

Summary
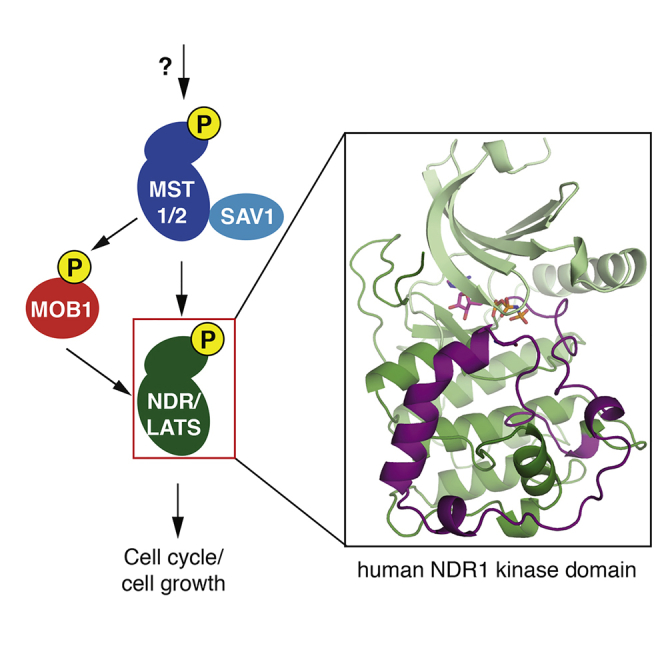
The human NDR family kinases control diverse aspects of cell growth, and are regulated through phosphorylation and association with scaffolds such as MOB1. Here, we report the crystal structure of the human NDR1 kinase domain in its non-phosphorylated state, revealing a fully resolved atypically long activation segment that blocks substrate binding and stabilizes a non-productive position of helix αC. Consistent with an auto-inhibitory function, mutations within the activation segment of NDR1 dramatically enhance in vitro kinase activity. Interestingly, NDR1 catalytic activity is further potentiated by MOB1 binding, suggesting that regulation through modulation of the activation segment and by MOB1 binding are mechanistically distinct. Lastly, deleting the auto-inhibitory activation segment of NDR1 causes a marked increase in the association with upstream Hippo pathway components and the Furry scaffold. These findings provide a point of departure for future efforts to explore the cellular functions and the mechanism of NDR1.
Keywords: protein kinase, X-ray crystallography, Hippo tumor suppressor pathway, NDR family, kinases, MOB1, FRYL, Furry-like
Graphical Abstract
Highlights
-
•
2.2 Å crystal structure of an inactive human NDR1 kinase domain is determined
-
•
An atypically long activation segment auto-inhibits the NDR1 kinase domain
-
•
MOB1 and the activation segment of NDR1 act through independent mechanisms
-
•
The activation segment of NDR1 influences interaction with MST1/2 and Furry
NDR family kinases play important roles in regulating cell cycle and growth from yeast to human. To understand how NDR family kinases are regulated, Xiong et al. determine the crystal structure of human NDR1 kinase domain in its non-phosphorylated inactive state.
Introduction
The NDR (Nuclear Dbf-2 Related) kinases, a sub-group of the AGC (cyclic AMP and cyclic GMP-dependent protein kinases and protein kinase C-related protein kinases) family, are conserved across eukaryotes (Hergovich et al., 2006, Pearce et al., 2010). Budding yeast (Saccharomyces cerevisiae) expresses three NDR members: Dbf2 and its 77% identical paralog Dbf20 control exit from mitosis, while the more divergent Cbk1 promotes cytokinesis and regulates morphogenesis (Bardin and Amon, 2001, Rock et al., 2013). Drosophila melanogaster's two NDR members have divergent functions: Tricornered regulates the integrity of epidermal outgrowth and dendritic tiling and branching, while Warts is a central player of the conserved Hippo pathway in which it directly phosphorylates the transcription coactivator Yorkie to regulate tissue homeostasis (Emoto et al., 2006, Huang et al., 2005). These divergent functions are maintained in humans: the Warts orthologs LATS1 and 2 are primarily studied in the context of the Hippo pathway (Hao et al., 2008, Lei et al., 2008, Zhao et al., 2007, Zhao et al., 2010), while the Tricornered-related NDR1 and NDR2 kinases (gene names STK38 and STK38L) have been ascribed a variety of functions, notably in the regulation of the G1/S phase transition (Cornils et al., 2011), T cell migration (Cornils et al., 2010, Tang et al., 2015), and centrosome duplication (Hergovich et al., 2009). Despite these apparently divergent roles, more recent studies have also outlined potentially overlapping functions between LATS and NDR family members, as human NDR1 and NDR2 were shown to phosphorylate the Yorkie ortholog YAP1 both in vitro and in vivo, allowing them to function as tumor suppressors in the intestinal epithelium (Tang et al., 2015, Zhang et al., 2015).
The NDR family kinases possess a conserved and characteristic domain architecture consisting minimally of an N-terminal MOB1 coactivator binding domain (MBD), a central protein kinase domain with an atypically long activation segment (ranging from 63 residues in NDR1/2 to 75 residues in LATS1/2), followed by a C-terminal hydrophobic motif (HM; Figure 1A). Regulation of NDR family catalytic function is mediated minimally at three key core levels: firstly by binding of MOB1 to the MBD (Hergovich et al., 2006), secondly by auto-phosphorylation on sites within the atypically long activation segment in the kinase domain (Ser281/282 in NDR1/2; Ser909/872 in LATS1/2; Chan et al., 2005, Stegert et al., 2004, Tamaskovic et al., 2003), and thirdly by phosphorylation of sites within the HM (Thr444/442 in NDR1/2; Thr1079/1041 in LATS1/2) by the upstream protein kinases MST1, MST2, and/or MST3 (Chan et al., 2005, Stegert et al., 2004, Stegert et al., 2005, Tamaskovic et al., 2003). In addition to these three core regulatory mechanisms, NDR family kinase activity is affected by scaffold-like proteins, including Tao3p in S. cerevisiae, which cooperates with the NDR ortholog Cbk1p to control cell polarity (Bogomolnaya et al., 2006, Nelson et al., 2003). Tao3p orthologs are also implicated in NDR function: Furry in D. melanogaster collaborates with Trc in dendritic branching and planar cell polarity (Emoto et al., 2004, Fang and Adler, 2010, Fang et al., 2010, He et al., 2005), and Furry and Furry-like (FRY and FRYL) in mammals participates with NDR1/2 in mitotic spindle and chromosome alignment (Chiba et al., 2009). How precisely the association of FRY/FRYL proteins with the mammalian NDR1/2 kinases is mediated and regulated remains an open question.
Figure 1.

Crystal Structure of the Protein Kinase Domain of Human NDR1 in Its Non-Phosphorylated State
(A) Domain architecture of NDR1 kinase highlighting an atypically long activation segment and two phosphoregulatory sites, S281 and T444. Constructs used in this study are highlighted above with black lines.
(B) Ribbon representation of the kinase domain of human NDR1.
X-ray crystallographic analysis of yeast Cbk1 bound to Mob2 (Gogl et al., 2015) provided a first view of the underlying structure of an NDR family kinase and its binding mode to an MOB protein (Figure S1A). This study also uncovered a non-canonical mechanism by which a phosphorylated HM engages the N lobe of an AGC family kinase domain to regulate catalytic function (Gogl et al., 2015). The MBD of Cbk1 consists of an α-helix (denoted αMOB) followed by an extended strand element (denoted N-linker) that together compose the primary docking site for the MOB protein. Recent crystal structures of the isolated MBD of human LATS1 and NDR2 bound to human MOB1 confirmed the general architecture of the MBD and its binding mode to MOB proteins (Kim et al., 2016, Kulaberoglu et al., 2017, Ni et al., 2015).
Interestingly, the kinase domain of Cbk1 establishes few direct contacts with its Mob2 activator. Instead, these two elements communicate indirectly through bridging interactions involving the C-terminal HM of Cbk1, which is nestled in a cleft between one surface of the MBD and the N lobe of the kinase domain. Notably, the mode of interaction between the HM and the N lobe of the kinase domain differs substantially from the canonical mode of interaction displayed by other AGC family members (Knighton et al., 1991, Xu et al., 2004, Yang et al., 2002).
The Cbk1-Mob2 complex structure, together with molecular dynamics simulations, supported a model in which Mob2 binding to the MBD induces conformational changes in the MBD that are communicated to the kinase domain in part by the HM. The structures of the complex between Mob2 and either the unphosphorylated Cbk1 or Cbk1 harboring a phosphomimetic mutation in the HM showed no major structural differences. Both co-crystal structures revealed a partially disordered activation segment and unresolved helix αC in the kinase domain of Cbk1, suggesting that neither the binding of Mob2 nor phosphomimetics were sufficient to force a fully active conformation on the Cbk1 kinase domain. While the functional significance of these features was not probed in detail, the former hinted at the possibility that the activation segment would play an auto-inhibitory role on kinase catalytic function (Bichsel et al., 2004).
Here, we report the crystal structure of the human NDR1 kinase domain at 2.2 Å resolution, revealing a completely resolved helix αC and entirely defined, elongated activation segment. The activation segment adopts a circuitous path that blocks potential substrate-binding surfaces in the immediate vicinity of the kinase active site and around helix αG, while additionally restricting helix αC to a non-productive conformation not visualized previously in other NDR family structures. We probed the involvement of the MBD and the elongated activation segment for their effects on regulating NDR1 protein kinase activity and dynamics by hydrogen-deuterium exchange (HDX) analysis. Our findings support a model in which the elongated activation segment operates independently of MOB1 binding to the MBD, which is consistent with their remote sites of action in the protein kinase domain structure. Furthermore, deletion of the auto-inhibitory region within the activation segment induced NDR1 association with multiple activating proteins in cells, and several of these interactions were further enhanced by serine/threonine phosphatase inhibition with okadaic acid. These findings reveal an unanticipated regulatory role for the activation segment of NDR1 in modulating not only the intrinsic catalytic function of the kinase domain but also the interactions with the Furry scaffold FRYL and upstream activating kinases, which may afford multiple opportunities to modulate NDR1/2 activity in the context of living cells.
Results
Structure of the NDR1 Protein Kinase Domain
To visualize the regulatory states that could shed light onto NDR1 kinase signaling, we set out to determine the structure of the isolated kinase domain of NDR1. To delimit a fragment of NDR1 suitable for crystallization, we first performed limited proteolysis on an NDR1 fragment (residues 12–418 encompassing the MBD and the kinase domain, denoted NDR1MBD−KD) that could be expressed and purified from bacteria (Figure 1A). While the MBD of this NDR1 construct was readily cleaved from the kinase domain, the atypically long activation segment of NDR1 was surprisingly resistant to proteolysis despite the presence of numerous predicted protease sites (Figure S2A). We next expressed and purified the isolated kinase domain of human NDR1 (residues 82–418, denoted NDR1KD; Figure 1A) from Escherichia coli. Intact mass analysis did not reveal significant auto-phosphorylation (Figure S2B). Analytical ultracentrifugation demonstrated that NDR1KD was a monomer in solution under all conditions tested (Figure S2C). We crystallized NDR1KD in the presence of the ATP analog β,γ-imidoadenosine-5′-triphosphate (AMP-PNP), obtaining well-diffracting crystals that enabled us to determine the crystal structure by molecular replacement (Rfactor/Rfree = 18.0%/21.4%, 2.2 Å, see STAR Methods and Table 1 for details). The crystal asymmetric unit contained two NDR1KD molecules, each bound to AMP-PNP, with all but the most N-terminal and C-terminal residues ordered, including the entire length of the 63-residue activation segment (Figure S3A). As the two chains were virtually identical (main-chain alpha-carbon root-mean-square deviation [RMSD] = 0.051 Å over 337 ordered residues), we focused our descriptions to a single chain A (Figure 1B, see Figure 2 for a structure-based sequence alignment of the catalytic domain of the NDR1/2 kinases).
Table 1.
Crystallographic Data Collection and Refinement Statistics for Human NDR1KD
| Hs NDR1 + AMP-PNP + Mg2+ | |
|---|---|
| Space group | C2 a = 117.2 α = 90° b = 118.0 β = 124.9° c = 95.0 γ = 90° |
| Molecules per a.u.(RMSD) | 2 (0.051) |
| Wavelength (Å) | 0.9788 |
| Resolution (Å) | 2.2 |
| Rsym (high resolution) | 0.109% (0.491%) |
| Total reflections | 220717 |
| Completeness (high resolution) | 93.7% (87%) |
| Redundancy | 3.5 |
| I/σ (high resolution) | 8.333 (1.632) |
| Refinement | |
| Resolution range (Å) | 50–2.2 |
| Reflections | 47,691 |
| Rfactor/Rfree | 18%/20% |
| Rms deviations | |
| Bonds (Å) | 0.02 |
| Angles (°) | 2.06 |
| Average B-factor (activation segment) | 31.2 (24.6) |
| Ramachandran Data | |
| Most favored | 95.6% |
| Additionally allowed | 4.4% |
| Disallowed | 0% |
Figure 2.

Structure-Based Sequence Alignment of the Kinase Domain of NDR1
Secondary structure elements are highlighted above the sequence. Degree of conservation highlighted by black (most) to white (least) shading. Species from top to bottom correspond to human (NP_009202.1, NP_055815.1), gorilla (NP_001075071.1), bovine (NP_001075071.1), eagle (XP_010582907.1), zebrafish (NP_998621.1), lizard (XP_003220400.1), turtle (XP_007063535.1), fly (NP_524170.2), worm (NP_508627.5), and budding yeast (NP_014238.3). Red star indicates the previously identified auto-phosphorylation site, Ser281. Blue stars indicate conserved basic residues implicated in the canonical stabilization of the phosphorylated activation segment in a productive conformation. Orange circles indicate serine and threonine residues in the activation segment that may possess phospho-regulation potential. The alignment was generated using CCP4 ALINE (Bond and Schuttelkopf, 2009).
Structure of the NDR1 Kinase Domain Reveals an Inactive Conformation
NDR1KD adopts the bilobal fold typical of protein kinases (Figure 1B). The most notable and distinguishing features of the structure included a fully ordered, atypically long activation segment that occupies established substrate-binding sites in other protein kinases (see below), and a non-productively oriented helix αC within the kinase N lobe suggestive of an inactive state (see below).
Commencing at the DFG motif, the elongated activation segment (residues 230–292, Figure 1B) emerged from the catalytic cleft and wrapped around the front face of the C lobe, circumnavigating helix αG, before re-directing toward the catalytic cleft and terminating in the APE motif. The elongated activation segment adopted an irregular extended conformation interspersed by three helical regions, giving rise to an alternation of extended and helical segments: EX1 (230–238), H1 (239–246), EX2 (247–256), H2 (257–261), EX3 (262–263), a kinked helix denoted H3 (264–280), and EX4 (281–293). The extensive interface between the activation segment and the kinase domain, covering 2,740 Å2, was mediated by a mixture of hydrogen bonds, salt bridges, and hydrophobic interactions (Figure S4). We note that only a small portion of the activation segment centered on helix H1 was involved in crystal packing interactions (contact surface = 694 Å2) (Figures S3B and S3C).
Comparison of NDR1 with a typical active-state protein kinase structure, such as that of phosphorylase kinase (PHK) (PDB: 2PHK), revealed an outwardly rotated position of helix αC (Figure 3A). As a result of this non-productive orientation, the conserved Glu137 side chain within helix αC was swung away from the catalytic cleft (≈100° relative to the corresponding residue E91 in active PHK) toward the solvent. This precluded formation of an essential salt bridge with Lys118 (within β3) that would enable Lys118 to productively coordinate the α- and β-phosphates of ATP for catalysis (Figure 3A top inset). The non-productive conformation of helix αC was stabilized in part by contacts with the extended region EX1 of the activation segment, and involved a salt bridge between the Mg2+-coordinating residue Asp230 of the DFG motif and Arg138 of helix αC. The inactive state of the kinase domain was further shown by a broken regulatory (R-) spine (Figure 3B) and the absence of Mg2+ (despite its presence in the crystallization buffer), both of which are critical for proper ATP coordination and catalysis (Figure 3; Endicott et al., 2012).
Figure 3.

Inactive State Features of the Kinase Domain of NDR1
(A) Superimposition of NDR1KD in green and the kinase domain of phosphorylase kinase (PHKKD) in gray bound to the MC peptide substrate in cyan (PDB: 1PHK; Lowe et al., 1997). Top right inset highlights the non-productive outward rotated (≈100°, as indicated by the red arrow) position of helix αC in NDR1KD. Bottom right inset highlights steric clash of the atypically long activation segment of NDR1KD with the MC peptide substrate bound to PHKKD.
(B) Hydrophobic catalytic and regulatory spine representation of NDR1KD.
Structural comparison of NDR1 with other protein kinases bound to either a peptide substrate (Figure 3A) or intact substrates (Figure S5) suggested that the elongated activation segment of NDR1 may block substrate binding at two levels: first at the primary phospho-acceptor binding site in the catalytic cleft (Figure 3A, bottom inset), and second at higher-order substrate-binding sites involving helix αG (Figure S5). The former is apparent by overlap of activation segment elements EX1 and EX4 of NDR1 with the Modified Cantley (MC) peptide substrate bound to PHK (Figure 3A bottom inset), while the latter is apparent by overlap of the activation segment elements H3, EX2, and EX1/H1 with the respective substrate components in the PKR-eIF2α (Dar et al., 2005, Dey et al., 2005), the ROCK1-RhoE (Komander et al., 2008), and the BRAF-MEK1 (Haling et al., 2014) enzyme-substrate complexes (Figure S5).
The Activation Segment of NDR1 Serves an Auto-inhibitory Role
Based on our crystallographic findings, we hypothesized that the structure of the NDR1 kinase domain reflected a downregulated state that was maintained in part by the inhibitory conformation of the elongated activation segment. To test this notion, we sought to introduce point mutations and deletions into NDR1, aimed at disrupting the activation segment from its contact surface on the kinase domain (see Figure S6A–S6C for schematic). We chose to introduce mutations into the longer NDR1MBD−KD construct because, unlike NDR1KD, it preserved many of the key regulatory properties of the full-length enzyme (Figure 4). As assessed by auto-phosphorylation and by phosphorylation of the previously reported substrate RAB3A-interacting protein (gene RAB3IP; also called Rabin8; Chiba et al., 2013, Ultanir et al., 2012), wild-type NDR1MBD−KD displayed repressed catalytic activity (Figures 4A–4C) that could be potently relieved by addition of MOB1 (at the level of kinase auto-phosphorylation and RAB3IP phosphorylation) and to a lesser extent by phospho-HM (at the level of RAB3IP phosphorylation). In contrast, NDR1KD displayed higher basal catalytic activity that was not responsive to the addition of MBD or phospho-HM (Figures 4B and 4C). These results were consistent with a model in which the MBD of NDR1 exerts an additional repressive effect on the adjacent kinase domain that can be relieved by the binding of NDR1 to MOB1 and to the HM peptide.
Figure 4.

Activation of NDR1 Kinase by MOB1 and/or a Phosphorylated HM Peptide
(A) In vitro kinase assay analysis of wild-type GST-NDR1MBD−KD in the presence and absence of MOB1 activator and/or a phospho-HM peptide in trans. The top two panels show the autoradiograph, and the bottom three panels show protein loading by Coomassie staining.
(B) In vitro auto-phosphorylation analysis of NDR1MBD−KD versus NDR1KD in the presence and absence of MOB1 and/or a phospho-HM peptide in trans. The top panel displays the autoradiograph, and the bottom two panels show protein loading by Coomassie staining.
(C) In vitro auto-phosphorylation and phosphorylation of RAB3IP substrate by NDR1MBD−KD and NDR1KD, in the presence and absence of MOB1 and/or a phospho-HM peptide in trans. The top two panels display autoradiographs for NDR1 auto-phosphorylation and RAB3IP phosphorylation, respectively. The bottom three panels show protein loading by Coomassie staining.
We next tested the effect of activation segment mutations on the kinase activity of NDR1MBD−KD. Individual mutations in NDR1MBD−KD of residues K267 (aliphatic portion of the side chain engaging a hydrophobic pocket formed by Y319, Y313, T342, P321, P320, and P321), W271 (binding a hydrophobic pocket formed by P215, L278, Y288, I311, and P320), K272 (binding to D181 and G319 backbone via hydrogen bonds), and Y331 (binding a hydrophobic pocket formed by Y246, H251, P291, F294, V334, and M335) (Figures S6D–S6F and S6H, respectively) stimulated RAB3IP phosphorylation by up to 10-fold (Figures 5A and S7). The WY271/331CA double mutation increased RAB3IP phosphorylation by 50-fold.
Figure 5.

Mutational Analysis of the Atypically Long Activation Segment of Human NDR1
See also accompanying Figure S6 for schematic of mutations tested.
(A) In vitro phosphorylation of RAB3IP by wild-type NDR1MBD−KD and the indicated point mutants without (left panel) and with (right panel) the addition of MOB1A. Loading controls are shown by Coomassie staining.
(B) In vitro phosphorylation of the NDR1 substrate RAB3IP by NDR1MBD−KD wild-type and the indicated activation segment deletion mutants without (left panel) and with (right panel) the addition of MOB1A; loading controls are shown by Coomassie staining. Quantification of the autoradiography signals is shown at the bottom (n = 3 independent experiments; error bars, SEM).
A previously reported five-site activating mutation within the activation segment of NDR1 (Bichsel et al., 2004), denoted 5A (K265A/R266A/K267A/K272A/R273A), resulted in a 50-fold increase in NDR1 phosphorylation of RAB3IP (Figures 5A, and see Figure S7 for quantification). The mechanism by which these mutations act was not previously discerned, but only residues K267 and K272 made appreciable contacts with the surface of the kinase domain in our crystal structure (Figures S6D, S6F, and S6G). We hypothesized that mutations at these two sites were the primary drivers of activation by causing dissociation of the inhibitory activation segment from its contact surface on the kinase domain. In partial support of this inference, the K267A and K272A mutations on their own, unlike the other single-site lysine mutations, displayed some ability to stimulate kinase activity (Figure 5A). However, robust activation was only observed when all five sites were mutated, suggesting that all five sites collaborate to relieve auto-inhibition.
The activation segment of NDR1 houses 10 serine or threonine sites with potential for phospho-regulation (Figure S8A). Ser281 was identified previously as an auto-phosphorylation site that, when mutated to alanine, compromised full-length enzyme function in vitro and in cells (Tamaskovic et al., 2003). We confirmed this finding using the NDR1KD crystallization construct (Figure S8B) and further showed that mutation of Ser281 to Glu restored catalytic function, but not as potently as a Y331A mutation, which disrupts a key contact of the activation segment insert with the kinase domain C lobe (Figure S8C). In the crystal structure of NDR1, Ser281 resides in a solvent-accessible position following helix H3. We reason that auto-phosphorylation on Ser281 may only weakly perturb the auto-inhibitory conformation of the activation segment and that any essential phospho-regulatory function derives from its ability to stabilize a productive conformation of the activation segment through canonical phosphate coordinating interactions with arginine 211 of the HRD motif and with histidine 133 of helix αC.
We further corroborated the auto-inhibitory function of the elongated activation segment by deletion analysis. We replaced stretches of 34, 38, and 40 residues in the elongated activation segment with a single glycine residue while maintaining the DFG motif, the APE motif, and the Ser 281 auto-phosphorylation regulatory site (Figure S6C). Compared with the wild-type NDR1MBD−KD, the Δ34G, Δ38G, and Δ40G deletion mutants displayed ∼200-fold, 500-fold, and 70-fold increases in RAB3IP phosphorylation, respectively (Figure 5B). For all mutants, including those displaying the strongest activation, kinase activity could be further potentiated by the addition of MOB1 (Figures 5, and see Figure S7 for quantification). This observation indicated that the regulation of NDR1 kinase activity by the activation segment and by MOB1 represent independent layers of control.
Similar activating effects brought by MOB1 binding and activating mutations were also observed when GST-YAP50−171 (YAP fragment encompassing amino acids 50–171) was used as an NDR1 substrate in vitro (Figure S9). Taken together, our data demonstrated that the conformation of the activation segment visualized in the NDR1KD crystal structure serves an auto-inhibitory role.
HDX Corroborates Independent Regulation of NDR1 by the Atypical, Elongated Activation Segment and MOB1A Binding
Comparison of the NDR1KD structure presented here and the Cbk1-Mob2 co-crystal structure reported previously revealed that the activation segment of NDR kinase engages the kinase domain remotely from the MOB-MBD site of contact (Figures 6 and S1C). The only point of convergence between the two could be helix αC, which is disordered in the Cbk1-Mob2 crystal structure. This, together with our enzymatic characterizations, provided support to the notion that the activation segment and MBD regulatory inputs might operate independently to regulate protein kinase activity. To investigate the impact of two regulatory inputs on the structure and dynamics of NDR1 free from the constraints of a crystal lattice, we employed HDX coupled mass spectrometry (HDX-MS). This method measures the rate of deuterium incorporation into a target protein upon the substitution of H2O by D2O. For this experiment, we chose to analyze the NDR1MBD−KD 5A mutant over the most potent Δ38G deletion mutant because it more closely preserved protein sequence and length in the activation segment for a more useful comparison with wild-type NDR1MBD−KD with or without MOB1A binding (we did not use full-length NDR1 because the construct could not be expressed in E. coli). Consistent with our structural and enzymatic inferences, the 5A mutations and the addition of MOB1A affected HDX in distinct regions of the kinase construct (Figure 6, graph numbers I–VIII), leaving other regions, with varying degrees of solvent accessibility, relatively unaffected (Figure 6, graph numbers IX–XI). MOB1A binding mostly affected the N terminus of the kinase construct, including the MBD, the MBD-kinase domain linker, and the area around the catalytic base (HRD peptide). MOB1A addition substantially reduced the rate of deuterium incorporation in the MBD and the MBD-kinase linker (Figure 6, graph numbers I, II, and III), consistent with the fact that complex formation would directly mask these regions from exchange. In contrast, MOB1A addition increased deuterium incorporation in the catalytic site in both NDR1 wild-type and 5A constructs (Figure 6, graph number IV). Conversely, the activating 5A mutation did not change the rate of deuterium incorporation in the kinase N terminus and only modestly increased D2O incorporation at the catalytic site (Figure 6, graph numbers I, II, III, and IV).
Figure 6.

Conformational and Dynamics Analysis of NDR1 by HDX Mass Spectrometry
HDX-MS was used to evaluate the solvent accessibility of NDR1MBD−KD upon addition of MOB1A or upon the introduction of the 5A activating mutations. Changes to the average deuterium incorporation are highlighted in the structural superimposition of NDR1KD and the Cbk1-Mob2 complex, with appended HDX plots for the indicated regions. For each plot corresponding to the indicated peptide region, HDX time course profiles are shown for NDR1MBD−KD wild-type alone (black), NDR1MBD−KD wild-type with the addition of MOB1A (blue), NDR1MBD−KD 5A alone (green), and NDR1MBD−KD 5A with the addition of MOB1A (purple). HDX plots for peptide elements labeled I–IV represent regions of NDR1 that were most greatly affected by MOB1A binding, as shown by large deviations between blue and black profiles and between purple and green profiles (note: in panel III, the trace for NDR1MBD−KD + MOB1 in blue overlaps with the trace for NDR1MBD−KD5A + MOB1 in pink). HDX plots VI–VIII represent regions of NDR1 most greatly affected by introduction of the 5A mutations, as shown by large deviations between green and black profiles and between purple and blue profiles. HDX plots in panel V represent the DFG motif of NDR1 that was greatly affected by both the addition of MOB1 and the introduction of the 5A mutations. HDX plots VIX–XI are controls representing regions of NDR1 that were not affected by either MOB1 binding or 5A mutations. HDX plots in panels IX, X, and XI represent the NDR1 helix αC, a C-terminal region of the kinase domain (denoted C′ tail peptide 1), and helix αF, respectively. Data points represent means of three replicates; error bars, SEM. WT, wild-type.
In contrast, the 5A mutation strongly affected peptides within the activation segment insert, the region around helix αG, and the activation segment region containing the APE motif, which led to increased HDX in these regions (Figure 6, graph numbers VI, VII, and VIII). HDX in these areas was only marginally affected by MOB1A binding. Lastly, only in the area around the DFG motif did we observe increased HDX for both the 5A mutation and MOB1A addition (Figure 6, graph number V). This peptide is notable in being centrally located between the sites of interaction in the MOB-MBD complex and the activation segment in the kinase domain core. Neither MOB1A binding nor 5A mutation stabilized helix αC in our HDX experiments, which might explain the disordered helix αC in the previous yeast Cbk1-Mob2 co-crystal structure (Figures S1A and 6, graph number IX). Overall, our HDX results are consistent with independent modes of action for kinase regulation by MOB1 engagement and by the activation segment.
Effect of Activating Mutations and MOB1 Binding on Enzyme Kinetics
We next examined how mutation of the elongated activation segment and binding to MOB1 affected the enzymatic parameters of NDR1MBD−KD phosphorylation of RAB3IP in vitro. A change in Km for substrate would reflect modulation of enzyme-substrate engagement, while a change in kcat would reflect modulation of enzyme catalytic efficiency. Several kinases, such as PKR, ROCK, and RAF, utilize helix αG to interact with their substrates (Dey et al., 2005, Haling et al., 2014; Figure S6; Komander et al., 2008). Since the elongated activation segment of NDR1 hinders access to helix αG, we predicted that activation segment mutants might cause a pronounced improvement in Km, if NDR1 also used helix αG to engage the substrate RAB3IP. Conversely, we predicted that MOB1 binding to NDR1 would cause a pronounced improvement on kcat, since the site of MOB1 binding to NDR1 is remote from the substrate recognition infrastructure in the kinase C lobe. In comparing the enzyme kinetic parameters of the NDR1MBD−KD Y331A and 5A mutants with the wild-type (as above, we chose these mutants over the most potent Δ38G deletion mutant because they more closely preserved protein sequence and length in the elongated activation segment for more meaningful comparison with the wild-type NDR1), we observed that the kcat for the phosphorylation of RAB3IP was substantially improved. by 40- and 200-fold respectively, while Km was not enhanced comparably (Figure 7). We also observed a further enhancement of kcat for the phosphorylation of RAB3IP by all three NDR1 constructs upon MOB1A binding. These results indicated that the activation segment of NDR1 and MOB1 both affect NDR1 kinase activity primarily by modulating the catalytic efficiency of the enzyme and not by modulating substrate engagement, at least in the case of the RAB3IP substrate. Our kinetic results raise the possibility that the phospho-acceptor region of RAB3IP may bind to the NDR1 active site in a way that differs to some degree from canonical kinase-substrate complexes and therefore may not be significantly impeded by the NDR1 activation segment as one may expect based on our structural findings.
Figure 7.

Michaelis-Menten Kinetic Analysis of the Phosphorylation of the NDR1 Substrate RAB3IP by NDR1MBD−KD
Phosphorylation kinetic analysis of NDR1MBD−KD wild-type (top panel), Y331A mutant (middle panel), and 5A mutant (bottom panel) in the absence of MOB1 (left column) or in the presence of MOB1 (right column). n = 3 experiments done in technical duplicate; error bars, SEM.
Effect of Activating Mutations on NDR1 Function in Cells
The murine NDR1 homolog Ndr2 was shown to phosphorylate YAP1 and influence Hippo pathway signaling in the intestinal epithelium (Zhang et al., 2015). We tried to exploit this finding to test NDR1 structure-function relationships in cells using a TEAD-luciferase reporter assay in SW480 colorectal cancer cells. However, although we observed reporter repression by NDR1, as has been seen for Ndr2, the effect of wild-type NDR1 was subtle (Figure 8A), and mutations expected to cause activation or impairment of NDR1 catalytic activity also had minimal effects (Figure 8A).
Figure 8.

Analysis of NDR1 Activating Mutations in Cells
(A) TEAD luciferase reporter analysis of NDR1 in Hippo signaling. SW480 cells were co-transfected with the indicated MYC-tagged NDR1 constructs or empty vector, a TEAD luciferase reporter, and a Renilla luciferase control reporter. Luciferase activities were normalized for Renilla luciferase control reporter activities and expressed relative to reporter activities in the absence of NDR1 (vector). The western blot illustrates NDR1 expression levels (β-actin was detected in parallel as a loading control). Asterisks indicate a band seen in the absence of MYC-NDR1 expression and in non-transfected cells, which likely represents endogenous c-MYC. n = 10 experiments (nine in technical duplicate, one singlet); error bars, SEM. Note that transiently expressed MYC-NDR1 Δ38G was consistently expressed at lower levels than other NDR1 constructs.
(B) Affinity purification coupled mass spectrometry analysis of NDR1 interactions in cells. Cells stably expressing BirA∗-FLAG-tagged wild-type NDR1 or NDR1 harboring a Δ38G deletion were induced with tetracycline (total 24 hr) and treated with 150 nM okadaic acid or DMSO for the last 2.5 hr of induction. Affinity purification coupled to mass spectrometry was performed in biological duplicates alongside negative controls, and high-confidence interactions were scored with SAINTexpress and visualized with ProHits-viz (see legend inset). Only interactions with previously reported or suspected regulators are depicted here; the complete list of high-confidence interactions is given in Table S1. OA, okadaic acid.
(C) Validation of the increased interactions between NDR1 Δ38G and FRYL by immunoprecipitation and immunoblotting. FLAG immunoprecipitates or total lysates were incubated with antibodies to FLAG or endogenous FRYL; the negative control consisted of the parental cell line.
As an alternative means to assess structure-function relationships in cells, we therefore took a systems-wide approach using interaction proteomics to analyze the consequences of the most potent Δ38G deletion mutation on the interactions established by NDR1. We generated stable cell lines inducibly expressing BirA∗-FLAG-tagged versions of NDR1 wild-type and Δ38G (the 5A mutant was expressed to markedly lower levels and thus could not be used in this study). A set of cells was treated with okadaic acid to inhibit PP2A-family phosphatases, a treatment that led to multiple changes in protein-protein interaction within the Hippo signaling pathway in our previous study (Couzens et al., 2013). FLAG affinity purification was performed in biological duplicates for each condition and results analyzed alongside negative controls by mass spectrometry (see STAR Methods for details). Spectral counts were further evaluated with the statistical tool SAINTexpress (Teo et al., 2014) to identify high-confidence interaction partners, and the resulting data were visualized with ProHits-viz (Knight et al., 2017).
Wild-type NDR1 purifications readily recovered MOB1A, and the Δ38G activation loop deletion did not abrogate this interaction, although okadaic acid treatment appeared to potentiate it (Figure 6A). A similar pattern was observed for MOB2, a paralog of MOB1 that can associate with NDR1 and NDR2 but not with LATS1 or LATS2 (Bichsel et al., 2004, Hergovich et al., 2005, Xiong et al., 2017). Interestingly, while the Δ38G mutant did not lose any interactors relative to the wild-type protein, it did yield many additional interactors upon treatment with okadaic acid. Among these were the upstream activator kinases STK3 and STK4 (also known as MST2 and MST1, respectively) and their partners in Hippo pathway signaling, SAV1 and RASSF2 (interactors previously linked to NDR kinases are shown in Figure 8B; all interactions are reported in Table S1). These observations provided evidence that, in the context of living cells, the extended activation segment of NDR1 may play an important regulatory role for the recruitment of kinase activators. Since MOB1A recruits these components in a phospho-dependent manner through direct interaction with an auto-phosphorylation site in STK3 and STK4 (Couzens et al., 2013, Couzens et al., 2017, Ni et al., 2015, Rock et al., 2013, Xiong et al., 2017), we were not surprised to see an okadaic acid-mediated induction. However, the absence of interaction in the context of the wild-type protein suggests an inhibitory role of the activation segment in enabling these associations.
Besides the Hippo pathway components, another protein selectively recruited to the Δ38G mutant was the less well-characterized activator Furry (FRYL), whose orthologs work as critical scaffolds for NDR kinases (Bogomolnaya et al., 2006, Chiba et al., 2009, Emoto et al., 2004, Fang and Adler, 2010, Fang et al., 2010, He et al., 2005, Nelson et al., 2003). FRYL association with NDR1 was further enhanced by phosphatase inhibition (Figures 8B and 8C). This provides evidence suggesting that the interaction between FRYL and NDR is a regulated event.
To validate our interaction proteomics observations for FRYL, cells expressing the BirA∗-FLAG tagged wild-type and Δ38G constructs as above were treated with okadaic acid or DMSO and used in an immunoprecipitation/immunoblotting experiment to detect the recovery of FRYL with an antibody to the endogenous protein. FRYL was recovered in the precipitates of wild-type NDR1 but not in the negative control, and this interaction was far more pronounced for the Δ38G protein (Figure 8C). Furthermore, inhibition of phosphatases by okadaic acid potentiated this interaction (Figure 8C). These findings validate our proteomics observations that the atypically long activation segment of NDR1 regulates its association with the known activator FRYL.
Discussion
NDR family kinases are regulated by distinct mechanisms, notably binding of the MOB proteins, auto-phosphorylation within its atypically long activation segment, transphosphorylation on the HM by upstream kinases, and association with scaffolding proteins (SAV1, RASSF2, Furry family). However, the relative contribution of these different regulatory steps toward kinase activation and signaling, as well as the potential for synergy, had not been fully elucidated.
Here, we described the high-resolution structure of an inactive NDR1 kinase domain, revealing a completely resolved and atypically long activation segment and fully ordered helix αC. Both in vitro kinase assays and HDX-MS experiments demonstrate that regulation of NDR1 kinase by MOB1 binding and by modulation of the elongated activation segment occur through independent mechanisms. Our in vitro kinase analyses also showed that disrupting the interaction of the elongated activation segment with the surrounding kinase domain dramatically increased NDR1 phosphorylation of RAB3IP primarily at the level of kcat and not Km. Whether other substrates of NDR1, which may use altogether different modes of engagement to the NDR1 kinase domain, would be similarly affected remains to be determined.
In addition to increasing the intrinsic kinase activity of NDR1, disruption of the atypically long activation segment drastically increased association of NDR1 with the upstream kinases STK3 and STK4, which target the NDR1 HM for phosphorylation, and with the Furry family scaffold, FRYL. This finding suggested that the activation segment of NDR1 may control NDR1 interaction with physiological regulators, although the basis for this effect, including how the auto-inhibitory conformation of the activation segment is relieved, remains to be determined. However, the finding that the binding of NDR1 to STK3, STK4, and FRYL was further stimulated by the inhibition of phosphatases with okadaic acid suggests that phosphorylation plays an integral role in the recruitment of these factors. For STK3 and STK4, we hypothesize that recruitment occurs through their direct interaction partner MOB1, which binds to the auto-phosphorylated forms of both protein kinases (Couzens et al., 2013, Couzens et al., 2017, Ni et al., 2015, Rock et al., 2013, Xiong et al., 2017). Why okadaic acid is not able to stimulate STK3 and STK4 binding to wild-type NDR1 remains an open question.
The NDR family kinase Warts was first identified in Drosophila as a tumor suppressor and subsequently found conserved in mammals with the two human orthologs (LATS1 and LATS2) playing a key role in regulating the Hippo tumor suppressor pathway. Despite the fast pace at which the Hippo pathway components have been identified and characterized for function, few studies have focused on the mechanistic details of how the catalytic output of LATS1/2 is regulated. Given the similarity between all NDR family kinases, our detailed studies on NDR1 may provide a useful model for understanding the general principles that govern the regulation of other NDR family members, such as LATS1/2.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Sheep anti-mouse IgG coupled to HRP | GE Healthcare | Cat#NA931; RRID:AB_772210 |
| Donkey anti-rabbit IgG coupled to HRP | GE Healthcare | Cat#NA934; RRID:AB_772206 |
| Mouse anti-FLAG M2 coupled to HRP | Sigma-Aldrich | Cat#A8592-.2MG; RRID:AB_439702 |
| Rabbit anti-FRYL antibody | Bethyl | Cat#A302-643A-M; RRID:AB_1058725 |
| Magnetic anti-FLAG M2 beads slurry | Sigma-Aldrich | Cat#M8823; RRID:AB_2637089 |
| Mouse anti-MYC 9E10 IgG | Thermo Fisher Scientific | Cat#MA1-81358; RRID:AB_930029 |
| Mouse anti-beta-actin IgG (AC-15) | Abcam | Cat#ab6276; RRID:AB_2223210 |
| Goat anti-mouse IgG coupled to DyLight 680 | Thermo Fisher Scientific | Cat#35518; RRID:AB_614942 |
| Bacterial and Virus Strains | ||
| E. coli BL21 (DE3) CodonPlus-RIL | Agilent | Cat#230280 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Okadaic acid | LC Labs | Cat#O-5857-1mg |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat#P8340 |
| ECL prime Western blotting detection reagent | GE Healthcare | Cat#45002401 |
| Lipofectamine 2000 transfection reagent | Thermo Fisher Scientific | Cat#11668019 |
| 32P-γ-ATP | Perkin Elmer | Cat#BLU002Z250UC |
| Adenosine 5′-(β,γ-imido)triphosphate lithium salt hydrate (AMP-PNP) | Sigma-Aldrich | Cat#A2647-5MG |
| Adenosine 5′-triphosphate (ATP) disodium salt hydrate (ATP) | Sigma-Aldrich | Cat#A26209-10G |
| Critical Commercial Assays | ||
| Dual-Luciferase® Reporter Assay System | Promega | Cat#E1910 |
| Deposited Data | ||
| Interaction proteomics dataset | Xiong et al., 2017; MassIVE | https://massive.ucsd.edu/ (MassIVE ID MSV00008179 ) |
| Interaction proteomics dataset | Xiong et al., 2017; ProteomeXchange | http://www.proteomexchange.org/ (ProteomeXchange ID PXD008416) |
| Protein Data Bank | This study | PDB ID: 6BXI |
| Experimental Models: Cell Lines | ||
| HEK293 Flp-In T-REx | Invitrogen | Cat#R780-07, RRID:CVCL_U427 |
| SW480 | gift from Chris Lord, ICR; originally obtained from ATCC | Cat#CCL-228 |
| Oligonucleotides | ||
| hsNDR1(nco,m12)f | ccgCTCGAGttatctatttcttttccaggtttc | Sigma |
| hsNDR1(i418,stp,kpn)r | catgCCATGGctatgagtaacc | Sigma |
| hsNDR1(k238a)f | cggGGTACCTTAaatatcagattctg | Sigma |
| hsNDR1(k238a)r | ctttgcacaggactggcaaaagcacataggac | Sigma |
| hsNDR1(h241a)f | gtcctatgtgcttttgccagtcctgtgcaaag | Sigma |
| hsNDR1(h241a)r | ggactgaaaaaagcagctaggacagaattttatagg | Sigma |
| hsNDR1(d256a)f | cctataaaattctgtcctagctgcttttttcagtcc | Sigma |
| hsNDR1(d256a)r | cagcctccccagtgctttcactttccag | Sigma |
| hsNDR1(w271c)f | ctggaaagtgaaagcactggggaggctg | Sigma |
| hsNDR1(w271c)r | ggaaagcagaaacctgtaaaagaaatagacg | Sigma |
| See Table S2 for the complete primer list | ||
| Recombinant DNA | ||
| pDEST-pcDNA5-BirA∗-FLAG N-term | Couzens et al., 2013 | Vector ID: V8164 (LTRI database) |
| pDEST-pcDNA5-BirA∗-FLAG N-term-NDR1 and derivatives | this study | N/A |
| pETM-30 | EMBL | https://www.embl.de/pepcore/pepcore_services/cloning/choice_vector/ecoli/embl/popup_emblvectors/ |
| pETM-30-Hs NDR1(82-418) and derivatives | this study | N/A |
| pETM-30-Hs NDR1(12-418) and derivatives | this study | N/A |
| 8xGTIIC-luciferase | obtained from Stefano Piccolo via Addgene (Dupont et al., 2011) | Cat#34615 |
| ptkRL Renilla luciferase control plasmid | Promega | N/A |
| pDNR-MCS SA | modified from Clontech pDNR Dual (Colwill et al., 2006) | N/A |
| pcDNA5 FRT TO-MYC-NDR1 and derivatives | this study | N/A |
| Software and Algorithms | ||
| MS data storage and analysis: ProHits | Liu et al., 2010 | N/A |
| MS data, Significance Analysis of INTeractome analysis: SAINT | Teo et al., 2014 | Version exp3.3 |
| MS data visualization: ProHits-viz | Knight et al., 2017 | N/A |
| HDX workbench | Scripps Institute Florida | http://hdx.florida.scripps.edu/hdx_workbench/Home.html |
| Coot | Emsley and Cowtan, 2004 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Afonine et al., 2012 | http://www.phenix-online.org/ |
| PyMol | Schrodinger | CCP4 program suite |
| Refmac | Winn et al., 2003 | CCP4 program suite |
| Prism 7 | GraphPad Software, Inc. | http://www.graphpad.com |
| Other | ||
| LTQ Orbitrap Elite / Velos | Thermo Fisher Scientific | N/A |
| Odyssey® Imaging System | LI-COR | N/A |
| Victor X5 Multilabel Plate Reader | PerkinElmer | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact Frank Sicheri (sicheri@lunenefeld.ca).
Experimental Model and Subject Details
For the TEAD reporter assays, SW480 colorectal adenocarcinoma cells (male) were obtained from Chris Lord (ICR, London), who purchased the cells from ATCC (see table above) and confirmed cell line identity in the expanded laboratory stock by STR typing (March 2016). The cell stock was also confirmed to be free of mycoplasma. The cells were grown at 37°C in DMEM containing 4,500 mg/L glucose, supplemented with 10% Fetal Bovine Serum and antibiotics (100 mg/L streptomycin sulfate, 60 mg/L benzylpenicillin).
For interaction proteomics, Flp-In T-REx 293 cell lines (female) were obtained from Invitrogen (see table above) and grown at 37°C in DMEM high glucose supplemented with 5% Fetal Bovine Serum, 5% Cosmic calf serum and 100 U/mL penicillin/streptomycin (growth media). The parental cell line is routinely monitored for mycoplasma contamination but has not been independently authenticated after purchasing from Invitrogen.
Method Details
Recombinant Protein Expression and Purification
Human wild-type NDR1KD (amino acids 82–418), NDR1MBD-KD (amino acids 12–418), LATS1KD (amino acids 698–1049), MOB1A (amino acids 2–216), RAB3A-interacting protein (RAB3IP, also known as Rabin8; amino acids 2–476) and all mutant variants were expressed in E. coli BL21 (DE3) CodonPlus-RIL cells as Tobacco Etch Virus (TEV) protease-cleavable, dual N-terminal 6xhistidine (His) and glutathione S-transferase (GST) fusion proteins using a modified pETM-30 vector. His-GST tagged proteins were purified in batch on glutathione-sepharose resin and eluted by cleavage with TEV protease. TEV protease was removed from the eluted protein by subtractive immobilized Ni-NTA affinity chromatography. Cleaved proteins were concentrated and further purified by size exclusion chromatography (SEC) using a Superdex 75 120 ml column pre-equilibrated in 25 mM HEPES-NaOH pH 7.5, 100 mM NaCl, and 3 mM DTT. Pooled fractions were concentrated to 50 mg/ml, flash-frozen in liquid nitrogen and stored at -80°C. For the expression and purification of GST-YAP50–171 (amino acids 50 to 171) and other GST-fusions as indicated, the TEV cleavage step was omitted, and proteins were eluted from glutathione-sepharose resin with glutathione.
Sedimentation Velocity Analytical Ultracentrifugation
Concentrations of NDR1, ranging from 5 μM to 40 μM, were tested minimally in duplicate in analytical ultracentrifugation buffer (15 mM HEPES-NaOH pH 7.0, 200 mM NaCl, 3 mM DTT). Samples were loaded to the two-channel charcoal/Epon60K sedimentation velocity cells, with approximately 350 μl per channel. Centrifugation was conducted with a Beckman ProteomeLab XL-I (Bechman Coulter, USA) at a speed of 25,000 RPM and a temperature of 20°C using an An-60Ti rotor. This produces a depletion of solute near the meniscus and the formation of a sharp boundary between the depleted region and the uniform concentration of sedimenting solutes. For the ultracentrifugation sedimentation velocity analysis, at least 200 scans of samples were measured at a wavelength of 280 nm for each experiment over 7.5 hours, a period of time sufficient for the major concentration boundary to migrate to the bottom of the cell. The test mode was based on the sedimentation velocity, and a continuous c(s) distribution was applied in the analysis mode. The derivative profiles were used to calculate the experimental sedimentation coefficient (S-values). The raw dataset of S-values was analyzed by Sedfit (Schuck, 2000), with the model c(s) destruction and proper parameters. Data were plotted in Prism (GraphPad).
Crystallization and Data Collection
Crystals of human NDR1KD bound to AMP-PNP (adenylyl-imidodiphosphate), of the space group C2 (a=117.19 Å, b=117.97 Å, c=95.04 Å, alpha=90°, beta=124.88°, gamma=90°) with two molecules in the asymmetric unit, were obtained using the hanging drop method by mixing equal volumes of 10 mg/ml protein solution (25 mM HEPES-NaOH pH 7.5, 100 mM NaCl, 3 mM DTT, and 5 mM MgCl2) with precipitant (4 M ammonium acetate, 0.1 M Bis-Tris propane pH 7.0) in a 1:1 ratio over a reservoir containing 20 μl of precipitant. NDR1KD crystals formed at 20°C after 48-72 hours. For X-ray diffraction experiments, crystals were flash-frozen in mother liquor supplemented with 20%–25% (v/v) ethylene glycol.
Structure Solution and Refinement
An X-ray diffraction data set was collected at the Advanced Photon Source (beamline NECAT-24-ID-C) and processed using the HKL2000 software suite (Otwinowski and Minor, 1997). The human NDR1KD structure was solved by molecular replacement with Phaser (McCoy, 2007) using the C-lobe of RSK-1 kinase domain (amino acids 148-333; PDB ID: 2Z7Q) as a search model. Model autobuilding was carried out by Buccaneer (Cowtan, 2006) with manual corrections using Coot (Emsley and Cowtan, 2004). Refinement was performed with Refmac (Winn et al., 2003). Structure representations were generated using PyMol (PyMOL Molecualr Graphics System, Version 2). Simulated annealing omit maps were generated by Phenix (Afonine et al., 2012).
Radiometric In Vitro Kinase Assay
Protein kinase assays were performed using [gamma-32P]-ATP (Perkin Elmer). For each 10 μl reaction, 1 μCi of radiolabeled ATP, 150 μM unlabeled ATP, 2 mM MgCl2, and 2 μM NDR1 or LATS1 kinases were used to assess kinase autophosphorylation activity at 30°C for 30 minutes. To assess NDR1 or LATS1 substrate phosphorylation, final concentrations of 6 μM RAB3IP or 20 μM GST-YAP121-133 were added. The reactions were stopped by the addition of Laemmli (sodium dodecylsulfate, SDS) sample loading buffer and incubation at 95°C for 10 minutes. Reactions were resolved by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by autoradiography using a Typhoon PhosphorImager (Molecular Dynamics). The resulting autoradiograph was analyzed by ImageQuant (GE Healthcare). The calculation of the kinetic parameters was performed by fitting the Michaelis-Menten function (y=ax/b+x) to the data presented in the Figure 7 using GraphPad-Prism 5.0. n=3 experiments done in technical duplicate; error bars, SEM.
Limited Proteolysis
20-μl reactions containing 500 ng/μl of NDR1MBD-KD were incubated with 0.1 ng/μl trypsin (Sigma-Aldrich) in 25 mM HEPES-NaOH pH 7.0, 200 mM NaCl, and 3 mM DTT at room temperature. A time course of partial proteolysis was carried out, and the reactions were stopped at 0 min, 15 min, 30 min, 60 min, 120 min, and 240 min by the addition of SDS sample buffer and heated to 95°C for 10 minutes. When ATP and MgCl2 were used during trypsin treatment, the kinase : ATP : MgCl2 concentration ratio was set at 1:5:50. Reaction samples of the partial proteolysis were resolved by SDS-PAGE and stained with Coomassie Blue as seen in Figure S2A.
N-terminal boundaries of the indicated protein species were determined by Edman degradation at the SPARC BioCentre at The Hospital for Sick Children, Toronto. Briefly, the reaction samples were resolved by SDS-PAGE; proteins were subsequently transferred to a polyvinylidene fluoride (PVDF) membrane and stained with Coomassie blue. Desired protein bands were excised from the stained membrane and analyzed using an Applied Biosystems Procise 491C Protein Sequencing System (Thermo Fisher Scientific & Perkin Elmer, USA). For detailed mass spectrometry services provided by SPARC BioCentre, please visit http://lab.research.sickkids.ca/sparc-molecular-analysis/services/amino-acid-analysis-and-edman-sequencing/edman-sequencing/.
Intact Mass Determination by Mass Spectrometry
The integrity and phosphorylation status of wild-type NDR1KD employed for crystallization studies was determined using ESI-LC/MS at the AIMS Mass Spectrometry Laboratory in the Department of Chemistry at the University of Toronto. 20 μl of 200 μM protein sample in 25 mM HEPES-NaOH pH 7.5, 100 mM NaCl, and 2 mM DTT was submitted for analysis. Sample was diluted in water 1000-fold prior to analysis using an Agilent 6538 Q-TOF interfaced to a 1290 ultrahigh pressure HPLC. A list of the ionic formulae was generated by the Molecular Formula Generation Algorithm as implemented in the MassHunter data processing software. The Molecular Formulae Generation Score takes into account the best match for the mass accuracies and relative abundances of the isotopomers identified for the target isotopic envelope. The standard m/z acquisition range for biomolecule analysis was set between 200–2000 Da; however, the mass spectrum was expanded to highlight the appropriate regions of interest. The reconstructed mass spectrum was generated by the Maximum Entropy algorithm in a plot of neutral mass (Da) versus intensity. Selected peaks were labeled with mass values rounded to one decimal place. Data processing was performed with the MassHunter BioConfirm software package.
Hydrogen-Deuterium Exchange Coupled Mass Spectrometry
Wild-type NDR1MBD-KD and its 5A activating mutant variant, without or with MOB1A at 1:1 molar ratio, were incubated on ice with a tenfold excess of AMP-PNP for 10 minutes prior to starting the time course. For each time course, the sample was diluted into 84.5% D2O (v/v), and the final concentration of NDR1 protein was 4.5 μM. The buffer conditions were kept constant with 25 mM HEPES-NaOH pH 7.5, 200 mM NaCl, 2 mM MgCl2 and 3 mM DTT. A set of standard peptides was spiked into the D2O buffer before adding the protein to estimate the methodical back exchange. At each desired time point, aliquots of 10 μL were removed from the pool and flash-frozen in liquid nitrogen. Quenching was done by adding an equal volume 50 mM ice-cold succinic/citric acid (1:1) directly before loading the sample onto the HPLC (Eksigent Tempo, AB SCIEX) system. Pepsin digestion was performed online at room temperature (21°C) on an in-house made fused silica column filled with immobilized pepsin (Poros Pepsin, Applied Biosciences). Trap (Poros R1, Applied Biosciences) and analytical column (Reprosil C18) were cast in-house as well and immersed in water/ice slurry during the analysis. The sample was loaded directly from the pepsin column onto the trap for 4 minutes. The gradient for the analytical column was 15 minutes going from 10% buffer A (H2O, 0.1% formic acid) to 60% buffer B (acetonitrile, 0.1% formic acid), followed by 10 minutes wash at 80% buffer B. The complete run time was 35 minutes. The analysis was followed by two 15-minutes wash gradients to ensure that there was no carry-over between samples. An Orbitrap XL was used as the back end and coupled to the HPLC via a nano-electrospray interface. HDX Workbench (Scripps Institute Florida) was used to analyze the data.
FLAG Affinity Purification Coupled with Mass Spectrometry (AP-MS) for NDR1 Mutants
Epitope-tagged constructs were generated via Gateway cloning into pDEST 5’BirA∗-FLAG pcDNA5 FRT TO. Point mutations and deletions were introduced by polymerase chain reaction-directed mutagenesis (the position of the mutated amino acids is indicated on the basis of the reference sequence). All constructs were sequence-verified. Stable cell lines were generated as Flp-In 293 T-REx cell pools as described (Kean et al., 2012), and expression was induced for 24 hours with tetracycline (1 μg/ml; see cell culture conditions above). Okadaic acid (OA; LC Labs, O-5857-1mg) was added at a concentration of 150 nM for 2.5 hours unless otherwise indicated, and DMSO was used as a negative control. In each case, one 15-cm plate was used per biological replicate, and two biological replicates were performed. Negative controls consisted of cells expressing BirA∗-FLAG alone that were processed in parallel to the true biological samples.
Cell pellets from one 15-cm plate treated as indicated (150 nM OA, or DMSO as vehicle control for 2.5 hours) were lysed in 50 mM HEPES-KOH pH 8.0, 100 mM KCl, 2 mM EDTA, 0.1% NP-40, and 10% glycerol containing PMSF (1 mM) and Sigma-Aldrich protease inhibitor P8340 (1:500) and affinity-purified with M2-FLAG magnetic beads and on-bead digest as described (Kean et al., 2012). Peptides were analyzed by nano-LCMS using a home-packed 0.75 μm x 10 cm C18 emitter tip (Reprosil-Pur 120 C18-AQ, 3 μm). A NanoLC-Ultra HPLC system (Eksigent) was coupled to an LTQ Orbitrap Elite or Velos (Thermo Fisher Scientific), and samples were analyzed in data-dependent acquisition mode. A 60,000 resolution MS scan was followed by 10 CID MS/MS ion trap scans on multiple charged precursor ions with a dynamic exclusion of 20 seconds. The LC gradient was delivered at 200 nl/minute and consisted of a ramp of 2-35% acetonitrile (0.1% formic acid) over 90 minutes, 35-80% acetonitrile (0.1% formic acid) over 5 milliseconds, 80% acetonitrile (0.1% formic acid) for 5 minutes, and then 2% acetonitrile for 20 minutes.
MS Data Analysis for FLAG AP-MS for NDR1 Mutants
Raw files were converted to mzXML and mgf files using ProteoWizard 3.0.4468 (Kessner et al., 2008) and analyzed using the iProphet pipeline (Shteynberg et al., 2011) implemented within ProHits (Liu et al., 2010) as follows. The database consisted of the human and adenovirus sequences in the RefSeq protein database (version 57) supplemented with “common contaminants” from the Max Planck Institute (http://141.61.102.106:8080/share.cgi?ssid=0f2gfuB) and the Global Proteome Machine (GPM; http://www.thegpm.org/crap/index.html). The search database consisted of forward and reverse sequences (labeled “gi|9999” or “DECOY”); in total, 72,226 entries were searched. Spectra were analyzed separately using Mascot (2.3.02; Matrix Science) and Comet [2012.01 rev.3 (Eng et al., 2013)] for trypsin specificity with up to two missed cleavages; deamidation (Asn or Gln) and oxidation (Met) as variable modifications; the mass tolerance of the precursor ion was set at +/-12 parts per million (ppm), the fragment ion tolerance at +/- 0.6 amu. The resulting Comet and Mascot results were individually processed by PeptideProphet (Keller et al., 2002) and combined into a final iProphet output using the Trans-Proteomic Pipeline (TPP; Linux version, v0.0 Development trunk rev 0, Build 201303061711). TPP options were as follows: general options were -p0.05 -x20 -d“gi|9999,” iProphet options were –ipPRIME, and PeptideProphet options were –OpdP. All proteins with a minimal iProphet probability of 0.05 were parsed to the relational module of ProHits. For analysis with SAINT, only proteins with an iProphet protein probability of >0.95 were considered. Hits were also restricted to those detected with a minimum of 2 unique peptides.
Validation of the Interaction with FRYL
Flp-In T-REx cells expressing BirA∗-FLAG-tagged versions of NDR1 wild-type or the Δ38G mutant were induced for expression, treated with okadaic acid and harvested and lysed in 1 ml lysis buffer as above. FLAG immunoprecipitation with 12.5 μl packed resin was performed for 2 hours at 4°C, and resin was washed thrice with lysis buffer prior to elution in boiling 2X Laemmli (SDS) sample buffer (50 μl). Immunoprecipitates and total lysates were run on precast Criterion gels (4-15%; Bio-Rad, 5671084), and proteins were transferred onto nitrocellulose membranes by Western blotting. Membranes were blocked in 5% milk in TBS-T; the high-molecular-weight area (> ∼120 kDa) of the membrane was incubated with rabbit anti-FRYL antibody (Bethyl, A302-643A-M; 1:500) while the lower- molecular-weight area was incubated with mouse anti-FLAG antibody (Sigma-Aldrich, F3165; 1:1000). Horseradish-peroxidase-coupled secondary antibodies were from GE Healthcare (anti-mouse IgG, NA931-1ML and ECL anti-rabbit IgG, NA934V), and chemiluminescence detection was performed using ECL prime Western blotting detection reagent (GE Healthcare, 45002401) on film (Clonex BioFlex MSI Film, 2479-MED-CLM5810).
TEAD Luciferase Reporter Assay
SW480 colorectal adenocarcinoma cells were plated on white 96-well tissue culture plates (30,000 cells/well) and after one day transfected with 15 ng/well of the 8xGTIIC-luciferase YAP/TAZ-responsive reporter plasmid (Addgene plasmid # 34615, obtained from Stefano Piccolo; Dupont et al., 2011), 2 ng/well of ptkRL (Renilla luciferase control plasmid, Promega) and 80 ng/well of pcDNA5 FRT TO-MYC-NDR1 constructs or the corresponding empty vector, filled up to a total of 100 ng of DNA/well with empty pDNR-MCS SA lacking a promoter (Colwill et al., 2006) as carrier DNA. Transfections were done using Lipofectamine 2000 (Thermo Fisher Scientific / Invitrogen) with a DNA:transfectant ratio of 1:3 in Opti-MEM II. Media were changed for DMEM with 10% FBS 4 hours after the addition of DNA complexes. Twenty hours after media change, samples were processed for luminescence readings using the Dual-Luciferase Reporter Assay system (Promega) and a Perkin Elmer VICTOR X5 plate reader (5 seconds integration time). Nine luciferase reporter experiments were performed in technical duplicate. Individual Firefly and Renilla Luciferase activity readings were first background-subtracted. The background-subtracted Firefly Luciferase readings were normalised for cell number and transfection efficiency using the background-corrected Renilla Luciferase readings. The resulting values from technical duplicates were averaged and expressed relative to the vector-only data. A third technical replicate (from transfections with the same master mix) was analysed for MYC-NDR1 expression, but more robust detection of overexpressed MYC-NDR1 by Western blotting (see Figure 8A) required larger sample volumes. Transfections for a tenth experiment were therefore set up in 6-well plates, using the same transfection master mix as for 96-well plates and scaling up the number of cells and quantity of reagents by a factor of 25. Small volumes of the lysate samples were analyzed for luciferase activity as before (from a single set of samples) while larger volumes of lysates from the same samples were analyzed by SDS-PAGE and Western blotting, using anti-MYC 9E10 (MA1-81358, Thermo Fisher Scientific, 1:1,000) and anti-β-actin (Abcam, AC-15, ab6276, 1:10,000) primary antibodies. Western blot signals, from a secondary goat-anti-mouse-DyLight680 antibody (35518, Thermo Fisher Scientific), were detected using an Odyssey infrared imaging system (LI-COR).
Quantification and Statistical Analysis
Kinase reactions were resolved by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by autoradiography using a Typhoon PhosphorImager (Molecular Dynamics). The resulting autoradiograph was analyzed by ImageQuant (GE Healthcare). The calculation of the kinetic parameters was performed by fitting the Michaelis-Menten function (y=ax/b+x) to the data presented in the Figure 7 using GraphPad-Prism 5.0. n=3 experiments done in technical duplicate; error bars, SEM.
HDX Workbench (Scripps Institute Florida) was used to analyze the Hydrogen-deuterium exchange coupled mass spectrometry experiments. For each set of experiments, the percentage of deuterium incorporation and the corresponding time points were extracted from the HDX workbench and plotted using simple non-linear regression. The results were presented in Figure 6 using GraphPad-Prism 5.0. n=3 experiments done in technical duplicate; error bars, SEM.
Luciferase reporter data were from nine experiments, each performed in technical duplicate, and a scaled-up tenth experiment without technical replicate performed for the parallel, robust detection of NDR1 expression levels (see Method Details). Raw Firefly Luciferase and Renilla Luciferase luminescence values were background-subtracted, and normalised reporter activity values calculated for each replicate by dividing the background-subtracted Firefly Luciferase by the corresponding Renilla Luciferase values for each sample. The normalised values from technical replicates were then averaged and used to calculate the relative reporter activity, as described in the Method Details. Data were expressed as means with SEM.
For the statistical analysis of AP-MS data (see above for the scoring of the mass spectrometry data), each bait was analyzed in biological duplicate from cells grown, treated, and processed at different times to maximize the variability and increase the robustness in the detection of true interactors. Cells expressing the BirA∗-FLAG tag alone were used as negative controls and processed in parallel to mitigate “batch effect” artifacts (Breitkreutz et al., 2010). The quality of each sample was assessed by manually aligning the runs for the biological replicates in ProHits. SAINTexpress version 3.6.1 was used with default options (Teo et al., 2014). 14 negative control experiments consisting of a) cells expressing FLAG alone treated with DMSO (3) or OA (3) or b) cells expressing BirA∗-FLAG alone treated with DMSO (4) or OA (4) were compressed to 7 virtual controls for SAINTexpress scoring to increase robustness (as in Mellacheruvu et al., 2013). Proteins with FDR (False Discovery Rate, Bayesian-estimation) of ≤1% were considered true positive interactions. Visualization of the interactions as dot plots was through prohits-viz.lunenfeld.ca (Knight et al., 2017) using the default options. Selecting the 1% FDR threshold first identifies all interactors that pass the cut-off with at least one bait; in a second step, all quantitative information (i.e. averaged spectral counts) for all these interactors across all baits is recovered as a matrix, with associated FDRs for each bait-prey pair. This second step prevents bias in which a protein detected with several spectral counts would pass the FDR cut-off with one bait, but miss it in the other (with would otherwise be shown as an erroneous “0” value). On these dot plots, the color intensity maps to the averaged spectral counts across both replicates (capped at a maximal value), while the size of the circles is proportional to the maximal spectral count value for the bait across all samples analyzed in parallel. The confidence score from SAINTexpress is mapped as the edge color.
Data and Software Availability
The coordinates of the structures together with the structure factors are deposited at the Protein Data Bank. Accession codes and homepages of the software are available in the Key Resources Table. The accession numbers for the NDR1 kinase domain structures reported in this paper are PDB code: 6BXI.
AP-MS data: This dataset consisting of 22 raw files and associated peak list and results files has been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV00008179 and PXD008416, ftp://massive.ucsd.edu/MSV000081798.
HDX workbench software is available at Scripps Institute Florida (http://hdx.florida.scripps.edu/hdx_workbench/Download_Info.html).
Acknowledgments
We are grateful to Hala Abdouni for technical assistance and to Brett Larsen for help with mass spectrometry. A.-C.G. and F.S. are the Canada Research Chairs (Tier 1) in Functional Proteomics and Structural Biology of Cell Signaling, respectively. Work in the Guettler laboratory is supported by The Institute of Cancer Research (ICR), Cancer Research UK through a Career Establishment Award to S.G. (C47521/A16217) and the Lister Institute of Preventive Medicine. Work in the Gingras and Sicheri laboratories was supported by the Canadian Institutes of Health Research (FDN 143301 and 143277, respectively) and from the Terry Fox Research Institute Program Project grant. Proteomics work was performed at the Network Biology Collaborative Center at the Lunenfeld-Tanenbaum Research Institute, a facility supported by Canada Foundation for Innovation funding, by the Ontarian Government and by Genome Canada and Ontario Genomics (OGI-139). Diffraction work conducted at the Northeastern Collaborative Access Team beamlines was funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403) and by an NIH-ORIP HEI grant (S10 RR029205). S.X. is supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Alexander Graham Bell Scholarship. S.G. was supported by postdoctoral fellowships from the Human Frontier Science Program (HFSP) and the European Molecular Biology Organization (EMBO).
Author Contributions
S.X. designed and performed biochemical, biophysical and structural analyses. K.L. designed and performed HDX-MS; A.L.C. designed and performed interaction proteomics; C.M.T. designed and performed the TEAD reporter assays. D.R. performed immunoprecipitation coupled to immunoblotting, S.G. and D.Y.L.M. designed and made all the clones used in this paper; S.G. performed limited proteolysis and initiated structure-function studies. Y.-C.J. contributed to the initial structural model building. D.C. performed the analytical ultracentrifugation experiments. I.K. collected X-ray diffraction data. S.G. supervised C.M.T. and initially S.X.; A.-C.G. supervised A.L.C. and D.R.; and F.S. supervised S.X., D.Y.L.M., Y.-C.J., and D.C. S.X., S.G., A.-C.G., and F.S. conceived the project and wrote the manuscript with input from all authors.
Declaration of Interests
The authors declare no competing interests.
Published: July 5, 2018
Footnotes
Supplemental Information includes nine figures and two tables and can be found with this article online at https://doi.org/10.1016/j.str.2018.05.014.
Contributor Information
Sebastian Guettler, Email: sebastian.guettler@icr.ac.uk.
Anne-Claude Gingras, Email: gingras@lunenfeld.ca.
Frank Sicheri, Email: sicheri@lunenfeld.ca.
Supplemental Information
References
- Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin A.J., Amon A. Men and sin: what's the difference? Nat. Rev. Mol. Cell Biol. 2001;2:815–826. doi: 10.1038/35099020. [DOI] [PubMed] [Google Scholar]
- Bichsel S.J., Tamaskovic R., Stegert M.R., Hemmings B.A. Mechanism of activation of NDR (nuclear Dbf2-related) protein kinase by the hMOB1 protein. J. Biol. Chem. 2004;279:35228–35235. doi: 10.1074/jbc.M404542200. [DOI] [PubMed] [Google Scholar]
- Bogomolnaya L.M., Pathak R., Guo J., Polymenis M. Roles of the RAM signaling network in cell cycle progression in Saccharomyces cerevisiae. Curr. Genet. 2006;49:384–392. doi: 10.1007/s00294-006-0069-y. [DOI] [PubMed] [Google Scholar]
- Bond C.S., Schuttelkopf A.W. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 2009;65:510–512. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
- Breitkreutz A., Choi H., Sharom J.R., Boucher L., Neduva V., Larsen B., Lin Z.Y., Breitkreutz B.J., Stark C., Liu G. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.H., Nousiainen M., Chalamalasetty R.B., Schafer A., Nigg E.A., Sillje H.H. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- Chiba S., Amagai Y., Homma Y., Fukuda M., Mizuno K. NDR2-mediated Rabin8 phosphorylation is crucial for ciliogenesis by switching binding specificity from phosphatidylserine to Sec15. EMBO J. 2013;32:874–885. doi: 10.1038/emboj.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S., Ikeda M., Katsunuma K., Ohashi K., Mizuno K. MST2- and Furry-mediated activation of NDR1 kinase is critical for precise alignment of mitotic chromosomes. Curr. Biol. 2009;19:675–681. doi: 10.1016/j.cub.2009.02.054. [DOI] [PubMed] [Google Scholar]
- Colwill K., Wells C.D., Elder K., Goudreault M., Hersi K., Kulkarni S., Hardy W.R., Pawson T., Morin G.B. Modification of the Creator recombination system for proteomics applications–improved expression by addition of splice sites. BMC Biotechnol. 2006;6:13. doi: 10.1186/1472-6750-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornils H., Kohler R.S., Hergovich A., Hemmings B.A. Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol. Cell. Biol. 2011;31:1382–1395. doi: 10.1128/MCB.01216-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornils H., Stegert M.R., Hergovich A., Hynx D., Schmitz D., Dirnhofer S., Hemmings B.A. Ablation of the kinase NDR1 predisposes mice to the development of T cell lymphoma. Sci. Signal. 2010;3:ra47. doi: 10.1126/scisignal.2000681. [DOI] [PubMed] [Google Scholar]
- Couzens A.L., Knight J.D., Kean M.J., Teo G., Weiss A., Dunham W.H., Lin Z.Y., Bagshaw R.D., Sicheri F., Pawson T. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Couzens A.L., Xiong S., Knight J.D.R., Mao D.Y., Guettler S., Picaud S., Kurinov I., Filippakopoulos P., Sicheri F., Gingras A.C. MOB1 Mediated Phospho-recognition in the core mammalian hippo pathway. Mol. Cell. Proteomics. 2017;16:1098–1110. doi: 10.1074/mcp.M116.065490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Dar A.C., Dever T.E., Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- Dey M., Cao C., Dar A.C., Tamura T., Ozato K., Sicheri F., Dever T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Emoto K., He Y., Ye B., Grueber W.B., Adler P.N., Jan L.Y., Jan Y.N. Control of dendritic branching and tiling by the Tricornered-kinase/Furry signaling pathway in Drosophila sensory neurons. Cell. 2004;119:245–256. doi: 10.1016/j.cell.2004.09.036. [DOI] [PubMed] [Google Scholar]
- Emoto K., Parrish J.Z., Jan L.Y., Jan Y.N. The tumour suppressor Hippo acts with the NDR kinases in dendritic tiling and maintenance. Nature. 2006;443:210–213. doi: 10.1038/nature05090. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Endicott J.A., Noble M.E., Johnson L.N. The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Eng J.K., Jahan T.A., Hoopmann M.R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Fang X., Adler P.N. Regulation of cell shape, wing hair initiation and the actin cytoskeleton by Trc/Fry and Wts/Mats complexes. Dev. Biol. 2010;341:360–374. doi: 10.1016/j.ydbio.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Lu Q., Emoto K., Adler P.N. The Drosophila Fry protein interacts with Trc and is highly mobile in vivo. BMC Dev. Biol. 2010;10:40. doi: 10.1186/1471-213X-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogl G., Schneider K.D., Yeh B.J., Alam N., Nguyen Ba A.N., Moses A.M., Hetenyi C., Remenyi A., Weiss E.L. The structure of an NDR/LATS kinase-mob complex reveals a novel kinase-coactivator system and substrate docking mechanism. PLoS Biol. 2015;13:e1002146. doi: 10.1371/journal.pbio.1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haling J.R., Sudhamsu J., Yen I., Sideris S., Sandoval W., Phung W., Bravo B.J., Giannetti A.M., Peck A., Masselot A. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 2014;26:402–413. doi: 10.1016/j.ccr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Hao Y., Chun A., Cheung K., Rashidi B., Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008;283:5496–5509. doi: 10.1074/jbc.M709037200. [DOI] [PubMed] [Google Scholar]
- He Y., Fang X., Emoto K., Jan Y.N., Adler P.N. The tricornered Ser/Thr protein kinase is regulated by phosphorylation and interacts with furry during Drosophila wing hair development. Mol. Biol. Cell. 2005;16:689–700. doi: 10.1091/mbc.E04-09-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A., Bichsel S.J., Hemmings B.A. Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol. Cell. Biol. 2005;25:8259–8272. doi: 10.1128/MCB.25.18.8259-8272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A., Kohler R.S., Schmitz D., Vichalkovski A., Cornils H., Hemmings B.A. The MST1 and hMOB1 tumor suppressors control human centrosome duplication by regulating NDR kinase phosphorylation. Curr. Biol. 2009;19:1692–1702. doi: 10.1016/j.cub.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Hergovich A., Stegert M.R., Schmitz D., Hemmings B.A. NDR kinases regulate essential cell processes from yeast to humans. Nat. Rev. Mol. Cell Biol. 2006;7:253–264. doi: 10.1038/nrm1891. [DOI] [PubMed] [Google Scholar]
- Huang J., Wu S., Barrera J., Matthews K., Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Kean M.J., Couzens A.L., Gingras A.C. Mass spectrometry approaches to study mammalian kinase and phosphatase associated proteins. Methods. 2012;57:400–408. doi: 10.1016/j.ymeth.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Kessner D., Chambers M., Burke R., Agus D., Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.Y., Tachioka Y., Mori T., Hakoshima T. Structural basis for autoinhibition and its relief of MOB1 in the Hippo pathway. Sci. Rep. 2016;6:28488. doi: 10.1038/srep28488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J.D.R., Choi H., Gupta G.D., Pelletier L., Raught B., Nesvizhskii A.I., Gingras A.C. ProHits-viz: a suite of web tools for visualizing interaction proteomics data. Nat. Methods. 2017;14:645–646. doi: 10.1038/nmeth.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton D.R., Zheng J.H., Ten Eyck L.F., Ashford V.A., Xuong N.H., Taylor S.S., Sowadski J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Komander D., Garg R., Wan P.T., Ridley A.J., Barford D. Mechanism of multi-site phosphorylation from a ROCK-I: RhoE complex structure. EMBO J. 2008;27:3175–3185. doi: 10.1038/emboj.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaberoglu Y., Lin K., Holder M., Gai Z., Gomez M., Assefa Shifa B., Mavis M., Hoa L., Sharif A.A.D., Lujan C. Stable MOB1 interaction with Hippo/MST is not essential for development and tissue growth control. Nat. Commun. 2017;8:695. doi: 10.1038/s41467-017-00795-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q.Y., Zhang H., Zhao B., Zha Z.Y., Bai F., Pei X.H., Zhao S., Xiong Y., Guan K.L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Zhang J., Larsen B., Stark C., Breitkreutz A., Lin Z.Y., Breitkreutz B.J., Ding Y., Colwill K., Pasculescu A. ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 2010;28:1015–1017. doi: 10.1038/nbt1010-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe E.D., Noble M.E., Skamnaki V.T., Oikonomakos N.G., Owen D.J., Johnson L.N. The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. EMBO J. 1997;16:6646–6658. doi: 10.1093/emboj/16.22.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J. Solving structures of protein complexes by molecular replacement with phaser. Acta Crystallogr. D Biol. Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson B., Kurischko C., Horecka J., Mody M., Nair P., Pratt L., Zougman A., McBroom L.D., Hughes T.R., Boone C. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol. Biol. Cell. 2003;14:3782–3803. doi: 10.1091/mbc.E03-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L., Zheng Y., Hara M., Pan D., Luo X. Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev. 2015;29:1416–1431. doi: 10.1101/gad.264929.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pearce L.R., Komander D., Alessi D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Rock J.M., Lim D., Stach L., Ogrodowicz R.W., Keck J.M., Jones M.H., Wong C.C., Yates J.R., 3rd, Winey M., Smerdon S.J. Activation of the yeast Hippo pathway by phosphorylation-dependent assembly of signaling complexes. Science. 2013;340:871–875. doi: 10.1126/science.1235822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D., Deutsch E.W., Lam H., Eng J.K., Sun Z., Tasman N., Mendoza L., Moritz R.L., Aebersold R., Nesvizhskii A.I. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.007690. M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegert M.R., Hergovich A., Tamaskovic R., Bichsel S.J., Hemmings B.A. Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20-like kinase MST3. Mol. Cell. Biol. 2005;25:11019–11029. doi: 10.1128/MCB.25.24.11019-11029.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegert M.R., Tamaskovic R., Bichsel S.J., Hergovich A., Hemmings B.A. Regulation of NDR2 protein kinase by multi-site phosphorylation and the S100B calcium-binding protein. J. Biol. Chem. 2004;279:23806–23812. doi: 10.1074/jbc.M402472200. [DOI] [PubMed] [Google Scholar]
- Tamaskovic R., Bichsel S.J., Rogniaux H., Stegert M.R., Hemmings B.A. Mechanism of Ca2+-mediated regulation of NDR protein kinase through autophosphorylation and phosphorylation by an upstream kinase. J. Biol. Chem. 2003;278:6710–6718. doi: 10.1074/jbc.M210590200. [DOI] [PubMed] [Google Scholar]
- Tang F., Gill J., Ficht X., Barthlott T., Cornils H., Schmitz-Rohmer D., Hynx D., Zhou D., Zhang L., Xue G. The kinases NDR1/2 act downstream of the Hippo homolog MST1 to mediate both egress of thymocytes from the thymus and lymphocyte motility. Sci. Signal. 2015;8:ra100. doi: 10.1126/scisignal.aab2425. [DOI] [PubMed] [Google Scholar]
- Teo G., Liu G., Zhang J., Nesvizhskii A.I., Gingras A.C., Choi H. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics. 2014;100:37–43. doi: 10.1016/j.jprot.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultanir S.K., Hertz N.T., Li G., Ge W.P., Burlingame A.L., Pleasure S.J., Shokat K.M., Jan L.Y., Jan Y.N. Chemical genetic identification of NDR1/2 kinase substrates AAK1 and Rabin8 Uncovers their roles in dendrite arborization and spine development. Neuron. 2012;73:1127–1142. doi: 10.1016/j.neuron.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D., Murshudov G.N., Papiz M.Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- Xiong S., Couzens A.L., Kean M.J., Mao D.Y., Guettler S., Kurinov I., Gingras A.C., Sicheri F. Regulation of protein interactions by Mps one binder (MOB1) phosphorylation. Mol. Cell. Proteomics. 2017;16:1111–1125. doi: 10.1074/mcp.M117.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.B., Chaudhary D., Olland S., Wolfrom S., Czerwinski R., Malakian K., Lin L., Stahl M.L., Joseph-McCarthy D., Benander C. Catalytic domain crystal structure of protein kinase C-theta (PKCtheta) J. Biol. Chem. 2004;279:50401–50409. doi: 10.1074/jbc.M409216200. [DOI] [PubMed] [Google Scholar]
- Yang J., Cron P., Thompson V., Good V.M., Hess D., Hemmings B.A., Barford D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell. 2002;9:1227–1240. doi: 10.1016/s1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- Zhang L., Tang F., Terracciano L., Hynx D., Kohler R., Bichet S., Hess D., Cron P., Hemmings B.A., Hergovich A. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. 2015;25:296–305. doi: 10.1016/j.cub.2014.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Li L., Tumaneng K., Wang C.Y., Guan K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Wei X., Li W., Udan R.S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.