

Abstract
Aptamers are chemically synthesized oligonucleotides or peptides with molecular recognition capabilities. We investigated recognition of substrate-tethered small-molecule targets, using neurotransmitters as examples, and fluorescently labeled DNA aptamers. Substrate regions patterned via microfluidic channels with dopamine or L-tryptophan were selectively recognized by previously identified dopamine or L-tryptophan aptamers, respectively. The on-substrate dissociation constant determined for the dopamine aptamer was comparable to, though slightly greater than the previously determined solution dissociation constant. Using pre-functionalized neurotransmitter-conjugated oligo(ethylene glycol) alkanethiols and microfluidics patterning, we produced multiplexed substrates to capture and to sort aptamers. Substrates patterned with L-DOPA, L-DOPS, and L-5-HTP enabled comparison of the selectivity of the dopamine aptamer for different targets via simultaneous determination of in situ binding constants. Thus, beyond our previous demonstrations of recognition by protein binding partners (i.e., antibodies and G-protein-coupled receptors), strategically optimized small-molecule-functionalized substrates show selective recognition of nucleic acid binding partners. These substrates are useful for side-by-side target comparisons, and future identification and characterization of novel aptamers targeting neurotransmitters or other important small-molecules.
Keywords: oligonucleotide, neurotransmitter, chemical patterning, microfluidics, self-assembled monolayers, molecular recognition, fluorescence microscopy
ToC GRAPHIC
INTRODUCTION
Aptamers are single-stranded oligonucleotides or peptides having selective recognition for a wide variety of targets.1–5 Isolation of aptamer sequences is most often carried out through the use of an in vitro selection method termed systematic evolution of ligands by exponential enrichment (SELEX).6,7 Commonly, SELEX involves covalent modification of stationary phase materials, e.g., sepharose, with targets.6–9 Target-modified materials are exposed to combinatorial libraries containing large numbers of nucleic acid sequences possessing random regions. Only small numbers of sequences recognizing targets are isolated and enriched over subsequent rounds of selection.
Compared to antibodies or other protein recognition elements, aptamers possess advantages associated with chemical synthesis and engineering for stability in biological environments.10 Aptamer affinities can be tuned by evolving or mutating recognition sequences.11–13 As such, aptamers have emerged as candidates for integration into diverse sensing platforms.14–16 Identifying new aptamers and understanding relationships between aptamer affinities, kinetics, and conformational dynamics will be critical for realizing the potential of these recognition elements for device architectures and diverse applications including in vivo sensing.17–19
Following isolation, the thermodynamic dissociation constant (Kd) that characterizes aptamer-target binding is typically determined in solution by fluorescence enhancement, quenching, or anisotropy measurements.20,21 A different approach would be to measure aptamer affinities in environments that more closely mimic those in which they are isolated (i.e., on substrates).22 Dissociation constants determined in solution can differ from on-substrate Kd values. Improvements in aptamer-based sensing applications will be facilitated by characterizing interactions between aptamers and targets immobilized on sensor surfaces. Thus, we developed the ability to determine on-substrate Kd values for aptamer-target recognition for comparison to in-solution determined values, and to multiplex target-functionalized substrates enabling side-by-side comparisons of binding affinities. One goal was to ascertain whether the substrates investigated here have the potential to be used for future SELEX aimed at identifying and characterizing novel aptamers for small molecules.
Previously, we developed small-molecule-functionalized substrates using neurotransmitters as exemplars.23–25 Optimization entailed (1) spacing small-molecules for improved recognition by larger biomolecules via dilute solution deposition of tethering thiols during the preparation of mixed self-assembled monolayers (SAMs) or through the insertion of tethers into naturally existing or engineered defects in SAMs.26–29 (2) We utilized the amino acid precursors of neurotransmitters for tethering via the additional carboxyl moiety to render amino groups more accessible for molecular recognition.23,24 And (3), we incorporated oligo(ethylene glycol)-terminated thiols as these had been shown by others to reduce nonspecific molecular recognition.30,31 The major difference between our substrates and those commonly used for SELEX is that we precisely control surface parameters, such as target densities, tethering chemistries, and spacing, to maximize small-molecule accessibility while minimizing nonspecific binding.
Using small-molecule tethered substrates, binding partners including antibodies25,27 and G-protein-coupled receptors,23,24 were selectively captured and sorted from mixtures to their respective targets. We developed soft-lithography patterning methods32,33 to create regions of target contrast, which enabled visualization and quantification via fluorophore-labeled binding partners relative to nonspecific binding on the same substrates.25,28,29,34 The capability to recognize and to sort native G-protein-coupled receptors with minimal nonspecific binding suggested to us that these substrates might also recognize artificial receptors in the form of aptamers, an idea that was previously untested.
Thus, our main goal was to determine whether small-molecule functionalized substrates, which incorporate all of the optimization strategies described above, recognize known aptamers identified by solid-phase SELEX. In the future, these small-molecule-functionalized substrates might enable improvements in isolating new high-affinity, high-selectivity aptamers for small molecules. Aptamers targeting small molecules have been difficult to identify by SELEX and we hypothesize that this is partly due to poorly controlled surface chemistries on traditional solid-phase column materials.
We investigated these ideas using previously identified DNA aptamers for the neurotransmitter dopamine (3,4-dihydroxyphenylalanine)35,36 or L-tryptophan.37 We determined that dopamine and L-tryptophan aptamers selectively and reversibly recognize target-functionalized substrates. We compared Kd values for aptamer recognition of dopamine in solution vs. surface-tethered dopamine. Using pre-functionalized neurotransmitter-conjugated oligoethylene alkanethiols,25 we produced multiplexed substrates with similarly structured neurotransmitters tethered in parallel. These substrates enabled simultaneous determination and comparison of in situ binding affinities and evaluation of aptamer specificity.
RESULTS AND DISCUSSION
Dopamine Aptamers Recognize Surface-Tethered Dopamine.
Our general strategy for substrate functionalization is shown in Scheme 1A. The amino group of dopamine had been functionalized to agarose columns for the original aptamer selection reported by others.38 Thus, for an initial demonstration of aptamer capture on our substrates, we also immobilized dopamine via its primary amine for amide bond formation with surface-tethered carboxyl groups.
Scheme 1.
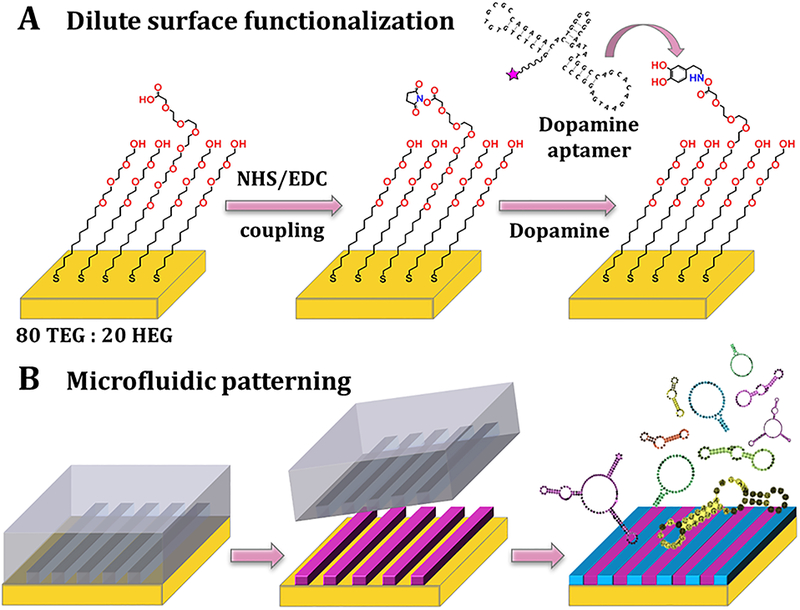
Schematics (not to scale) of dilute target functionalization and patterning strategies. (A) The Au substrates were incubated in ethanolic solutions of 80% hydroxyl tri(ethylene glycol)-terminated undecanethiol (TEG) and 20% carboxyl hexa(ethylene glycol)-terminated undecanethiol (HEG). Following N-hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N′ethylcarbodiimide hydrochloride (EDC) activation, HEG carboxyl groups were conjugated with small-molecule neurotransmitters, such as dopamine, and tested for recognition with aptamers. (B) Microfluidics devices were used to pattern substrates with tethered small molecules. After self-assembly of 80/20 TEG/HEG and formation of activated esters, devices were sealed onto substrates and target solutions (purple) were injected into the channels for functionalization restricted to the channel areas. After device removal, substrates were backfilled with additional TEG (blue) to minimize non-specific binding, particularly in the unfunctionalized regions that had been in contact with devices. Aptamer recognition was evaluated on patterned substrates via fluorescence microscopy.
The density of surface-tethered dopamine was optimized for aptamer recognition by altering ratios of hydroxyl-terminated (background molecules) vs. carboxyl-terminated (tether molecules) thiols to minimize target crowding (Figure S1).
Microfluidic channels were used to pattern substrates so that substrates had alternating dopamine functionalized and unfunctionalized regions enabling relative fluorescence quantification (Scheme 1B).25,28,29,34 Nonspecific binding was minimized through the use of hydroxyl tri(ethylene glycol)-terminated undecanethiol (TEG) in the functionalized and unfunctionalized regions.27,30,31 We have previously characterized small-molecule-functionalized substrates using infrared spectroscopy,26,27 X-ray photoelectron spectroscopy, and electrochemistry.25 Nucleic acids were conjugated with fluorophores on their 5’-ends (Figure 1). The “correct” dopamine aptamer sequence was a 57-base DNA homolog35 of an RNA aptamer originally isolated by SELEX by Mannironi and co-workers (Figure 1A).38
Figure 1.
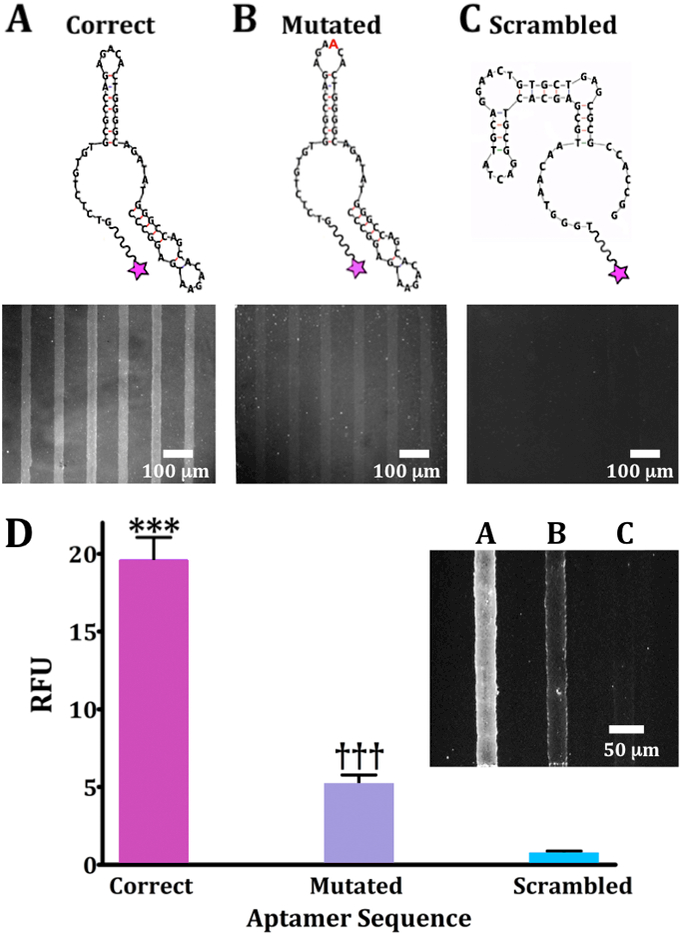
Oligonucleotide binding on dopamine-patterned substrates. Representative fluorescence images are from N=3 substrates per sequence for (A) a 57-base correct dopamine aptamer sequence, (B) a mutated sequence with an extra adenine (red) in a recognition loop at position 21, and (C) a scrambled sequence with the same nucleotides as the correct sequence but a different primary sequence to generate a different secondary structure. Substrates were imaged at an emission wavelength of 605 nm for AlexaFluor® 546 (excitation at 556 nm). (D) Relative fluorescence intensities for patterned dopamine substrates exposed to the three aptamer sequences (20 μM) captured on the same substrate but in different channels (inset; representative image). Error bars are standard errors of the means for N=3 substrates. Group means were significantly different [F(2,15)=719; P<0.001]; ***P<0.001 vs. mutated or scrambled; †††P<0.001 vs. correct or scrambled.
To investigate dopamine-specific recognition by the DNA aptamer on our planar substrates, we quantified relative fluorescence binding of the correct sequence vs. two incorrect (control) sequences. The first, a “mutated” 58-base sequence contained an additional adenine at position 21. The extra base lies in a recognition loop of the “correct” sequence and was therefore hypothesized to interfere with recognition. This mutated sequence was first reported by Walsh and DeRosa when describing the DNA homolog of the RNA dopamine aptamer (Figure 1B).35 The second sequence was a scrambled sequence, which we designed to have the same numbers of each nucleotide from the correct 57-base aptamer but a primary sequence predicted to generate a different secondary structure (Figure 1C). Two-dimensional secondary structures shown in Figure 1 were obtained using the Mfold program, which generates thermodynamically favorable conformations contingent on base-sequences and external constraints, such as temperature and ionic conditions.39
Recognition of surface-tethered dopamine by the correct aptamer sequence produced the highest fluorescence contrast between regions conjugated with dopamine and unfunctionalized regions (Figure 1A). The mutated sequence showed a large reduction in dopamine-associated fluorescence (Figure 1B), while the scrambled sequence showed minimal aptamer capture and no detectable pattern (Figure 1C).
We quantified binding of the correct, mutated, and scrambled sequences side-by-side on dopamine-functionalized substrates (Figure 1D, inset). Differences in recognition of surface-tethered dopamine by the different sequences were similar to those determined from separate substrates. These results highlight a key advantage of patterned substrates—target recognition by different binding partners can be directly compared on the same substrates. Additionally, these results further exemplify that a single base change in a key target-recognition region significantly alters aptamer binding.13
Next, we carried out measurements on substrates where each dopamine-functionalized channel was exposed to a different aptamer concentration (1–50 μM) to determine the on-substrate dissociation constant (Kd) (Figure 2A,B). A Langmuir isotherm (saturation curve) was generated by plotting increasing aptamer concentrations vs. mean relative fluorescence intensities (normalized to the background fluorescence of each of the six substrates tested). The half-saturation point corresponded to a Kd=6.8 ± 1 μM (Figure 2C). The on-substrate Kd value was one order of magnitude greater than the Kd previously reported for this aptamer recognizing dopamine in solution (0.7 μM).35 Differences in the Kd values determined on-substrate vs. in-solution suggest that steric hindrance associated with surface-bound targets may result in modest increases in apparent dissociation constants (i.e., lower affinity) for tethered small-molecules. These findings further imply that in-solution Kd values may overestimate affinities for aptamers selected using target-modified column materials.
Figure 2.
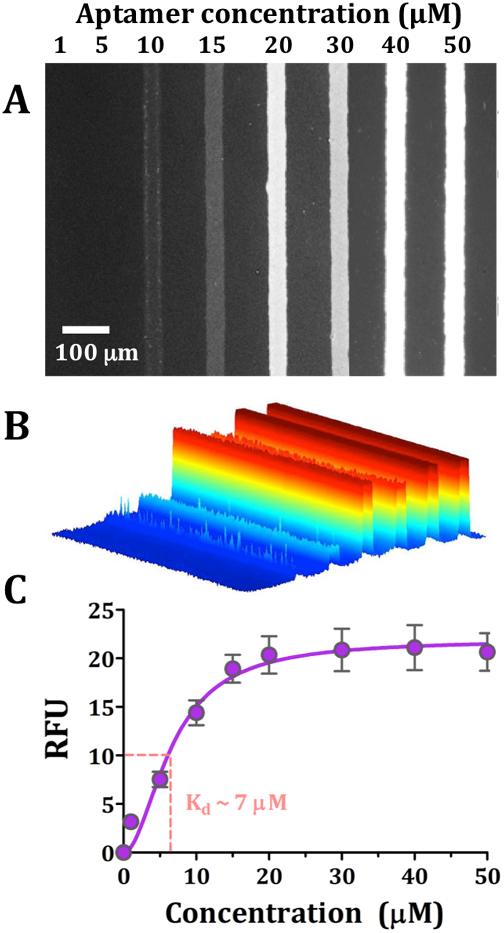
Dopamine aptamer dissociation constant for surface-tethered dopamine. (A) Representative fluorescence image from N=6 substrates imaged at a florescence emission wavelength of 605 nm for AlexaFluor® 546 (excitation at 556 nm). Dopamine aptamer recognition was visualized for increasing concentrations of aptamer (1–50 μM). (B) A false-colored perspective plot for the fluorescence image shown in (A). In (C), the substrate binding affinity was obtained (6.8 ± 1 μM) by plotting relative fluorescence intensities vs. increasing aptamer concentrations. Error bars represent standard errors of the means and are too small to be visualized at the lowest concentration tested.
Aptamers make use of noncovalent intermolecular interactions (i.e., hydrogen bonding, hydrophobic interactions, electrostatic interactions, and π-π-stacking) for target recognition. When selecting aptamers for immobilized targets, access to target functional groups may be somewhat reduced due to fewer degrees of freedom and/or steric hindrance associated with aptamer diffusion to substrates, both of which can affect binding affinities.40 This situation emphasizes challenges associated with selecting aptamers for small-molecule targets, which have limited functional groups for recognition (and tethering, vide infra).41
Aptamer Kd values were previously shown to differ depending on the methods by which they are determined.42 Dissociation constants obtained in three independent laboratories by different techniques were compared for an aptamer targeting anaphylactic allergen β-conglutin.43 Use of surface plasmon resonance, filter-binding assay, or microscale thermophoresis resulted in Kd values that were within an order of magnitude of each other (24 nM, 1.8 nM, and 12 nM, respectively). Similarly, the substrate-determined dissociation constant varied significantly from the solution-phase Kd value for a thrombin aptamer measured by surface plasmon resonance.44
Together, previous and current findings suggest that relative differences between Kd values determined on-substrate vs. in-solution are comparable within reasonable methodological expectations but may differ somewhat for surface-tethered vs. solution dopamine due to surface constraints associated with the former. Notably, we optimize aptamer recognition by diluting tethered targets, which are attached to flexible ethylene(glycol) alkanethiols. The current results support the general assertion that the surface functionalization strategies employed here enable reasonable mimicry of free (in-solution) small-molecules.
Competitive Displacement.
We investigated the reversibility of aptamer binding to surface-tethered dopamine by including dopamine in solution (Scheme 1). Patterned substrates were incubated with 20 μM dopamine aptamer, a concentration at which surface-tethered dopamine binding was saturated (Figure 2C), as well as different concentrations of free dopamine to vary the degree of displacement (Scheme 2). The concentration range for solution dopamine was based on the on-substrate Kd value and spanned approximately one order of magnitude above and below this dissociation constant.
Scheme 2.
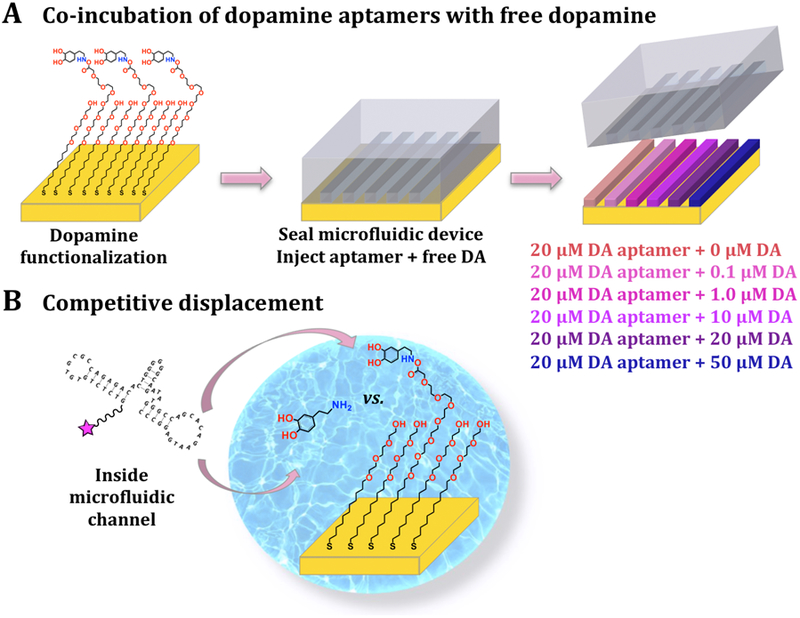
Schematics (not to scale) of competitive displacement in microfluidic channels. (A) Here, dopamine (DA) was first functionalized uniformly across substrates. Microfluidic devices were then sealed on the substrates and a different concentration of dopamine in solution was co-incubated with a constant concentration of dopamine aptamer in each channel. (B) Free dopamine vs. surface-tethered dopamine competed for aptamer binding.
Representative fluorescence images for competitive displacement experiments are shown in Figure S2. The target-response curve was sigmoidal for increasing logarithmic concentrations of solution dopamine (Figure 3A). The inhibitory constant, Ki, was calculated to be 1.4 ± 0.5 μM, using the on-substrate Kd value and the fixed aptamer concentration.45 Thus, at ~1 μM free dopamine, half maximal inhibition of aptamer binding to surface-tethered dopamine occurred. If the probability of aptamer binding to surface-tethered vs. solution dopamine were equal, then the Ki would equal the Kd. The fact that the Ki was within an order of magnitude of the Kd value determined on-substrate (~7 μM) further indicates that surface-tethered dopamine generally mimics dopamine in solution. However, the slightly lower Ki value vs. the on-substrate Kd also supports the idea that the lower degree of freedom of on-substrate targets slightly reduces aptamer affinity.
Figure 3.
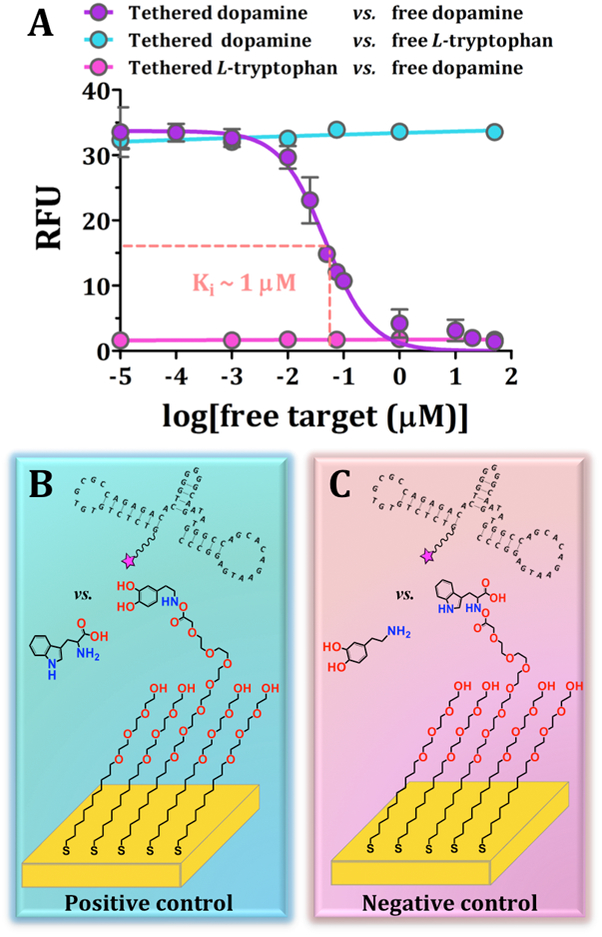
Competitive displacement indicates reversible recognition of surface-tethered dopamine by the dopamine aptamer. (A) Inhibitory dose-response curve for surface-tethered dopamine vs. free dopamine. Error bars are standard errors of the means for N=4 substrates for tethered vs. in solution dopamine and N=2 for control conditions and are too small to be visualized in some cases. Schematics (not to scale) for (B) the positive control with incorrect target, L-tryptophan, in solution and (C) the negative control with the incorrect target, L-tryptophan, tethered.
We carried out two competitive displacement control experiments. For the positive control condition (Figure 3A,B), substrates were functionalized with dopamine but co-incubated with free L-tryptophan in the presence of the dopamine aptamer. Here, maximal fluorescence was observed across the range of L-tryptophan concentrations, suggesting negligible competition of L-tryptophan with substrate-bound dopamine for dopamine-aptamer binding. For the negative control condition (Figure 3A,C), L-tryptophan was surface-tethered instead of dopamine and free dopamine was co-incubated with the dopamine aptamer. In this case, we observed negligible fluorescence suggesting that the dopamine aptamer did not recognize surface-bound L-tryptophan. The aptamer likely preferentially recognized free dopamine and, in any case, was removed during post-incubation wash steps. Together, these findings indicate the selectivity of this dopamine aptamer in differentiating the amino acid L-tryptophan (the biological precursor of the neurotransmitter serotonin) from dopamine.
Multiplexed Substrates to Investigate Aptamer Selectivities.
Yang and co-workers identified an aptamer that recognizes L-tryptophan immobilized via its amino group on epoxy-activated Sepharose beads (Kd=1.8 μM).37 We hypothesized that tethering L-tryptophan by the same functional group would enable recognition by this L-tryptophan aptamer on our substrates. We tethered L-tryptophan and dopamine on the same substrates in alternating patterned channels (Figure 4A). Dopamine and L-tryptophan aptamers were labeled with different fluorophores and substrates were incubated with mixtures of these aptamers. Substrates were imaged at different wavelengths corresponding to the non-overlapping emission spectra for each fluorophore.
Figure 4.
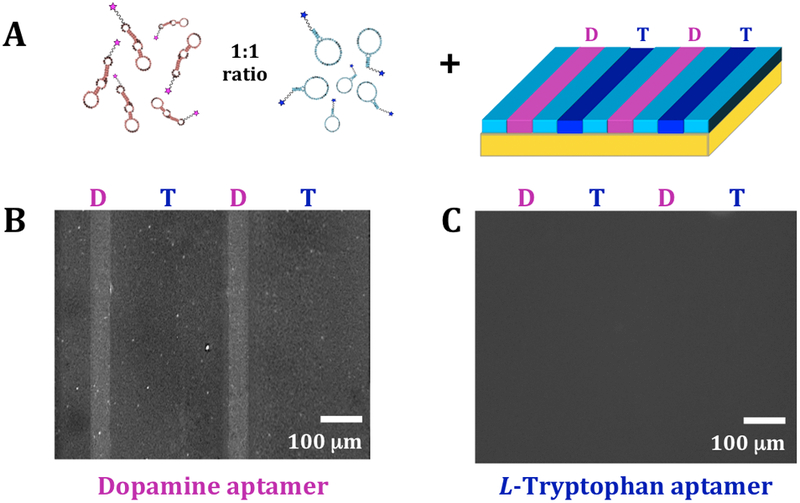
Bi-functionalization of dopamine and L-tryptophan. (A) Schematic (not to scale) of incubation of multiplexed substrates with 1:1 mixtures of dopamine and L-tryptophan aptamers. Representative fluorescence images of the same substrate at (B) an emission wavelength of 605 nm for AlexaFluor® 546 (excitation at 556 nm) to image bound dopamine aptamers and (C) an emission wavelength of 525 nm for AlexaFluor® 488 (excitation at 490 nm) to visualize bound L-tryptophan aptamers. Selective binding was observed for the dopamine aptamer, while negligible binding was detected for the L-tryptophan aptamer (N=3 substrates).
Under imaging conditions for the dopamine aptamer, we observed fluorescence only in the dopamine-functionalized lanes (Figure 4B). This finding further exemplifies that this dopamine aptamer recognizes substrate-immobilized dopamine, while negligibly recognizing surface-tethered L-tryptophan. However, using imaging conditions for the L-tryptophan aptamer, fluorescence patterns were not detectable for L-tryptophan-functionalized channels (Figure 4C).
Capture and sorting of the dopamine aptamer but not the L-tryptophan aptamer suggested a number of possibilities. For one, the L-tryptophan aptamer may be unable to recognize L-tryptophan under the functionalization conditions used here. However, prior to investigating multiplexed substrates, recognition of surface-tethered L-tryptophan by the L-tryptophan aptamer was verified (Figure S3). The selectivities of the L-tryptophan and dopamine aptamers individually against dopamine of L-tryptophan, respectively were also tested (Figure S4). An alternate explanation is that the multiplexing strategy used above may have resulted in insufficient surface-tethered L-tryptophan.
L-Tryptophan requires reaction for ~48 h at pH 9.5 for optimal functionalization (Figure S3).37 By contrast, dopamine self-polymerizes after only 3 h in solution, depositing polydopamine layers on substrates that can be tens of nanometers thick.46 Here, dopamine was dissolved in buffer solutions at pH 6.7, which retards but does not completely inhibit polymerization. In our experiments, microfluidic devices were used for patterning purposes only and were not connected to a flow system (i.e., targets were injected by hand into each channel). For the experiments shown in Figure 4, targets were sequentially injected into all microfluidic channels; devices were removed after 3-h incubation periods to prevent dopamine polymerization. Entire substrates were then rinsed to remove unfunctionalized targets. Under these conditions, incubation times were likely insufficient for L-tryptophan functionalization.
Generally, multiplexed on-substrate chemistries present difficulties for tethering multiple targets due to the various chemistries/conditions required for covalent modification of individual species.24,25,29,47 To circumvent these difficulties and to enable simultaneous on-substrate determination of Kd values for multiple targets, we utilized pre-functionalized neurotransmitter-conjugated oligoethylene alkanethiols, which we previously synthesized to enable multiplexed patterning of small molecules.25 Using pre-functionalized thiols, aptamer recognition and binding does not depend on the efficiencies of on-substrate coupling chemistries.
As shown in Scheme 3, we first used microfluidic devices to pattern different pre-functionalized thiols in different channels. We then used microfluidic devices to create aptamer concentration gradients orthogonal to the original thiol patterns to obtain simultaneous Kd determinations for targets in situ. We self-assembled thiols pre-functionalized with L-3,4-dihydroxyphenylalanine (L-DOPA), L-threo-dihydroxyphenylserine (L-DOPS), or L-5-hydroxytryptophan (L-5-HTP), which are precursors to the neurotransmitters dopamine, norepinephrine, and serotonin, respectively (Figure 5A, 5B). By tethering amino-acid neurotransmitter precursors via their (extra) carboxyl groups, both the catechol/indole and amino moieties remained available for aptamer recognition.23,24 When we designed these pre-functionalized thiols, we hypothesized that the availability of all functional groups, despite tethering, might enable eventual selection of aptamers with improved target recognition.
Scheme 3.
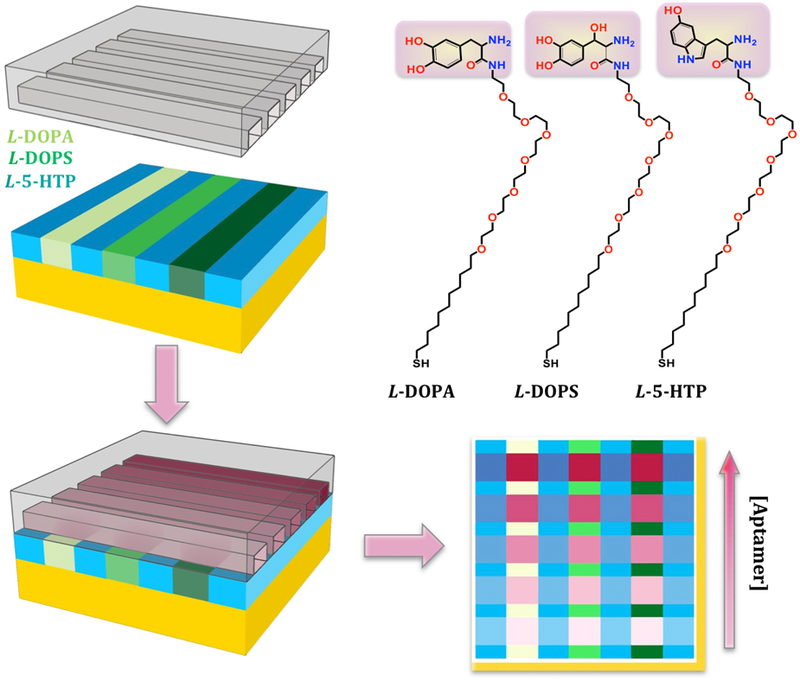
Schematics (not to scale) of multiplexed patterning of three pre-functionalized thiols (i.e., pre-functionalized with L-DOPA, L-DOPS, or L-5-HTP). After pre-functionalized thiol self-assembly, substrates were backfilled with TEG (blue) to minimize non-specific binding. A new microfluidic device was sealed over all three molecules at a 90° angle to the initial channel orientation and an aptamer concentration gradient was incubated across the different channels of each substrate. This enabled simultaneous Kd determinations for all three tethered targets. The structures of pre-functionalized L-DOPA, L-DOPS, and L-5-HTP are shown on the top right.
Figure 5.
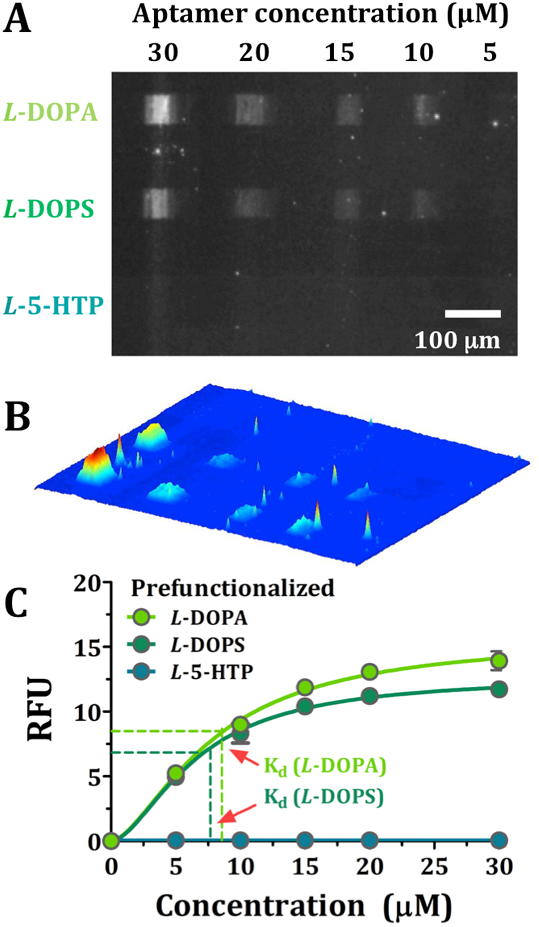
(A) Representative fluorescence image at an emission wavelength of 605 nm for AlexaFluor® 546 (excitation at 556 nm) to visualize different concentrations of dopamine aptamer binding to each of three pre-functionalized thiols (i.e., L-DOPA, L-DOPS, and L-5-HTP). (B) A false-colored perspective plot of the fluorescence image shown in (A). In (C), on-substrate binding affinities were obtained simultaneously for all three targets by plotting fluorescence intensities at increasing aptamer concentrations. Error bars represent standard errors of the means for N=4 substrates and are too small to be visualized in most cases.
Fluorescence intensities were comparable at each concentration of dopamine aptamer incubated with tethered L-DOPA or L-DOPS (Figure 5C). The Kd values were 8.3 ± 0.8 μM and 6.8 ± 0.9 μM, respectively, and were indistinguishable [(t(6)=1.8, P>0.05] (Figure 5C). These Kd values were similar to the Kd for dopamine-aptamer recognition of dopamine tethered by its primary amine further indicating that this particular aptamer primarily recognizes the catechol moiety. Cross-reactivity of the dopamine aptamer for norepinephrine, for which L-DOPS is the precursor, has been previously reported and is presumably due to aptamer recognition largely involving the catechol moiety present in these highly similar targets.35,38,48,49 Minimal relative fluorescence was observed for the L-5-HTP pre-functionalized thiol, similar to the specificity of this dopamine aptamer for tethered dopamine vs. L-tryptophan (Figures 3, 4).
Mannironi et al. used structure-activity relationships to determine the mechanism of dopamine recognition by this particular dopamine aptamer.38 Norepinephrine was 60% effective in eluting dopamine-bound aptamers. Other catechols, i.e., L-DOPA, catechol, also displaced the dopamine aptamer from dopamine-functionalized affinity columns, albeit with reduced potency. Replacing the 4-hydroxyl group with a methoxy group further reduced displacement to ~15%. Phenethylamine targets lacking the catechol moiety were unable to displace the dopamine aptamer. These findings led Mannironi et al. to hypothesize that the mechanism of dopamine aptamer recognition involved hydrogen bonding between the hydroxyl group at position 3 of dopamine and an acceptor in the aptamer binding pocket. Further, they posited that both the benzene ring and the proximal region of the aliphatic chain may be embedded in the aptamer binding pocket.
Ferapontova and coworkers recently asserted that the DNA homolog of the original RNA dopamine aptamer is not an aptamer due to poor selectivity for dopamine vs. structurally related catecholamines.50 We observed similar cross-reactivity of this DNA aptamer with surface-tethered dopamine vs. norepinephrine, with Kd values for these two targets being indistinguishable. However, we demonstrated that a single-base change in the correct dopamine aptamer sequence resulted in significantly reduced recognition of dopamine, and a scrambled sequence showed no dopamine binding (Figure 1), indicating sequence-specific dopamine recognition. Furthermore, substrate-tethered L-tryptophan (Figure 4,S4) and L-5-HTP (Figure 5) were differentiated from dopamine by the DNA dopamine aptamer. Thus, while lacking the ability to differentiate catecholamines, this DNA dopamine aptamer nonetheless exhibits specific recognition of surface-bound dopamine with respect to indoleamines.
Dopamine aptamer cross-reactivity highlights the challenge of isolating aptamers that differentiate target molecules that differ by as little as a single functional group (in this case, the β-hydroxyl group of norepinephrine).51 Even so, chemical intuition, carefully designed nucleic acid libraries, and stringent counter-selection protocols can lead to aptamers that distinguish physiologically important yet structurally similar small molecules.20,21,52–54 Multiplexed small-molecule-tethered substrates that enable relative comparisons of dissociation constants for aptamer binding to specific vs. nonspecific or closely structured counter-targets reduces measurement variabilities across different substrates. In the future, substrates patterned with many different targets may have the potential to be used for multiplexed counter-selection to discover or to refine identification of highly selective aptamers.
Aptamer Elution from Multiplexed Substrates.
In SELEX, captured nucleic acid sequences are eluted prior to reverse transcription, in the case of RNA, and amplification by polymerase chain reaction before subsequent rounds of selection.6,7 Tightly bound DNA sequences are typically eluted using strong base.55,56 Deprotonation disrupts intramolecular G/C and T/A hydrogen-bonded networks, which disorders nucleic-acid secondary structure and interrupts target interactions enabling removal of bound sequences.57
We investigated elution by 0.5 M potassium hydroxide (KOH) of aptamers bound to dopamine-functionalized substrates (Figure 6A). Substrates (8.75 cm2) were unpatterned and functionalized nominally with 20% dopamine (80:20 TEG:HEG solution deposition for SAM formation followed by covalent modification of HEG with dopamine). Prior to substrate incubation, solutions contained 1 × 10−9 mol of dopamine aptamer, calculated from the solution volume and the aptamer concentration measured by ultraviolet visible spectroscopy (20 ± 0.4 μM), and in agreement with the calculated concentration. Eluents contained 3 × 10−10 moles of dopamine aptamer (3 × 10−11 mol/cm2), indicating that a measurable amount of aptamer was captured from solution and recovered from substrates. (Figure 6B). The density of oligo(ethylene glycol)-terminated alkanethiols on Au substrates is on the order of ~1014 molecules/cm2.58 Assuming 100% yields for dopamine surface functionalization and aptamer recognition, aptamers eluted from substrates would be ~1013 molecules/cm2 or ~2 × 10−11 mol/cm2. This calculated density agrees well with the measured aptamer density.
Figure 6.
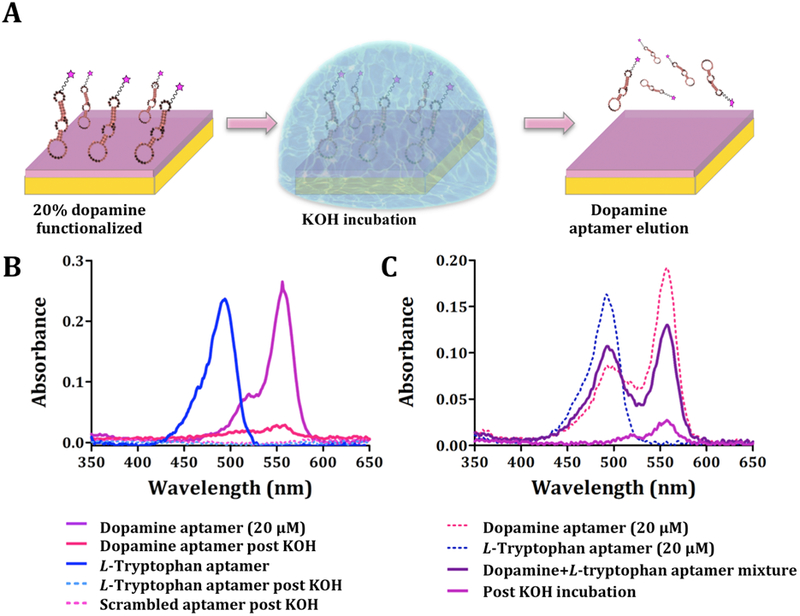
Elution of aptamers following substrate capture. (A) Schematic (not to scale) of incubation of dopamine-functionalized substrates with dopamine aptamers. Unbound sequences were rinsed from substrates. Captured aptamers were eluted by treatment with KOH. (B) Representative UV-vis spectra (N=3) showing the absorbance spectra of eluted aptamers based on AlexaFluor® 546 and AlexaFluor® 488 emission wavelengths for dopamine (correct and scrambled) and L-tryptophan aptamers, respectively. Control experiments with the L-tryptophan aptamer and the scrambled dopamine aptamer showed negligible nucleic acid elution. (C) Representative UV-vis spectra (N=3) for selective elution of dopamine aptamers from mixtures with L-tryptophan aptamers (1:1 ratio).
We conducted two negative control elution experiments using the scrambled dopamine aptamer or the L-tryptophan aptamer. When dopamine-functionalized substrates were incubated with either of these sequences, we were unable to detect quantifiable amounts of fluorescently labeled nucleic acids in the eluents (Figure 6B), indicating minimal nonspecific sequence recognition and the potential to identify sequences that selectively recognize surface-tethered targets of interest.
A possible route to novel aptamers that recognize small molecules, including neurotransmitters, will be to capture aptamer candidates from combinatorial libraries and to elute them from the types of target-modified substrates described here, followed by amplification and subsequent rounds of capture. As a simple example of sequence capture when substrates were exposed to multiple sequences, we investigated dopamine and L-tryptophan aptamers co-incubated on dopamine-functionalized substrates. Each aptamer was tagged with a different fluorophore. Dopamine-functionalized substrates were incubated with 1:1 aptamer mixtures (20 μM) and 0.5 M KOH was used to elute the captured sequences. We were unable to visualize or to quantify an emission peak for the Alexa488®-L-tryptophan aptamer. However, 2 × 10−10 mol of the dopamine aptamer were detected in the eluents (Figure 6C). The amount of dopamine aptamer eluted from the mixed aptamer exposure was comparable to the amount recovered from dopamine-functionalized substrates when the dopamine aptamer was incubated by itself (Figure 6B). The ability to capture aptamers and to recover them selectively from small-molecule-functionalized substrates illustrates possible use of these substrates in the future for selecting and/or characterizing novel aptamers.
Dissociation constants of previously identified aptamers for small-molecule targets are typically in the μM range and these aptamers often show cross-reactivity with similarly structured molecules (e.g. the dopamine aptamer investigated here).35,36,48,49 These difficulties have been attributed to the complexity in targeting molecules with low molecular mass and rotatable bonds.59,60 Recent advances in solution-phase SELEX methods that obviate tethering targets have enabled identification of aptamers that recognize small-molecule, low-epitope targets.21,61–64 Solution-phase selection and/or the use of the target-modified substrates described herein for SELEX are expected to lead to increased opportunities for high-affinity small-molecule aptamer discovery. Small-molecule-functionalized substrates have advantages associated with comparisons of binding affinities in parallel and in environments that may be more predictive of aptamer binding characteristics in on-substrate applications (e.g., implantable biosensors).
Future applications could involve incorporation of microfluidics with flow on multiplexed substrates to enable high-throughput screening of oligonucleotide libraries. Scaling up is feasible since each round of selection involves amplifying the small numbers of sequences captured. The time to run highly multiplexed assays would not increase substantially since all targets are interrogated simultaneously. One key advantage of on-substrate selection compared to conventional solid-phase SELEX is the capacity to extract multiple binding affinities simultaneously while interrogating aptamer selectivity to similarly structured targets.
CONCLUSIONS
Nucleic acid aptamers are capable of recognizing small-molecule targets on substrates having highly controlled surface chemistries and optimized tethering. Patterned small molecules enable quantification of relative fluorescence associated with specific binding. The current findings show that surface-tethered small-molecule neurotransmitters are selectively and reversibly recognized by previously identified dopamine and L-tryptophan aptamers. On-substrate determination of equilibrium dissociation constants enabled comparisons with aptamer Kd values measured in solution. Small differences were attributed to on-substrate environments with lower degrees of freedom due to surface functionalization.
Pre-functionalized thiols that display more consistent biomolecule recognition,25 were utilized to create multiplexed substrates. Multiplexed substrates enabled measurements of Kd values for multiple targets in situ. We evaluated aptamer cross-reactivity for structurally similar targets (e.g., dopamine vs. norepinephrine). Finally, we demonstrated that aptamers captured on small-molecule-functionalized substrates could be selectively eluted and quantified. Together, these findings suggest the potential of patterned small-molecule functionalized substrates to identify and/or to characterize novel high affinity, highly selective aptamers, which have been problematic to isolate via conventional solid-phase SELEX.
METHODS
Materials.
The Si substrates with Au films (100 nm) overlaying Ti (10 nm) were purchased from Platypus Technologies (Madison, WI). Hydroxyl tri(ethylene glycol)-terminated undecanethiol (TEG) and carboxyl hexa(ethylene glycol)-terminated undecanethiol (HEG) were purchased from Toronto Research Chemicals Inc. (Toronto, Canada). N-Hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 4-methylpiperidine, and dopamine hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). The SYLGARD® 184 silicone elastomer kits were from Ellsworth Adhesives (Germantown, WI). Absolute, 200-proof ethanol was purchased from Decon Laboratories, Inc. (King of Prussia, PA) and 95% hexane was purchased from Fisher Scientific (Hampton, NH). Deionized water (~18 MΩ) was obtained from a Millipore water purifier (Billerica, MA).
The following fluorescently labeled aptamer sequences were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA): AlexaFluor® 546-conjugated single-stranded correct dopamine aptamer: 5’/5Alex546N/-GTC TCT GTG TGC GCC AGA GAC ACT GGG GCA GAT ATG GGC CAG CAC AGA ATG AGG CCC 3’ with molecular weight 18,790.9 g/mol and melting point 74.1 °C, AlexaFluor® 546-conjugated single-stranded mutated dopamine aptamer (additional base underlined): 5’/5Alex546N/-GTC TCT GTG TGC GCC AGA GAA CAC TGG GGC AGA TAT GGG CCA GCA CAG AAT GAG GCC C 3’ with molecular weight 18,926.0 g/mol and melting point 74.2 °C, AlexaFluor® 546-conjugated single-stranded scrambled dopamine aptamer: 5’/5Alex546N/-TGG GTA ACA ATG CGA GCA CTG CGG ACT ATG CAG GAA CTG TGC TGA GCG CGC CAC CGG 3’ with molecular weight 18,790.9 g/mol and melting temperature 75.0 °C, AlexaFluor® 488-conjugated single-stranded L-tryptophan aptamer: 5’/5Alex488N/-AGC ACG TTG GTT AGG TCA GGT TTG GGT TTC GTG C 3’ with molecular weight 11,262.5 g/mol and melting point 67.2 °C, and AlexaFluor® 488-conjugated single-stranded scrambled L-tryptophan aptamer: 5’/5Alex488N/-TCG AGG CTG GAT TTC ATT CGG GAT TTC GGT GGG T 3’ with molecular weight 11,262.5 g/mol and melting point 67.6 °C. Aptamer solutions (100 μM in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8.0) as received from Integrated DNA Technologies, Inc.) were stored at -20 °C and were diluted to specific concentrations as needed prior to each experiment.
Microfluidic Device Fabrication.
Polydimethylsiloxane (PDMS) microfluidic devices were prepared by mixing a 10:1 mass ratio of SYLGARD® 184 silicone elastomer base and curing agent. The mixture was degassed under vacuum until bubbles were no longer visible. The PDMS mixture was then cast onto a silicon master substrate having twelve 70-μm-wide channels with 30-μm interchannel spacings patterned by photolithography. Each elastomeric mixture was cured at 60 °C for ~20 h. Polymerized PDMS microfluidic devices were removed from the silicon master and soaked in fresh 95% hexane for 1.5 h three times. These soaking steps removed unpolymerized residues on devices preventing them from being deposited on substrates, thereby decreasing nonspecific binding.65 Finally, devices were rinsed with 50/50 deionized H2O/ethanol for 15 min and dried with nitrogen gas.
Self-Assembled Monolayer Preparation and Patterning.
The Au substrates were annealed with a hydrogen flame and submerged in 0.5 mM TEG and 0.125 mM HEG (80:20 mol fraction) in ethanol for ~16 h to produce mixed self-assembled monolayers.24 The use of TEG in monolayers minimizes nonspecific binding of biomolecules.23,24,26,31 Following self-assembly, substrates were rinsed thoroughly with ethanol and blown dry with nitrogen gas. For on-substrate functionalization, TEG/HEG SAM-modified Au substrates were incubated for 1 h with an aqueous solution of 35 mM NHS and 35 mM EDC. This step converts the terminal carboxyl groups of HEG to NHS-ester-activated moieties in preparation for amide bond formation (Scheme 1A).23 Substrates were then rinsed thoroughly with deionized H2O and blown dry.
Hexane-treated microfluidic devices were then sealed to substrates and either dopamine or L-tryptophan solutions (35 mM) were injected by hand into individual channels and incubated for 3 h or 48 h, respectively (Scheme 1B). Microfluidic devices were not connected to a continuous flow system but were used principally to address targets to specific substrate regions and to confine incubation solutions. Dopamine stock solutions were freshly prepared in phosphate buffer (10 mM KH2PO4, 40 mM K2HPO4, pH 6.7) and used immediately for each experiment to avoid dopamine polymerization. L-Tryptophan stock solutions were also freshly prepared in phosphate buffer (0.73 mM KH2PO4, 499 mM K2HPO4, pH 9.5) and used immediately. For multiplexed on-substrate patterning, solutions of dopamine or L-tryptophan were injected into alternating microfluidic channels and incubated for 3 h.
Following small-molecule conjugation, microfluidic devices were immediately removed from substrates while substrates were submerged in H2O to minimize exposure of unfunctionalized regions to the dopamine or L-tryptophan solutions in the channels. Substrates were then immersed in fresh deionized H2O for 10 min to hydrolyze unreacted NHS-activated carboxyl groups on HEG.66,67 Substrates were subsequently incubated with 0.5 mM TEG for 1 h to replenish TEG in the substrate areas that had been in contact with microfluidic devices and where TEG may have been removed and ultimately, to minimize nonspecific binding in these areas.28
As an alternate route to multiplexed substrates, we self-assembled oligoethyleneglycol alkanethiols pre-functionalized with neurotransmitters; the synthesis of these heterobifunctional disulfides is described in detail elsewhere.25 Here, Au surfaces were flame-annealed and hexane-treated PDMS devices were sealed to substrates. Solutions containing 0.125 mM oligoethyleneglycol alkanethiols pre-functionalized with L-3,4-dihydroxyphenylalanine, L-threo-3,4-dihydroxyphenylserine, or L-5-hydroxytryptophan and 0.5 mM TEG (20/80 ratio) were injected into separate channels and incubated for 15 min.
Following self-assembly and microfluidic device removal, substrates were incubated with 0.5 mM TEG for 1 h for self-assembly in the areas surrounding the pre-functionalized thiols and unpatterned regions. The Fmoc groups used to protect amino moieties during the chemical synthesis of pre-functionalized thiols were removed by immersing substrates in 20% 4-methylpiperidine in deionized H2O for 15 min. All substrates were thoroughly washed in deionized H2O and blown dry in preparation for aptamer capture, which occurred immediately thereafter.
Aptamer Capture on Small-Molecule-Patterned Substrates.
Aptamers were diluted 1:100 in binding buffer (10 mM Na2HPO4, 2 mM KH2PO4, 2.7 mM KCl, 5 mM MgCl2, 500 mM NaCl, pH 7.4) followed by heating at 95 °C in a water bath for 5 min.68 Aptamer solutions were then cooled in an ice bath for 20 s before equilibration at room temperature for 10 min. This standard procedure of heating and cooling removes misfolds in aptamer structures and maximizes correctly folded secondary structures. Substrates were incubated with aptamers (20 μM except where otherwise noted) for 1 h at room temperature in the dark to prevent fluorophore photobleaching.
For binding affinity measurements, different concentrations of dopamine aptamers (1–50 μM) were incubated in different channels of microfluidic devices. For competitive displacement experiments, microfluidic devices were sealed on unpatterned dopamine-functionalized substrates and in each channel, the dopamine aptamer (20 μM) was co-incubated with different concentrations of free dopamine (0.1–50 μM).
For selectivity determinations, substrates functionalized with alternating dopamine and L-tryptophan patterned regions were incubated with heat-treated mixtures of both aptamers. To create aptamer concentration gradients (1–30 μM) on multiplexed pre-functionalized thiols substrates, new hexane-treated microfluidic devices were placed on substrates 90° to the original channel orientations and dopamine aptamers were injected sequentially into channels. Substrates were imaged via fluorescence microscopy immediately following 1 h aptamer incubation unless aptamer elution was carried out.
Aptamer Elution from Small-Molecule-Patterned Substrates.
Following aptamer incubation, substrates were rinsed to remove unbound sequences and incubated in 0.5 M KOH for 15 min to elute bound sequences. Ultraviolet-visible (UV-vis) spectra of eluents (2 μL) containing fluorophore-labeled aptamers were obtained using a Thermo Scientific NanoDrop 2000 spectrophotometer. Concentrations of aptamers before and after elution were calculated from measured peak absorbances using Beer’s Law (A=εlc). The extinction coefficients (ε) for AlexaFluor® 488 (L-tryptophan) and AlexaFluor® 546 (dopamine) were 73,000 L/mol•cm and 112,000 L/mol•cm, respectively, and the path length was 1.0 mm.
Image Analysis and Statistics.
Immediately after aptamer binding and rinsing, substrates were carefully removed from the last wash so as to keep surfaces wet while glass coverslips were mounted. An inverted fluorescence microscope (Axio Observer.D1) with an AxioCam MRm charged-coupled device camera (Carl Zeiss MicroImaging, Inc., Thornwood, NY) was used to visualize aptamer capture on substrates. The microscope was equipped with two fluorescence filter sets. One had excitation and emission wavelengths at 470 ± 20 nm and 525 ± 25 nm, respectively (38 HE/high efficiency); the other had excitation and emission wavelengths at 550 ± 12 nm and 605 ± 35 nm (43 HE/high efficiency). Substrates were imaged using a 10× objective lens.
Fluorescence intensities were determined by line scans encompassing functionalized and unfunctionalized regions at 30-pixel scanning widths using AxioVs40 version 4.7.1.0 software (Carl Zeiss MicroImaging, Inc., Thornwood, NY). On average, five line scans were acquired per substrate. All substrates from the same experiment were imaged using the same exposure times to standardize contrast and brightness. Mean fluorescence intensities for target-functionalized regions were divided by mean fluorescence values from unpatterned regions on a per substrate basis and are reported as relative fluorescence units (RFU; unitless) (N=2 for control conditions; N=4–6 for experimental conditions).25,34
The tunable contrast of fluorescence only influences RFU values at the extremes. At low levels of brightness, fluorescence for unfunctionalized regions is not detectable. At the opposite extreme, fluorescence intensities for unfunctionalized regions are detectable but intensities for functionalized regions are saturated. All data herein were collected under conditions where fluorescence for unfunctionalized regions was detectable (albeit low) and fluorescence for functionalized regions was not saturated. Under these conditions, RFU values are independent of brightness and are only related to differences in the numbers of bound aptamers. Fluorescence images shown in the figures are those that most closely represented mean fluorescence intensities.
Statistics were computed using GraphPad Prism (GraphPad Software Inc., San Diego). All data are reported/graphed as means ± standard errors of the means with probabilities P<0.05 considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Wei-Ssu Liao for guidance and expertise in microfluidics, Dr. John M. Abendroth and Professor Milan N. Stojanović for helpful discussions, Professor André Nel for fluorescence microscope use at the UCLA Center for Environmental Implications of Nanotechnology, and Dr. Hongyan Yang for assistance with graphics.
Funding
This work was supported by the Cal-Brain Neurotechnology Program, the National Institute on Drug Abuse (DA045550), the National Institutes of Health Director’s New Innovator Award Program (DP2-OD008533), the National Science Foundation (1509794, CMMI-1536136), and the Merkin Family Foundation.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Surface density optimization for surface-tethered dopamine, aptamer competitive displacement experiments, selectivity of dopamine and L-tryptophan aptamers, and the patterning scheme and visualization for L-tryptophan aptamer capture are available free of charge on the ACS Publications website at DOI:
The authors declare no competing financial interest.
REFERENCES
- (1).Ni X; Castanares M; Mukherjee A; Lupold SE Nucleic Acid Aptamers: Clinical Applications and Promising New Horizons. Curr. Med. Chem 2011, 18, 4206–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Reverdatto S; Burz DS; Shekhtman A Peptide Aptamers: Development and Applications. Curr. Top. Med. Chem 2015, 15, 1082–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kumar PK R. Monitoring Intact Viruses Using Aptamers. Biosensors-Basel 2016, 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Soldevilla MM; Villanueva H; Pastor F Aptamers: A Feasible Technology in Cancer Immunotherapy. J. Immunol. Res 2016, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ruscito A; McConnell EM; Koudrina A; Velu R; Mattice C; Hunt V; McKeague M; DeRosa MC In Vitro Selection and Characterization of DNA Aptamers to a Small Molecule Target. Curr. Protoc. Chem. Biol 2017, 9, 233–268. [DOI] [PubMed] [Google Scholar]
- (6).Ellington AD; Szostak JW In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- (7).Tuerk C; Gold L Systematic Evolution of Ligands by Exponential Enrichment – RNA Ligands to Bacteriophage-T4 DNA-Polymerase. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (8).Drolet DW; Jenison RD; Smith DE; Pratt D; Hicke BJ A High Throughput Platform for Systematic Evolution of Ligands by Exponential Enrichment (SELEX). Comb. Chem. High T. Scr 1999, 2, 271–278. [PubMed] [Google Scholar]
- (9).Stoltenburg R; Reinemann C; Strehlitz B SELEX – A (R)Evolutionary Method to Generate High-Affinity Nucleic Acid Ligands. Biomol. Eng 2007, 24, 381–403. [DOI] [PubMed] [Google Scholar]
- (10).Kuai HL; Zhao ZL; Mo LT; Liu H; Hu XX; Fu T; Zhang XB; Tan WH Circular Bivalent Aptamers Enable in Vivo Stability and Recognition. J. Am. Chem. Soc 2017, 139, 9128–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Vallee-Belisle A; Ricci F; Plaxco KW Engineering Biosensors with Extended, Narrowed, or Arbitrarily Edited Dynamic Range. J. Am. Chem. Soc 2012, 134, 2876–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Imaizumi Y; Kasahara Y; Fujita H; Kitadume S; Ozaki H; Endoh T; Kuwahara M; Sugimoto N Efficacy of Base-Modification on Target Binding of Small Molecule DNA Aptamers. J. Am. Chem. Soc 2013, 135, 9412–9419. [DOI] [PubMed] [Google Scholar]
- (13).Ricci F; Valee-Belisle A; Simon AJ; Porchetta A; Plaxco KW Using Nature’s “Tricks” to Rationally Tune the Binding Properties of Biomolecular Receptors. Acc. Chem. Res 2016, 49, 1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhou WZ; Huang PJJ; Ding JS; Liu J Aptamer-Based Biosensors for Biomedical Diagnostics. Analyst 2014, 139, 2627–2640. [DOI] [PubMed] [Google Scholar]
- (15).Labib M; Green B; Mohamadi RM; Mepham A; Ahmed SU; Mahmoudian L; Chang IH; Sargent EH; Kelley SO Aptamer and Antisense-Mediated Two-Dimensional Isolation of Specific Cancer Cell Subpopulations. J. Am. Chem. Soc 2016, 138, 2476–2479. [DOI] [PubMed] [Google Scholar]
- (16).Lu DQ; He L; Zhang G; Lv AP; Wang RW; Zhang XB; Tan WH Aptamer-Assembled Nanomaterials for Fluorescent Sensing and Imaging. Nanophotonics 2017, 6, 109–121. [Google Scholar]
- (17).Andrews AM The BRAIN Initiative: Toward a Chemical Connectome. ACS Chem. Neurosci 2013, 4, 645–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nakatsuka N; Andrews AM Neurochips Enable Nanoscale Devices for High-Resolution in Vivo Neurotransmitter Sensing. Neuropsychopharmacol 2016, 41, 378–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Arroyo-Curras N; Somerson J; Vieira PA; Ploense KL; Kippin TE; Plaxco KW Real-Time Measurement of Small Molecules Directly in Awake, Ambulatory Animals. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang KA; Pei RJ; Stefanovic D; Stojanovic MN Optimizing Cross-Reactivity with Evolutionary Search for Sensors. J. Am. Chem. Soc 2012, 134, 1642–1647. [DOI] [PubMed] [Google Scholar]
- (21).Yang KA; Barbu M; Halim M; Pallavi P; Kim B; Kolpashchikov DM; Pecic S; Taylor S; Worgall TS; Stojanovic MN Recognition and Sensing of Low-Epitope Targets via Ternary Complexes with Oligonucleotides and Synthetic Receptors. Nat. Chem 2014, 6, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McKeague M; McConnell EM; Cruz-Toledo J; Bernard ED; Pach A; Mastronardi E; Zhang XR; Beking M; Francis T; Giamberardino A; Cabecinha A; Ruscito A; Aranda-Rodriguez R; Dumontier M; DeRosa MC Analysis of in Vitro Aptamer Selection Parameters. J. Mol. Evol 2015, 81, 150–161. [DOI] [PubMed] [Google Scholar]
- (23).Vaish A; Shuster MJ; Cheunkar S; Singh YS; Weiss PS; Andrews AM Native Serotonin Membrane Receptors Recognize 5-Hydroxytryptophan-Functionalized Substrates: Enabling Small-Molecule Recognition. ACS Chem. Neurosci 2010, 1, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liao WS; Cao HH; Cheunkar S; Shuster MJ; Altieri SC; Weiss PS; Andrews AM Small-Molecule Arrays for Sorting G-Protein-Coupled Receptors. J. Phys. Chem. C 2013, 117, 22362–22368. [Google Scholar]
- (25).Cao HH; Nakatsuka N; Deshayes S; Yang H; Abendroth JM; Kasko AM; Weiss PS; Andrews AM Small-Molecule Patterning via Pre-Functionalized Alkanethiols. In Revision 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shuster MJ; Vaish A; Szapacs ME; Anderson ME; Weiss PS; Andrews AM Biospecific Recognition of Tethered Small Molecules Diluted Is Self-Assembled Monolayers. Adv. Mater 2008, 20, 164–167. [Google Scholar]
- (27).Shuster MJ; Vaish A; Cao HH; Guttentag AI; McManigle JE; Gibb AL; Martinez MM; Nezarati RM; Hinds JM; Liao WS; Weiss PS; Andrews AM Patterning Small-Molecule Biocapture Surfaces: Microcontact Insertion Printing vs. Photolithography. Chem. Commun 2011, 47, 10641–10643. [DOI] [PubMed] [Google Scholar]
- (28).Cao HH; Nakatsuka N; Serino AC; Liao WS; Cheunkar S; Yang HY; Weiss PS; Andrews AM Controlled DNA Patterning by Chemical Lift-Off Lithography: Matrix Matters. ACS Nano 2015, 9, 11439–11454. [DOI] [PubMed] [Google Scholar]
- (29).Cao HH; Nakatsuka N; Liao WS; Serino AC; Cheunkar S; Yang HY; Weiss PS; Andrews AM Advancing Biocapture Substrates via Chemical Lift-Off Lithography. Chem. Mater 2017, 29, 6829–6839. [Google Scholar]
- (30).Harder P; Grunze M; Dahint R; Whitesides GM; Laibinis PE Molecular Conformation in Oligo(Ethylene Glycol)-Terminated Self-Assembled Monolayers on Gold and Silver Surfaces Determines Their Ability to Resist Protein Adsorption. J. Phys. Chem. B 1998, 102, 426–436. [Google Scholar]
- (31).Xu FJ; Li HZ; Li J; Teo YHE; Zhu CX; Kang ET; Neoh KG Spatially Well-Defined Binary Brushes of Poly(Ethylene Glycol)s for Micropatterning of Active Proteins on Anti-Fouling Surfaces. Biosens. Bioelectron 2008, 24, 773–780. [DOI] [PubMed] [Google Scholar]
- (32).Mullen TJ; Srinivasan C; Hohman JN; Gillmor SD; Shuster MJ; Horn MW; Andrews AM; Weiss PS Microcontact Insertion Printing. Appl. Phys. Lett 2007, 90, 1–3. [Google Scholar]
- (33).Liao WS; Cheunkar S; Cao HH; Bednar HR; Weiss PS; Andrews AM Subtractive Patterning via Chemical Lift-Off Lithography. Science 2012, 337, 1517–1521. [DOI] [PubMed] [Google Scholar]
- (34).Abendroth JM; Nakatsuka N; Ye M; Kim D; Fullertor EE; Andrews AM; Weiss PS Analyzing Spin Selectivity in DNA-Mediated Charge Transfer via Fluorescence Microscopy. ACS Nano 2017, 11, 7516–7526. [DOI] [PubMed] [Google Scholar]
- (35).Walsh R; DeRosa MC Retention of Function in the DNA Homolog of the RNA Dopamine Aptamer. Biochem. Biophys. Res. Commun 2009, 388, 732–735. [DOI] [PubMed] [Google Scholar]
- (36).Holahan MR; Madularu D; McConnell EM; Walsh R; DeRosa MC Intra-Accumbens Injection of a Dopamine Aptamer Abates MK-801-Induced Cognitive Dysfunction in a Model of Schizophrenia. PLoS One 2011, 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yang XJ; Bing T; Mei HC; Fang CL; Cao ZH; Shangguan DH Characterization and Application of a DNA Aptamer Binding to L-Tryptophan. Analyst 2011, 136, 577–585. [DOI] [PubMed] [Google Scholar]
- (38).Mannironi C; DiNardo A; Fruscoloni P; Tocchini Valentini GP In Vitro Selection of Dopamine RNA Ligands. Biochemistry 1997, 36, 9726–9734. [DOI] [PubMed] [Google Scholar]
- (39).Zuker M Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wilson C; Szostak JW Isolation of a Fluorophore-Specific DNA Aptamer with Weak Redox Activity. Chem. Biol 1998, 5, 609–617. [DOI] [PubMed] [Google Scholar]
- (41).McKeague M; Derosa MC Challenges and Opportunities for Small Molecule Aptamer Development. J. Nucleic Acids 2012, 2012, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).McKeague M; De Girolamo A; Valenzano S; Pascale M; Ruscito A; Velu R; Frost NR; Hill K; Smith M; McConnell EM; DeRosa MC Comprehensive Analytical Comparison of Strategies Used for Small Molecule Aptamer Evaluation. Anal. Chem 2015, 87, 8608–8612. [DOI] [PubMed] [Google Scholar]
- (43).Rubio MJ; Svobodova M; Mairal T; Schubert T; Kunne S; Mayer G; O’Sullivan CK Beta-Conglutin Dual Aptamers Binding Distinct Aptatopes. Anal. Bioanal. Chem 2016, 408, 875–884. [DOI] [PubMed] [Google Scholar]
- (44).Daniel C; Roupioz Y; Gasparutto D; Livache T; Buhot A Solution-Phase vs Surface-Phase Aptamer-Protein Affinity from a Label-Free Kinetic Biosensor. PLoS One 2013, 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Brandt RB; Laux JE; Yates SW Calculation of Inhibitor Ki and Inhibitor Type from the Concentration of Inhibitor for 50-Percent Inhibition for Michaelis-Menten Enzymes. Biochem. Med. Metab. B 1987, 37, 344–349. [DOI] [PubMed] [Google Scholar]
- (46).Zangmeister RA; Morris TA; Tarlov MJ Characterization of Polydopamine Thin Films Deposited at Short Times by Autoxidation of Dopamine. Langmuir 2013, 29, 8619–8628. [DOI] [PubMed] [Google Scholar]
- (47).Xia N; Xing Y; Wang GF; Feng QQ; Chen QQ; Feng HM; Sun XL; Liu L Probing of EDC/NHSS-Mediated Covalent Coupling Reaction by the Immobilization of Electrochemically Active Biomolecules. Int. J. Electrochem. Sc 2013, 8, 2459–2467. [Google Scholar]
- (48).Li BR; Hsieh YJ; Chen YX; Chung YT; Pan CY; Chen YT An Ultrasensitive Nanowire-Transistor Biosensor for Detecting Dopamine Release from Living PC12 Cells under Hypoxic Stimulation. J. Am. Chem. Soc 2013, 135, 16034–16037. [DOI] [PubMed] [Google Scholar]
- (49).Kim J; Rim YS; Chen HJ; Cao HH; Nakatsuka N; Hinton HL; Zhao CZ; Andrews AM; Yang Y; Weiss PS Fabrication of High-Performance Ultrathin In2O3 Film Field-Effect Transistors and Biosensors Using Chemical Lift-Off Lithography. ACS Nano 2015, 9, 4572–4582. [DOI] [PubMed] [Google Scholar]
- (50).Alvarez-Martos I; Ferapontova EE A DNA Sequence Obtained by Replacement of the Dopamine RNA Aptamer Bases Is Not an Aptamer. Biochem. Biophys. Res. Commun 2017, 489, 381–385. [DOI] [PubMed] [Google Scholar]
- (51).Nakatsuka N; Andrews AM Differentiating Siblings: The Case of Dopamine and Norepinephrine. ACS Chem. Neurosci 2017, 8, 218–220. [DOI] [PubMed] [Google Scholar]
- (52).Catherine AT; Shishido SN; Robbins-Welty GA; Diegelman-Parente A Rational Design of a Structure-Switching DNA Aptamer for Potassium Ions. FEBS Open Bio 2014, 4, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Neves MAD; Shoara AA; Reinstein O; Abbasi Borhani O; Martin TR; Johnson PE Optimizing Stem Length to Improve Ligand Selectivity in a Structure-Switching Cocaine-Binding Aptamer. ACS Sens 2017, 2, 1539–1545. [DOI] [PubMed] [Google Scholar]
- (54).Yang KA; Chun H; Zhang Y; Pecic S; Nakatsuka N; Andrews AM; Worgall TS; Stojanovic MN High-Affinity Nucleic-Acid-Based Receptors for Steroids. ACS Chem. Biol 2017, 12, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Poeckh T; Lopez S; Fuller AO; Solomon MJ; Larson RG Adsorption and Elution Characteristics of Nucleic Acids on Silica Surfaces and Their Use in Designing a Miniaturized Purification Unit. Anal. Biochem 2008, 373, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Liu QZ; Aroonyadet N; Song Y; Wang XL; Cao X; Liu YH; Cong S; Wu FQ; Thompson ME; Zhou CW Highly Sensitive and Quick Detection of Acute Myocardial Infarction Biomarkers Using In2O3 Nanoribbon Biosensors Fabricated Using Shadow Masks. ACS Nano 2016, 10, 10117–10125. [DOI] [PubMed] [Google Scholar]
- (57).Verdolino V; Cammi R; Munk BH; Schlegel HB Calculation of pKa Values of Nucleobases and the Guanine Oxidation Products Guanidinohydantoin and Spiroiminodihydantoin Using Density Functional Theory and a Polarizable Continuum Model. J. Phys. Chem. B 2008, 112, 16860–16873. [DOI] [PubMed] [Google Scholar]
- (58).Love JC; Estroff LA; Kriebel JK; Nuzzo RG; Whitesides GM Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem Rev 2005, 105, 1103–1169. [DOI] [PubMed] [Google Scholar]
- (59).Pfeffer P; Gohlke H Drugscore(RNA) – Knowledge-Based Scoring Function to Predict RNA-Ligand Interactions. J. Chem. Inf. Model 2007, 47, 1868–1876. [DOI] [PubMed] [Google Scholar]
- (60).Park H; Paeng IR Development of Direct Competitive Enzyme-Linked Aptamer Assay for Determination of Dopamine in Serum. Anal. Chim. Acta 2011, 685, 65–73. [DOI] [PubMed] [Google Scholar]
- (61).Martini L; Meyer AJ; Ellefson JW; Milligan JN; Forlin M; Ellington AD; Mansy SS In Vitro Selection for Small-Molecule-Triggered Strand Displacement and Riboswitch Activity. ACS Synth. Biol 2015, 4, 1144–1150. [DOI] [PubMed] [Google Scholar]
- (62).Spiga FM; Maietta P; Guiducci C More DNA-Aptamers for Small Drugs: A Capture-SELEX Coupled with Surface Plasmon Resonance and High-Throughput Sequencing. ACS Comb. Sci 2015, 17, 326–333. [DOI] [PubMed] [Google Scholar]
- (63).Gotrik MR; Feagin TA; Csordas AT; Nakamoto MA; Soh HT Advancements in Aptamer Discovery Technologies. Acc. Chem. Res 2016, 49, 1903–1910. [DOI] [PubMed] [Google Scholar]
- (64).Kim J; Olsen TR; Zhu J; Hilton JP; Yang KA; Pei R; Stojanovic MN; Lin Q Integrated Microfluidic Isolation of Aptamers Using Electrophoretic Oligonucleotide Manipulation. Sci. Rep 2016, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Schwartz JJ; Hohman JN; Morin EI; Weiss PS Molecular Flux Dependence of Chemical Patterning by Microcontact Printing. ACS Appl. Mater. Inter 2013, 5, 10310–10316. [DOI] [PubMed] [Google Scholar]
- (66).Grumbach IM; Veh RW Sulfo-N-Hydroxysuccinimide Activated Long-Chain Biotin – a New Microtitre Plate Assay for the Determination of Its Stability at Different pH Values and Its Reaction-Rate with Protein-Bound Amino-Groups. J. Immunol. Methods 1991, 140, 205–210. [DOI] [PubMed] [Google Scholar]
- (67).Nojima Y; Iguchi K; Suzuki Y; Sato A The pH-Dependent Formation of PEGylated Bovine Lactoferrin by Branched Polyethylene Glycol (PEG)-N-Hydroxysuccinimide (NHS) Active Esters. Biol. Pharm. Bull 2009, 32, 523–526. [DOI] [PubMed] [Google Scholar]
- (68).Sefah K; Shangguan D; Xiong XL; O’Donoghue MB; Tan WH Development of DNA Aptamers Using Cell-SELEX. Nat. Protoc 2010, 5, 1169–1185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.