):43–48. doi: 10.1002/ardp.201100094. [DOI] [PubMed] [Google Scholar]
- Sondhi S. M., Singh J., Rani R., Gupta P., Agrawal S., Saxena A. Eur. J. Med. Chem. 2010;45(2):555–563. doi: 10.1016/j.ejmech.2009.10.042. [DOI] [PubMed] [Google Scholar]
- Song Y., Shao Z., Dexheimer T. S., Scher E. S., Pommier Y., Cushman M. J. Med. Chem. 2010;53(5):1979–1989. doi: 10.1021/jm901649x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel E., Ludwig M., Lathia C., Robert C., Ropert S., Soria J.-C., Armand J.-P. Cancer Chemother. Pharmacol. 2011;68(1):53–61. doi: 10.1007/s00280-010-1423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Kim H., Yu H., Chung J. Y., Oh C.-H., Yoo K. H., Sim T., Hah J.-M. Bioorg. Med. Chem. Lett. 2010;20(5):1573–1577. doi: 10.1016/j.bmcl.2010.01.064. [DOI] [PubMed] [Google Scholar]
- Rajitha C., Dubey P., Sunku V., Piedrafita F. J., Veeramaneni V. R., Pal M. Eur. J. Med. Chem. 2011;46(10):4887–4896. doi: 10.1016/j.ejmech.2011.07.045. [DOI] [PubMed] [Google Scholar]
- Yu H., Jung Y., Kim H., Lee J., Oh C.-H., Yoo K. H., Sim T., Hah J.-M. Bioorg. Med. Chem. Lett. 2010;20(12):3805–3808. doi: 10.1016/j.bmcl.2010.04.039. [DOI] [PubMed] [Google Scholar]
- Niculescu-Duvaz D., Niculescu-Duvaz I., Suijkerbuijk B. M., Ménard D., Zambon A., Nourry A., Davies L., Manne H. A., Friedlos F., Ogilvie L. Bioorg. Med. Chem. 2010;18(18):6934–6952. doi: 10.1016/j.bmc.2010.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthou C., Tozer G. M. Int. J. Exp. Pathol. 2009;90(3):284–294. doi: 10.1111/j.1365-2613.2009.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M., Rigoreau L., Dowding S., Parker P. J. Chem. Biol. Drug Des. 2009;73(6):599–610. doi: 10.1111/j.1747-0285.2009.00817.x. [DOI] [PubMed] [Google Scholar]
- Hait W. N. Cancer Res. 2009;69(4):1263–1267. doi: 10.1158/0008-5472.CAN-08-3836. [DOI] [PubMed] [Google Scholar]
- Jordan A., Hadfield J. A., Lawrence N. J., McGown A. T. Med. Res. Rev. 1998;18(4):259–296. doi: 10.1002/(sici)1098-1128(199807)18:4<259::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Wang L. G., Liu X. M., Kreis W., Budman D. R. Cancer Chemother. Pharmacol. 1999;44(5):355–361. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- Simoni D., Romagnoli R., Baruchello R., Rondanin R., Rizzi M., Pavani M. G., Alloatti D., Giannini G., Marcellini M., Riccioni T. J. Med. Chem. 2006;49(11):3143–3152. doi: 10.1021/jm0510732. [DOI] [PubMed] [Google Scholar]
- Zhou J., Giannakakou P. Curr. Med. Chem.: Anti-Cancer Agents. 2005;5(1):65–71. doi: 10.2174/1568011053352569. [DOI] [PubMed] [Google Scholar]
- Carlson R. O. Expert Opin. Invest. Drugs. 2008;17(5):707–722. doi: 10.1517/13543784.17.5.707. [DOI] [PubMed] [Google Scholar]
- Budman D. R. Cancer Invest. 1997;15(5):475–490. doi: 10.3109/07357909709047587. [DOI] [PubMed] [Google Scholar]
- Katsumata N. Br. J. Cancer. 2003;89:S9–S15. doi: 10.1038/sj.bjc.6601495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath K., Jordan M. A. Cancer Res. 2003;63(18):6026–6031. [PubMed] [Google Scholar]
- Li W.-T., Hwang D.-R., Song J.-S., Chen C.-P., Chuu J.-J., Hu C.-B., Lin H.-L., Huang C.-L., Huang C.-Y., Tseng H.-Y. J. Med. Chem. 2010;53(6):2409–2417. doi: 10.1021/jm901501s. [DOI] [PubMed] [Google Scholar]
- Li W.-T., Hwang D.-R., Song J.-S., Chen C.-P., Chen T.-W., Lin C.-H., Chuu J.-J., Lien T.-W., Hsu T.-A., Huang C.-L. Invest. New Drugs. 2012;30(1):164–175. doi: 10.1007/s10637-010-9547-7. [DOI] [PubMed] [Google Scholar]
- Kamal A., Balakrishna M., Nayak V. L., Shaik T. B., Faazil S., Nimbarte V. D. ChemMedChem. 2014;9(12):2766–2780. doi: 10.1002/cmdc.201402310. [DOI] [PubMed] [Google Scholar]
- Stepanov A. I., Astrat'ev A. A., Sheremetev A. B., Lagutina N. K., Palysaeva N. V., Tyurin A. Y., Aleksandrova N. S., Sadchikova N. P., Suponitsky K. Y., Atamanenko O. P. Eur. J. Med. Chem. 2015;94:237–251. doi: 10.1016/j.ejmech.2015.02.051. [DOI] [PubMed] [Google Scholar]
- Semenova M. N., Kiselyov A., Semenov V. V. BioTechniques. 2006;40(6):765. doi: 10.2144/000112193. [DOI] [PubMed] [Google Scholar]
- Semenova M. N., Kiselyov A. S., Titov I. Y., Raihstat M. M., Molodtsov M., Grishchuk E., Spiridonov I., Semenov V. V. Chem. Biol. Drug Des. 2007;70(6):485–490. doi: 10.1111/j.1747-0285.2007.00591.x. [DOI] [PubMed] [Google Scholar]
- Kiselyov A. S., Semenova M. N., Chernyshova N. B., Leitao A., Samet A. V., Kislyi K. A., Raihstat M. M., Oprea T., Lemcke H., Lantow M. Eur. J. Med. Chem. 2010;45(5):1683–1697. doi: 10.1016/j.ejmech.2009.12.072. [DOI] [PubMed] [Google Scholar]
- Gesner T. G., Harrison S. D. Annu. Rep. Med. Chem. 2002;37:115–124. [Google Scholar]
- Sharma V., Hupp C. D., Tepe J. J. Curr. Med. Chem. 2007;14(10):1061–1074. doi: 10.2174/092986707780362844. [DOI] [PubMed] [Google Scholar]
- Li Q., Zhu G.-D. Curr. Top. Med. Chem. 2002;2(9):939–971. doi: 10.2174/1568026023393318. [DOI] [PubMed] [Google Scholar]
- Zhao H., Watkins J. L., Piwnica-Worms H. Proc. Natl. Acad. Sci. U. S. A. 2002;99(23):14795–14800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Chen Z., Gunasekera A. H., Sowin T. J., Rosenberg S. H., Fesik S., Zhang H. J. Biol. Chem. 2003;278(24):21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- Zhao B., Bower M. J., McDevitt P. J., Zhao H., Davis S. T., Johanson K. O., Green S. M., Concha N. O., Zhou B.-B. S. J. Biol. Chem. 2002;277(48):46609–46615. doi: 10.1074/jbc.M201233200. [DOI] [PubMed] [Google Scholar]
- Takahashi I., Kobayashi E., Asano K., Yoshida M., Nakano H. J. Antibiot. 1987;40(12):1782–1784. doi: 10.7164/antibiotics.40.1782. [DOI] [PubMed] [Google Scholar]
- Graves P. R., Yu L., Schwarz J. K., Gales J., Sausville E. A., O'Connor P. M., Piwnica-Worms H. J. Biol. Chem. 2000;275(8):5600–5605. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- Bunch R. T., Eastman A. Clin. Cancer Res. 1996;2(5):791–797. [PubMed] [Google Scholar]
- Busby E. C., Leistritz D. F., Abraham R. T., Karnitz L. M., Sarkaria J. N. Cancer Res. 2000;60(8):2108–2112. [PubMed] [Google Scholar]
- Wang Q., Fan S., Eastman A., Worland P. J., Sausville E. A., O'connor P. M. J. Natl. Cancer Inst. 1996;88(14):956–965. doi: 10.1093/jnci/88.14.956. [DOI] [PubMed] [Google Scholar]
- Sausville E. A., Arbuck S. G., Messmann R., Headlee D., Bauer K. S., Lush R. M., Murgo A., Figg W. D., Lahusen T., Jaken S. J. Clin. Oncol. 2001;19(8):2319–2333. doi: 10.1200/JCO.2001.19.8.2319. [DOI] [PubMed] [Google Scholar]
- Falck J., Mailand N., Syljuåsen R. G., Bartek J., Lukas J. Nature. 2001;410(6830):842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- Takai H., Naka K., Okada Y., Watanabe M., Harada N., Saito S., Anderson C. W., Appella E., Nakanishi M., Suzuki H. EMBO J. 2002;21(19):5195–5205. doi: 10.1093/emboj/cdf506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A., Cheung A., Duncan G., Girard P.-M., Elia A. J., Wakeham A., Okada H., Sarkissian T., Wong J. A., Sakai T. Mol. Cell. Biol. 2002;22(18):6521–6532. doi: 10.1128/MCB.22.18.6521-6532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z.-J., Barsanti P., Brammeier N., Diebes A., Poon D. J., Ng S., Pecchi S., Pfister K., Renhowe P. A., Ramurthy S. Bioorg. Med. Chem. Lett. 2006;16(12):3121–3124. doi: 10.1016/j.bmcl.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Arienti K. L., Brunmark A., Axe F. U., McClure K., Lee A., Blevitt J., Neff D. K., Huang L., Crawford S., Pandit C. R. J. Med. Chem. 2005;48(6):1873–1885. doi: 10.1021/jm0495935. [DOI] [PubMed] [Google Scholar]
- McClure K. J., Huang L., Arienti K. L., Axe F. U., Brunmark A., Blevitt J., Breitenbucher J. G. Bioorg. Med. Chem. Lett. 2006;16(7):1924–1928. doi: 10.1016/j.bmcl.2005.12.096. [DOI] [PubMed] [Google Scholar]
- Neff D. K., Lee-Dutra A., Blevitt J. M., Axe F. U., Hack M. D., Buma J. C., Rynberg R., Brunmark A., Karlsson L., Breitenbucher J. G. Bioorg. Med. Chem. Lett. 2007;17(23):6467–6471. doi: 10.1016/j.bmcl.2007.09.098. [DOI] [PubMed] [Google Scholar]
- Skibo E. B., Schulz W. G. J. Med. Chem. 1993;36(21):3050–3055. doi: 10.1021/jm00073a002. [DOI] [PubMed] [Google Scholar]
- Skibo E. B., Islam I., Heileman M. J., Schulz W. G. J. Med. Chem. 1994;37(1):78–92. doi: 10.1021/jm00027a010. [DOI] [PubMed] [Google Scholar]
- Boruah R. C., Skibo E. B. J. Med. Chem. 1994;37(11):1625–1631. doi: 10.1021/jm00037a013. [DOI] [PubMed] [Google Scholar]
- El-Naem S. I., El-Nzhawy A., El-Diwani H., Abdel Hamid A. Arch. Pharm. 2003;336(1):7–17. doi: 10.1002/ardp.200390005. [DOI] [PubMed] [Google Scholar]
- Ramla M. M., Omar M. A., Tokuda H., El-Diwani H. I. Bioorg. Med. Chem. 2007;15(19):6489–6496. doi: 10.1016/j.bmc.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Abdel-Mohsen H. T., Ragab F. A., Ramla M. M., El Diwani H. I. Eur. J. Med. Chem. 2010;45(6):2336–2344. doi: 10.1016/j.ejmech.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Ramla M. M., Omar M. A., El-Khamry A.-M. M., El-Diwani H. I. Bioorg. Med. Chem. 2006;14(21):7324–7332. doi: 10.1016/j.bmc.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Shaharyar M., Abdullah M., Bakht M., Majeed J. Eur. J. Med. Chem. 2010;45(1):114–119. doi: 10.1016/j.ejmech.2009.09.032. [DOI] [PubMed] [Google Scholar]
- Luo Y., Xiao F., Qian S., Lu W., Yang B. Eur. J. Med. Chem. 2011;46(1):417–422. doi: 10.1016/j.ejmech.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Refaat H. M. Eur. J. Med. Chem. 2010;45(7):2949–2956. doi: 10.1016/j.ejmech.2010.03.022. [DOI] [PubMed] [Google Scholar]
- Ng R. A., Guan J., Alford V. C., Lanter J. C., Allan G. F., Sbriscia T., Lundeen S. G., Sui Z. Bioorg. Med. Chem. Lett. 2007;17(4):955–958. doi: 10.1016/j.bmcl.2006.11.047. [DOI] [PubMed] [Google Scholar]
- Gowda N. T., Kavitha C., Chiruvella K. K., Joy O., Rangappa K. S., Raghavan S. C. Bioorg. Med. Chem. Lett. 2009;19(16):4594–4600. doi: 10.1016/j.bmcl.2009.06.103. [DOI] [PubMed] [Google Scholar]
- Hranjec M., Pavlović G., Marjanović M., Kralj M., Karminski-Zamola G. Eur. J. Med. Chem. 2010;45(6):2405–2417. doi: 10.1016/j.ejmech.2010.02.022. [DOI] [PubMed] [Google Scholar]
- Neochoritis C. G., Zarganes-Tzitzikas T., Tsoleridis C. A., Stephanidou-Stephanatou J., Kontogiorgis C. A., Hadjipavlou-Litina D. J., Choli-Papadopoulou T. Eur. J. Med. Chem. 2011;46(1):297–306. doi: 10.1016/j.ejmech.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Demirayak S., Kayagil I., Yurttas L. Eur. J. Med. Chem. 2011;46(1):411–416. doi: 10.1016/j.ejmech.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Chen A. Y., Yu C., Gatto B., LIu L. F. Proc. Natl. Acad. Sci. U. S. A. 1993;90(17):8131–8135. doi: 10.1073/pnas.90.17.8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. Y., Yu C., Bodley A., Peng L. F., Liu L. F. Cancer Res. 1993;53(6):1332–1337. [PubMed] [Google Scholar]
- Kraut E. H., Fleming T., Segal M., Neidhart J. A., Behrens B. C., MacDonald J. Invest. New Drugs. 1991;9(1):95–96. doi: 10.1007/BF00194556. [DOI] [PubMed] [Google Scholar]
- Tolner B., Hartley J. A., Hochhauser D. Mol. Pharmacol. 2001;59(4):699–706. doi: 10.1124/mol.59.4.699. [DOI] [PubMed] [Google Scholar]
- Beerman T., McHugh M., Sigmund R., Lown J., Rao K., Bathini Y. Biochim. Biophys. Acta, Gene Struct. Expression. 1992;1131(1):53–61. doi: 10.1016/0167-4781(92)90098-k. [DOI] [PubMed] [Google Scholar]
- Ozkay Y., Isikdag I., Incesu Z., Akalın G. Eur. J. Med. Chem. 2010;45(8):3320–3328. doi: 10.1016/j.ejmech.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Wang X.-Q., Liu L.-X., Li Y., Sun C.-J., Chen W., Li L., Zhang H.-B., Yang X.-D. Eur. J. Med. Chem. 2013;62:111–121. doi: 10.1016/j.ejmech.2012.12.040. [DOI] [PubMed] [Google Scholar]
- Singh K., Verma V., Yadav K., Sreekanth V., Kumar D., Bajaj A., Kumar V. Eur. J. Med. Chem. 2014;87:150–158. doi: 10.1016/j.ejmech.2014.09.055. [DOI] [PubMed] [Google Scholar]
- Copp B. R., Fairchild C. R., Cornell L., Casazza A. M., Robinson S., Ireland C. M. J. Med. Chem. 1998;41(20):3909–3911. doi: 10.1021/jm980294n. [DOI] [PubMed] [Google Scholar]
- Hassan W., Edrada R., Ebel R., Wray V., Berg A., van Soest R., Wiryowidagdo S., Proksch P. J. Nat. Prod. 2004;67(5):817–822. doi: 10.1021/np0305223. [DOI] [PubMed] [Google Scholar]
- Newman D. J., Cragg G. M. Curr. Med. Chem. 2004;11(13):1693–1713. doi: 10.2174/0929867043364982. [DOI] [PubMed] [Google Scholar]
- Alvi K., Crews P., Loughhead D. G. J. Nat. Prod. 1991;54(6):1509–1515. [Google Scholar]
- Alvi K., Peters B. M., Lisa H. M., Phillip C. Tetrahedron. 1993;49(2):329–336. [Google Scholar]
- Pectasides D., Yianniotis H., Alevizakos N., Bafaloukos D., Barbounis V., Varthalitis J., Dimitriadis M., Athanassiou A. Br. J. Cancer. 1989;60(4):627. doi: 10.1038/bjc.1989.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawwa A. F., Millership J. S., Collier P. S., Vandenbroeck K., McCarthy A., Dempsey S., Cairns C., Collins J., Rodgers C., McElnay J. C. Br. J. Clin. Pharmacol. 2008;66(4):517–528. doi: 10.1111/j.1365-2125.2008.03248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephy P., Palcic B., Skarsgard L. Br. J. Cancer. 1981;43(4):443. doi: 10.1038/bjc.1981.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varia M. A., Calkins-Adams D. P., Rinker L. H., Kennedy A. S., Novotny D. B., Fowler W. C., Raleigh J. A. Gynecol. Oncol. 1998;71(2):270–277. doi: 10.1006/gyno.1998.5163. [DOI] [PubMed] [Google Scholar]
- Tageja N., Nagi J. Cancer Chemother. Pharmacol. 2010;66(3):413–423. doi: 10.1007/s00280-010-1317-x. [DOI] [PubMed] [Google Scholar]
- Hartmann M., Zimmer C. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1972;287(3):386–389. doi: 10.1016/0005-2787(72)90282-1. [DOI] [PubMed] [Google Scholar]
- Kath R., Blumenstengel K., Fricke H., Höffken K. J. Cancer Res. Clin. Oncol. 2001;127(1):48–54. doi: 10.1007/s004320000180. [DOI] [PubMed] [Google Scholar]
- Yang Y., Jin C., Li H., He Y., Liu Z., Bai L., Dou K. Exp. Ther. Med. 2012;3(2):299–303. doi: 10.3892/etm.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E. C., Davis L. E. Clin. Ther. 2003;25(11):2669–2708. doi: 10.1016/s0149-2918(03)80327-2. [DOI] [PubMed] [Google Scholar]
- Thomas X., Elhamri M. Biol.: Targets Ther. 2007;1:415–424. [PMC free article] [PubMed] [Google Scholar]
- Raats J. I., Falkson G., Falkson H. C. J. Clin. Oncol. 1992;10(1):111–116. doi: 10.1200/JCO.1992.10.1.111. [DOI] [PubMed] [Google Scholar]
- Beck D. E., Agama K., Marchand C., Chergui A., Pommier Y., Cushman M. J. Med. Chem. 2014;57(4):1495–1512. doi: 10.1021/jm401814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. L., Kao H. F., Wang J. Y., Wei G. T. J. Chin. Chem. Soc. 2014;61(7):763–769. [Google Scholar]
- Nishie K., Waiss Jr. A. C., Keyl A. C. Toxicol. Appl. Pharmacol. 1969;14:301–307. doi: 10.1016/0041-008x(69)90111-2. [DOI] [PubMed] [Google Scholar]
- Norregaard A., Jensen S. S., Kolenda J., Aaberg-Jessen C., Christensen K. G., Jensen P. H., Schrøder H. D., Kristensen B. W. Neurotoxic. Res. 2012;22(1):43–58. doi: 10.1007/s12640-011-9300-9. [DOI] [PubMed] [Google Scholar]
- Singh K., Verma V., Yadav K., Sreekanth V., Kumar D., Bajaj A., Kumar V. Eur. J. Med. Chem. 2014;87:150–158. doi: 10.1016/j.ejmech.2014.09.055. [DOI] [PubMed] [Google Scholar]
- Balderas-Renteria I., Gonzalez-Barranco P., Garcia A., Banik B. K., Rivera G. Curr. Med. Chem. 2012;19(26):4377–4398. doi: 10.2174/092986712803251593. [DOI] [PubMed] [Google Scholar]
- Caraglia M., De Rosa G., Salzano G., Santini D., Lamberti M., Sperlongano P., Lombardi A., Abbruzzese A., Addeo R. Curr. Cancer Drug Targets. 2012;12(3):186–196. doi: 10.2174/156800912799277421. [DOI] [PubMed] [Google Scholar]