

Abstract
Stress-activated MAPKs (SAPKs) respond to a wide variety of stressors. In most cases, the pathways through which specific stress signals are transmitted to the SAPKs are not known. In this study, we delineate the intracellular signaling pathway by which the trivalent toxic metalloid arsenite [As(III)] activates the yeast SAPK Hog1. We demonstrate that, to activate Hog1, As(III) must enter the cell through the glycerol channel Fps1 and must be metabolized to methyl arsenite [MAs(III)] by the dimeric methyltransferase Mtq2:Trm112. We found that Mtq2:Trm1 displays SAM-dependent methyltransferase activity toward both As(III) and MAs(III). Additionally, we present genetic and biochemical evidence that MAs(III), but not As(III), is a potent inhibitor of the protein tyrosine phosphatases (Ptp2 and Ptp3) that normally maintain Hog1 in an inactive state. Inhibition of Ptp2 and Ptp3 by MAs(III) results in elevated Hog1 phosphorylation without activation of the protein kinases that act upstream of the SAPK and raises the possibility that other Hog1-activating stressors act intracellularly at different points along the canonical Hog1 activation pathway. Finally, we show that arsenate [As(V)], a pentavalent form of arsenic, also activates Hog1, but through a pathway that is distinct from that of As(III) and involves activation of the Hog1 MEK Pbs2.
INTRODUCTION
Arsenic is among the most common poisons found in the environment (Rosen and Liu, 2009). Human exposure to arsenic is mainly through food, water, and air, and contamination of groundwater is a worldwide health problem (Smedley and Kinniburgh, 2002). Inorganic aqueous arsenic exists mainly as oxyanions of trivalent arsenite [As(III)] and pentavalent arsenate [As(V)]. Chronic exposure to inorganic arsenic is associated with cardiovascular disease and hypertension, diabetes mellitus, and various forms of cancer (Abernathy et al., 2003; Beane Freeman et al., 2004). Despite these health effects, As(III) is in current use as a highly effective treatment for acute promyelocytic leukemia (Rehman and Naranmandura, 2012). Therefore, it is important to understand the various mechanisms by which arsenic is transported and metabolized by eukaryotic cells, as well as the cellular responses mobilized by arsenic-induced stress.
In mammalian cells, uptake of As(V), which is an analog of inorganic phosphate, is mediated by the high-affinity phosphate transporter NaPi-IIb, whereas As(III) enters cells through the aquaglyceroporins and the glucose permeases (Maciaszczyk-Dziubinska et al., 2012). Once inside the cell, As(V) is reduced to As(III) by Cdc25 protein phosphatases/arsenate reductases (Bhattacharjee et al., 2010). As(III) is metabolized in mammals principally by As(III) S-adenosylmethionine (SAM) methyltransferase (AS3MT), which catalyzes the transfer of methyl groups from SAM to As(III) to produce methyl arsenite [MAs(III)] and dimethyl arsenite [DMAs(III)] (Ren et al., 2011; Cullen, 2014; Dheeman et al., 2014; Dong et al., 2015), both of which are more toxic than inorganic As(III) (Dong et al., 2015).
In the yeast Saccharomyces cerevisiae, As(V) uptake takes place similarly through the phosphate transporters (Wysocki and Tamas, 2010), whereas As(III) enters the cell principally through the aquaglyceroporin Fps1, a bidirectional channel that normally functions to transport glycerol (Wysocki et al., 2001; Maciaszczyk-Dziubinska et al., 2012). As(V) is reduced to As(III) in yeast by the arsenate reductase Acr2 (Mukhopadhyay and Rosen, 1998), and As(III) is actively transported out of the yeast cell through the plasma membrane metalloid/H+ antiporter Acr3 (Maciaszczyk-Dziubinska et al., 2011), but also flows out of the cell through Fps1 when produced intracellularly by reduction of As(V). Alternatively, As(III) can be conjugated to glutathione and sequestered in the yeast vacuole through the ABC family transporters Ycf1 and Vmr1 (Maciaszczyk-Dziubinska et al., 2012). Methylation of As(III) has not been reported in yeast, but a candidate As(III) methyltransferase, Mtq2, has been identified on the basis of increased As(III) tolerance of an mtq2Δ mutant (Ren et al., 2011).
With regard to signaling, As(III) stimulates the mammalian stress-activated MAPK (SAPK) p38 (Elbirt et al., 1998; Verma et al., 2002) and both SAPKs of yeast—Hog1 of the high osmolarity glycerol (HOG) pathway (Sotelo and Rodríguez-Gabriel, 2006; Thorsen et al., 2006) and Mpk1 (Slt2) of the cell wall integrity pathway (Ahmadpour et al., 2016). As(V) also activates Mpk1 (Matia-Gonzalez and Rodriguez-Gabriel, 2011), but has not been shown to activate Hog1. In all cases, there is evidence that the activated SAPKs play significant roles in tolerance to these metalloids. In yeast, both As(III) and As(V) activate transcription of arsenic-specific detoxification genes by the AP-1-like transcription factor Acr1 (also known as Yap8; Wysocki et al., 2004). Although the mechanism by which arsenic induces transcription is not fully understood, in the case of As(III) stress, this cysteine-reactive toxin binds directly to Acr1 to induce transcription (Kumar et al., 2016). However, its ability to activate Acr1-driven transcription is partially dependent upon Hog1 (Sotelo and Rodríguez-Gabriel, 2006). Another Hog1-dependent response to As(III) is the closure of Fps1, consistent with the glycerol channel serving as an adventitious port for As(III) entry (Thorsen et al., 2006). Here again, however, the mechanism by which this regulation occurs is not known.
The HOG pathway of yeast has been well characterized with regard to its regulation by hyperosmotic stress (Saito and Posas, 2012). However, Hog1 is also stimulated by a wide array of unrelated stress signals, including heat shock (Winkler et al., 2002), cold shock (Panadero et al., 2006), citric and acetic acid (Lawrence et al., 2004; Mollapour and Piper, 2006), oxidative stress (Bilsland et al., 2004), methylglyoxal (Aguilera et al., 2005), bacterial lipopolysaccharide (Marques et al., 2006), glucose starvation (Piao et al., 2012), curcumin (Azad et al., 2014), cadmium (Jiang et al., 2014), and arsenite (Sotelo and Rodríguez-Gabriel, 2006; Thorsen et al., 2006). Mammalian p38 SAPK is the functional ortholog of Hog1 (Han et al., 1994), and is similarly activated by most of these stress-inducing agents (Ono and Han, 2000; de Nadal et al., 2002). This raises two important and related questions. First, do these various stresses activate the SAPK through a common pathway, or through alternative inputs? This is especially important because the cell surface osmosensors of the HOG pathway are the only known inputs to Hog1. Second, how does an activated SAPK mount a specific response appropriate to the particular stress experienced? We have begun to address these questions with an analysis of HOG pathway signaling in response to arsenic-induced stress.
In this study, we demonstrate that As(III) activates Hog1 indirectly through an intracellular mechanism that requires its metabolism to MAs(III) by the dimeric methyltransferase Mtq2:Trm112. MAs(III) activates Hog1 through inhibition of the protein tyrosine phosphatases (Ptp2 and Ptp3) that normally maintain Hog1 in an inactive state, rather than through activation of the canonical HOG signaling pathway stimulated by cell surface osmosensors. Intracellular activation of SAPKs through different input nodes in response to various stressors may have broad implications for consequent SAPK target specificity.
RESULTS
Hog1 is known to be activated by As(III), but not by As(V). To investigate further the activation of Hog1 by these metalloids, we used antibodies that specifically recognize dually phosphorylated (on Thr and Tyr) Hog1 (α-phospho-p38). We chose 1 mM As(III) and 3 mM As(V) for treatments because these doses induced slight growth impairment (Supplemental Figure S1). We found that, similarly to As(III), As(V) can activate Hog1 (Figure 1A), and under these conditions, we observed stronger activation by As(V) than by As(III). Activation of Hog1 by both stressors was quite rapid, peaking within 5–10 min after treatment and declining by 30 min after treatment (Figure 1A). Because As(V) is converted to As(III) by the arsenate reductase Acr2 (Mukhopadhyay and Rosen, 1998), we asked whether As(III) generated by Acr2 was responsible for As(V) activation of Hog1. An acr2∆ mutant displayed slightly higher basal Hog1 phosphorylation than the wild type, but was not appreciably impaired for Hog1 activation by As(V) (Figure 1B), suggesting that activation by As(V) does not require its conversion to As(III). Both As(III) and As(V) stimulate arsenic-protective gene expression, including the ACR2 and ACR3 genes, through the AP-1-like transcription factor Acr1 (Wysocki et al., 2004). In the case of As(III), this transcriptional induction is partially dependent on Hog1 despite the fact that As(III) does not induce nuclear accumulation of Hog1 (Sotelo and Rodríguez-Gabriel, 2006; Thorsen et al., 2006). Therefore, we investigated the requirement for Hog1 in As(V)-induced transcription of ACR3. Unlike As(III), As(V) induction of ACR3 was completely independent of Hog1 (Figure 1C). However, similarly to As(III), As(V) treatment failed to induce nuclear translocation of Hog1 (Figure 2A) and failed to induce transcription of the osmoprotective gene GPD1, which is induced by Hog1 in response to hyperosmotic shock (Figure 2B).
FIGURE 1:

As(III) and As(V) activate Hog1. (A) Activation of Hog1 by As(III) and As(V). Wild-type yeast cells (DL3187) expressing Hog1-HA (from p3225) were treated as indicated and extracts were separated by SDS–PAGE prior to examination by immunoblot for activated (phospho)-Hog1 and total Hog1-HA. Fold activation values are relative to the untreated sample and normalized to the Hog1-HA input. Molecular mass markers (in kDa) are on the right. An α-Hog1 antibody was used for all subsequent immunoblots to detect endogenous Hog1. (B) Activation of Hog1 by As(V) does not require its conversion to As(III). Wild-type or acr2Δ cells (DL4341) were treated as indicated with 1 mM As(III) or 3 mM As(V) and examined for activation of endogenous Hog1. Fold activation values are relative to the untreated wild-type sample and normalized to the Hog1 input. Note that the acr2Δ mutant displays slightly higher basal Hog1 phosphorylation than the wild type. (C) Induction of the arsenic protection gene ACR3 by As(III), but not by As(V), is partially dependent upon HOG1. ACR3 expression was measured by quantitative real-time PCR in wild-type cells and hog1Δ cells (DL3158) after treatment with 1 mM As(III) or 3 mM As(V) for 5 min. Each data point represents the mean and SD of three biological replicates.
FIGURE 2:

Hog1 output in response to As(III) or As(V) treatment and requirement for As(III) to enter cell for Hog1 activation. (A) Neither As(III) nor As(V) induces nuclear accumulation of Hog1. Wild-type cells (DL3187), expressing Hog1-GFP from a centromeric plasmid (p3177), were treated as indicated and visualized by fluorescence microscopy and differential interference contrast (DIC) within 5 min to detect nuclear accumulation of Hog1. (B) The osmoresponsive gene GPD1 is induced by treatment with sorbitol, but not with As(III) or As(V). GPD1 expression was measured by quantitative real-time PCR in wild-type cells (DL3187) after treatment with 1 M sorbitol (to induce hyperosmotic shock), 1 mM As(III), 3 mM As(V), or no treatment “C” for 5 min. Each data point represents the mean and SD of three biological replicates. (C) As(III), but not As(V), treatment induces eviction of Rgc2 from Fps1. CoIP of Rgc2-HA and Fps1-Myc in wild-type cells (DL3187) treated with 1 mM As(III) or 3 mM As(V) for the indicated times. Anti-Myc immuneprecipitates (IPs) were separated by SDS–PAGE and subjected to immunoblot analysis. Molecular mass markers (in kDa) are shown on the right. (D) As(III) fails to activate Hog1 in fps1Δ or rgc1Δrgc2Δ mutants, which are blocked for As(III) entry through the glycerol channel, Fps1. Extracts from wild-type (DL3187), fps1Δ (DL3226), and rgc1Δrgc2Δ cells (DL3207) were examined by immunoblot for activation of Hog1 after treatment with 1 mM As(III), 3 mM As(V), or no treatment “C” for 5 min.
Because As(III) enters yeast cells through the glycerol channel Fps1 and this channel is closed in a Hog1-dependent manner in response to As(III) treatment (Thorsen et al., 2006), we examined the effect of both As(III) and As(V) treatment on Fps1 closure. Hog1-catalyzed eviction of the glycerol channel regulator, Rgc2, from Fps1 occurs in response to hyperosmotic shock and is responsible for the closure of Fps1 (Lee et al., 2013). We found through coimmuneprecipitation (coIP) experiments that As(III) treatment similarly induced rapid eviction of Rgc2 from Fps1, whereas As(V) did not (Figure 2C). This makes sense from a physiological perspective, because As(V) does not enter the cell through Fps1. However, we do not currently have a mechanistic explanation for this differential behavior.
Intracellular activation of Hog1 by As(III)
Previous reports indicated that upstream signaling components of the HOG pathway are required for activation of Hog1 by As(III) and for tolerance to As(III) (Sotelo and Rodríguez-Gabriel, 2006; Thorsen et al., 2006), the implication being that Hog1 is activated through signal propagation from the cell surface. However, we made an observation that suggested that As(III) must enter the cell to activate Hog1. Because, as noted above, As(III) entry to yeast cells is mainly through Fps1, we assessed the ability of As(III) to activate Hog1 in either an fps1∆ mutant or an rgc1∆rgc2∆ mutant, which is impaired for Fps1 channel activity and displays tolerance to As(III) similar to that of an fps1∆ mutant (Beese et al., 2009). In these settings, As(III) failed to activate Hog1, whereas As(V), which enters through the phosphate transporters, was able to activate Hog1 (Figure 2D).
If As(III) activates Hog1 through an intracellular mechanism, rather than through activation of the canonical HOG pathway, it must do so in one of two ways. First, As(III) might activate another signaling pathway that stimulates the HOG pathway at a point below the cell surface osmosensors. However, this explanation seems unlikely because the HOG pathway is composed of two independent branches (from the Sln1 and Sho1 osmosensors) that feed into Pbs2, the MEK of the HOG pathway (Figure 3A), and both branches must be blocked to prevent activation of Hog1 by As(III) (Sotelo and Rodríguez-Gabriel, 2006), thus suggesting that As(III) stress would have separate inputs to both branches. The second possibility is that As(III) treatment might amplify basal signal that flows through the pathway to Hog1. In this case, upstream pathway components would be required to provide that basal signal, but would not be activated by As(III) treatment.
FIGURE 3:

As(III) stimulates Hog1 through an intracellular mechanism that acts below Pbs2. (A) The HOG pathway showing known inputs from the cell surface osmosensors. The protein tyrosine phosphatases Ptp2 and Ptp3 maintain Hog1 in an inactive state under nonstress conditions. (B) As(III) activates Hog1 in strains in which the MAPK is mutationally severed from its upstream activators, but in which the basal signal is restored below the blockage. Strains bearing deletions in all three MEKK genes of the HOG pathway (ssk2∆ ssk22∆ ste11∆; DL2344) or deletions in each of the upstream inputs (ssk1∆ sho1∆; DL4306) and plasmids expressing constitutively active forms of Pbs2 (pPBS2-DD), Ste11 (pSTE11-∆N) at low level under repressing control of the GAL1 promoter, or empty vector, were examined for activation of Hog1 after treatment with 1 mM As(III) or 3 mM As(V) for the indicated times. Strains were grown in standard SD medium to minimize expression levels of the constitutive protein kinases. Fold activation values measured from immunoblots are relative to the untreated samples and normalized to the Hog1 input. (C) As(III) treatment does not activate Pbs2. In vitro protein kinase assay for Pbs2-TAP using Hog1-His6 as substrate. Pbs2 was isolated from untreated wild-type (DL3187) cells (Control) or cells treated with 1 mM As(III), 3 mM As(V), or 1 M sorbitol for 5 min and tested by immunoblot assay for its ability to phosphorylate Hog1. Molecular mass markers (in kDa) are on the right.
We tested this possibility in two ways. First, we examined As(III) activation of Hog1 in a pair of HOG pathway mutants in which Hog1 had been mutationally severed from its upstream activators, but with basal signal restored by a constitutive pathway mutation. In one case, both pathway branches were blocked by deletion mutations in upper-pathway components (ssk1∆ sho1∆; Figure 3A). Basal signal to Hog1 was restored in this strain by low-level expression of a constitutive allele of STE11 (STE11-∆N; Cairns et al., 1992), which encodes one of three HOG pathway MEKKs. The other mutant strain carries deletion mutations in all three MEKK-encoding genes of the HOG pathway (ssk2∆ssk22∆ste11∆). In this strain, basal signal was restored below the MEKKs by low-level expression of a constitutive phosphomimetic form of PBS2 (PBS2-DD) (Wurgler-Murphy et al., 1997). Although high-level expression of constitutive STE11 or PBS2 alleles results in strong Hog1 activity and cell cycle arrest (Cairns et al., 1992; Wurgler-Murphy et al., 1997), low-level expression from the GAL1 promoter under repressing conditions (glucose) did not activate Hog1 and did not interfere with cell growth. We found that As(III) activated Hog1 in both mutant strains, but only when basal signal was restored to the deletion mutants (Figure 3B). This is in contrast to As(V), which failed to activate Hog1 appreciably in either of these strains, suggesting that As(V) activates Hog1 through a different pathway than As(III). These results demonstrate that activation of Hog1 in response to As(III) treatment is through an intracellular mechanism that impacts the HOG pathway below the level of Pbs2, yet requires a basal signal from upstream components.
The above results suggested a second test for intracellular activation of Hog1 by As(III) treatment. If As(III) activates Hog1 by amplification of the basal signal from Pbs2, rather than by active signaling of the HOG pathway through Pbs2, we should not detect Pbs2 activation by As(III) treatment. To test this, we used an in vitro protein kinase assay to detect activation of Pbs2 (Pbs2-TAP) immuneprecipitated (IP) from cells that were treated for 5 min with 1 mM As(III), 3 mM As(V), or 1M sorbitol as a positive control for HOG pathway activation by hyperosmotic shock. Inactive Hog1 (Hog1-His6), purified from yeast cells lacking the MEKKs of the HOG pathway (ssk2∆, ssk22∆, ste11∆), was used as a substrate. In contrast to the sorbitol treatment, which activated Pbs2, we detected no Pbs2 activation by the As(III) treatment (Figure 3C), supporting the conclusion that As(III) activates Hog1 by amplification of basal signal, rather than by HOG pathway kinase cascade activation. Similar to hyperosmotic shock, treatment with As(V) activated Pbs2, confirming the conclusion that the pathways to Hog1 activation by As(III) and As(V) are distinct.
These results suggested the possibility that As(III) activates Hog1 by inhibition of the protein phosphatases that maintain Hog1 in an inactive state in the absence of stress. Three phosphatases have been implicated in the regulation of Hog1. These are the Tyr-specific phosphatases Ptp2 and Ptp3 and the Ser/Thr phosphatase Ptc1 (Saito and Posas, 2012). Deletion of PTP2 and PTP3 together, or all three of these protein phosphatase–encoding genes, greatly elevated basal Hog1 phosphorylation, with no detectable difference between the double mutant and the triple mutant (Figure 4A). However, activation of Hog1 by As(III) was completely blocked in these mutants (Figure 4B). In contrast to this, hyperosmotic shock by treatment with 0.4 M NaCl activated Hog1 further in both the double and triple phosphatase deletion mutants, supporting the hypothesis that As(III) activates Hog1 through inhibition of its protein phosphatases rather than by signal activation through the protein kinase cascade. Moreover, the similar behavior of the ptp2Δptp3Δptc1Δ mutant to the ptp2Δptp3Δ mutant suggests that Ptc1 does not play a significant role in the activation of Hog1 in response to As(III) treatment.
FIGURE 4:
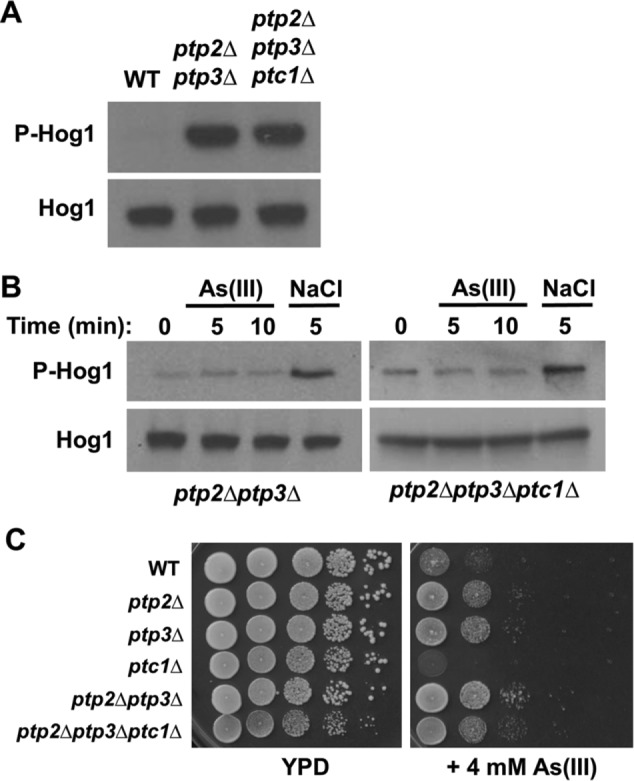
As(III) acts at the level of the Ptp2 and Ptp3 tyrosine phosphatases. (A) The basal phosphorylation of Hog1 is elevated strongly in the absence of the protein phosphatases that down-regulate it. Comparison of wild-type (DL2772), ptp2∆ ptp3∆ (DL4299), and a ptp2∆ ptp3∆ ptc1∆ mutant (DL4309) for Hog1 phosphorylation status. (B) As(III) fails to activate Hog1 in protein phosphatase mutants. The same mutant strains as above were tested for activation of Hog1 by 1 mM As(III) or 400 mM NaCl to induce hyperosmotic shock for the indicated times. (C) Mutations in PTP2 and PTP3 confer tolerance of As(III). Wild-type cells (DL2772), ptp2∆ (DL4295), ptp3∆ (DL4296), ptc1∆ (DL4297), ptp2∆ ptp3∆ (DL4299), and ptp2∆ ptp3∆ ptc1∆ (DL4309) were spotted onto YPD plates with or without 4 mM As(III) at serial 10-fold dilutions (from left to right). Plates were incubated at 30°C for 2 d.
If As(III) activates Hog1 by inhibiting the tyrosine-specific phosphatases that normally maintain it in a silent state, mutants in these genes might display increased tolerance to As(III) by preadaptation to As(III) stress. Therefore, we examined the effect of deletion mutations of the three phosphatase genes, individually and in combination, on As(III) tolerance. We found that both the ptp2∆ and ptp3∆ mutations conferred modest tolerance to As(III) and that a double ptp2∆ptp3∆ mutant displayed further tolerance (Figure 4C). However, the ptc1∆ mutation caused As(III) sensitivity, which may reflect a multitude of other functions attributed to this phosphatase, particularly on Mkk1, a MEK of the cell wall integrity signaling pathway (Tatjer et al., 2016).
A methylated metabolite of As(III), MAs(III), inhibits the Hog1 tyrosine phosphatases
As(III) is methylated in mammals to the more toxic metabolites methyl arsenite [MAs(III)] and dimethyl arsenite [DMAs(III); Cullen, 2014; Dong et al., 2015]. Additionally, it has been reported that, in contrast to As(III), these methylated metabolites are capable of inhibiting the mammalian tyrosine-specific phosphatases PTPB1 and CD45 by reaction with active-site cysteine residues (Rehman et al., 2012). The results presented to this point suggest that As(III) activates Hog1, directly or indirectly, through inhibition of the tyrosine phosphatases that normally maintain the SAPK in a low-activity state. Therefore, we tested the ability of As(III) and MAs(III) to inhibit the yeast tyrosine phosphatase Ptp3. GST-Ptp3 was isolated from yeast cells and used in an in vitro phosphatase assay with activated Hog1-His6 isolated from hyperosmotically shocked yeast cells. In the absence of inhibitor, dephosphorylation of Hog1 was nearly complete after treatment with Ptp3 for 1 h (Figure 5A). In contrast to this, a catalytically inactive form of Ptp3 (GST-Ptp3-C804A; Jacoby et al., 1997), isolated in parallel with GST-Ptp3, displayed very little phosphatase activity against phospho-Hog1 after treatment for 1 h (Figure 5B), indicating that the observed activity is intrinsic to GST-Ptp3. Under these conditions, MAs(III) completely inhibited Ptp3 activity at 10 μM, whereas As(III) failed to inhibit Ptp3 even at 100 μM (Figure 5C), supporting the conclusion that As(III) methylation is essential for its inhibition of the Ptps and consequent activation of Hog1. Additional inhibition experiments revealed that MAs(III) inhibits Ptp3 in the low micromolar range (Figure 5D).
FIGURE 5:

MAs(III) is the active metabolite of As(III) for inhibition of tyrosine phosphatase Ptp3 and consequent activation of Hog1. (A) In vitro protein phosphatase assay for GST-Ptp3 using phospho-Hog1-His6 as substrate showing time course of phospho-Hog1 dephosphorylation by Ptp3. GST-Ptp3 was isolated from wild-type cells (DL3187) and incubated for the indicated times with purified Hog1-His6 that had been activated by hyperosmotic shock in wild-type cells (DL3187). Values given were measured from the immunoblots and represent the fraction of phosphorylated Hog1 remaining normalized to the Hog1-His6 input. (B) Catalytically inactive Ptp3 does not dephosphorylate phospho-Hog1. Equivalent amounts of GST-Ptp3 or GST-Ptp3-C804A isolated from a ptp3Δ strain (DL4296) were tested for phosphatase activity by incubation for 1 h with phospho-Hog1-His6, as above. (C) MAs(III), but not As(III), is a potent inhibitor of tyrosine phosphatase Ptp3. The same conditions as in A were used in a protein phosphatase inhibition experiment. GST-Ptp3 was incubated for 1 h with phospho-Hog1-His6 in the presence of the indicated concentrations of As(III) or MAs(III). (D) Inhibition of Ptp3 by MAs(III). The proteins in C were used in inhibition assays testing the indicated concentrations of MA(III). Note the nearly complete inhibition of phosphatase activity by 5 µM MAs(III).
The yeast SAM-dependent methyltransferase Mtq2, which is responsible for methylation of the translation release factor eRF1 (Heurgue-Hamard et al., 2005; Poleveda et al., 2006), is a candidate to catalyze the methylation of As(III) in this species. An mtq2∆ mutant displayed increased tolerance to As(III), but not to its more toxic metabolite MAs(III) (Ren et al., 2011; Figure 6A), suggesting that it might be responsible for conversion of As(III) to MAs(III). If this is the case, we would expect Mtq2 to be required for activation of Hog1 by As(III). We explored this possibility by first testing the ability of As(III) and MAs(III) to activate Hog1 in an mtq2∆ mutant. Although basal Hog1 activity was elevated slightly in this mutant, we detected no further increase in Hog1 activation by As(III) treatment (Figure 6B). In contrast to this, we detected Hog1 activation by MAs(III) treatment both in wild-type cells and in the mtq2∆ mutant, consistent with the dual conclusions that As(III) must be converted to MAs(III) for Hog1 activation and that Mtq2 is likely responsible for this conversion. Additionally, the mtq2∆ mutation did not affect the ability of As(V) to stimulate Hog1 (Figure 6C), providing further confirmation that As(V) activates Hog1 through a different pathway from that of As(III).
FIGURE 6:
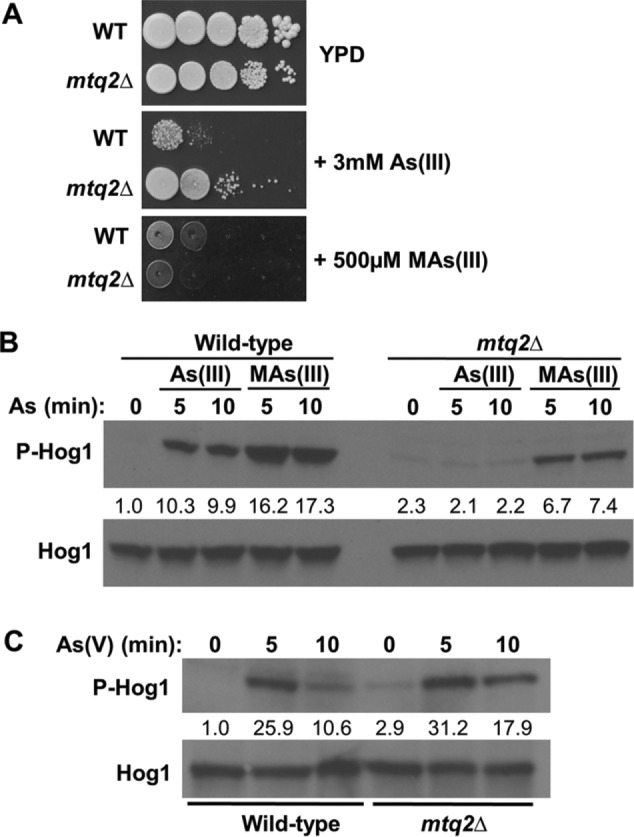
The methyltransferase encoded by MTQ2 is required for activation of Hog1 by As(III), but not by As(V). (A) An mtq2Δ mutation confers tolerance of As(III), but not of MAs(III). Wild-type (DL3187) cells and an mtq2Δ mutant (DL4313) were spotted at serial 10-fold dilutions (left to right) onto YPD plates with 3 mM As(III) or 500 µM MAs(III) and incubated at 30°C for 2 d. (B) An mtq2Δ mutation blocks Hog1 activation by As(III), but not by MAs(III). The same strains as above were treated with 1 mM As(III) or 500 µM MAs(III) for the indicated times and tested for Hog1 phosphorylation. Fold activation values measured from the immunoblot are relative to the untreated wild-type control and normalized to Hog1 input. Note that the mtq2Δ mutant displays slightly higher basal Hog1 phosphorylation than the wild type. (C) An mtq2Δ mutation fails to block Hog1 activation in response to As(V) treatment. The same strains were tested for Hog1 phosphorylation in response to 3 mM As(V) for the indicated times. Fold activation values were determined as above.
As a final test of the hypothesis that Mtq2 converts As(III) to MAs(III), we measured SAM-dependent methyltransferase activity of Mtq2 using As(III) as substrate. The active eRF1 methyltransferase is a heterodimer of Mtq2 with zinc-finger protein Trm112 (Heurgue-Hamard et al., 2006). Affinity-purified preparations of overexpressed Mtq2 from yeast without coexpression of Trm112 yield only weak methyltransferase activity against eRF1 (Heurgue-Hamard et al., 2005). Therefore, we coexpressed C-terminally TAP-tagged Mtq2 with Trm112 in yeast and isolated Mtq2-TAP by affinity purification. Overexpressed Mtq2-TAP in the absence of coexpressed Trm112 served as a control for this experiment. We detected a low level of As(III) methyltransferase activity from affinity-purified Mtq2-TAP alone, which was increased ∼15-fold for comparable amounts of Mtq2-TAP isolated from cells that also overexpressed Trm112 (Figure 7A). The detected Mtq2:Trm112 methyltransferase activity was SAM-dependent and additionally capable of using MAs(III) as a substrate (Figure 7B). Taken together, these results demonstrate that As(III) is converted to MAs(III) by Mtq2:Trm1 and that MAs(III) is the metabolite responsible for activation of Hog1 through inhibition of its tyrosine-specific phosphatases.
FIGURE 7:

Mtq2 is a SAM-dependent As(III) and MAs(III) methyltransferase. (A) SAM-dependent methyltransferase activity of Mtq2:Trm112 using As(III) as substrate was measured in a coupled luminescence assay (see Materials and Methods). Mtq2 was affinity-purified from cells overexpressing Mtq2-TAP alone or coexpressing Trm112. Inset shows an immunoblot of affinity-purified Mtq2-TAP from the two samples. Output is in relative luminscence units (RLU). (B) Mtq2:Trm112 methyltransferase activity against As(III) and MAs(III) is SAM-dependent. The same preparations as in A were used to assess substrate specificity of Mtq2:Trm112. Each data point represents the mean and SD for three samples.
DISCUSSION
There is a tendency to view SAPK signaling pathways through the same lens as growth factor signaling pathways in which signals are initiated from the cell surface. With the large and growing list of diverse stressors that activate a relatively small number of SAPKs, it seems increasingly likely that many of these stressors activate signaling through intracellular mechanisms, rather than by top-down signaling through their canonical signaling pathways. This is particularly true for the yeast S. cerevisiae, which possesses only two SAPK activation pathways, both of which are well characterized from their plasma membrane sensors to their kinase cascades and transcription factors. Yet both of these SAPKs are activated by a wide range of seemingly unrelated stressors. In this study, we found that trivalent inorganic arsenic, As(III), must enter yeast cells to stimulate the SAPK Hog1, which led us to investigate the intracellular signaling pathway responsible for Hog1 activation by this toxic metalloid. We demonstrated that, in contrast to hyperosmotic stress, which signals to Hog1 by stimulation of cell surface osmosensors connected to the HOG pathway kinase cascade, As(III) treatment activates Hog1 through inhibition of the protein tyrosine phosphatases Ptp2 and Ptp3, which normally maintain Hog1 in a low-activity state. Previous reports that indicated a requirement for HOG pathway components upstream of Hog1 for its activation by As(III) (Sotelo and Rodríguez-Gabriel, 2006; Thorsen et al., 2006) can be explained by our finding that basal signal through the HOG pathway, rather than activation of upstream components, is necessary for Hog1 activation by this stressor. We found that Hog1 that was mutationally severed from the signaling components known to act upstream of the SAPK could still be activated in response to As(III) treatment if a basal level of protein kinase activity was restored to its activating MEK, Pbs2, by phosphomimetic mutations. We demonstrated that this basal signal is amplified in response to As(III) treatment through the inhibition of Ptp2 and Ptp3. The further demonstration that As(III) treatment failed to activate wild-type Pbs2 supports the conclusion that Hog1 is activated in response to As(III) without stimulation of its immediately upstream protein kinase.
MAs(III) is the As(III) metabolite responsible for activation of Hog1
As(III) is metabolically activated by mammalian methyltransferases to the more toxic forms MAs(III) and DMAs(III) (Cullen 2014; Dong et al., 2015). Intriguingly, these metabolites, but not As(III), are potent inhibitors of the mammalian tyrosine-specific phosphatases PTP1 and CD45 (Rehman et al., 2012). On the basis of a single report by Ren et al. (2011), we suspected that As(III) was similarly metabolized in yeast. These authors demonstrated that deletion of yeast MTQ2, which encodes a SAM-dependent methyltransferase required for the methylation of translation release factor eRF1 (Heurgue-Hamard et al., 2005; Poleveda et al., 2006), confers resistance to As(III), but not to MAs(III), suggesting the possibility that Mtq2 converts As(III) to MAs(III). They showed additionally that overexpression of a mammalian orthologue of MTQ2, N-6 adenine-specific DNA methyltransferase (N6AMT1), in human urothelial cells catalyzed the secondary methylation of MAs(III) to DMAs(III). However, three lines of evidence support our conclusion that yeast Mtq2 is required for the conversion of As(III) to MAs(III), which is responsible for activation of Hog1 via Ptp inhibition. First, we found that Hog1 activation by As(III) was blocked in an mtq2∆ mutant and that the block was bypassed by MAs(III) treatment, suggesting that metabolic conversion of As(III) to MAs(III) is blocked in this mutant. Second, we found that although As(III) failed to inhibit Ptp3 in vitro at concentrations as high as 100 µM, MAs(III) was a potent inhibitor of this phosphatase at low micromolar concentrations, supporting the conclusion that As(III) must be converted to MAs(III) for the inhibition of Ptp2 and Ptp3 and the consequent activation of Hog1. The basis for the inhibitory activity of MAs(III) versus As(III) is not clear, but may reflect an ability of the methylated arsenical to access the active site cysteines of these enzymes. Finally, we demonstrated in vitro that the dimeric Mtq2:Trm112 methyltransferase was capable of SAM-dependent methyltransferase activity using As(III) as a substrate. We also detected methyltransferase activity for this enzyme using MAs(III) as substrate, consistent with the observed activity of its mammalian ortholog. However, we regard MAs(III), rather than DMAs(III), as the critical metabolite required for activation of Hog1 because MTQ2 was not required for Hog1 activation by MAs(III) (Figure 6B). Thus, As(III) enters the cell through Fps1 and is metabolized by Mtq2:Trm112 to MAs(III), which inhibits Ptp2 and Ptp3, changing the balance of phosphorylated versus unphosphorylated Hog1 to favor the accumulation of active Hog1. This scheme is set out in Figure 8.
FIGURE 8:
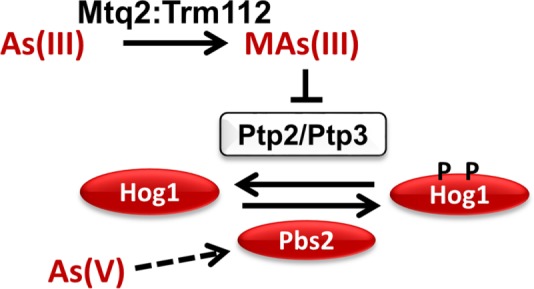
Model for the pathway by which As(III) activates Hog1. As(III) is metabolized by the methyltransferase Mtq2:Trm112 to MAs(III), which inhibits the Ptp2 and Ptp3 tyrosine phosphatases that modulate the phosphorylation state of Hog1. This results in activation of Hog1 by shifting the balance toward the phosphorylated state. As(V) activates Hog1 through a different pathway that involves the activation of Pbs2 (dashed arrow).
This activation mechanism suggests the possibility that other Hog1-activating stressors may also act intracellularly at different points along the pathway. This would provide an explanation as to how so many distinct stressors activate Hog1, which is known to be connected only to the cell surface osmosensors of the HOG pathway. Detailed examination of the pathways by which other stressors activate Hog1 may reveal a multitude of intracellular inputs to this SAPK. Along these lines, we found that Hog1 is also activated in response to As(V) treatment, but that this activation is through a separate pathway from that described for As(III). Three lines of evidence support this conclusion. First, Hog1 stimulation by As(V) did not require its reduction to As(III) by the arsenate reductase Acr2. Nor did it require conversion to MAs(III) by Mtq2, which argues against the involvement of an unknown arsenate reductase. Second, As(V) failed to activate Hog1 in either of two engineered strains in which Hog1 responded to As(III) without a regulated signal from upstream pathway components, suggesting that the As(V) signal either emanates from the cell surface or enters the HOG pathway through an intracellular input upstream of the HOG pathway MEKKs. Third, unlike As(III), As(V) treatment stimulated Pbs2 protein kinase activity toward Hog1 as measured in vitro, supporting the conclusion that As(V) treatment activates signaling of at least some components of the HOG pathway that function above Hog1.
Hog1 activated by different stresses drives divergent outputs
In this study, we considered the behavior of Hog1 activated by three different stresses: hyperosmotic shock, As(III), and As(V). When Hog1 is activated in response to hyperosmotic shock, it translocates rapidly to the nucleus and drives transcription of genes important for osmoregulation (Saito and Posas, 2012). Among these are GPD1 and GPP2, which are important for glycerol biosynthesis in response to hyperosmotic stress. In contrast to this, Hog1 activated by As(III) does not translocate to the nucleus (Thorsen et al., 2006) and does not drive GPD1 expression (Sotelo and Rodríguez-Gabriel, 2006). However, Hog1 activated by As(III) contributes to arsenic-responsive transcription by Acr1 through an unknown mechanism (Sotelo and Rodríguez-Gabriel, 2006). Our finding that As(V) also activates Hog1 provides another example of a stress-specific fate for the SAPK. Similarly to activation of Hog1 by As(III), As(V) treatment induced arsenic-responsive transcription, but did not induce nuclear translocation of Hog1 or transcriptional induction of GPD1. However, in contrast to As(III) treatment, Hog1 did not impact the arsenic-responsive transcription induced by As(V) treatment.
Although the bulk of Hog1 translocates to the nucleus in response to hyperosmotic shock, some is recruited to the plasma membrane glycerol channel Fps1, where it phosphorylates and evicts the positive regulators Rgc1 and Rgc2 from the Fps1 C-terminus to induce channel closure and allow glycerol accumulation (Lee et al., 2013). Fps1 is also a key portal for entry of As(III) into yeast cells and Hog1 is important for the closure of this channel in response to As(III) treatment as an adaptation by which the cell restricts entry of As(III) (Thorsen et al., 2006; Lee et al., 2013). We found that As(III) treatment, similarly to hyperosmotic shock, induces the eviction of Rgc2 from Fps1. The differential output of Hog1 activated by these stresses is critical to cell survival. As noted above, in response to hyperosmotic shock, Hog1 induces glycerol biosynthesis concomitant with Fps1 closure to cause a rapid increase in intracellular osmolarity. When As(III) stress is considered, this combined action of Hog1 in the absence of high external osmolarity would cause elevated turgor and consequent cell wall stress (Beese et al., 2009). Thus, As(III) stress mobilizes a Hog1 response that selectively closes Fps1 to block As(III) entry without activating a full hyperosmotic stress response.
Another point of divergence between the responses to As(III) and As(V) is that As(V) treatment did not drive Rgc2 eviction from Fps1 despite its ability to activate Hog1. This differential behavior makes sense, considering that As(V) does not enter the cell through Fps1, but it leaves open two important questions with regard to As(V) activation of Hog1. First, what is the function of Hog1 activated by As(V) if it does not regulate either arsenic-protective gene expression or the activity of Fps1? One possibility is that Hog1 regulates As(V) entry though the phosphate transporters, although we are unaware of evidence to support this idea. Second, how is Rgc2 protected from eviction by Hog1 that has been activated by As(V)? We found that Hog1 activation occurs equally rapidly in response to As(III) and As(V) treatment (within 5 min; Figure 1), implying that Hog1 activation kinetics is not responsible for its divergent behavior in response to these stressors. Nor can the relative levels of Hog1 activation by these agents explain the inability of As(V) to drive Rgc2 eviction from Fps1, as it induced stronger Hog1 activation than As(III).
Perhaps additional, as yet unknown, intracellular targets of As(III) or MAs(III) contribute to the differential regulation of Fps1 in response to trivalent versus pentavalent arsenic. For example, trivalent arsenic species are highly reactive with Cys residues in proteins (Shen et al., 2013), suggesting the potential for selective regulation of Hog1 output through As(III) or MAs(III) modification of Hog1 targets, or proteins responsible for its nucleocytoplasmic shuttling. Indeed, As(III) was shown recently to bind directly to the arsenic-responsive transcription factor, Acr1, and to induce a conformational change that activates transcription (Kumar et al., 2016). Therefore, the activated SAPK may cooperate with factors modified in a stress-specific manner to generate specific responses.
A second factor likely to be important in dictating the stress-specific output of Hog1 is the specific mechanism of its activation. Different activating inputs might result in different protein–protein interactions. Active Hog1 that forms different complexes depending upon its mechanism of activation may be directed to different targets. For example, Hog1 that is activated through inhibition of the Ptp phosphatases, rather than by activation of its MEK Pbs2, may engage in different interactions not only with Pbs2, which functions as a scaffold for the HOG pathway (Posas and Saito, 1997; Murakami et al., 2008), but with other pathway components, as well. It is perhaps significant in this regard that we have found that As(III) treatment induces dissociation of Ptp3 from Hog1, a finding that contrasts with their continued association after hyperosmotic shock (unpublished data). In any case, how the variety of stress signal input mechanisms contribute to the specificity of SAPK output is an important question and may be one key to understanding the full range of SAPK signaling in yeast and in humans.
MATERIALS AND METHODS
Strains, growth conditions, transformations, and gene deletions
The S. cerevisiae strains used in this study were all derived from Research Genetics background S288c and are listed in Table 1. Yeast cultures were grown in YPD (1% Bacto yeast extract, 2% Bacto Peptone, 2% glucose) or minimal selective medium, SDM (0.67% yeast nitrogen base, 2% glucose), supplemented with the appropriate nutrients to select for plasmids. Yeast cultures were transformed according to Gietz et al. (1995).
TABLE 1:
Yeast strains.
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| DL2344 | MATa S288c ssk2∆::leu2 ssk22∆::leu2 ste11∆::his3 | Posas and Saito, 1997 |
| DL2772 | MATα S288c (BY4742) his3∆ leu2∆ ura3∆ lys2∆ | Research Genetics |
| DL3158 | MATa S288c hog1∆::KanMX | Research Genetics |
| DL3187 | MATa S288c (BY4741) his3∆ leu2∆ ura3∆ lys2∆ | Research Genetics |
| DL3207 | MATa S288c rgc1∆::KanMX rgc2∆::KanMX | Beese et al., 2009 |
| DL3226 | MATa S288c fps1∆::KanMX | Research Genetics |
| DL4295 | MATα S288c ptp2∆::KanMX | Research Genetics |
| DL4296 | MATα S288c ptp3∆::KanMX | Research Genetics |
| DL4297 | MATα S288c ptc1∆::KanMX | Research Genetics |
| DL4299 | MATα S288c ptp2∆::KanMX ptp3∆::HPHMX4 | This study |
| DL4304 | MATa S288c ssk1∆::KanMX | This study |
| DL4306 | MATa S288c ssk1∆::KanMX sho1∆::HPHMX4 | This study |
| DL4309 | MATα S288c ptp2∆::KanMX ptp3∆::HPHMX4 ptc1∆::HIS3 | This study |
| DL4313 | MATa S288c mtq2∆::HPHMX4 | This study |
| DL4341 | MATa S288c acr2∆::HPHMX4 | This study |
Chromosomal deletion of the single ACR2, MTQ2, and SSK1 genes was carried out by homologous recombination. The hygromycin-resistance gene hphMX4 from pAG32 (Goldstein and McCusker, 1999) or the kanamycin-resistance gene KanMX from pFA6a-KanMX (Longtine et al., 1998) was amplified by high-fidelity PCR (Phusion; ThermoFisher) using primers containing the upstream region (40 base pairs immediately before the starting ATG) and downstream region (40 base pairs immediately after the stop codon) of the target gene. The PCR products were integrated into the genome of the wild-type strain by homologous recombination. Integrates were selected on plates containing hygromycin B, or G418, yielding acr2∆::HPHMX4 (DL4341), mtq2::HPHMX4 (DL4313) or ssk1∆::KanMX (DL4304). Deletion of SHO1 in the ssk1∆ background was generated using same method, resulting in ssk1∆::KanMX sho1∆::HPHMX4 (DL4306).
Deletion of PTP3 in the ptp2∆::KanMX strain (DL4295) was generated using the hphMX4 marker (ptp2∆::KanMX ptp3∆::HPHMX4; DL4299). Additional deletion of PTC1 in DL4299 was generated using the HIS3 as a selectable marker. The HIS3 gene from pFA6a-His3MX6 was amplified by PCR, as above. Integration into the genome of DL4299 was selected for on the plates without histidine, yielding ptp2∆::KanMX ptp3∆::HPHMX4 ptc1∆::HIS3 (DL4309). All gene replacements were validated by PCR analysis across both integration junctions.
Chemicals
Sodium arsenite (NaAsO2) and sodium arsenate (Na2HAsO4) were purchased from Sigma, and methyl arsenite (CH5AsO2) was purchased from ChemCruz.
Plasmid construction
The HOG1 gene was epitope-tagged on its C-terminus with the 3xHA epitope and expressed under its natural promoter for detection of Hog1 in Figure 1. The promoter region of HOG1 (from position -489) and the entire HOG1 gene without the stop codon were amplified from genomic yeast DNA by high-fidelity PCR (Phusion) using the primers designed with a XhoI site (upstream) and with a NotI site (downstream) and cloned into pRS426-3HA-ADH1T (p3150) to yield pRS426-HOG1-3HA (p3225). For subsequent experiments, endogenous Hog1 was detected using α-Hog1 antibodies.
For in vitro protein kinase assays, HOG1 was tagged at its C-terminus with 6xHis (His6) and expressed under the control of the MET25 promoter. The HOG1 coding sequence was amplified by high-fidelity PCR from genomic yeast DNA using a forward primer that contained an XbaI site immediately before the start codon and a reverse primer that introduced a His6 tag followed by a stop codon and an XhoI site. This fragment was digested with Xba1 and Xho1 and inserted into pUT36 (p2415) to yield pUT36-MET25P-HOG1-HIS6 (p3454).
For in vitro protein phosphatase assays, a catalytically inactive form of Ptp3 (Ptp3-C804A) was produced from parental plasmid GAL1P-GST-PTP3 (p3402) using Quick Change mutagenesis (Agilent Technologies) to yield GAL1P-GST-ptp3-C804A (p3464).
For in vitro methylation assays, TRM112 was overexpressed under the control of the MET25 promoter. The TRM112 coding sequence was amplified by high-fidelity PCR from genomic yeast DNA using a forward primer that contained an XbaI site immediately before the start codon and a reverse primer that introduced a stop codon and a SmaI site. This fragment was digested with Xba1 and Sma1 and inserted into pYEP181-MET25P-FPS1-Myc (p3121), from which FPS1-Myc was removed by digestion with the same enzymes, yielding pYEP181-MET25P-TRM112 (p3460). The plasmids used in this study are listed in Table 2.
TABLE 2:
Plasmids.
| Plasmid | Description | Source or reference |
|---|---|---|
| p115 | pRS316 | Sikorski and Hieter, 1989 |
| p1106 | pRS426 | Sikorski and Hieter, 1989 |
| p1230 | pYES2-GAL1P-STE11-N∆ | Posas and Saito, 1997 |
| p2415 | pUT36 | Millson et al., 2005 |
| p2501 | pUT36-MET25P-RGC2-HIS6 | Beese et al., 2009 |
| p2823 | pAG32 | Goldstein and McCusker, 1999 |
| p3121 | pYEP181-MET25P-FPS1-Myc | Lee et al., 2013 |
| p3147 | pRS315-3HA-ADH1T | Lee et al., 2013 |
| p3150 | pRS426-3HA-ADH1T | Lee et al., 2013 |
| p3151 | pRS316-MET25P-RGC2-3HA | Lee et al., 2013 |
| p3177 | pRS416-HOG1-GFP | Ferrigno et al., 1998 |
| p3225 | pRS426-HOG1-3HA | This study |
| p3261 | pFA6a-KanMX | Longtine et al., 1998 |
| p3263 | pFA6a-His3MX | Longtine et al., 1998 |
| p3370 | pYES2-GAL1P-PBS2DD | Wurgler-Murphy et al., 1997 |
| p3396 | GAL1P-PBS2-TAP (6HIS-HA-ZZ) | Open Biosystems |
| p3402 | GAL1P-GST-PTP3 | Open Biosystems |
| p3454 | pUT36-MET25P-HOG1-HIS6 | This study |
| p3459 | GAL1P-MTQ2-TAP (6HIS-HA-ZZ) | Open Biosystems |
| p3460 | pYEP181-MET25P-TRM112 | This study |
| p3464 | GAL1P-GST-PTP3-C804A | This study |
Protein extraction
Protein extraction was carried out as described previously (Kamada et al., 1995) for coIP or using the rapid boiling method (Kushnirov, 2000) for direct immunoblot experiments.
Cultures for coIP experiments with Rgc2-HA and Fps1-Myc were grown to mid–log phase in selective medium and starved for methionine for 2 h to induce expression of Rgc2 and Fps1 under the control of the conditional MET25 promoter. Cultures were treated with 1 mM As(III) or 3 mM As(V) for up to 60 min. Protein extraction and coIP were carried out as described previously (Lee et al., 2013). Extracts (100 µg of protein) were incubated with mouse monoclonal α-Myc antibody (1 µg, 9E10; Pierce), for 1 h at 4°C and precipitated with protein A affinity beads for 1 h at 4°C. Samples were washed with IP buffer three times and boiled in SDS–PAGE buffer.
SDS–PAGE and immunoblot analysis
Proteins were separated by SDS–PAGE (7.5% gels) followed by immunoblot analysis using mouse monoclonal α-Myc antibody (9E10; Santa Cruz), α-HA (16B12; Covance), α-GST (B-14; Santa Cruz), α-tetra His (Qiagen), or goat polyclonal α-Hog1 (yC-20; Santa Cruz) at a dilution of 1:10,000. Rabbit polyclonal α-phospho-p38 (T180/Y182, Cell Signaling) was used at a dilution of 1:2000 to detect phosphorylated Hog1. Secondary goat anti-mouse (Amersham) and donkey anti-goat (Santa Cruz) antibodies were used at a dilution of 1:10,000. Secondary donkey anti-rabbit (Amersham) was used at a dilution of 1:2000. All results involving immunoblot analyses were replicated at least once, and representative blots are shown.
Quantitative real-time PCR
Quantitative real-time PCR was conducted to quantify the levels of mRNA for GPD1 or ACR3 under arsenic stress or hyperosmotic shock. Wild-type cells were treated with 1 mM As(III), 3 mM As(V), or 1 M sorbitol for 5 min. Whole-cell mRNA was prepared with a NucleoSpin RNA kit (Macherey-Nagel), as directed by the manufacturer. Complementary DNA was generated using the Superscript III (Invitrogen) first-strand synthesis system for RT-PCR. Real-time PCR was performed using a Bio-Rad CFX96 Real-Time system with Bio-Rad SYBR green mix. The data from biological triplicates were plotted using Microsoft Excel.
In vitro protein kinase assay
An in vitro protein kinase assay with Pbs2 was performed using Hog1 as substrate. Pbs2 was prepared from a wild-type strain (DL3187) containing a plasmid expressing C-terminally TAP-tagged Pbs2 under the control of the GAL1 promoter (p3396). Cells were grown in selective medium containing 2% glucose and transferred to selective medium containing 2% raffinose and grown to mid–log phase. Galactose was added to these cultures at a final concentration of 2% to induce expression of Pbs2-TAP for 2 h. Cultures of 10 ml were then stressed with 1 mM As(III), 3 mM As(V), or 1 M sorbitol for 5 min, collected, and lysed with 20 mM HEPES buffer (pH 7.5) containing 0.5% Triton, protease inhibitors, and phosphatase inhibitors. The lysates (500 µl) were incubated with immunoglobulin G–sepharose 6 Fast Flow (50 µl; GE Healthcare) for 1 h at 4°C. The beads with Pbs2-TAP were washed with IP buffer three times and used for protein kinase assays.
The unphosphorylated form of Hog1 was prepared from a strain deleted for all of the HOG pathway MEKKs, ste11∆ssk2∆ssk22∆ (DL2344), which does not activate Hog1. His-tagged Hog1 was expressed in this strain from pUT36-MET25P-HOG1-HIS6 (p3454), which was grown in 50 ml of culture to mid–log phase in selective medium, starved for methionine to induce expression of Hog1, collected and lysed using 20 mM HEPES buffer containing 0.5% triton and protease inhibitors. The protein extract (1.5 ml) was incubated with HisPur Ni-NTA Superflow Agarose (200 µl; Thermo Scientific). The resin was washed with 20 mM HEPES binding buffer three times and the unphosphorylated form of Hog1-His6 was eluted with 250 mM imidazole. The Hog1-His6 eluate was dialyzed by Slide-A-Lyzer MINI Dialysis Device 10k MWCO (Thermo Scientific) against 50 mM Tris-HCl to a final volume of 1 ml.
Kinase reactions contained dialyzed Hog1-His6 eluate (100 µl) in protein kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 2 mM dithiothreitol [DTT]) with phosphatase inhibitors (PhosSTOP; Roche) and 10 µl of Pbs2-Tap resin. Reactions were initiated with ATP at a final concentration of 300 µM. Reaction mixtures were incubated at 30°C for 20 min and were stopped by adding SDS–PAGE buffer, followed by boiling for 5 min. Immunoblot analysis was performed as described above.
In vitro protein phosphatase assay
An in vitro protein phosphatase assay with Ptp3 was performed using phosphorylated Hog1 as substrate to test for inhibition of Ptp3 by As(III) and MAs(III). GST-Ptp3 was isolated from wild-type cells (DL3187) expressing GST-Ptp3 from a plasmid under the control of the GAL1 promoter (p3402). Cells were grown in selective medium containing 2% glucose and transferred to selective medium containing 2% raffinose for further growth to mid–log phase. Galactose was added to 100 ml of culture at a final concentration of 2% to induce expression of GST-Ptp3 for 2 h. Cells were lysed in extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton) containing protease inhibitors and the extract (1.5 ml) was incubated with glutathione-superflow resin (200 µl; Clontech) for 1 h at 4°C. The resin was washed three times with binding buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl). GST-Ptp3 was eluted with 20 mM reduced glutathione and dialyzed by Slide-A-Lyzer MINI Dialysis 10k MWCO against a phosphatase reaction buffer (25 mM imidazole, pH 7.5, 2.5 mM ETDA, 50 mM NaCl, 5 mM DTT) to a final volume of 1 ml.
The phosphorylated form of Hog1-His6 was prepared from wild-type cells (DL3187) after hyperosmotic shock. A 100-ml culture of cells containing pUT36-MET25P-HOG1-HIS6 (p3454) was grown to mid–log phase and osmotically shocked with 0.5 M NaCl. Cells were collected and lysed in extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton) containing protease inhibitors and phosphatase inhibitors. The extract (1.5 ml) was incubated with HisPur Ni-NTA Superflow Agarose (200 µl; Thermo Scientific). The resin was washed with binding buffer three times and Hog1-His6 was eluted with 250 mM imidazole buffer. Eluate was dialyzed as above against phosphatase reaction buffer and protease inhibitors were added to dialyzed eluates to a final volume of 1 ml. As(III) or MAs(III) was added to affinity purified GST-Ptp3 in reaction buffer and purified phosphorylated Hog1-His6 was added to initiate the reaction. Phosphatase reactions were initiated by mixing 40 µl GST-Ptp3 and 40 µl Hog1-His6 and were terminated by addition of SDS–PAGE buffer followed by boiling for 5 min. Immunoblot analysis was performed as described above.
In vitro arsenite methyltransferase assay
An in vitro arsenite methylation assay was performed using Mtq2 overexpressed alone in yeast or Mtq2 cooverepxressed with Trm112. Cultures were prepared from a wild-type strain (DL3187) bearing a plasmid that expresses C-terminally TAP-tagged (6HIS-HA-ZZ) Mtq2 under the control of the GAL1 promoter (p3459), with or without a second plasmid that expresses untagged Trm112 under the control of the MET25 promoter (p3460). Cells were grown in selective medium containing 2% glucose and transferred to selective medium containing 2% raffinose for growth to mid–log phase. Cultures (25 ml) were transferred to selective medium without methionine (to induce Trm112 expression) and with 2% galactose (to induce Mtq2-TAP expression) for 3 h. Cells were then collected and lysed with 20 mM HEPES buffer (pH 7.5) containing 0.5% Triton, protease inhibitors, and phosphatase inhibitors. The protein extract (1 ml) was precleared with agarose resin (200 µl; Thermo Scientific) for 1 h at 4°C and incubated with HisPur Ni-NTA Superflow Agarose (200 µl; Thermo Scientific) for 1 h at 4°C. The resin was washed three times with 20 mM HEPES (pH 7.5) buffer and Mtq2-TAP was eluted with 1 ml of 250 mM imidazole. The Mtq2-TAP eluate was dialyzed by Spin-X 5k MWCO (Corning) against 50 mM Tris-HCl and concentrated to a volume of ∼100 µl. Protein concentrations were measured by DC Protein Assay (Bio-Rad). Approximately 300–500 ng of total protein, corresponding to equivalent amounts of Mtq2-TAP based on immunoblot analysis, was used for in vitro methylation assays comparing Mtq2 alone or Mtq2 with copurified Trm112 with 100 µM As(III) or MAs(III) as substrate. Arsenite methylation assays were performed using the MTase-Glo Methyltransferase assay kit (Promega). The basis of this assay is the conversion of methyl donor S-adenosyl methionine (SAM) to S-adenosyl homocysteine (SAH) through the action of a methyltransferase. Coupled reactions converted SAH to ATP, which was detected by a luciferase reaction. Luminescence from triplicate samples was measured by a Microplate Luminometer (Veritas).
Florescence microscopic detection of Hog1
Haploid wild-type strain (DL3187) was transformed with plasmid pRS416-HOG1-GFP (p3177). Transformants were grown in selective medium and visualized with a Zeiss Axio Observer Z1 fitted with a GFP filter using a 100× objective. For detection of Hog1 nuclear localization after stress, 1 mM As(III), 3 mM As(V), or 1 M sorbitol was added to the cells immediately before live-cell imaging.
Notes on reproducibility
All protein kinase, protein phosphatase, and arsenite methyltransferase assays were reproduced at least once in independent experiments, representatives of which are shown. Similarly, all immunoblots and coIPs were reproduced at least once in independent experiments with representative images shown. Quantitation of signals from immunoblots was done using ImageJ software.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01 GM48533) to D.E.L. We thank Francesc Posas for plasmids.
Abbreviations used:
- As(III)
arsenite
- As(V)
arsenate
- AS3MT
SAM methyltransferase
- ABC
ATP-binding cassette
- ATP
adenosine triphosphate
- coIP
coimmuneprecipitation
- DIC
differential interference contrast
- DMAs(III)
dimethyl arsenite
- DTT
dithiothreitol
- GST
glutatione- S-transferase
- HOG
high osmolarity glycerol
- MAs(III)
methyl arsenite
- MEK
MAP kinase kinase
- MEKK
MAP kinase kinase kinase
- SAH
S-adenosyl homocysteine
- SAM
As(III) S-adenosylmethionine
- SAPK
stress-activated MAP kinase
- SDM
synthetic defined medium
- TAP
tandem affinity purification
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-03-0185) on May 30, 2018.
REFERENCES
- Abernathy CO, Thomas DJ, Calderon RL. (2003). Health effects and risk assessment of arsenic. J Nutr , 1536S–1538S. [DOI] [PubMed] [Google Scholar]
- Aguilera J, Rodrígues-Vargas S, Prieto JA. (2005). The HOG MAP kinase pathway is required for the induction of methylglyoxal-responsive genes and determines methylglyoxal resistance in Saccharomyces cerevisiae . Mol Microbiol , 228–239. [DOI] [PubMed] [Google Scholar]
- Ahmadpour D, Maciaszczyk-Dziubinska E, Babazadeh R, Dahal S, Migocka M, Andersson M, Wysocki R, Tamás MJ, Hohmann S. (2016). The mitogen-activated protein kinase Slt2 modulates arsenite transport through the aquaglyceroporin Fps1. FEBS Lett , 3649–3659. [DOI] [PubMed] [Google Scholar]
- Azad GK, Singh V, Thakare MJ, Barawal S, Tomar RS. (2014). Mitogen-activated protein kinase Hog1 is activated in response to curcumin exposure in the budding yeast Saccharomyces cerevisiae . BMC Microbiol , 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beane Freeman LE, Dennis LK, Lynch CF, Thorne PS, Just CL. (2004). Toenail arsenic content and cutaneous melanoma in Iowa. Am J Epidemiol , 679–687. [DOI] [PubMed] [Google Scholar]
- Beese SE, Negishi T, Levin DE. (2009). Identification of positive regulators of the yeast Fps1 glycerol channel. PLoS Genetics , e1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee H, Sheng J, Ajees AA, Mukhopadhyay R, Rosen BP. (2010). Adventitious arsenate reductase activity of the catalytic domain of the human Cdc25B and Cdc25C phosphatases. Biochemistry , 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland E, Molin C, Swaminathan S, Ramne A, Sunnerhagen P. (2004). Rck1 and Rck2 MAPKAP kinases and the HOG pathway are required for oxidative stress resistance. Mol Micro , 1743–1756. [DOI] [PubMed] [Google Scholar]
- Cairns BR, Ramer SW, Kornberg RD. (1992) Order of action of components in the yeast pheromone response pathway revealed with a dominant allele of the STE11 kinase and the multiple phosphorylation of the STE7 kinase. Genes Dev , 1305–1318. [DOI] [PubMed] [Google Scholar]
- Cullen WR. (2014). Chemical mechanism of arsenic biomethylation. Chem Res Toxicol , 457–461. [DOI] [PubMed] [Google Scholar]
- de Nadal E, Alupez PM, Posas F. (2002). Dealing with osmostress through MAP kinase activation. EMBO Rep , 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheeman SD, Packianathan C, Pillai JK, Rosen BP. (2014). Pathway of human AS3MT arsenic methylation. Chem Res Toxicol , 1979–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Madegowda M, Nefzi A, Houghten RA, Giulianotti MA, Rosen BP. (2015). Identification of small molecule inhibitors of human As(III) S-adenosylmethionine methyltransferase (AS3MT). Chem Res Toxicol , 2419–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbirt KK, Whitmarsh AJ, Davis RJ, Bonkovsky HL. (1998). Mechanism of sodium arsenite-mediates induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J Biol Chem , 8922–8931 [DOI] [PubMed] [Google Scholar]
- Ferrigno P, Posas F, Koepp D, Saito H, Silver PA. (1998). Regulated nucleocytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO J , 5606–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. (1995). Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast , 355–360. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae . Yeast , 1541–1553. [DOI] [PubMed] [Google Scholar]
- Han J, Lee J-D, Bibbs L, Ulevitch RJ. (1994). A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science , 808–811. [DOI] [PubMed] [Google Scholar]
- Heurgue-Hamard V, Champ S, Mora L, Merkoulova-Rainon T, Kisselev LL, Buckingham RH. (2005). The glutamine residue of the conserved GCQ motif in Saccharomyces cerevisiae release factor eRF1 is methylated by the product of the YDR140w gene. J Biol Chem , 2439–2445. [DOI] [PubMed] [Google Scholar]
- Heurgue-Hamard V, Graille M, Scrima N, Ulryck N, Champ S, van Tilbeurgh H, Buckingham RH. (2006). The zinc finger protein Ynr046w is plurifunctional and a component of the eRF1 methyltransferase in yeast. J Biol Chem , 36140–36148. [DOI] [PubMed] [Google Scholar]
- Jacoby T, Flanagan H, Faykin A, Seto AG, Mattison C, Ota I. (1997). Two protein –tyrosine phosphatases inactivate the osmotic stress response pathway in yeast by targeting the mitogen-activated protein kinase, Hog1. J Biol Chem , 17749–17755. [DOI] [PubMed] [Google Scholar]
- Jiang L, Cao C, Zhang L, Lin W, Xia J, Xu H, Zhang Y. (2014). Cadmium-induced activation of high osmolarity pathway through its Sln1 branch is dependent on the MAP kinase kinase kinase Ssk2, but not its paralog Ssk22, in budding yeast. FEMS Yeast Res , 1263–1272. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Jung US, Piotrowski J, Levin DE. (1995). The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev , 1559–1571. [DOI] [PubMed] [Google Scholar]
- Kumar NV, Yang J, Pillai JK, Rawat S, Solano C, Kumar A, Grøtli M, Stemmler TL, Rosen BP, Tamás MJ. (2016). Arsenic directly binds to and activates the yeast AP-1-like transcription factor Yap8. Mol Cell Biol , 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. (2000). Rapid and reliable protein extraction from yeast. Yeast , 857–860. [DOI] [PubMed] [Google Scholar]
- Lawrence CL, Botting CH, Antrobus R, Coote PJ. (2004). Evidence of a new role for the high-osmolarity glycerol mitogen-activated protein kinase pathway in yeast: regulating adaptation to citric acid stress. Mol Cell Biol , 3307–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Reiter W, Dohnal I, Gregori C, Beese-Sims S, Kuchler K, Ammerer G, Levin DE. (2013). MAPK Hog1 closes the S. cerevisiae glycerol channel Fps1 by phosphorylating and displacing its positive regulators. Genes Dev , 2590–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and economic PCR-based gene deletion and modification in Saccharomyces cerevisiae . Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- Maciaszczyk-Dziubinska E, Migocka M, Wysocki R. (2011). Acr3p is a plasma membrane antiporter that catalyzes As(III)/H+ and Sb(III)/H+ exchange in Saccharomyces cerevisiae . Biochim Biophys Acta , 1855–1859. [DOI] [PubMed] [Google Scholar]
- Maciaszczyk-Dziubinska E, Wawrzycka D, Wysocki R. (2012). Arsenic and antimony transporters in eukaryotes. Int J Mol Sci , 3527–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques JM, Rodrigues RJ, de Magalhães-Sant’ana AC, Gonçalves T. (2006). Saccharomyces cerevisiae Hog1 protein phosphorylation upon exposure to bacterial endotoxin. J Biol Chem , 24687–24694. [DOI] [PubMed] [Google Scholar]
- Matia-González AM, Rodríguez-Gabriel MA. (2011). Slt2 MAPK pathway is essential for cell integrity in the presence of arsenate. Yeast , 9–17. [DOI] [PubMed] [Google Scholar]
- Millson SH, Truman AW, King V, Prodromou C, Pearl LH, Piper PW. (2005). A two-hybrid screen of the yeast proteome for Hsp90 interactors uncovers a novel Hsp90 chaperone requirement in the activity of a stress-activated mitogen-activated protein kinase, Slt2p (Mpk1p). Eukaryot Cell , 1914–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Piper PW. (2006). Hog1p mitogen-activated protein kinase determines acetic acid resistance in Saccharomyces cerevisiae . FEMS Yeast Res , 1274–1280. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R, Rosen BP. (1998). Saccharomyces cerevisiae ACR2 gene encodes an arsenate reductase. FEMS Microbiol Lett , 127–136. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Tatebayashi K, Saito H. (2008). Two adjacent docking sites in the yeast Hog1 Mitogen-Activated Protein (MAP) kinase differentially interact with the Pbs2 MAP kinas kinase and the Ptp2 protein tyrosine phosphatase. Mol Cell Biol , 2481–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Han J. (2000). The p38 signal transduction pathway activation and function. Cell Signaling , 1–13. [DOI] [PubMed] [Google Scholar]
- Panadero J, Pallotti C, Rodrígues-Vargas S, Randez-Gil F, Prieto JA. (2006). A downshift in temperature activates the high osmolarity glycerol (HOG) pathway, which determines freeze tolerance in Saccharomyces cerevisiae . J Biol Chem , 638–4645. [DOI] [PubMed] [Google Scholar]
- Piao H, MacLean Freed J, Mayinger P. (2012). Metabolic activation of the HOG MAP kinase pathway by Snf1/AMPK regulates lipid signaling at the Golgi. Traffic , 1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleveda B, Span L, Sherman F. (2006). The yeast translation release factors Mrf1 and Sup45p (eRF1) are methylated, respectively, by the methyltransferases Mtq1p and Mtq2p. J Biol Chem , 2562–2571. [DOI] [PubMed] [Google Scholar]
- Posas F, Saito H. (1997). Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science , 1702–1705. [DOI] [PubMed] [Google Scholar]
- Rehman K, Chen Z, Wang WW, Wang YW, Sakamoto A, Zhang YF, Naranmandura H, Suzuki N. (2012). Mechanisms underlying the inhibitory effects of arsenic compounds on protein tyrosine phosphatase (PTP). Toxicol Appl Pharmacol , 273–280. [DOI] [PubMed] [Google Scholar]
- Rehman K, Naranmandura H. (2012). Arsenic metabolism and thioarsenicals. Metallomics , 881–892. [DOI] [PubMed] [Google Scholar]
- Ren X, Aleshin M, Jo WJ, Dills R, Kalman DA, Vulpe CD, Smith MT, Zhang L. (2011). Involvement of N-6 adenine-specific DNA methyltransferase (N6AMT1) in arsenic biomethylation and its role in arsenic-induced toxicity. Env Health Persp , 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen BP, Liu Z. (2009). Transport pathways for arsenic and selenium: a minireview. Environ Int , 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Posas F. (2012). Response to hyperosmotic stress. Genetics , 289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Li X-F, Cullen WR, Weinfeld M, Le XC. (2013). Arsenic binding to proteins. Chem Rev , 7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics , 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley PL, Kinniburgh DG. (2002). A review of the source, behavior and distribution of arsenic in natural waters. Applied Geochem , 517–568. [Google Scholar]
- Sotelo J, Rodríguez-Gabriel MA. (2006). Mitogen-activated protein kinase Hog1 is essential for the response to arsenite in Saccharomyces cerevisiae . Eukaryot Cell , 1826–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatjer L, Sacristán-Reviriego A, Casado C, González A, Rodríguez-Porrata B, Palacios L, Canadell D, Serra-Cardona A, Martín H, Molina M, Ariño J. (2016). Wide-ranging effects of the yeast Ptc1 protein phosphatase acting through the MAPK kinase Mkk1. Genetics , 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsen M, Di Y, Tängemo C, Morillas M, Ahmadpour D, Van der Does C, Wagner A, Johansson E, Boman J, Posas F, et al. (2006). The MAPK Hog1p modulates Fps1p-dependent arsenite uptake and tolerance in yeast. Mol Biol Cell , 4400–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Mohindru M, Deb DK, Sassano A, Kambhampati S, Ravandi F, Minucci S, Kalvakolanu DV, Platanias LC. (2002). Cutting edge: activation of the p38 mitogen-activated protein kinase signaling pathway mediates cytokine-induced hemopoietic suppression in aplastic anemia. J Immunol , 5984–5988. [DOI] [PubMed] [Google Scholar]
- Winkler A, Arkind C, Mattison CP, Burkholder A, Knoche K, Ota I. (2002). Heat stress activates the yeast high-osmolarity glycerol mitogen-activated protein kinase pathway, and protein tyrosine phosphatases are essential under heat stress. Eukaryot Cell , 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurgler-Murphy SM, Maeda T, Witten EA, Saito H. (1997). Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol , 1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki R, Chéry CC, Wawrzycka D, van Hulle M, Cornelis R, Thevelein JM, Tamás MJ. (2001). The glycerol channel Fps1p mediates the uptake of arsenite and antimonite in Saccharomyces cerevisiae . Mol Micro , 1391–1401. [DOI] [PubMed] [Google Scholar]
- Wysocki R, Fortier PK, Maciaszczyk E, Thorsen M, Leduc A, Odhagen A, Owsianik G, Ulaszewski S, Ramotar D, Tamás MJ. (2004). Transcriptional activation of metalloid tolerance genes in Saccharomyces cerevisiae requires the AP-1-like proteins Yap1p and Yap8p. Mol Biol Cell , 2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki R, Tamás MJ. (2010). How Saccharomyces cerevisiae copes with toxic metals and metalloids. FEMS Microbiol Rev , 925–951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.