

Abstract
Summary
Cyclic GMP-AMP synthase (cGAS) recognition of cytosolic DNA is critical for immune responses to pathogen replication, cellular stress, and cancer. Existing structures of the mouse cGAS-DNA complex provide a model for enzyme activation, but do not explain why human cGAS exhibits severely reduced levels of cyclic GMP-AMP (cGAMP) synthesis compared to other mammals. Here we discover that enhanced DNA-length specificity restrains human cGAS activation. Using reconstitution of cGAMP signaling in bacteria, we mapped the determinant of human cGAS regulation to two amino acid substitutions in the DNA-binding surface. Human-specific substitutions are necessary and sufficient to direct preferential detection of long DNA. Crystal structures reveal why removal of human substitutions relaxes DNA-length specificity, and explain how human-specific DNA interactions favor cGAS oligomerization. These results define how DNA-sensing in humans adapted for enhanced specificity, and provide a model of the active human cGAS-DNA complex to enable structure-guided design of cGAS therapeutics.
eTOC Blurb
The structure of the human cGAS–DNA complex reveals regulatory adaptations that balance enzymatic activity with DNA-length sensitivity, and additional features important for drug design.
Keywords: cGAS, STING, innate immunity, structural biology
Introduction
Aberrant localization of DNA into the cytosol of human cells induces activation of a potent immune response. In addition to DNA released during pathogen replication, accurate detection of cytosolic self-DNA is critical for the cellular responses to mitochondrial stress, abnormal chromosomal segregation, and cancer (Bakhoum et al., 2018; Dou et al., 2017; Gluck et al., 2017; Harding et al., 2017; Mackenzie et al., 2017; Woo et al., 2014; Yang et al., 2017). A major component of DNA sensing is controlled by the cytosolic enzyme cyclic GMP-AMP synthase (cGAS) (Sun et al., 2013). cGAS is a direct innate immune sensor that binds doublestranded DNA and catalyzes production of the second messenger 2–5′ / 3–5′ cyclic GMP-AMP (2′3′ cGAMP) (Ablasser et al., 2013; Civril et al., 2013; Diner et al., 2013; Gao et al., 2013a; Kranzusch et al., 2013; Li et al., 2013; Sun et al., 2013; Zhang et al., 2014). 2′3′ cGAMP then activates the receptor Stimulator of Interferon Genes (STING) to initiate a downstream transcription cascade and type I interferon signaling (Wu and Chen, 2014). The ability of cGAS to catalyze multi-turnover production of 2′3′ cGAMP enables dramatic signal amplification, allowing cells to sensitively detect and respond to small amounts of DNA. However, the enzymatic potential of cGAS necessitates strict regulatory control, and human mutations that disrupt normal tolerance to self-DNA result in severe autoimmunity (Crow and Manel, 2015). Recent experiments have highlighted a role for post-translational modification (Chen et al., 2016; Seo et al., 2018; Seo et al., 2015; Xia et al., 2016) and auxiliary binding partners (Liang et al., 2014; Yoh et al., 2015) in regulating cGAS activation. However, the ability of human cGAS to synthesize 2′3′ cGAMP is severely reduced compared to other mammalian homologs, suggesting the existence of additional unknown layers of regulation controlling primary enzymatic function.
A major limitation in our understanding of cGAS function is that no structural information exists for activated human cGAS (hcGAS) bound to DNA. All structural understanding of DNA recognition is instead derived from structures of the inactive human enzyme (Kato et al., 2013; Kranzusch et al., 2013; Li et al., 2013; Zhang et al., 2014) and studies of mouse cGAS (mcGAS) and mammalian homologs (Andreeva et al., 2017; Civril et al., 2013; Gao et al., 2013a; Li et al., 2013; Zhang et al., 2014). cGAS is one of the most rapidly diverging genes in the human genome (George et al., 2011; Hancks et al., 2015), and the human and mouse enzymes share <60% amino acid identity. The high variability in cGAS primary sequence limits the ability of existing homolog cGAS–DNA structures to guide design of small-molecules targeting the active human enzyme. While structures of mammalian homolog cGAS–DNA complexes provide a critically important model for DNA recognition and enzyme activation, they cannot explain the importance or function of human-specific cGAS variations.
Here we define an unexpected mode of cGAS regulation that increases the specificity of immune activation, and determine the structure of hcGAS in an active conformation bound to DNA. A critical feature of human cGAS-STING signaling is the preferential response of human cells to long (>45 bp) cytosolic DNA (Andreeva et al., 2017; Karayel et al., 2009; Luecke et al., 2017; Stetson and Medzhitov, 2006). Using a genetic approach, we take advantage of the altered levels of human and mouse cGAS 2′3′ cGAMP synthesis to map the molecular determinant of human-specific regulatory control. Two substitutions in the cGAS DNA-binding surface, hcGAS K187 and L195, are necessary and sufficient to dramatically enhance the ability of cGAS to discriminate short and long DNAs. High-resolution crystal structures of the active hcGAS–DNA complex reveal why human K187 and L195 substitutions are necessary for stringent DNA-length specificity, and explain how human-specific substitutions remodel DNA interactions to favor higher-order cGAS–DNA oligomerization. Together, these data explain a mechanism regulating cytosolic DNA sensing where cGAS activation in human cells is restrained by enhanced DNA selectivity. Our results identify a functional role of human-specific divergence in cGAS sequence in balancing DNA-sensing specificity and sensitivity, and provide structural insight into human cGAS DNA-recognition and enzyme activation.
Results
A rapid genetic assay in bacteria maps the molecular determinant of human-specific cGAS regulation to two substitutions
Previous results demonstrate that hcGAS produces less 2′3′ cGAMP than mcGAS (Sun et al., 2013). To better understand human-specific regulation of cGAS activation, we reconstituted cGAS activation in vitro with purified components, and directly measured 2′3′ cGAMP production by thin-layer chromatography. Purified hcGAS exhibits markedly reduced 2′3′ cGAMP production compared to mcGAS (Figure 1A), demonstrating that altered regulation is an intrinsic feature of the hcGAS protein not shared with mammalian homologs.
Figure 1. A rapid cGAS activity assay in bacteria reveals the molecular determinants of human-specific cGAS regulation.
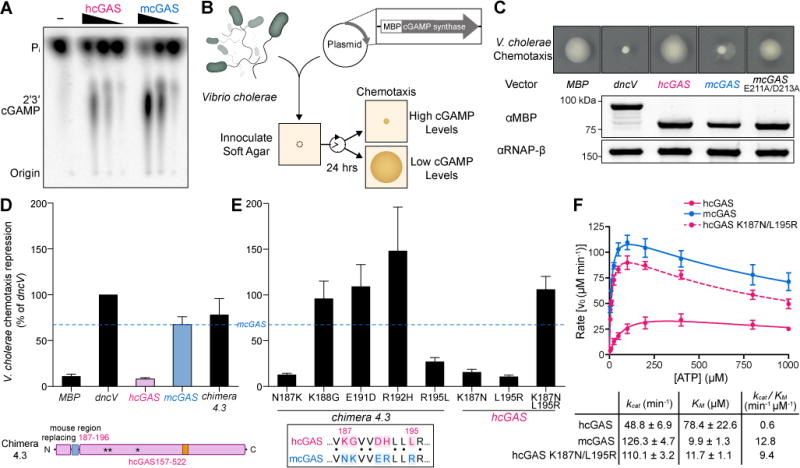
(A) cGAS production of 2′3′ cGAMP in vitro with purified components. A concentration gradient of recombinant hcGAS and mcGAS was activated with 45 bp double-stranded DNA and 2′3′ cGAMP formation was monitored by incorporation of [α-32P] ATP. Reactions were visualized by treating with alkaline phosphatase and separating by thin-layer chromatography.
(B) Schematic of a rapid, genetic cGAS activity assay. V. cholerae harboring an overexpression plasmid encoding an MBP fusion protein are inoculated onto chemotaxis agar. As bacteria grow and consume nutrients, they swim outward towards fresh medium (chemotaxis). cGAMP inhibits chemotaxis, which is visualized and quantified as the area of motile bacteria.
(C) Chemotaxis of V. cholerae strains overexpressing indicated cGAMP synthases. Genes were codon optimized and expressed as N-terminal MBP fusion proteins. Below, expression of each fusion protein 1 h post induction at log phase, visualized by Western blot. Images are representative of at least three independent experiments.
(D, E) Quantification of V. cholerae chemotaxis repression (see Star Methods) for strains overexpressing the synthases indicated. Dotted line represents mcGAS level of repression for reference. Data are represented as mean ± SEM for at least three independent experiments. Below, schematic of hcGAS Chimera 4.3 and alignment of the mcGAS N172–R181 region replacing hcGAS K187–R196. See also Figures S1, S2, and S3.
(F) Analysis of hcGAS, mcGAS and hcGAS K187N/L195R enzyme kinetics. cGAS enzyme activity was measured as a function of varying ATP concentration, and 2′3′ cGAMP product formation was quantified and fit according to Michaelis-Menten kinetics accounting for substrate inhibition. Data are represented as mean ± SD of three independent experiments. See also Figures S1, S2, and S3.
The human gene encoding cGAS (CGAS, MB21D1) is under intense positive selection (George et al., 2011; Hancks et al., 2015), and there are 116 amino acid differences between the hcGAS and mcGAS enzymatic domains (Figure S1). The resulting low sequence homology, and a complete lack of structural information for the active hcGAS–DNA complex, prevents rational determination of the key residues responsible for altered hcGAS function. To overcome this limitation, we developed a bacterial assay in Vibrio cholerae that monitors 2′3′ cGAMP synthesis and allows rapid determination of relative cGAS enzymatic activity (Figure 1B). The V. cholerae enzyme dinucleotide cyclase in Vibrio (DncV) is a structural homolog of hcGAS that synthesizes a chemically-related second messenger 3–5′ / 3–5′ cGAMP (3′3′ cGAMP) to control host colonization and bacterial chemotaxis (Davies et al., 2012; Kranzusch et al., 2014; Zhu et al., 2014). Increased levels of 3′3′ cGAMP in V. cholerae arrest chemotaxis, and we hypothesized that ectopic cGAS expression and 2′3′ cGAMP production may also induce this phenotype. As expected, overexpression of dncV inhibits chemotaxis, while the negative control E. coli maltose-binding protein (MBP) has no effect (Figure 1C). In agreement with our hypothesis that 2′3′ cGAMP can agonize the bacterial 3′3′ cGAMP pathway, overexpression of mcGAS in V. cholerae potently arrested chemotaxis and phenocopied dncV. However, no loss of chemotaxis is observed upon expression of hcGAS (Figure 1C). The V. cholerae genome is the likely source of cGAS-activating DNA in the bacterial cytosol, and we confirmed in vitro that V. cholerae genomic DNA is capable of stimulating both hcGAS and mcGAS 2′3′ cGAMP synthesis (Figure S2A). In agreement with synthetic DNA experiments (Figure 1A), mcGAS also produces more 2′3′ cGAMP than hcGAS in response to bacterial genomic DNA (Figure S2A). Additionally, hcGAS and mcGAS proteins are stable and express to similar levels, indicating that chemotaxis inhibition is most likely due to higher levels of 2′3′ cGAMP produced by mcGAS compared to hcGAS (Figure 1C). Mutations to the mcGAS active-site that disrupt catalytic magnesium coordination (E211A/D213A) relieve chemotaxis inhibition, confirming that the V. cholerae phenotype observed with mcGAS is due to elevated 2′3′ cGAMP production (Figure 1C).
We used V. cholerae as a sensitive platform to rapidly screen cGAS activity, and applied a chimera approach to map the genetic determinant of human-specific cGAS regulation. Based on the apo structure of hcGAS (PDB 4KM5) (Kranzusch et al., 2013), we designed a construct encoding the first half of the mcGAS enzymatic domain fused to the second half from hcGAS (Chimera 1: mcGAS P147–K372, hcGAS E385–F522). This chimeric cGAS construct exhibited potent inhibition of V. cholerae chemotaxis equal to that of wildtype mcGAS, demonstrating that human–mouse cGAS chimeras retain enzymatic activity (Figure S2B). The inverse construct encoding the mcGAS C-terminal half of the enzymatic domain (Chimera 2: hcGAS D157–K384, mcGAS Y373–L507) resulted in significantly weaker chemotaxis inhibition, indicating that the critical determinant of hcGAS regulation resides in the first ~220 amino acids of the catalytic domain. Through iterative rounds of structure-guided chimera design, we mapped the location of the human-specific regulatory element to a minimal region of 10 residues that contains five human-specific amino acid substitutions (Chimera 4.3: hcGAS, with K187–R196 replaced with mcGAS N172–R181) (Figure 1D and Figure S2B).
Using the highly active hcGAS Chimera 4.3 as a background, we next reverted each of the unique amino acid substitutions individually back to the wildtype human sequence and measured activity in V. cholerae. Wildtype human sequence at hcGAS position K187 and L195 results in loss of chemotaxis inhibition, while individual substitutions at the remaining three positions have no effect (Figure 1E). Neither K187N nor L195R alone are sufficient to enhance hcGAS activity. Instead, a K187N/L195R double-mutant potently inhibited V. cholerae chemotaxis, demonstrating that both amino acid substitutions are required and together are sufficient to elevate levels of 2′3′ cGAMP synthesis (Figure 1E). To confirm these results, we purified recombinant cGAS chimera enzymes from each stage of the genetic analysis and directly quantified 2′3′ cGAMP synthesis in vitro (Figure S3). Measurement of the kinetics and catalytic efficiency of 2′3′ cGAMP synthesis reveals that hcGAS is ~20-fold less efficient than mcGAS. Strikingly, the engineered hcGAS K187N/L195R variant exhibits elevated 2′3′ cGAMP synthesis and enzyme kinetics nearly equal to mcGAS, indicating that these residues control the major regulatory difference between human and mouse enzyme activation (Figure 1F). Together, these data demonstrate that two unique substitutions K187 and L195 in the N-terminus of hcGAS are responsible for human-specific control of 2′3′ cGAMP synthesis.
Structural basis of how K187 and L195 substitutions control hcGAS activity
We dissected the impact of hcGAS K187 and L195 substitution on enzyme activity using a panel of eight recombinant cGAS enzymes and direct measurement of 2′3′ cGAMP synthesis. Analysis of all possible combinations of K187/N187 and L195/R195 substitutions in both hcGAS and mcGAS backgrounds revealed a clear pattern showing that human-specific substitutions reduce total 2′3′ cGAMP output (Figure 2A). The presence of K187 and L195 results in reduced enzyme activity (as compared to N187/R195) in both the hcGAS and mcGAS backgrounds, and a double K187/L195 mutation fully restrains mcGAS 2′3′ cGAMP synthesis to human levels. We next sought to determine the structural basis of how K187 and L195 control enzyme function. All previous efforts in the field to determine the structure of hcGAS bound to activating DNA have been unsuccessful. However, using hcGAS K187N/L195R and a 17 bp activating DNA, we were now able to identify conditions that allowed growth of crystals suitable for structure determination. We used phase information obtained through molecular replacement with the apo structure of hcGAS, and determined the structure of the hcGAS–DNA binary complex to 2.3 Å and the hcGAS–DNA–ATP ternary complex to 2.8 Å (Table S1).
Figure 2. Structural basis of how K187 and L195 substitutions control hcGAS activity.
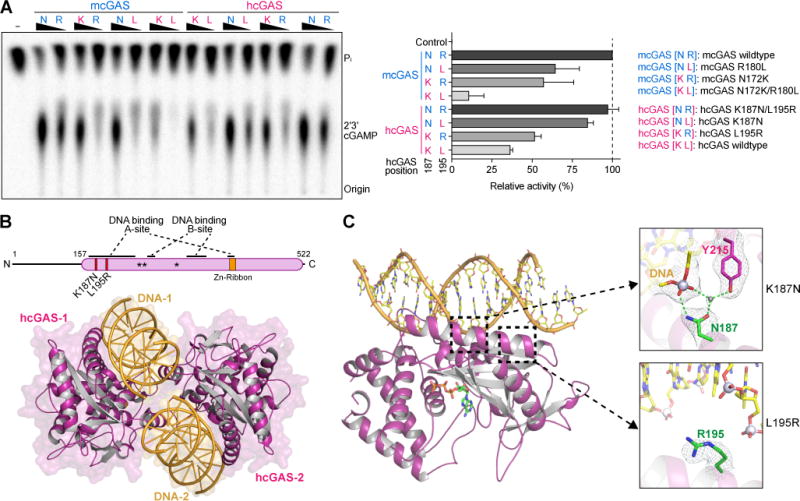
(A) In vitro analysis of the role of hcGAS K187 and L195 variation in 2′3′ cGAMP synthesis regulation. Human and mouse amino acid sequences at 187 (human K187 and mouse-equivalent N187, denoted K or N) and 195 (denoted L or R) were analyzed in both hcGAS and mcGAS backgrounds. Enzymes were stimulated with 45 bp DNA, and 2′3′ cGAMP synthesis was measured as in Figure 1A and quantified relative to maximal activity observed with mcGAS. Data are represented as mean ± SD of three independent experiments.
(B) Schematic and overview of the hcGAS–DNA complex. hcGAS forms a 2:2 complex with DNA where each cGAS monomer has two distinct DNA-binding surfaces (DNA A-Site and DNA B-Site). Stars in the schematic denote the enzyme metal-coordinating active-site residues, schematic not to scale.
(C) Overview of a single 1:1 cGAS–DNA unit in the hcGAS–DNA–ATP complex. Zoom-in cutaways of the locations of K187 and L195 human-specific cGAS substitutions in the DNA A-site. The water molecule coordinated by the K187N mutation and Y215 is depicted as a grey sphere. See also Table S1 and Figure S4.
The structure of hcGAS K187N/L195R bound to activating DNA reveals a minimal 2:2 complex of two molecules of cGAS embracing two molecules of double-stranded DNA (Figure 2B). The overall complex topology is similar to previous mcGAS–and Sus scrofa cGAS–DNA structures (Civril et al., 2013; Gao et al., 2013a; Li et al., 2013; Zhang et al., 2014), including two separate DNA-binding surfaces on each enzyme monomer designated the DNA “A-site” and “B-site,” and a conserved mechanism of structural rearrangement induced by DNA-binding that allows competent active-site formation and initiation of 2′3′ cGAMP synthesis (Figure 2B, Figure S4). hcGAS K187 and L195 map to a long alpha-helix designated the “spine” that braces the back half of the enzyme nucleotidyltransferase domain and forms part of the A-site DNA-binding surface. The engineered hcGAS mutation K187N makes direct contact with the DNA phosphate backbone. Likewise, L195R further stabilizes DNA interactions and increases the overall positive charge of the A-site DNA-binding surface (Figure 2C). Surprisingly, in both cases, the normal human-specific K187 and L195 substitutions must therefore remodel DNA contacts and alter the interaction between hcGAS and DNA. Additionally, the K187N substitution repositions Y215 and stabilizes a loop in the enzyme active site that is required for nucleotide coordination (Figure 2C). Loss of Y215 stabilization in hcGAS provides a further structural explanation for why overall 2′3′ cGAMP synthesis is restrained in hcGAS compared to the mouse enzyme. These results demonstrate that unique substitutions modify the hcGAS DNA-binding surface and create an unexpected layer of regulation that controls 2′3′ cGAMP synthesis.
Mechanism of human-specific cGAS–DNA recognition
The structure of the hcGAS K187N/L195R–DNA complex reveals that the human substitutions K187 and L195 specifically alter the DNA-binding surface. These two hcGAS substitutions occurred ~10–15 million years ago, and are only present in humans and closely related hominoid species (Figure 3A) (Scally et al., 2012), further highlighting that human cGAS–DNA recognition is controlled by distinct contacts not shared with other mammalian cGAS homologs. We next measured the impact of K187 and L195 on the ability of hcGAS and mcGAS to interact in vitro with a 45 bp immunostimulatory DNA sequence (Stetson and Medzhitov, 2006). Human cGAS quickly aggregates in the presence of DNA, however, we adapted a technique from the nucleosome assembly field (Lee and Narlikar, 2001), and used gradient dialysis to specifically generate and monitor stable cGAS–DNA complexes (Figure S5A). We observed that wildtype mcGAS forms two types of DNA complexes: (1) a lower molecular weight minimal cGAS–DNA complex, and (2) multiple slowly migrating protein-DNA complexes that are stabilized by increasing cGAS concentration and indicate higher-order oligomerization (Figure 3B, Top). Notably, introduction of the human K187 and L195 mutations into mcGAS ablated all detectable formation of the lower-molecular-weight complex, but still permitted formation of higher-order oligomeric interactions (Figure 3B, Top). Wildtype hcGAS preferentially forms higher-order oligomeric complexes that run near the top of the gel similar to the highest migrating mcGAS–DNA species (Figure 3B, Bottom). We verified that hcGAS and mcGAS robustly synthesize 2′3′ cGAMP under these conditions used for cGAS–DNA complex formation, demonstrating that higher-order migration is due to functional cGAS–DNA oligomerization and not non-specific aggregation (Figure S5B). Intriguingly, hcGAS–DNA higher-order complex formation is unaffected in the K187N/L195R variant, suggesting that these mutations do not impact cGAS–DNA oligomerization. Instead, introduction of the K187N and L195R mutations restores the ability of hcGAS to form a stable lower molecular weight protein–DNA complex (Figure 3B, Bottom). These results reveal that human-specific substitutions have a greater impact on minimal cGAS–DNA complex formation than on the ability of cGAS to oligomerize on a long DNA.
Figure 3. Mechanism of human-specific cGAS–DNA recognition.
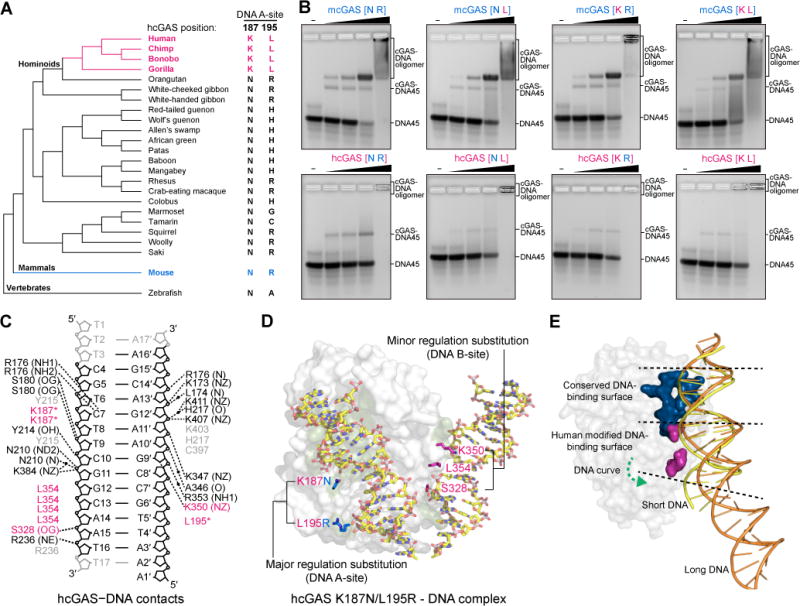
(A) Cladogram depicting evolution of hcGAS DNA A-site K187 and L195 positions in primates and relevant vertebrates. Human-specific substitutions are denoted in magenta, and the mouse cGAS sequence is denoted in blue for reference.
(B) Electrophoretic mobility shift assay measurement of in vitro cGAS–DNA complex formation. hcGAS and mcGAS variants (labeled as in Figure 2A) were incubated with 45 bp DNA, assembled using gradient dialysis, and the resulting stable complexes were resolved on a 2% agarose gel.
(C) Schematic map of protein–DNA contacts in the hcGAS–DNA complex. Human-specific contacts are highlighted in magenta, and black dots denote interactions bridged by water molecules. Black labels indicate contacts directly observed in the hcGAS–DNA complex, grey labels indicate water-mediated contacts potentially conserved with mcGAS but not observed in the hcGAS–DNA complex. See also Figure S4B.
(D) Overview of the hcGAS–DNA complex highlighting the location of human-specific DNA A-site and B-site substitutions. A-site substitutions have a major role in enzyme regulation, and B-site substitutions play an additional minor role (See Figure S5). Human-specific substitutions are shown as sticks in magenta, and the mouse-like K187N and L195R DNA A-site mutations are denoted in blue. One cGAS protein monomer from the 2:2 complex is omitted for clarity.
(E) Cartoon model of the hcGAS bound to short DNA (yellow) overlaid with the path of long DNA (orange) derived from the mcGAS-39 bp DNA structure (PDB 5N6I). Human-specific cGAS substitutions (magenta) weaken interactions with DNA in a portion of the DNA-binding surface that is not required for recognition of long DNA. Short and long DNA share identical interactions in the top conserved portion of the cGAS DNA A-site (blue), but assembly of an oligomerized cGAS–DNA complex causes long DNA to curve away and no longer make contacts with the bottom portion of the cGAS DNA A-site where the human-specific substitutions K187 and L195 are located. See also Figure S5.
A defining feature of human cGAS-STING signaling is that activation is dependent on DNA length, and DNA >45 bp is required for efficient cGAS recognition and 2′3′ cGAMP synthesis (Andreeva et al., 2017; Karayel et al., 2009; Luecke et al., 2017; Stetson and Medzhitov, 2006). To explain why hcGAS substitutions may specifically impact length-dependent DNA recognition, we compared all DNA contacts in our hcGAS–DNA structure with the DNA contacts in previously determined mcGAS–DNA structures. Human-specific substitutions S328, K350 and L354 in the DNA B-site surface form additional altered DNA contacts that can function cooperatively with the major K187/L195 substitutions to further regulate DNA recognition and 2′3′ cGAMP synthesis (Figure 3C, D, Figure S5). A recent structure of mcGAS bound to a 39 bp DNA revealed that recognition of long DNA involves formation of a higher-order cGAS–DNA “ladder” complex where long DNA adopts a curved conformation between adjacent cGAS dimers (Andreeva et al., 2017). DNA curving in the mcGAS–39 bp structure reduces the overall contact between cGAS and DNA, indicating that fewer protein–DNA interactions are required in the context of long DNA recognition. Human K187, L195, and B-site substitutions are located within the small patch of the cGAS DNA-binding surface that corresponds to where long DNA begins to curve away and no longer contacts cGAS (Figure 3E). Strikingly, these results suggest that human substitutions specifically weaken a portion of the cGAS DNA-binding surface that is not required during recognition of long DNAs, and suggest a mechanism where human cGAS DNA recognition is precisely altered to limit recognition of short DNAs. Together, structural and biochemical analysis of hcGAS–DNA complex formation reveals that unique alterations to the DNA-binding surface specifically inhibit minimal cGAS–DNA complex formation and underlie a key regulation controlling 2′3′ cGAMP synthesis in human cells.
Human cGAS adaptations re-shape DNA-length specificity
Our biochemical and structural data suggest that human-specific cGAS regulation controls enzyme activation by biasing cGAS–DNA interactions away from a minimal 2:2 complex and towards higher-order protein–DNA oligomerization. An important prediction of this model is that hcGAS K187 and L195 adaptations should specifically alter the ability of cGAS to respond to DNA in a length-dependent manner. To directly test this prediction, we compared the ability of cGAS variants to respond to 17 and 45 bp DNA. Wildtype hcGAS robustly synthesizes 2′3′ cGAMP in response to increasing concentrations of 45 bp DNA, but remains inactive in the presence of 17 bp DNA. In contrast, mcGAS recognizes both 45 bp and 17 bp DNA with near equal efficiency, consistent with ability of mcGAS to stably form the minimal 2:2 complex required for short DNA recognition and enzyme activation (Figure 4A, C) (Andreeva et al., 2017). Strikingly, the contrasting ability of hcGAS and mcGAS to respond to short DNA is completely reversed through human-specific K187/L195 substitution. The hcGAS K187N/L195R variant exhibits full enzyme activation in the presence of 17 bp DNA, while the ability of humanized mcGAS K187/L195 to recognize 17 bp DNA is abolished (Figure 4B, C). We observed the same phenotype using full-length recombinant hcGAS including the unstructured N-terminal tail, further confirming the critical importance of positions K187 and L195 in specifically controlling discrimination of long and short DNAs (Figure S6). These data provide a mechanistic rationale for how human-specific cGAS adaptation allows enhanced control of length-dependent DNA recognition and immune surveillance.
Figure 4. Human cGAS adaptations re-shape DNA-length specificity.
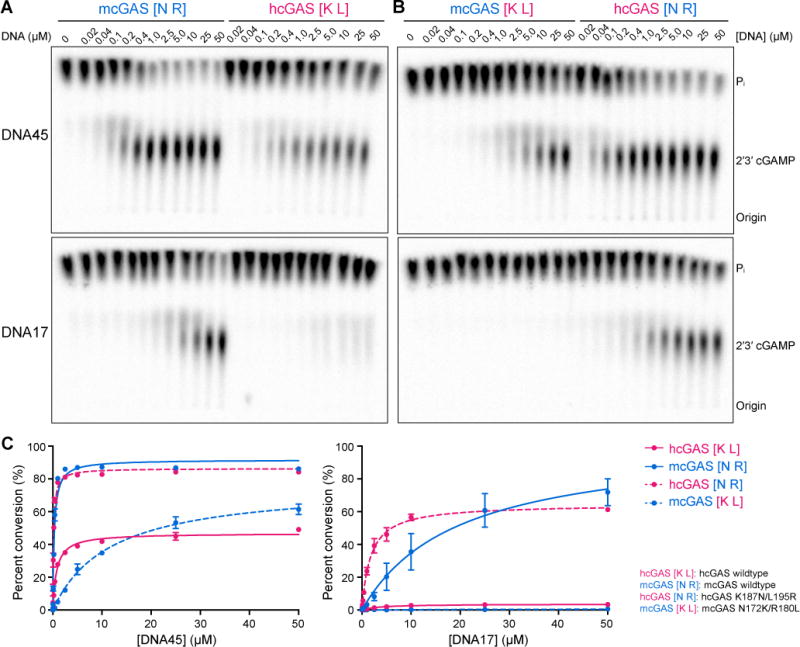
(A) In vitro analysis of cGAS DNA-length specificity. Purified hcGAS and mcGAS enzymes were stimulated with increasing concentration of 45 bp (top) or 17 bp (bottom) DNA. Enzyme activation was analyzed as in Figure 1A. Unlike mcGAS, hcGAS is only able to activate 2′3′ cGAMP synthesis in the presence of long 45 bp DNA.
(B) Identical experiment as in A, using mcGAS with human-like K187 and L195 substitutions or hcGAS with mouse-like N187 and R195 substitutions. Human-specific K187 and L195 substitutions are necessary and sufficient for cGAS length-dependent DNA discrimination.
(C) Quantification of cGAS DNA length-dependent activation experiments in A and B. 2′3′ cGAMP synthesis was quantified as in Figure 1A and graphed as total conversion of ATP to 2′3′ cGAMP. Data are represented as mean ± SD of three independent experiments. See also Figure S6.
The structure of hcGAS in an active conformation provides a key template to understand cGAS mutations and guide therapeutic intervention
The crystallized hcGAS–DNA complex contains no engineered mutations other than K187N and L195R in the DNA-binding surface, and presents the first opportunity to analyze the human enzyme in an active conformation. cGAS mutations are frequently observed in cancers (Cerami et al., 2012). Consistent with the role of cGAS-STING signaling in antitumor immunity (Bakhoum et al., 2018; Dou et al., 2017; Harding et al., 2017; Woo et al., 2014), tumor-associated mutations have been predicted to impact cGAS function, but their exact structural role has remained unclear. Mapping these residues onto the active hcGAS–DNA structure reveals that 37 of the 60 tumor-associated mutations impact key positions involved in DNA-binding, nucleotide coordination, and overall protein stability (Figure 5A,B). These results establish a molecular explanation for how tumor-associated mutations impede cGAS function, and provide a structural correlate to recent experimental work confirming that many tumor-associated mutations negatively impact cGAS activity in cells (Konno et al., 2018).
Figure 5. The hcGAS–DNA structure provides insight into tumor-associated mutations and small-molecule inhibitor design.
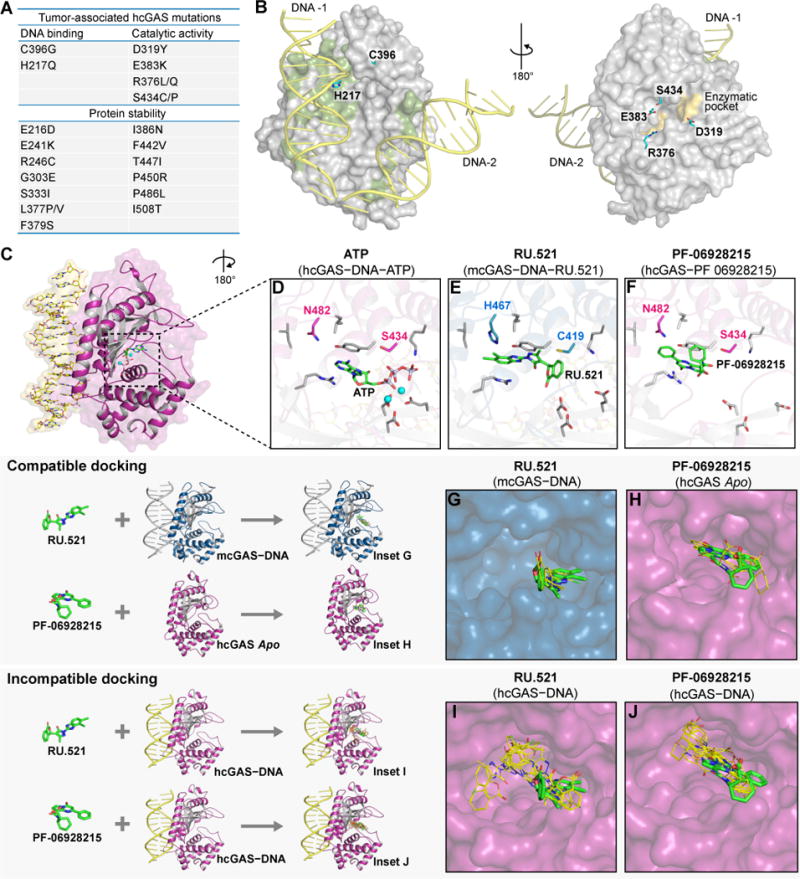
(A) Tumor-associated mutations in cGAS, and the structural role of each mutated residue predicted by the hcGAS–DNA–ATP ternary complex.
(B) Highlights of the tumor-associated mutations in hcGAS on the hcGAS–DNA–ATP ternary complex.
(C) Overview of a single 1:1 cGAS-unit in the hcGAS–DNA–ATP ternary complex with (D,E,F) zoom-in cutaways of the cGAS active-site showing protein residues in contact with ATP and small-molecule inhibitors. Nucleotide substrate and inhibiting compounds are shown in green, human-specific cGAS substitutions are in magenta, corresponding mcGAS residues are in blue, and conserved active-site amino acids are in grey.
(G–J) Molecular docking analysis of the compatibility of existing cGAS inhibitors with the active hcGAS–DNA complex. All top docked inhibitor poses of the mcGAS inhibitor RU.521 (PDB 5XZG) and hcGAS inhibitor PF-06928215 (PDB 5V8N) shown in orange lines, are compatible with the mcGAS–DNA complex and apo hcGAS structure respectively, and agree with the experimentally derived crystallographic binding poses shown in green sticks for reference. In contrast, the docked inhibitor poses with the active hcGAS–DNA complex structure are distinct, further confirming that the hcGAS active site differs from previously observed structures.
We next used the active human cGAS–DNA structure to analyze existing small-molecule inhibitor data and provide insight into the specificity of cGAS inhibition. Recently, reports of small-molecules targeting cGAS demonstrated that a pocket above the ATP donor site is critical for therapeutic inhibition of 2′3′ cGAMP synthesis (Hall et al., 2017; Vincent et al., 2017). In addition to the DNA-induced structural rearrangements that distinguish the hcGAS–DNA structure from the apo enzyme conformation, the hcGAS–DNA–ATP ternary complex reveals that human-specific residues S434 and N482 brace both sides of the inhibitor pocket and create an enzyme active-site that is structurally distinct from previously observed mouse cGAS–DNA structures (Figure 5C–F). Structure-based molecular docking and virtual screening has become a critical component of drug development and high-throughput screening, but this analysis has not previously been possible for the active hcGAS conformational state. To understand how unique features of the hcGAS active site may impact interactions of currently available cGAS inhibitors, we analyzed the mcGAS inhibitor RU.521 (Vincent et al., 2017) and the hcGAS inhibitor PF-06928215 (Hall et al., 2017) with molecular docking. We first performed a validation experiment using RU.521 and the mcGAS–DNA complex (PDB 5XZG), or PF-06928215 and the previous inactive hcGAS complex (PDB 5V8N) confirming that the top small-molecule poses scored during molecular docking correctly identified the observed location and orientation within the inhibitor binding site (Figure 5G, H). However, molecular docking analysis of these inhibitors against the active hcGAS–DNA complex revealed that no accepted poses were compatible with previously observed binding conformations (Figure 5I–J). In each case, our analysis demonstrates that the hcGAS–DNA complex active-site conformation is structurally distinct from previous apo hcGAS and mcGAS–DNA structures, and indicates incompatibilities that can may explain differences in hcGAS-specific targeting and inhibitor potency.
To validate the impact of our findings on inhibitor design, we directly assessed the human-specific substitutions in the inhibitor binding pocket on cGAS-inhibitor specificity. RU.521 more potently inhibits mcGAS, while PF-06928215 is a more potent inhibitor against hcGAS, demonstrating clear species-specificity of inhibitor interactions (Figure 6A and Figure S7). Importantly, we overcame the reduced enzymatic activity of wildtype hcGAS, a significant barrier to drug design (Vincent et al., 2017), by using the engineered hcGAS K187N/L195R enzyme discovered in our Vibrio genetic assay (hcGAS*). hcGAS* behaves similarly to wild type hcGAS but exhibits > 15-fold enhanced 2′3′ cGAMP synthesis with activity comparable to wildtype mcGAS (Figure S7). Our docking analysis of RU.521 suggested that the human residues at 434 and 482 play a crucial role in protein–inhibitor interactions and we hypothesized that these positions might explain the species-specificity of RU.521. Strikingly, the species-specificity of RU.521 is completely dependent on the hcGAS S434 and N482 substitutions. Purified mcGAS with humanizing C419S/H467N mutations is strongly resistant to RU.521, while hcGAS* with mouse-like S434C/N482H mutations gains susceptibility to RU.521 inhibition (Figure 6B,C). These results reveal that specific substitutions in the enzyme active site are necessary and sufficient to control susceptibility to the RU.521 cGAS inhibitor, and demonstrate the importance of the hcGAS–DNA structure to informing small-molecule design. Together with our discovery of the regulatory role of hcGAS K187 and L195 in controlling DNA-length specificity, these results reveal the structural basis of hcGAS activation and provide a critical model to guide development of small-molecules targeting cGAS activity (Figure 7).
Figure 6. The molecular basis of cGAS inhibitor specificity.
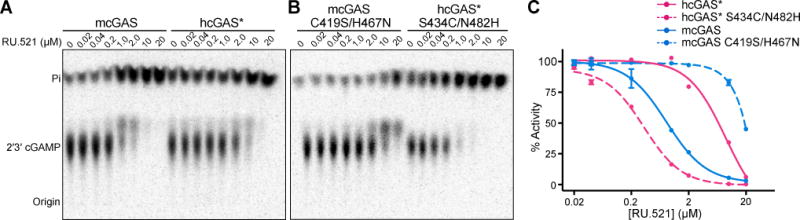
(A) In vitro analysis of mcGAS and hcGAS K187N/L195R (hcGAS*) 2′3′ cGAMP synthesis with increasing concentration of the mcGAS inhibitor RU.521. Enzyme activation was analyzed as in Figure 1A.
(B) Identical experiment as in A, using mcGAS with humanizing C419S/H467N mutations or hcGAS* with mouse-like S434C/N482H mutations in the inhibitor binding pocket. Mouse-specific C419 and H467 substitutions are necessary and sufficient for susceptibility to RU.521.
(C) Quantification of cGAS inhibition by RU.521. 2′3′ cGAMP synthesis was quantified as in Figure 1A and was normalized to the DMSO control (set as 100%). Data are represented as mean ± SD of two independent experiments. See also Figure S7.
Figure 7. Determination of the hcGAS–DNA structure reveals that human-specific substitutions enhance regulation of cGAS activation.
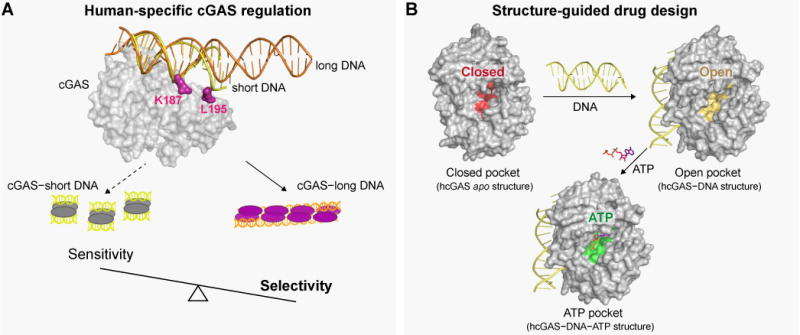
(A) Human-specific substitutions increase the DNA-length specificity of cGAS. Control of DNA sensing must be balanced to maintain sensitivity to pathogen-or stress-derived DNA, and allow accurate tolerance of self-DNA. Human substitutions in cGAS re-shape this balance, and reduce 2′3′ cGAMP synthesis in order to favor enhanced DNA selectivity.
(B) Binding of DNA to cGAS induces a large conformational change, resulting in an “open” active-site conformation that is competent to associate with nucleotides for 2′3′ cGAMP synthesis. Structures of active human cGAS will be critical to guide drug development and analysis of human disease-related cGAS polymorphisms.
Discussion
Our results reveal a layer of regulation in cGAS-STING signaling, where human-specific adaptations in cGAS allow enhanced control of DNA-sensing. Key substitutions in the DNA-binding surface refine protein–DNA interactions and directly augment the ability of hcGAS to discriminate long DNAs. Human K187 and L195 substitutions are required for altered enzyme regulation, and are sufficient to confer human-like regulation to mcGAS. Mechanistically, we demonstrate that K187 and L195 substitutions map to a unique surface on cGAS that is critical for recognition of short DNA, but not required during higher-order cGAS–DNA recognition. Human-specific cGAS substitutions have two functional consequences. First, they restrain total 2′3′ cGAMP production and therefore immune activation. Second, they dramatically enhance the ability of cGAS to discriminate DNA in a length-dependent manner (Figure 7A). Given the affinity of STING for 2′3′ cGAMP is as strong as ~4 nM (Gao et al., 2013b; Zhang et al., 2013), the threshold of 2′3′ cGAMP required for immune activation is extremely low and we hypothesize that the major outcome of human-specific cGAS substitutions is the ability to accurately recognize long DNAs and tolerate shorter DNA fragments. Our results demonstrate that in addition to post-translational modifications and binding partners (Chen et al., 2016; Liang et al., 2014; Seo et al., 2018; Seo et al., 2015; Xia et al., 2016; Yoh et al., 2015), intrinsic substitutions in the hcGAS protein sequence play a key role in regulation of cGAS-STING signaling in human cells.
Human-specific DNA A-site substitutions are evolutionarily more recent than the DNA B-site substitutions (Figure S5C), suggesting current human regulation has evolved as the result of at least two separate events. Interestingly, some of the hcGAS substitutions mapped in this study, including the major DNA A-site substitution L195, were previously demonstrated to be under strong positive selection (dN/dS > 1), a genetic signature that is associated with rapid evolution (Hancks et al., 2015). Unlike traditional examples of positive selection, such as an “arms race” at a host interface antagonized by a pathogen effector (Elde and Malik, 2009), our results demonstrate that positive selection might also mark protein surfaces that modify regulation of innate immune signaling. The distribution of K187 and L195 suggests a genetic bottleneck in hominoid evolution where tolerance to smaller fragments of DNA in the cytosol provided a major selective advantage during a catastrophic event. Although tolerance to small fragments of self-DNA could decrease the occurrence of autoimmune disease and chronic inflammation, these do not seem consistent with the extreme penetrance of the key K187/L195 cGAS substitutions. Alternatively, DNA-length discrimination may improve resistance to pathogens or disease tolerance. Intriguingly, hcGAS variation mirrors a similar phenomenon in STING where emergence of a human-specific R232H mutation in the cyclic dinucleotide binding pocket increases specificity of STING for the cGAS product 2′3′ cGAMP and limits recognition of bacterial-derived 3′3′ linked cyclic dinucleotides (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013b; Kranzusch et al., 2015; Zhang et al., 2013). The evolutionary pressures driving selection of human-specific mutations that reduce the affinity of STING for 3′3′ linked cyclic dinucleotides while maintaining recognition of 2′3′ cGAMP has been a topic for speculation (Danilchanka and Mekalanos, 2013; Kranzusch et al., 2015; Margolis et al., 2017) and potentially the same pressures have selected for the human allele that alters the responsiveness of cGAS to DNA fragment length. Substitutions in both cGAS and STING allow cells to fine-tune a balance between specificity and sensitivity in innate immune signaling.
In addition to recognizing pathogen DNA, cGAS-STING immunity plays a key role in controlling responses to cellular stress and cancer. cGAS is critical for recognition of DNA released during mitochondrial damage (Rongvaux et al., 2014; White et al., 2014), aberrant chromosomal segregation (Dou et al., 2017; Gluck et al., 2017; Harding et al., 2017; Mackenzie et al., 2017; Yang et al., 2017), and during the immune response to cancer cells and checkpoint blockade therapy (Bakhoum et al., 2018; Dou et al., 2017; Harding et al., 2017; Woo et al., 2014). Importantly, the structure of hcGAS in an active conformation removes the previous reliance on mammalian homologs to understand enzyme activation, the mutational burden in cancer, and therapeutic design (Figure 7B). Efforts to develop small-molecule agonists and antagonists of cGAS-STING immunity have already identified 2′3′ cGAMP analogues as adjuvants for cancer immunotherapy (Corrales et al., 2015; Fu et al., 2015), and small-molecule cGAS inhibitors for the treatment of autoimmunity (Hall et al., 2017; Vincent et al., 2017). Current cGAS inhibitors target a pocket above the ATP donor position in the enzyme active-site. Notably, the hcGAS–DNA–ATP ternary complex reveals that additional human-specific substitutions in the enzyme active-site line both sides of this pocket and alter the inhibitor specificity and potency (Figure 5 and 6). Our structure of the active human cGAS–DNA complex, and discovery of key human-specific adaptations, now provides a missing template required to guide development of cGAS therapeutics and establish a complete model of DNA sensing in human cells.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philip J. Kranzusch (Philip_kranzusch@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Vibrio cholerae Strains
V. cholerae strains were all derivatives of streptomycin resistant C6706 dncV::Tn (to remove endogenous 3′3′ cGAMP synthesis) (Davies et al., 2012). V. cholerae were cultured at 37°C on LB media (1% tryptone, 0.5% yeast extract, 0.5% NaCl w/v), stored at −80°C in LB supplemented with 30% glycerol. Where appropriate, antibiotics and supplemented nutrients were used at the following concentrations: streptomycin (100 µg ml−1), carbenicillin (100 µg ml−1), and diaminopimelic acid (300 µg ml−1).
Escherichia coli Strains
Recombinant cGAS enzymes were expressed in E. coli BL21-RIL DE3 (Agilent) bacteria harboring a pRARE2 tRNA plasmid. Transformations and starter cultures were grown in MDG media (0.5% glucose, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.25% aspartic acid, 100 µg ml−1 ampicillin, 34 µg ml−1 chloramphenicol, and trace metals) overnight at 37°C and used to seed 1 L cultures grown in M9ZB media (0.5% glycerol, 1% Cas-Amino Acids, 47.8 mM Na2HPO4, 22 mM KH2PO4, 18.7 mM NH4C 85.6 mM NaCl, 2 mM MgSO4, 100 µg ml−1 ampicillin, 34 µg ml−1 chloramphenicol, and trace metals) (Studier, 2005). M9ZB cultures were cultivated at 37°C until OD600 of ~1.5–2.5, cooled on ice for 20 min, and then recombinant protein synthesis was induced by supplementation with 0.5 mM IPTG. Cultures were incubated at 16°C with shaking for ~1 6 h before harvest.
METHOD DETAILS
Protein Expression and Purification
Human cGAS (encoding residues 157–522) and mouse cGAS truncations (encoding residues 147–507) were PCR amplified from previously described plasmids (Diner et al., 2013; Kranzusch et al., 2013) and cloned into a custom pET vector for expression of an N-terminal 6×His-SUMO2 fusion protein in E. coli (Table S2). E. coli BL21-RIL DE3 (Agilent) bacteria harboring a pRARE2 tRNA plasmid were transformed with a pET cGAS plasmid, and starter cultures were grown in MDG media. E. coli from large-scale M9ZB cultures (typically 2 L for each construct) were pelleted, washed with 1× PBS, and flash-frozen in liquid nitrogen and stored at −80°C.
Recombinant cGAS was purified as previously described (Kranzusch et al., 2014) with modifications specific to SUMO2 tag removal. Bacterial pellets from 2× 1 L M9ZB cultures were re-suspended in lysis buffer (20 mM HEPES-KOH pH 7.5, 400 mM NaCl, 30 mM imidazole, 10% glycerol, 1 mM DTT) chilled on ice and lysed by sonication. Lysate was clarified by centrifugation and subsequent filtration through glass-wool. 6×His-SUMO2-cGAS was purified from clarified lysate by binding to Ni-NTA (Qiagen) and gravity chromatography. Resin was washed with lysis buffer supplemented to 1 M NaCl, and 6×His-SUMO2-cGAS was eluted with lysis buffer supplemented to 300 mM imidazole. The elution fraction was supplemented with ~250 µg of human SENP2 protease (fragment D364–L589 with M497A mutation), and dialyzed overnight at 4°C in dialysis buffer (20 mM HEPES-KO H pH 7.5, 300 mM NaCl, 1 mM DTT). Untagged cGAS was further purified by binding to 2×5 ml Heparin HP ion-exchange columns connected in tandem (GE Healthcare) and eluting with a gradient of 300–1000 mM NaCl followed by subsequent size-exclusion chromatography using a 16/600 Superdex S75 column (GE Healthcare) equilibrated with storage buffer (20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM TCEP). Final cGAS protein was concentrated to ~10 mg ml−1, flash-frozen in liquid nitrogen, and stored at −80°C for cry stallography and biochemical experiments. Mutant cGAS variants were generated using standard cloning techniques. Full-length hcGAS and cGAS variants were purified as described above, except initial chimera cGAS screen variants in Figure S3 that were dialyzed after Ni-NTA purification without SUMO2 tag removal for direct use in biochemistry experiments.
In vitro cGAS 2′3′ cGAMP Synthesis Assays
cGAS activation and 2′3′ cGAMP synthesis was performed in vitro using purified components and measured with thin-layer chromatography as previously described (Kranzusch et al., 2014). Briefly, 1 µM cGAS was incubated with a 45 bp double-stranded interferon stimulatory DNA (Stetson and Medzhitov, 2006) in the 20 µl reaction buffer containing 50 mM Tris-HCl pH 7.5, 35 mM KCl, 5 mM Mg(OAc)2, 1 mM DTT, 25 µM ATP, 25 µM GTP, and [α-32P] ATP (~1 µCi) at 37°C for 30 min. Where indicated, cGAS enzyme titrations were as follows: 1, 0.2, 0.04 µM for Fig. 1A; 1, 0.2 µM for 2A; 1, 0.2, 0.04 µM for Fig. S2A; 1, 0.2 µM cGAS for Fig. S3B; 1, 0.2 µM for Fig. S5D). Reactions were terminated by heating at 95°C for 3 min, and subsequently incubated with 4 U of alkaline phosphatase (New England Biolabs) at 37°C for 30 min to hydrolyze unreacted NTPs. One microliter of each reaction was spotted on a PEI-Cellulose F thin-layer chromatography plate (EMD Biosciences) developed with 1.5 M KH2PO4 (pH 3.B) as a running buffer. Radiolabeled products were detected by Typhoon Trio Variable Mode Imager system (GE Healthcare) and quantified with ImageQuant 5.2. The relative activity of cGAS was determined based on the ratio of radiolabeled 2′3′ cGAMP to the total radiolabeled products. cGAS activation in response to bacterial DNA was tested using wildtype V. cholerae genomic DNA isolated with a DNA Extraction Kit (DNeasy Blood & Tissue, Qiagen), and supplemented in reactions as above using a final DNA concentration of 27.5 ng µl−1 (equivalent to 1 µM of 45 bp DNA).
cGAS enzyme kinetics were measured as a function of varying ATP concentration, and 2′3′ cGAMP product formation was quantified with phosphorimaging and fit according to Michaelis-Menten kinetics. Briefly, ATP/GTP titrations (0, 5, 10, 25, 50, 100, 200, 400, 800, and 1000 µM) were performed in the presence of fixed cGAS (1 µM) and 45 bp dsDNA concentration (1.5 µM) in 10 µl of reaction buffer containing 50 mM Tris-HCl pH 7.5, 35 mM KCl, 5 mM Mg(OAc)2, 1 mM DTT, and [α-32P] ATP (~1 µCi) at 37°C for 5 min. Reactions were terminated and analyzed using thin layer chromatography as indicated above. The ATP-dependent cGAS enzyme kinetics were plotted and fitted according to Michaelis-Menten substrate inhibition analysis in GraphPad Prism (version 7.0c).
V. cholerae Chemotaxis Assay
hcGAS (D157-F522) and mcGAS (P147-L507) were codon optimized for bacterial expression (Genscript and IDT respectively), and all cGAS enzymes and control genes were overexpressed from a custom plasmid pBAD24T, a conjugation-proficient version of pBAD24 that harbors an arabinose inducible promoter and a strong ribosomal binding site that was further modified to include a multiple cloning site (a generous gift of Florence Caro, Harvard Medical School). All genes overexpressed in V. cholerae were encoded with MBP N-terminal fusions except the MBP alone negative control vector (Table S2).
Plasmids were introduced into V. cholerae by conjugation using MFDIpir E. coli as the plasmid donor and diaminopimelic acid auxotrophy for counter selection, and V. cholerae chemotaxis assays were performed as previously described (Davies et al., 2012). Briefly, chemotaxis medium (1% tryptone, 0.5% NaCl, 0.3% agar w/v) was prepared, autoclaved, and stored at 2× concentration; and chemotaxis plates were prepared 2-24 h prior to inoculation. 2× medium was melted, diluted to 1× with sterile water, and supplemented with 0.2% (w/v) arabinose and carbenicillin. Deep-well plates (OmniTray w/ Lid, Thermo Fisher) were poured at 40 ml for each plate. Overnight, stationary phase cultures (16–24 h post inoculation) were diluted 10-fold with fresh LB, arrayed in a 96-well plate, then applied to the surface of a chemotaxis plate via pinning with a 96-pin, floating pin replicator (VP 408FH, V&P Scientific). Alternatively, 1 µl of diluted culture was applied to the surface of chemotaxis agar by pipetting. Chemotaxis plates were incubated at 30°C for 16–24 h.
Chemotaxis was quantified by imaging plates at 16–24 h post inoculation and digitally measuring the area of motile bacteria using ImageJ. Chemotaxis area was normalized to MBP alone vector for each experiment for expression of “chemotaxis as a % of MBP”. Values were transformed into “chemotaxis repression” by calculating (chemotaxis as a % of MBP)−1, then normalizing this value to dncV. Chemotaxis repression was determined and used to simplify a direct relationship between cGAMP levels and plotted values.
V. cholerae cGAS Expression Analysis
Log phase cultures of V. cholerae growing in LB medium were induced for 1 h with 0.2% (w/v) arabinose, and cultures were harvested by centrifugation at 8,000 × g for 5 min. Pellets were re-suspended in 1× LDS loading buffer plus reducing agent (Life Technologies) equivalent to an OD of 2.5, and boiled for 10 min. Samples were separated by SDS-PAGE using 4–12 % bis-tris gels (Life Technologies). Proteins were transferred to nitrocellulose membranes and probed with primary antibodies: rabbit anti-MBP at 1:5,000 (polyclonal, catalog #AB3596, Millipore Sigma) and mouse anti-RNA polymerase beta at 1:5,000 (monoclonal 8RB13, catalog #MA1-25425, Thermo Fisher Scientific). Secondary antibodies: IRDye 800CW goat anti-rabbit IgG at 1:10,000 (polyclonal, catalog #925-32211, LiCor), IRDye 680LT goat anti-mouse IgG at 1:10,000 (polyclonal, catalog #926-68020, LiCor). Membranes were imaged using a Licor Odyssey CLx imager.
Crystallization and Structure Determination
For crystallization, purified human cGAS K187N/L195R (residues 157–522) was mixed with 17 bp DNA in a molar ratio of 1:1.25 protein:DNA in a buffer containing 20 mM HEPES-KOH pH 7.5, 125 mM KCl, 5 mM ATP, and 10.5 mM MgCl2, incubated at room-temperature for 30 min, and centrifuged to pellet precipitation prior to crystallization trials. Crystals were obtained with hanging drop vapor diffusion in drops mixed 1:1 over a reservoir of 0.1 M HEPES-NaOH pH 7.0, 1.4 M sodium citrate after 3 days of growth at 18°C. Crystals were cryo-protected using reservoir solution supplemented with 10% glycerol and flash-frozen in LiN2. Sodium citrate in the crystallization buffer chelated Mg2+ and prevented NTP incorporation in the cGAS active-site. To overcome this problem, fully-grown crystals were transferred to a soaking solution without citrate (0.1 M sodium succinate, 35% PEG-5000 MME, 3 mM ATP, 0.3 mM Gpcpp, 6.3 mM MgCl2, 10% glycerol) for 10 min and then harvested and flash-frozen as above. Crystallization drops additionally included 0.5 mM nonhydrolyzable GTP, but no clear density was observed.
X-ray diffraction data were collected at the Advanced Photon Source (Beamlines 24-IE-E and 24-IE-C) and then processed with XDS and AIMLESS (Kabsch, 2010) using the SSRL autoxds script (A. Gonzalez, Stanford SSRL). Crystals for both complexes were indexed according to the hexagonal spacegroup P 61 2 2, and contain one copy of the hcGAS–DNA complex in the asymmetric unit. Phases were determined with molecular replacement using Phaser in PHENIX (Adams et al., 2010) and the apo human cGAS structure (PDB 4KM5) as a search model. Structure determination was completed with iterative model building and refinement using Coot (Emsley and Cowtan, 2004) and PHENIX, respectively. Data collection and refinement statistics are listed in Table S1.
In vitro cGAS–DNA Complex Formation Analysis
cGAS–DNA complexes were stably assembled using a method adapted from a nucleosome assembly gradient dialysis protocol (Lee and Narlikar, 2001), and complex formation was measured by electrophoretic mobility shift analysis. Briefly, a concentration gradient of recombinant cGAS (0.2, 0.4, 2, 10 µM) was mixed with 2 µM 45 bp dsDNA in a high-salt buffer containing 20 mM HEPES-KOH pH 7.5, 400 mM KCl, 1 mM DTT. The reconstitution mixture was then transferred into 8–10 kDa molecular weight cut-off dialysis tubing (Spectrum Labs), and placed into a beaker of the high-salt buffer. The KCl concentration was reduced gradually by slowly replacing high-salt buffer with low-salt buffer (20 mM HEPES-KOH pH 7.5, 50 mM KCl, 1 mM DTT) with a peristaltic pump (Bio-Rad) at a rate of 200 µl min−1 for ~48 h at 4°C. After gradient dialysis, reactions were supple mented to 5% glycerol, and subsequently separated on a 2% agarose gel in 0.5× TBE buffer at 4°C. Agarose gels were stained in a solution containing 10 µg ml−1 ethidium bromide and visualized with a ChemiDoc MP Imaging System (Bio-Rad). Alternatively, where indicated in Figure S5A and S5C, cGAS–DNA complexes were assembled by incubating directly in reaction buffer containing 20 mM HEPES-NaOH pH 7.8, 75 mM KCl, 1 mM DTT at 4°C for 20 min and analyzed by electrophoretic mobility shift analysis as indicated above.
Molecular Docking Analysis
The crystal structures of RU.521 bound to the mcGAS–DNA complex (PDB 5XZG), PF-06928215 bound to inactive hcGAS (PDB 5V8N), and the active hcGAS–DNA complex were prepared using the Protein Preparation Wizard in Maestro (Maestro v.11.5.011). Default settings were used, except that all crystallographic water molecules >5 Å from heteroatom groups were removed. The docking receptor grid was created using the Receptor Grid Generation module in Glide (Maestro v.11.5.011). The grid box and center were set to default by using the active site ligand, with the active site ligand excluded from the grid. The ligands were prepared using the LigPrep module with OPLS3 force field and default settings (Maestro v.11.5.011). The docking poses were generated using the LigandDocking protocol as implemented in Schrödinger Suite 2018-1. All default settings were used, except that the Standard Precision (SP) or Extra Precision (XP) scoring functions were used with flexible ligand sampling. Briefly, the grid box and center were set at default using the active site ligand, and no constraints were defined. The default set of ligand poses with the lowest Glide SP score is shown for hcGAS–DNA docking of RU.521 and PF-06928215, eight and four respectively, and two ligand poses each for mcGAS-DNA docking of RU.521 (XP score) and inactive hcGAS (XP and SP score). Alignment of the mcGAS–DNA bound RU.521 (PDB 5XZG) and inactive hcGAS bound PF-06928215 (PDB 5V8N) to hcGAS–DNA in Figure 5G and H was generated in PyMOL (The PyMOL Molecular Graphics System, version 1.8.6.0 Schrödinger, LLC).
Small-Molecule cGAS Inhibition Assays
The cGAS small-molecule inhibitors PF-06928215 (Sigma) (Hall et al., 2017) and RU.521 (Aobious) (Vincent et al., 2017) were prepared as ~10 mM and ~20 mM stocks respectively in 100% dimethyl sulfoxide (DMSO). Liquid chromatography, mass-spectrometry analysis of the RU.521 manufacturer preparation revealed the additional presence of lower-molecular weight byproducts, and the effective RU.521 stock concentration was adjusted accordingly. cGAS inhibition was measured as a function of varying small-molecule concentration compared to DMSO alone. Briefly, small-molecule titrations (0.02-100 µM) were performed in the presence of cGAS (1 µM) and 45 bp dsDNA concentration (1 µM) in 20 µl of reaction buffer containing 50 mM Tris-HCl pH 7.5, 35 mM KCl, 10 mM Mg(OAc)2, 1 mM DTT, 100 µM ATP, 100 µM GTP, [α-32P] ATP (~1 µCi), and 1% final DMSO concentration at 37°C for 30 min. Reactions were terminated and analyzed using thin layer chromatography as indicated above. 2′3′ cGAMP product formation was quantified with phosphorimaging and the inhibitor dose response curves for RU.521 and PF-06928215 were plotted and fit according to standard “log(inhibitor) vs. response – Variable slope (four parameters)” equation in GraphPad Prism (version 7.0c).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for each experiment can be found in the figure legends, and outlined in the corresponding methods details section. V. cholerae chemotaxis data are plotted with error bars representing the standard error of the mean (SEM), all other data are plotted with error bars representing as the standard deviation of the mean (SD).
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the hcGAS–DNA and hcGAS–DNA–ATP crystal structure data reported in this paper have been deposited in the Protein Data Bank under ID codes PDB: 6CT9 and 6CTA.
Supplementary Material
Alignment of human and mouse cGAS amino acid sequences with known alpha-helix (magenta) and beta-strand (grey) secondary structure depicted below. Conserved residues are shaded in grey, residues mediating cGAS–DNA contacts are boxed in black, and the metal-coordinating active-site residues are marked with a red asterisk. Human-specific substitutions in DNA A-site and B-site binding surfaces are labeled in magenta and corresponding mouse residues are labeled in blue.
(A) In vitro analysis of hcGAS and mcGAS activation in response to bacterial genomic DNA. 2′3′ cGAMP synthesis in the presence of purified V. cholerae genomic DNA was visualized by thin-layer chromatography as in Figure 1A and quantified relative to maximal activity observed with mcGAS. Data are represented as mean ± SD of three independent experiments.
(B) Quantification of V. cholerae chemotaxis repression (see Star Methods) for strains overexpressing the synthases indicated. Left, schematic representation of chimeric and mutant cGAS constructs. Metal coordinating residues in the active-site are represented as asterisks, the Zn-ribbon is represented in orange, amino acid numbers are colored magenta for hcGAS and blue for mcGAS. Schematic is not to scale. Reference dotted lines represent mcGAS (in blue) or hcGAS (in magenta) levels of repression. Data are represented as mean ± SEM for at least three independent experiments.
(A) Purified hcGAS and mcGAS enzymes used in this study. Purified recombinant cGAS (~1 µg each) was resolved on a denaturing SDS-PAGE gel and visualized with Coomassie G-250 stain. Initial chimera screening was conducted with 6×His-SUMO2-tagged cGAS variants purified with Ni-NTA affinity. All subsequent biochemistry was performed with untagged cGAS variants purified with Ni-NTA, heparin ion-exchange, and Superdex S75 size-exclusion chromatography.
(B) In vitro analysis of hcGAS chimera 2′3′ cGAMP synthesis. The relative 2′3′ cGAMP synthesis levels of key active hcGAS chimeras (see Figure 1 and Figure S2) were visualized by thin-layer chromatography as in Figure 1A, and compared to relative levels of wildtype hcGAS and mcGAS activity. The minimal element required for human-like cGAS regulation maps to chimera 4.3 (Chi 4.3). Data are representative of at least three independent experiments.
(A) 2Fo–Fc electron density map of the 2.3 Å hcGAS–DNA complex (contoured at 1.0 σ). hcGAS protein is shown as a ribbon (magenta) and the double-stranded DNA is shown as sticks (yellow).
(B) Schematic map of protein–DNA contacts in the hcGAS–DNA and mcGAS–DNA (PDB 4K98) complexes. Human-specific contacts are highlighted in magenta as in Figure 3C, and mouse-specific contacts are highlighted in blue. Black dots denote interactions bridged by water molecules. Black labels indicate contacts directly observed in the hcGAS–DNA complex, grey labels indicate water-mediated contacts potentially conserved with mcGAS but not observed in the hcGAS–DNA complex.
(C) Superposition of apo hcGAS (PDB 4KM5, grey) and the hcGAS–DNA complex (magenta).
(D) Superposition of the hcGAS–DNA complex (grey) and hcGAS–DNA–ATP ternary complex (magenta). The mechanism of hcGAS structural activation upon DNA-binding is conserved with mammalian cGAS homologs (Civril et al., 2013; Gao et al., 2013a; Li et al., 2013; Zhang et al., 2014). Double-stranded DNA recognition causes a conformational change in cGAS resulting in remodeling of the N-terminal “spine helix” (shown in inset of C) and repositioning of an active-site loop that allows metal coordination and nucleotide binding (depicted in inset of D).
(A) In vitro electrophoretic mobility shift assay measurement of wildtype mcGAS–DNA and hcGAS K187N/L195R (NR)–DNA complex formation. Complexes were assembled directly in low salt (75 mM KCl) and directly visualized on a 2% agarose gel. Alternatively, cGAS–DNA complexes were assembled during gradient dialysis (400 to 50 mM KCl, over 48 h) to specifically monitor stable complex formation.
(B) In vitro analysis of hcGAS, mcGAS, and hcGAS K187N/L195R (NR) 2′3′ cGAMP synthesis, confirming that the elevated protein concentration conditions used for the electrophoretic mobility shift assay analysis support functional enzyme activation. The relative 2′3′ cGAMP synthesis levels were visualized by thin-layer chromatography as in Figure 1A, and compared to relative levels of wildtype mcGAS activity. Data are representative of at least two individual experiments.
(C) Cladogram depicting evolution of hcGAS DNA A-site and B-site substitutions in primates. Human-specific substitutions are denoted in magenta, and the mouse cGAS sequence is denoted in blue for reference. Human positions L195 and K350 were previously identified to be under positive selection (Hancks et al., 2015).
(D) In vitro analysis of the role of hcGAS DNA B-site variation in 2′3′ cGAMP synthesis regulation. Human and mouse amino acid sequences at S328, K350, and L354 (human S328/K350/L354 and mouse-equivalent G/T/R positions are denoted SKL or GTR respectively) were analyzed in both hcGAS and mcGAS backgrounds. Enzymes were stimulated with 45 bp DNA, and 2′3′ cGAMP synthesis was measured as in Figure 1A and quantified relative to maximal activity observed with mcGAS. Data are represented as mean ± SD of at least two independent experiments.
(E) In vitro electrophoretic mobility shift assay measurement of cGAS–DNA complex formation. hcGAS and mcGAS variants (labeled as in Figure S5C) were incubated with 45 bp DNA in the presence of low salt and the resulting complexes were directly resolved on a 2% agarose gel without gradient dialysis.
(A) Purified full-length hcGAS enzymes used in this study. Full-length variants were expressed as 6×His-SUMO2-tagged fusions and purified with Ni-NTA, Heparin ion-exchange, and Superdex S200 size-exclusion chromatography. Each protein (~1 µg each) was resolved on a denaturing SDS-PAGE gel and visualized with Coomassie G-250 stain.
(B) In vitro analysis of full-length hcGAS and hcGAS K187N/L195R (NR) 2′3′ cGAMP synthesis and DNA-length specificity. Purified full-length hcGAS variants were stimulated with increasing concentration of 45 bp (top) or 17 bp (bottom) DNA, and enzyme activation was visualized as in Figure 1A and analyzed as in Figure 4. Human-specific K187 and L195 cGAS substitutions prevent activation of 2′3′ cGAMP synthesis in the presence of short 17 bp DNA.
(C) Quantification of cGAS DNA length-dependent activation experiments in B. 2′3′ cGAMP synthesis was quantified as in Figure 1A and graphed as total conversion of ATP to 2′3′ cGAMP. Data are represented as mean ± SD of three independent experiments.
(A,B) In vitro analysis of hcGAS, mcGAS, and hcGAS K187N/L195R (hcGAS*) 2′3′ cGAMP synthesis with increasing concentration of cGAS inhibitor RU.521 (A) or PF-06928215 (B). Enzyme activation was analyzed as in Figure 1A, and inhibition was quantified and normalized to the DMSO control (set as 100%). Data are represented as mean ± SD of two independent experiments. RU.521 more potently blocks mouse cGAS and the PF-06928215 compound more potently blocks human cGAS. hcGAS* exhibits similar inhibitor sensitivities as wildtype hcGAS, and was selected for further inhibition analysis due to elevated levels of 2′3′ cGAMP synthesis.
(C) Cladogram depicting evolution of hcGAS S434/N482 in primates. Human-like substitutions are denoted in magenta, and the mouse cGAS sequence is denoted in blue for reference.
(D) SDS-PAGE and Coomassie G-250 analysis of purified mcGAS and hcGAS* inhibitor binding site variants used in this study (~1 µg each).
(Chen et al., 2010; Karplus and Diederichs, 2012; Weiss, 2001)
Table S2. Plasmid and insert sequences, Related to STAR Methods.
Highlights.
cGAS DNA-sensing in humans is adapted for enhanced specificity
A bacterial genetic assay allows rapid mapping of human cGAS regulatory determinant
Human cGAS–DNA structures reveal altered contacts favor DNA-length discrimination
cGAS active site variation explains species-specificity of small-molecule inhibitors
Acknowledgments
The authors are grateful to Amy Lee and Kozak Chat (Brandeis University), Russell Vance (U.C. Berkeley) and members of the Kranzusch and Mekalanos labs for helpful comments and discussion. The authors also acknowledge Olga Danilchanka and Elena Del Tordello for initial observations in Vibrio cholerae. The work was funded by the Claudia Adams Barr Program for Innovative Cancer Research (P.J.K.), Richard and Susan Smith Family Foundation (P.J.K.), Charles H. Hood Foundation (P.J.K.), a Cancer Research Institute CLIP Grant (P.J.K.), NIAID (AI-01845) (J.J.M.), and NIH grant NCI R01CA214608 (E.S.F.). A.T.W. is supported as a fellow of The Jane Coffin Childs Memorial Fund for Medical Research, C.O.M. is supported as a Cancer Research Institute / Eugene V. Weissman Fellow, and B.R.M. is supported by the NIH T32 Cancer Immunology training grant (5T32CA207021-02). X-ray data were collected at the Northeastern Collaborative Access Team beamlines, which are funded by the NIGMS (P41 GM103403) and an NIH-ORIP HEI grant (S10 RR029205), and used resources of the DOE Argonne National Laboratory Advanced Photon Source, (under Contract No. DE-AC02-06CH11357).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Experiments were designed by W.Z., A.T.W., J.J.M., and P.J.K. All structural and biochemical experiments were conducted by W.Z. with assistance from B.R.M., C.O.M., and P.J.K. A.T.W. performed and analyzed all bacteria chemotaxis assays. Molecular docking analysis was performed by R.P.N., E.S.F., and N.S.G. The manuscript was written by W.Z., A.T.W., and P.J.K., and all authors contributed to editing the manuscript and support the conclusions.
Supplemental Information
Supplemental Information includes seven figures and two tables and can be found with this article online at
Declaration of Interests
The Dana-Farber Cancer Institute and Harvard Medical School have patents pending for human cGAS technologies on which the authors are inventors.
Accession Numbers
Coordinates of human cGAS–DNA binary complex and human cGAS–DNA–ATP ternary complex have been deposited in the RCSB Protein Data Bank under accession numbers 6CT9 and 6CTA.
Literature Cited
- Ablasser A, Goldeck M, Caviar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva L, Hiller B, Kostrewa D, Lassig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M, Leonhardt H, Hornung V, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. 2017;549:394–398. doi: 10.1038/nature23890. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–472. doi: 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang RF, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol Cell. 2016;64:105–119. doi: 10.1016/j.molcel.2016.08.025. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962–970. doi: 10.1016/j.cell.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BW, Bogard RW, Young TS, Mekalanos JJ. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell. 2012;149:358–370. doi: 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Malik HS. The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol. 2009;7:787–797. doi: 10.1038/nrmicro2222. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra252. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013a;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell. 2013b;154:748–762. doi: 10.1016/j.cell.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George RD, McVicker G, Diederich R, Ng SB, MacKenzie AP, Swanson WJ, Shendure J, Thomas JH. Trans genomic capture and sequencing of primate exomes reveals new targets of positive selection. Genome Res. 2011;21:1686–1694. doi: 10.1101/gr.121327.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–1070. doi: 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, Aulabaugh A, Aivazian D, Castro D, Chen M, et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12:e0184843. doi: 10.1371/journal.pone.0184843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC. Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet. 2015;11:e1005203. doi: 10.1371/journal.pgen.1005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayel E, Burckstummer T, Bilban M, Durnberger G, Weitzer S, Martinez J, Superti-Furga G. The TLR-independent DNA recognition pathway in murine macrophages: Ligand features and molecular signature. Eur J Immunol. 2009;39:1929–1936. doi: 10.1002/eji.200939344. [DOI] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Ishii R, Goto E, Ishitani R, Tokunaga F, Nureki O. Structural and functional analyses of DNA-sensing and immune activation by human cGAS. PLoS One. 2013;8:e76983. doi: 10.1371/journal.pone.0076983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, Barber GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. 2018 doi: 10.1038/s41388-017-0120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013;3:1362–1368. doi: 10.1016/j.celrep.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee ASY, Wilson SC, Solovykh MS, Vance RE, Berger JM, Doudna JA. Structure-guided reprogramming of human cGAS dinucleotide linkage specificity. Cell. 2014;158:1011–1021. doi: 10.1016/j.cell.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Wilson SC, Lee AS, Berger JM, Doudna JA, Vance RE. Ancient Origin of cGAS-STiNg Reveals Mechanism of Universal 2′,3′ cGAMP Signaling. Mol Cell. 2015;59:891–903. doi: 10.1016/j.molcel.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Narlikar G. Assembly of nucleosomal templates by salt dialysis. Current protocols in molecular biology. 2001 doi: 10.1002/0471142727.mb2106s54. Chapter 21, Unit 21.26. [DOI] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner KP, Ha T, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–238. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke S, Holleufer A, Christensen MH, Jonsson KL, Boni GA, Sorensen LK, Johannsen M, Jakobsen MR, Hartmann R, Paludan SR. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18:1707–1715. doi: 10.15252/embr.201744017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SR, Wilson SC, Vance RE. Evolutionary Origins of cGAS-STING Signaling. Trends Immunol. 2017;38:733–743. doi: 10.1016/j.it.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, et al. Insights into hominid evolution from the gorilla genome sequence. Nature. 2012;483:169–175. doi: 10.1038/nature10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun. 2018;9:613. doi: 10.1038/s41467-018-02936-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, Jung JU. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015;13:440–449. doi: 10.1016/j.celrep.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750. doi: 10.1038/s41467-017-00833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MS. Global indicators of X-ray data quality. Journal of Applied Crystallography. 2001;34:130–135. [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, Fan Z. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol. 2016;17:369–378. doi: 10.1038/ni.3356. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, De Jesus PD, Ruan C, de Castro E, Ruiz PA, et al. PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell. 2015;161:1293–1305. doi: 10.1016/j.cell.2015.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, Chen ZJ. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014;6:421–430. doi: 10.1016/j.celrep.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Wang L, Shang G, Liu X, Zhu J, Lu D, Wang L, Kan B, Zhang JR, Xiang Y. Structural biochemistry of a Vibrio cholerae dinucleotide cyclase reveals cyclase activity regulation by folates. Mol Cell. 2014;55:931–937. doi: 10.1016/j.molcel.2014.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignment of human and mouse cGAS amino acid sequences with known alpha-helix (magenta) and beta-strand (grey) secondary structure depicted below. Conserved residues are shaded in grey, residues mediating cGAS–DNA contacts are boxed in black, and the metal-coordinating active-site residues are marked with a red asterisk. Human-specific substitutions in DNA A-site and B-site binding surfaces are labeled in magenta and corresponding mouse residues are labeled in blue.
(A) In vitro analysis of hcGAS and mcGAS activation in response to bacterial genomic DNA. 2′3′ cGAMP synthesis in the presence of purified V. cholerae genomic DNA was visualized by thin-layer chromatography as in Figure 1A and quantified relative to maximal activity observed with mcGAS. Data are represented as mean ± SD of three independent experiments.
(B) Quantification of V. cholerae chemotaxis repression (see Star Methods) for strains overexpressing the synthases indicated. Left, schematic representation of chimeric and mutant cGAS constructs. Metal coordinating residues in the active-site are represented as asterisks, the Zn-ribbon is represented in orange, amino acid numbers are colored magenta for hcGAS and blue for mcGAS. Schematic is not to scale. Reference dotted lines represent mcGAS (in blue) or hcGAS (in magenta) levels of repression. Data are represented as mean ± SEM for at least three independent experiments.
(A) Purified hcGAS and mcGAS enzymes used in this study. Purified recombinant cGAS (~1 µg each) was resolved on a denaturing SDS-PAGE gel and visualized with Coomassie G-250 stain. Initial chimera screening was conducted with 6×His-SUMO2-tagged cGAS variants purified with Ni-NTA affinity. All subsequent biochemistry was performed with untagged cGAS variants purified with Ni-NTA, heparin ion-exchange, and Superdex S75 size-exclusion chromatography.
(B) In vitro analysis of hcGAS chimera 2′3′ cGAMP synthesis. The relative 2′3′ cGAMP synthesis levels of key active hcGAS chimeras (see Figure 1 and Figure S2) were visualized by thin-layer chromatography as in Figure 1A, and compared to relative levels of wildtype hcGAS and mcGAS activity. The minimal element required for human-like cGAS regulation maps to chimera 4.3 (Chi 4.3). Data are representative of at least three independent experiments.
(A) 2Fo–Fc electron density map of the 2.3 Å hcGAS–DNA complex (contoured at 1.0 σ). hcGAS protein is shown as a ribbon (magenta) and the double-stranded DNA is shown as sticks (yellow).
(B) Schematic map of protein–DNA contacts in the hcGAS–DNA and mcGAS–DNA (PDB 4K98) complexes. Human-specific contacts are highlighted in magenta as in Figure 3C, and mouse-specific contacts are highlighted in blue. Black dots denote interactions bridged by water molecules. Black labels indicate contacts directly observed in the hcGAS–DNA complex, grey labels indicate water-mediated contacts potentially conserved with mcGAS but not observed in the hcGAS–DNA complex.
(C) Superposition of apo hcGAS (PDB 4KM5, grey) and the hcGAS–DNA complex (magenta).
(D) Superposition of the hcGAS–DNA complex (grey) and hcGAS–DNA–ATP ternary complex (magenta). The mechanism of hcGAS structural activation upon DNA-binding is conserved with mammalian cGAS homologs (Civril et al., 2013; Gao et al., 2013a; Li et al., 2013; Zhang et al., 2014). Double-stranded DNA recognition causes a conformational change in cGAS resulting in remodeling of the N-terminal “spine helix” (shown in inset of C) and repositioning of an active-site loop that allows metal coordination and nucleotide binding (depicted in inset of D).
(A) In vitro electrophoretic mobility shift assay measurement of wildtype mcGAS–DNA and hcGAS K187N/L195R (NR)–DNA complex formation. Complexes were assembled directly in low salt (75 mM KCl) and directly visualized on a 2% agarose gel. Alternatively, cGAS–DNA complexes were assembled during gradient dialysis (400 to 50 mM KCl, over 48 h) to specifically monitor stable complex formation.
(B) In vitro analysis of hcGAS, mcGAS, and hcGAS K187N/L195R (NR) 2′3′ cGAMP synthesis, confirming that the elevated protein concentration conditions used for the electrophoretic mobility shift assay analysis support functional enzyme activation. The relative 2′3′ cGAMP synthesis levels were visualized by thin-layer chromatography as in Figure 1A, and compared to relative levels of wildtype mcGAS activity. Data are representative of at least two individual experiments.
(C) Cladogram depicting evolution of hcGAS DNA A-site and B-site substitutions in primates. Human-specific substitutions are denoted in magenta, and the mouse cGAS sequence is denoted in blue for reference. Human positions L195 and K350 were previously identified to be under positive selection (Hancks et al., 2015).
(D) In vitro analysis of the role of hcGAS DNA B-site variation in 2′3′ cGAMP synthesis regulation. Human and mouse amino acid sequences at S328, K350, and L354 (human S328/K350/L354 and mouse-equivalent G/T/R positions are denoted SKL or GTR respectively) were analyzed in both hcGAS and mcGAS backgrounds. Enzymes were stimulated with 45 bp DNA, and 2′3′ cGAMP synthesis was measured as in Figure 1A and quantified relative to maximal activity observed with mcGAS. Data are represented as mean ± SD of at least two independent experiments.
(E) In vitro electrophoretic mobility shift assay measurement of cGAS–DNA complex formation. hcGAS and mcGAS variants (labeled as in Figure S5C) were incubated with 45 bp DNA in the presence of low salt and the resulting complexes were directly resolved on a 2% agarose gel without gradient dialysis.
(A) Purified full-length hcGAS enzymes used in this study. Full-length variants were expressed as 6×His-SUMO2-tagged fusions and purified with Ni-NTA, Heparin ion-exchange, and Superdex S200 size-exclusion chromatography. Each protein (~1 µg each) was resolved on a denaturing SDS-PAGE gel and visualized with Coomassie G-250 stain.
(B) In vitro analysis of full-length hcGAS and hcGAS K187N/L195R (NR) 2′3′ cGAMP synthesis and DNA-length specificity. Purified full-length hcGAS variants were stimulated with increasing concentration of 45 bp (top) or 17 bp (bottom) DNA, and enzyme activation was visualized as in Figure 1A and analyzed as in Figure 4. Human-specific K187 and L195 cGAS substitutions prevent activation of 2′3′ cGAMP synthesis in the presence of short 17 bp DNA.
(C) Quantification of cGAS DNA length-dependent activation experiments in B. 2′3′ cGAMP synthesis was quantified as in Figure 1A and graphed as total conversion of ATP to 2′3′ cGAMP. Data are represented as mean ± SD of three independent experiments.
(A,B) In vitro analysis of hcGAS, mcGAS, and hcGAS K187N/L195R (hcGAS*) 2′3′ cGAMP synthesis with increasing concentration of cGAS inhibitor RU.521 (A) or PF-06928215 (B). Enzyme activation was analyzed as in Figure 1A, and inhibition was quantified and normalized to the DMSO control (set as 100%). Data are represented as mean ± SD of two independent experiments. RU.521 more potently blocks mouse cGAS and the PF-06928215 compound more potently blocks human cGAS. hcGAS* exhibits similar inhibitor sensitivities as wildtype hcGAS, and was selected for further inhibition analysis due to elevated levels of 2′3′ cGAMP synthesis.
(C) Cladogram depicting evolution of hcGAS S434/N482 in primates. Human-like substitutions are denoted in magenta, and the mouse cGAS sequence is denoted in blue for reference.
(D) SDS-PAGE and Coomassie G-250 analysis of purified mcGAS and hcGAS* inhibitor binding site variants used in this study (~1 µg each).
(Chen et al., 2010; Karplus and Diederichs, 2012; Weiss, 2001)
Table S2. Plasmid and insert sequences, Related to STAR Methods.