

Abstract
Lafora disease (LD, OMIM 254780) is a rare disorder characterized by epilepsy and neurodegeneration leading patients to a vegetative state and death, usually within the first decade from the onset of the first symptoms. In the vast majority of cases LD is related to mutations in either the EPM2A gene (encoding the glucan phosphatase laforin) or the EPM2B gene (encoding the E3-ubiquitin ligase malin). In this work, we characterize the mutations present in the EPM2A gene in a patient displaying a slow progression form of the disease. The patient is compound heterozygous with Y112X and N163D mutations in the corresponding alleles. In primary fibroblasts obtained from the patient, we analyzed the expression of the mutated alleles by quantitative real time PCR and found slightly lower levels of expression of the EPM2A gene respect to control cells. However, by Western blotting we were unable to detect endogenous levels of the protein in crude extracts from patient fibroblasts. The Y112X mutation would render a truncated protein lacking the phosphatase domain and likely degraded. Since minute amounts of laforin-N163D might still play a role in cell physiology, we analyzed the biochemical characteristics of the N163D mutation. We found that recombinant laforin N163D protein was as stable as wild type and exhibited near wild type phosphatase activity towards biologically relevant substrates. On the contrary, it showed a severe impairment in the interaction profile with previously identified laforin binding partners. These results lead us to conclude that the slow progression of the disease present in this patient could be either due to the specific biochemical properties of laforin N163D or to the presence of alternative genetic modifying factors separate from pathogenicity.
1.- INTRODUCTION
Lafora progressive myoclonus epilepsy (LD, OMIM 254780) is a fatal, autosomal recessive, glycogen storage disease and neurodegenerative disorder (Gentry et al., 2018). A hallmark of LD is the presence of intracellular inclusions of poorly branched, hyperphosphorylated forms of glycogen called Lafora bodies (LBs) in the brain and peripheral tissues [(Lafora and Glueck, 1911), (Sakai et al., 1970)]. Patients with LD have mutations in one of two genes: Epilepsy, Progressive Myoclonus 2A EPM2A [(Minassian et al., 1998), (Serratosa et al., 1999)] or Epilepsy, Progressive Myoclonus 2B EPM2B (Chan et al., 2003). EPM2A is mutated in approximately 60% of LD cases (Serratosa et al., 1995). It encodes laforin, a dual-specificity phosphatase (DSP) of 331 amino acids with a functional carbohydrate binding module (CBM) at the N-terminus [(Minassian et al., 2000), (Wang et al., 2002)]. A second gene, EPM2B, was found to be mutated in the majority of the remaining LD patients [(Chan et al., 2003), (Gómez-Abad et al., 2005)]. It encodes malin, an E3 ubiquitin ligase of 395 amino acids with a RING finger domain at the N-terminus and six NHL repeats in the C-terminal region [(Lohi et al., 2005), (Gentry et al., 2005)].
We and others have recently described that laforin forms a functional complex with malin [(Lohi et al., 2005), (Gentry et al., 2005), (Solaz-Fuster et al., 2008)]. Both the formation of a laforin-malin complex and the observation that patients with mutations in laforin or malin are neurologically and histologically indistinguishable (Gómez-Abad et al., 2005), strongly suggest that the two LD proteins may participate in similar pathways in cell physiology. So far, by expressing laforin and malin in cell cultures multiple groups have found that malin ubiquitinates proteins involved in glycogen metabolism, and laforin is required for this modification [(Solaz-Fuster et al., 2008), (Vilchez et al., 2007), (Cheng et al., 2007), (Worby et al., 2008), (Rubio-Villena et al., 2013)]. Thus, the proposed model is that laforin recruits specific substrates to be ubiquitinated by malin. Most of these substrates are involved in the regulation of glycogen biosynthesis, such as the muscle isoform of glycogen synthase (GS) (Vilchez et al., 2007), glycogen debranching enzyme (AGL) (Cheng et al., 2007), and the R5/PTG and R6 that are both glycogen targeting subunits of protein phosphatase type 1 (PP1) [(Solaz-Fuster et al., 2008), (Worby et al., 2008), (Rubio-Villena et al., 2013)]. However, in vivo experiments using malin knock out mice indicates that the protein levels of these putative substrates were not increased in the target tissues, with the exception of glycogen synthase [(DePaoli-Roach et al., 2010), (Turnbull et al., 2010), (Valles-Ortega et al., 2011)]. Thus, the definitive function of malin in regulating glycogen metabolism is still unclear.
In addition to the recruitment role of laforin in the laforin-malin complex, it has been shown that laforin dephosphorylates carbohydrates rather than proteins and that its physiological substrate is glycogen [(Worby et al., 2006), (Tagliabracci et al., 2007)]. Thus, laforin acts as a glycogen phosphatase and its function is necessary for the maintenance of glycogen particles with normal phosphate content [(Gentry et al., 2007), (Tagliabracci et al., 2008), (Roach et al., 2012), (Roach, 2015)]. Taken together, these results suggest a role for both laforin and malin in the regulation of glycogen homeostasis that is consistent with the presence of intracellular polyglucosan inclusions (Lafora bodies, LBs), as one of the histological determinants of LD.
LD is a fatal disorder that occurs worldwide, but is relatively more frequent in Mediterranean countries. LD initially manifests during adolescence with multiple types of seizures (generalized tonic-clonic seizures, myoclonus, absences), drop attacks and visual hallucinations. The disease progresses with dementia, inability to perform purposive actions (apraxia), inability to understand or express speech (aphasia) and visual loss, eventually leading to patients in a vegetative state and death, usually within a decade of initial seizure onset [(Berkovic et al., 1986), (Ganesh et al., 2006), (Turnbull et al., 2016)]. However, in some cases there is a late-onset and/or slower progression of the disease. An emerging theme is that the malin or laforin mutations in these cases only affect partially the function of the corresponding protein. Alternatively, genetic and/or environmental modifiers may influence the regular progression of the disease [(Franceschetti et al., 2006), (Striano et al., 2008), (Guerrero et al., 2011), (Ferlazzo et al., 2014), (Kecmanovic et al., 2016)]. In this work we describe a case of late-onset and slow progression of Lafora disease and analyze the consequences of the mutations in the EPM2A gene present in the patient.
2.- MATERIAL AND METHODS
2.1.- Culture conditions
Escherichia coli DH5α strain was used as the host strain for plasmid constructions and protein production. Cells were grown in LB (1% peptone, 0.5% yeast extract, 1% NaCl, pH 7.5) medium supplemented with 50 mg/L ampicillin to OD 0.5. Protein expression was induced by the addition of 0.1 mM IPTG and 2% ethanol, after which cells were grown overnight at 20°C and then harvested by centrifugation. Yeast strain used in this work was THY-AP4 (MATa, ura3, leu2, lexA::lacZ::trp1, lexA::HIS3, lexA::ADE2). Yeast transformation was carried out using the lithium acetate protocol (Ito et al., 1983). Yeast cultures were grown in synthetic complete (SC) medium lacking the corresponding supplements to maintain selection for plasmids (Rose et al., 1990).
Human embryonic kidney (HEK293) cells (from HEPA Culture Collections cat. N° 85120602) were grown in DMEM (Lonza, Barcelona, Spain) supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine, 10% inactivated fetal bovine serum (GIBCO, Madrid, Spain) in a humidified atmosphere at 37°C with 5% CO2. Cells were transfected with the appropriated amount of each plasmid using X-treme GENE HP transfection reagent (Roche Diagnostics, Barcelona, Spain), according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were scraped on ice in lysis buffer [25 mM Tris-HCl pH 7.4, 15 mM EDTA, 50 mM NaF, 15 mM Na4P2O7, 0.6 M sucrose, 1% nonidet P40, 10 mM NaCl, 1 mM PMSF] and protease inhibitor cocktail (Roche Diagnostics, Barcelona, Spain). Cells were lysed by repeated passage through a 25Gx5/8” needle and clarified by centrifugation at 13,000 × g for 10 min at 4°C. Proteins in the soluble fraction of cell lysates were analyzed by SDS-PAGE and Western blotting using appropriated antibodies.
Primary LD fibroblasts were obtained from patient EPM2A Y112X/N163D (a gift from Dr. Salas, Neurology Dept. Hospital Vall Hebron, Barcelona, Spain). Primary LD fibroblasts from a homozygous EPM2A R241X/R241X patient (a gift from Dr. José Serratosa, Fundación Jimenez Diaz, Madrid, Spain) were also used in this work. The corresponding informed consents were obtained from patients or their families for experimentation. Control fibroblasts were matched by sex and age. All experiments were carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki). Experiments were performed in human fibroblasts at passage number 10-14 to avoid culture aging effects. Fibroblasts were grown in DMEM medium as above and incubated at 37°C in a humidified atmosphere at 5% (v/v) CO2.
2.2.- Plasmids and site directed mutagenesis
pET21b+-laforin N163D was obtained by site directed mutagenesis using the Quick Change kit (Stratagene), plasmid pET21b+-laforin (Dukhande et al., 2011) as template, and the corresponding mutagenic oligonucleotides (N163D-1: 5′-CATTATTCAAGAATTCTACCAGATATCTGGCTGGGTAGCTGCCC-3′; N163D-2: 5′-GGGCAGCTACCCAGCCAGATATCTGGTAGAATTCTTGAATAATG-3′). Mutant plasmid was sequenced to ensure that additional mutations were not introduced during the mutagenesis procedure. Similarly, plasmid pCMV-HA-laforin N163D was obtained by site directed mutagenesis using the Quick Change kit (Stratagene), plasmid pCMV-HA-laforin (Vernia et al., 2009) as template and the corresponding mutagenic oligonucleotides aforementioned. Plasmid pGADT7-laforin N163D was obtained by subcloning a SfiI/XhoI fragment from pCMV-HA-laforin N163D into pGADT7 (Clontech). Plasmid pBTM116-Laforin N163D was obtained by subcloning a BamHI/XhoI fragment from plasmid pGADT7-laforin N163D into the BamHI/SalI sites of plasmid pBTM116. Other plasmids used in this study were pBTM-laforin, pGAD-laforin, pACT2-malin, pACT2-R5/PTG (Solaz-Fuster et al., 2008) pACT2-R6 (Garcia-Haro et al., 2010) and p28b-laforin W32G (Raththagala et al., 2015).
2.3.- Immunoprecipitation and Western blot analyses
Crude extracts from primary cultures of control, EPM2A Y112X/N163D and R241X/R241X patient fibroblasts, were obtained as in (Roma-Mateo et al., 2015). One mg of proteins from the clarified extracts were immunoprecipitated as in (Sherwood et al., 2013a), using anti-laforin antibody #113. Proteins in the samples were denatured using sample buffer (125 mM Tris-HCl pH 6.8,, 4% SDS, 20% glycerol, 31mg/mL DTT, 0.01% bromophenol blue) and heating to 95°C for 5 min. The samples were subjected to SDS-PAGE (12% acrylamide) and transferred onto PVDF membranes (Millipore, Madrid, Spain). Membranes were blocked with 5% skimmed milk in TBS-Tween for 1 h and slices of these membranes were incubated with the following specific antibodies: anti-HA (H9658, 1:5000; Sigma Aldrich, Madrid, Spain), anti-laforin (MABN606, 1:1000; Millipore, Madrid, Spain), anti-p62 (ab56416, 1:1000; Abcam, Cambridge, UK), anti-GAPDH (sc32233, 1:10000; Santa Cruz Biotech, USA) and anti-actin (A2066, 1:1000; Sigma Aldrich, Madrid, Spain). Thereafter, blots were washed again with TBS-Tween and further incubated for 1 h with the corresponding secondary antibody conjugated with horseradish peroxidase. Finally, membranes were washed (3×5 min) with TBS-Tween and analyzed by chemiluminiscence (ECL Western Blotting Detection Reagents, GE Healthcare, UK) using an image reader LAS-4000 (GE Healthcare, UK).
2.4.- RNA purification and quantitative real-time PCR analyses
Total RNA from primary cultures of control and EPM2A Y112X/N163D and R241X/R241X patient fibroblasts was isolated with the TriPure Isolation Reagent (Roche, Barcelona, Spain) following the manufacturer’s protocol. One μg RNA was reverse transcribed using the Expand Reverse transcriptase (Roche, Barcelona, Spain) following the protocol provided by the supplier. The cDNA was used as template for qRT-PCR according to Universal Probe Library (UPL) database (Roche, Barcelona, Spain). The universal probes used were #20 for EPM2A, #22 for HPRT and #60 for GAPDH genes. Analyses were performed in triplicate for each gene using ABI PRISM 7500 Fast. PCR thermal cycling parameters were: one step of 95°C for 20 sec, and 40 cycles of 95°C for 3 sec and 60°C for 30 sec. The geometric mean values of two house-keeping genes, hypoxanthine-guanine phosphoribosyltranferase (HPRT) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), were used as internal controls for normalization. Reaction master mix was made with Real-time PCR Master mix (TaqMan Fast Universal PCR Master Mix; Applied Biosystems, Madrid, Spain), probes, primers and template according to the instruction manuals. Samples were analyzed with the double-delta cycle threshold (ΔΔCT) method. Results were analyzed with Student’s t-test when required. The significance level was set at * p < 0.05 and ** p < 0.01.
2.5.- Phosphatase activity on complex carbohydrates
Glycogen dephosphorylation assays were performed using a malachite green assay and glycogen purified from rabbit muscle as previously described [(Tagliabracci et al., 2007), (Sherwood et al., 2013b), (Raththagala et al., 2015)]. Assays were performed at room temperature in triplicate 100 μL reactions containing phosphatase buffer (100 mM sodium acetate, 50 mM bis-Tris, 50 mM Tris-HCl, pH 6.5, and 2 mM DTT), 1 mg glycogen, and 2.5 μg protein. Proteins were incubated with glycogen for 30 minutes, and the PiColorLock Gold system (Innova Biosciences, Cambridge, UK) was used to quench the reaction and measure inorganic phosphate release.
Site-specific phosphate release was determined using starch labeled with 33P at either the C3- or C6-position [(Santelia et al., 2011), (Meekins et al., 2015)]. Phosphate-free starch purified from the Arabidopsis sex1-3 mutant (Yu et al., 2001) was labeled at the C6-position by incubation with glucan water dikinase and [β-33P]-ATP (Hartmann Analytic, Brunswick, Germany), followed by washing, incubation with phospho-glucan water dikinase and unlabeled ATP, and washing. C3-labeled substrate was generated by incubation with glucan water dikinase and unlabeled ATP, followed by washing, incubation with phospho-glucan water dikinase and [β-33P]-ATP, and washing. Dephosphorylation reactions were performed in phosphatase buffer supplemented with 1mg/mL BSA, and 0.05% Triton, with C6- or C3-labeled starch (3 mg/mL) and 50 ng of enzyme for a total volume of 150 μL, and incubated on a rotating wheel for 2.5 minutes at 25°C, parameters which are within the linear range of the assay. Reactions were terminated by the addition of 50 μL 10% SDS, centrifuged at 16,000 × g for 5 minutes, and 33P release in 150 μL of the supernatant was determined using a 1900 TR liquid scintillation counter (Packard). Reactions were performed in triplicate to determine specific activity.
2.6.- Thermal stability
Differential scanning fluorimetry (DSF) was used to determine the stability of laforin WT and N163D proteins and to quantify oligosaccharide binding based on conditions previously described (Raththagala et al., 2015). Proteins were dissolved in DSF buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 2 mM DTT) with 5x SYPRO Orange Protein Gel stain (Invitrogen) for a final concentration of 2 μM. Melting was monitored in triplicate 40 μL reactions using a CFX96 Real-Time PCR system (BioRad) from 20 to 90 °C at a rate of 1 °C/50 s. Melting temperature (Tm) was determined by calculating the first derivative of the melting curve and using a Gaussian fit to find the peak maximum. Prism analytical software (Graphpad) was used for analysis. To quantify oligosaccharide binding, maltoheptaose (cat# GLU317) and DP24 (cat# GLU310) were purchased from Elicityl (Crolles, France) and added to reactions for a final concentration of 10 mM.
2.7.- Yeast two-hybrid analyses
Saccharomyces cerevisiae THY-AP4 strain was transformed with the corresponding plasmids. Transformants were selected in SC+2% glucose plates lacking tryptophan and leucine, and were subsequently screened for β-galactosidase activity using a filter lift assay (Yang et al., 1992). The strength of the interaction was determined by measuring β-galactosidase activity in permeabilized yeast cells and expressed in Miller units as described by Ludin and collaborators (Ludin et al., 1998).
2.8.- Statistical analyses
For statistical analyses, results are shown as mean and standard deviation. At least three independent samples were analyzed in each case. Differences between paired samples were analyzed by two-tailed Student’s t-tests using Graph Pad Prism version 5.0 statistical software. P values have been considered in all figures as *p<0.05, **p<0.01 and ***p<0.001.
3.- RESULTS
3.1.- Clinical characteristics
A 28-year-old woman was received at the Neurology Service of the Hospital Vall d’Hebron (Barcelona, Spain), because she had a first seizure at the age of 16 years characterized by loss of consciousness with generalized tonic-clonic convulsions, which appeared on awakening. Several weeks before, she suffered myoclonic jerks on awakening. Neurological and intellectual examinations were normal. EEG showed generalized spike and poly-spike discharges that increased during intermittent photic stimulation. In addition, on the EEG recordings there was epileptiform activity on both occipital regions. Nevertheless, the patient did not suffer partial epileptic seizures with visual semiology. Familial and personal antecedents were unremarkable. At that stage, juvenile myoclonic epilepsy was suspected and she showed a good control of seizures during the following two years with sodium valproate. However, at the age of 18 she continued with clonic-tonic-clonic seizures on awakening or during sleep. Myoclonic jerks and eye-lid myoclonus were observed and a cognitive decline appeared. EEG showed a slow background activity with very frequent spike and poly-spike and wave generalized discharges (Supplementary file S1). Somatosensory evoked potentials were normal. MRI was normal. EEG during sleep showed a difficult distinction among different sleep stages with no activation of epileptiform discharges. At that age, axillary skin biopsy was performed, showing Lafora bodies. A genetic study confirmed the diagnosis of Lafora disease, indicating compound heterozygous mutations with two different mutations in the alleles of the EPM2A gene: exon 2 c335-336 ins A (TAC > TAA), pY(Tyr)112 fs and exon 3 c487 A>G p.Asn163 > Asp. At present, the patient is 28 years old and during the latter years the evolution has been stationary. She suffers a mean of one or two clonic-tonic-clonic seizures every month. She has some myoclonic jerks on upper limbs and eye-lid myoclonus. She still maintains significant autonomy and cognitive capacity, but has shown a progressive intellectual decline since first diagnosed with mild dysarthria but with no ataxia or visual impairment. EEG shows the same initial findings with no more deterioration (Fig. 1A). Presently, she is on sodium valproate 1800 mg/d, brivaracetam 150 mg/d and clonazepam 2 mg/d to control seizures.
Fig. 1. Phenotypic and genotypic characterization of an unusual case of LD.
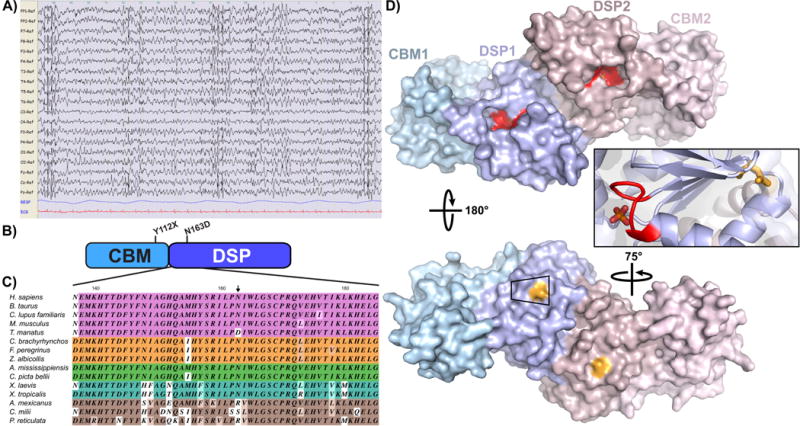
A) EEG analysis of the patient when she was 28 years old. The EEG shows a slow background activity with bilateral spike and spike and wave discharges. B) Domain structure of the EPM2A gene encoding laforin. Y112X maps to the end of the CBM, while N163D maps to the DSP domain. C) Representative partial alignment of DSP domain sequences, colored by vertebrate class. 43% of residues are strictly conserved based on our analysis (See supplemental file S2 for full alignment) while N163 is substituted with an aspartate, serine or arginine. D) The position of the mutations present in the LD patient were mapped in the laforin structure described recently (Raththagala et al., 2015) (PDB 4RKK) using PyMol software (DeLano Scientific LLC, USA). The two subunits of the dimer are shown in shades of blue and pink; the CBM is shown as a lighter shade than the DSP domain. The classic DSP catalytic motif (CX5R) is shown in red on the front face of the dimer, and the bound phosphate is partially obscured from view. N163 (shown in light orange) maps to the back of the dimer, opposite to the catalytic cleft (red) and bound phosphate.
3.2.- Characteristics of the EPM2A mutations present in the patient
Although clinically LD is often considered as a homogeneous disorder, different courses of the disease have been described, from more severe to late-onset and slow progression of the disease (Turnbull et al., 2016). This patient carries compound heterozygous EPM2A Y112X and N163D mutations and presents with this slower progression. In order to understand why the disease progresses slowly in this patient, we analyzed several parameters related to the functionality of the corresponding laforin Y112X and N163D mutations.
We first mapped the position of the mutations on the EPM2A gene encoding laforin. The Y112X mutation maps at the end of the carbohydrate binding module (Fig. 1B). As it is a nonsense mutation, this alteration generates a premature termination codon (PTC) and results in a premature stop in the translation of the protein. PTC mutations either activate the nonsense-mediated mRNA decay mechanism (Brogna and Wen, 2009) or result in truncated proteins that are degraded by the proteasome, both resulting in no laforin protein being formed. In addition, the laforin Y112X mutation has already been described in two patients, homozygous for the Y112X mutation, who presented a regular progression of the disease (Poyrazoglu et al., 2015). So, the slow phenotype presented in the patient Y112X/N163D is likely not due to the Y112X mutation, but due to the laforin N163D mutation. To our knowledge, laforin N163D is a novel LD mutation and no biochemical analysis has been performed on this mutant. It is a missense mutation affecting an asparagine residue in the dual specificity phosphatase domain (DSP) (Fig. 1B-D). The DSP domain is 82% similar and 67% identical among vertebrates and 43% of residues in this region are strictly conserved throughout vertebrates (Supplementary file S2). N163 is largely conserved in mammals, birds, reptiles, and amphibians, with the exception of an aspartate substitution in manatees; in fish it is substituted with either an arginine or serine (Fig. 1C and Supplementary file S2). These data demonstrate that an asparagine is not strictly needed at this position and suggest that conservative substitution at this position is likely not deleterious to the biochemical function of laforin. The laforin crystal structure was recently described (Raththagala et al., 2015) and N163 maps to the back face of the catalytic cleft and is partially surface exposed (Fig. 1D, colored in orange; the catalytic CX5R motif is shown in red).
3.3.- Expression of the mutated laforin forms in primary patient fibroblasts
Next, we checked the expression of the corresponding laforin mutated forms in primary fibroblasts from the patient. To our surprise, we were unable to detect any laforin protein in crude extracts from patient fibroblasts by Western blotting (Fig. 2A, second panel from the top). In order to inhibit protein degradation, we treated cells with 25 μM MG132 for 18 hours, but we did not observe any accumulation of the mutated laforin forms (Fig. 2A, second panel from the top). However, this treatment increased the levels of the autophagy receptor p62 (Galluzzi et al., 2017), indicating that the treatment had impaired protein degradation. Next, we immunoprecipitated the laforin mutated forms following a recently described bioassay for endogenous laforin levels (Sherwood et al., 2013a). In Fig. 2A (top panel) we observed an enrichment of the levels of endogenous laforin in control samples, but we did not observe mutated laforin forms in the immunoprecipitates from patient fibroblasts. No laforin mutated forms were detected either in samples from a homozygous patient carrying R241X/R241X mutations, as in this case the mutation introduces a premature stop codon. These results are in agreement with a nonsense-mediated mRNA decay mechanism that would lead to the absence of protein being synthesized. Therefore, our results suggest that, by Western blotting, fibroblasts from Y112X/N163D patient express undetectable levels of the laforin mutated forms.
Fig. 2. Expression of laforin mutated forms in primary fibroblasts from the Y112X/N163D patient.
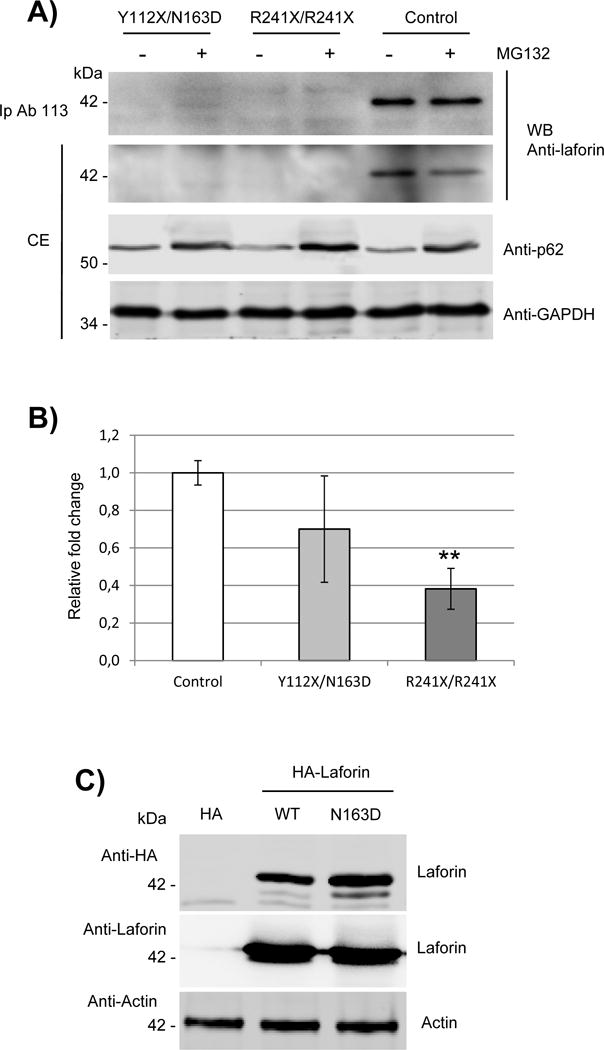
A) Western blot analysis of endogenous levels of laforin in primary fibroblasts. Crude extracts (CE, 60 μg) from primary cultures of control, EPM2A Y112X/N163D and R241X/R241X fibroblasts were analyzed by western blotting using the indicated antibodies. When indicated, samples were previously treated with 25 μM MG132 for 18h to block protein degradation. In the top panel, one mg of proteins from the clarified extracts were immunoprecipitated as in (Sherwood et al., 2013a) using anti-laforin antibody #113, and the immunoprecipitates analyzed as above. Images are representative western blots of at least three independent experiments. B) Quantitative real-time PCR analyses of the expression of EPM2A gene in primary cultures of control, Y112X/N163D and R241X/R241X fibroblasts. Gene expression was analyzed as described in Materials and Methods. The mean values of two house-keeping genes, hypoxanthine-guanine phosphoribosyltranferase (HPRT) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), were used as internal controls for normalization. Values are means of three independent experiments. Bars indicate standard deviation. Differences between paired samples were analyzed by two-tailed Student’s t-tests using Graph Pad Prism version 5.0 statistical software; **P< 0.01. C) HEK293 cells were transfected with plasmids pCMV-HA (empty), pCMV-HA-Laforin or pCMV-HA-Laforin N163D. Twenty-four hours after transfection, 40 μg of crude extracts were analyzed by Western blot using anti-HA and anti-laforin antibodies. Anti-Actin was used as loading control. Images are representative western blots of at least three independent experiments.
In order to define the cause of this defect, we analyzed the expression of the EPM2A gene in primary fibroblasts. As shown in Fig. 2B, we detected significantly lower levels of mRNA from the R241X/R241X patient derived fibroblasts, which was in agreement with the aforementioned nonsense-mediated mRNA decay mechanism present in this sample. In the case of the Y112X/N163D sample we detected intermediate levels of mRNA between R241X/R241X and control samples. As the expression of the Y112X allele is also likely subjected to nonsense-mediated mRNA decay, the observed mRNA levels should be due to the expression of the N163D allele.
As the defect in expression of laforin N163D in the patient was not likely due to altered transcription, we analyzed the stability of the protein by expressing the laforin N163D under the control of a strong promoter. In Fig. 2C we show that the expression of laforin N163D in HEK293 cells rendered similar levels of protein as wild type. We utilized both an antibody against an HA-tag present in the construct and the anti-laforin antibody used above. We observed that both antibodies detected similar levels of laforin for wild type and N163D. These results indicate that the N163D mutation does not affect the epitope that is recognized by the anti-laforin antibody, and also that the N163D protein is as stable as wild type once it has been translated.
Taking all these results together, we suggest that the undetectable levels of laforin in fibroblasts from the Y112X/N163D patient could be the result of a defective process not related to gene expression or stability of the protein.
3.4.- The novel laforin N163D mutant has similar phosphatase activity against complex phosphorylated carbohydrates and similar ability to bind carbohydrates as wild type
Since minute amounts of protein might still play a role in cell physiology, we decided to analyze the biochemical characteristics of laforin N163D. First, we expressed laforin wild type and N163D in bacteria and, as laforin has a functional carbohydrate binding module (CBM), we purified the proteins using an amylose sepharose column based on our previous protocol [(Dukhande et al., 2011), (Sanchez-Martin et al., 2013)]. The yield of the purification of laforin N163D was similar to wild type, indicating that the mutation did not alter the ability of the protein to bind to carbohydrates. Second, we analyzed the ability of laforin N163D to dephosphorylate phosphorylated complex carbohydrates. As shown in Fig. 3A, laforin N163D had a similar specific activity as laforin wild type when glycogen was used as substrate. Glycogen can be phosphorylated at the C2-, C3-, and C6-hydroxyls and LBs have increased phosphorylation at these positions [(Tagliabracci et al., 2011), (Nitschke et al., 2013), (DePaoli-Roach et al., 2015), (Contreras et al., 2016)]. We previously showed that laforin can remove phosphate from both the C3- and C6-position from complex carbohydrates (Meekins et al., 2015). Therefore, we tested if laforin N163D showed any change in site-specific phosphate release. We found that laforin N163D displayed a similar specific activity toward both the C3- and C6-position as wild type laforin (Fig. 3B). As a percentage of total dephosphorylation, the ratio of C3- versus C6-phosphate release for N163D was indistinguishable from wild type (Fig. 3C). Therefore, the ability to dephosphorylate complex carbohydrates is preserved in the laforin N163D mutant. Additionally, we previously demonstrated that if carbohydrate binding is impinged then carbohydrate phosphatase activity is also diminished [(Meekins et al., 2015), (Raththagala et al., 2015), (Emanuelle et al., 2016), (Gentry et al., 2016)]. Therefore, the results of the phosphatase assays also demonstrate that laforin N163D binds glycogen similar to laforin wild type.
Fig. 3. Laforin N163D has a similar carbohydrate phosphatase activity and similar ability to bind carbohydrates as wild type.
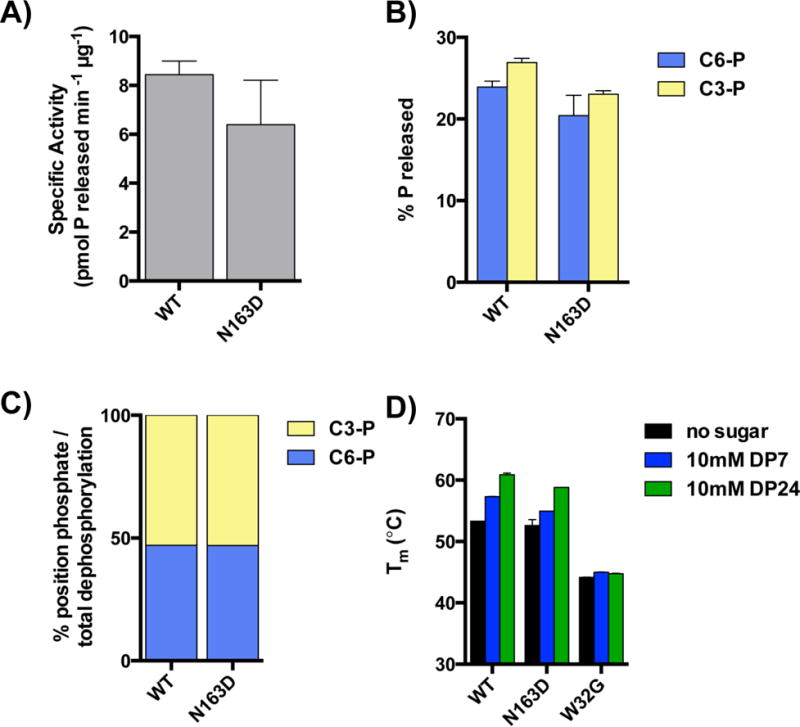
Recombinant laforin wild type and N163D were purified from bacteria. Phosphatase activities of laforin wild type and N163D toward glycogen (A) and substrate radiolabeled at either the C3- or C6-position (B) were assayed as described in Material and Methods. The C3 vs. C6-phosphate release as a percentage of total dephosphorylation is shown in (C). At least three independent samples were analyzed in each case. D) The thermal stability and oligosaccharide binding of purified laforin wild type, N163D and W32G was assessed by differential scanning fluorimetry, as described in Material and Methods. At least three independent samples were analyzed in each case.
Next, we analyzed the stability of laforin N163D using differential scanning fluorimetry (DSF) to quantify the protein’s melting temperature (Tm). As shown in Fig. 3D, purified recombinant laforin N163D has a similar Tm as wild type laforin again indicating that the protein is properly folded (black bars). When incubated with oligosaccharides of various lengths, laforin exhibits an increased Tm, indicative of oligosaccharide binding (Raththagala et al., 2015). Laforin N163D exhibited similar increased Tm shifts and no differences in binding was observed for maltoheptoase (DP7, blue bars) or longer oligosaccharides (average length of 24 glucose units, or DP24, green bars), indicating this mutant binds to oligosaccharides. Conversely, the disease-causing mutation W32G has been previously shown to be destabilized and lacks the ability to bind carbohydrates [(Wang et al., 2002), (Wang and Roach, 2004), (Raththagala et al., 2015)]. Indeed, laforin W32G exhibited a lowered Tm as previously reported and no increase in Tm in the presence of DP7 or DP24 (Fig. 3D).
3.5.- Laforin N163D has an impairment in the ability to interact with recognized partners
We and others have described that laforin forms a functional complex with malin, which is involved in the ubiquitination of glycogenic substrates [(Solaz-Fuster et al., 2008), (Vilchez et al., 2007)]. In this complex, laforin recruits specific substrates to be ubiquitinated by the E3-ubiquitin ligase activity of malin. As discussed in the introduction, the exact physiological role of the laforin-malin interaction is currently disputed, but the proteins do interact. In order to analyze the interaction pattern of laforin N163D, we performed yeast two-hybrid analyses with laforin itself [as laforin also forms dimers (Raththagala et al., 2015)], malin (Solaz-Fuster et al., 2008) and the glycogenic substrates R5/PTG (Solaz-Fuster et al., 2008) and R6 (Rubio-Villena et al., 2013)]. We observed that laforin N163D displays a reduced ability to interact with itself in the yeast two-hybrid system (Fig. 4, left panel), and a reduced capacity to interact with malin, R5/PTG, or R6 (Fig. 4, right panel), in spite of the protein being expressed in yeast at similar levels as wild type.
Fig. 4. Laforin N163D loses the ability to interact with recognized partners.
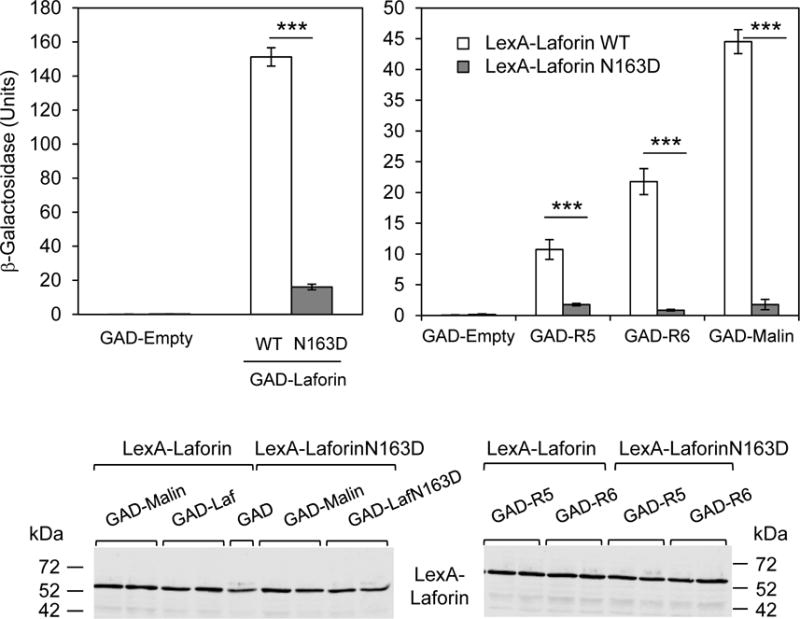
Yeast strain THY-AP4 was transformed with the following combination of plasmids: in the left panel, plasmids expressing LexA-laforin wild type were combined with plasmids pGAD-empty or pGAD-laforin WT, in order to check dimerization; similarly plasmid expressing LexA-laforin N163D was combined with pGAD-empty or pGAD-laforin N163D. In the right panel, plasmids expressing LexA-laforin wild type or N163D were combined with and plasmids pGAD-empty, pGAD-R5, pGAD-R6 and pGAD-malin respectively. The strength of the interaction was determined by measuring β-galactosidase activity as indicated in Material and Methods. Results are the mean of at least six independent transformants in each case (bars indicate standard deviation; ***p<0.001). A representative Western blot of the proteins expressed by the different transformants (two samples of each combination) is shown in the lower panel. Western blot was analyzed with anti-LexA antibodies.
Taking together, these results indicate that laforin N163D, although it has a functional carbohydrate binding module and a functional DSP domain, has reduced ability to interact with its corresponding partners. Thus we propose that laforin N163D is less competent in forming a functional laforin-malin complex and interacting with specific partners (R5/PTG, R6) or with itself.
4.- DISCUSSION
Most consider that Lafora disease is a clinically homogeneous disorder (Turnbull et al., 2016). It starts with seizures in the second decade of life and follows with a progressive dementia, refractory status epilepticus and the eventual death of the patient within around a decade from the first symptoms. However, in some cases there is a late-onset or a slower progression of the disease. In these cases it is assumed that either the mutation present in the EPM2A or EPM2B genes only partially affects the function of the corresponding protein, or that genetic or environmental modifiers may influence the regular progression of the disease [(Franceschetti et al., 2006), (Striano et al., 2008), (Guerrero et al., 2011), (Ferlazzo et al., 2014), (Kecmanovic et al., 2016)]. In this work, we describe a case of late-onset and slow progression Lafora disease. The affected patient is at present 28 years old and still has an independent life. She was diagnosed at the Hospital Vall d’Hebron in Barcelona (Spain) and the genetic analyses revealed a compound heterozygosity in the EPM2A gene: Y112X/N163D.
In order to establish a possible correlation between the clinical phenotype and the observed genetic alterations, we searched in the literature for the information about both laforin mutations. First, the Y112X mutation has been described recently in two affected patients from Turkey (Poyrazoglu et al., 2015). They were homozygous for the Y112X mutation and presented the common rapid progression of the disease. The Y112X mutation belongs to the nonsense or premature termination codon mutation class. In these mutations the nonsense-mediated mRNA decay mechanism (Brogna and Wen, 2009) degrades the corresponding mRNA, so presumably no protein is formed. Therefore, the Y112X allele present in our patient should not be responsible for the slow progression of the disease. Conversely, the laforin N163D missense mutation is novel.
To our surprise, when we analyzed the endogenous levels of the mutated laforin forms in primary fibroblasts from the patient we were not able to detect them by Western blotting. We analyzed gene expression of the EPM2A alleles and observed intermediate levels between control and R241X/R241X fibroblasts, being the latter an example of expression of genes subjected to nonsense-mediated mRNA decay mechanisms. We also analyzed the stability of the laforin N163D protein by expressing it in HEK293 cells and observed a similar pattern as wild type. This latter result prompted us to biochemically analyze the consequences of this mutation, in order to understand the slow progression of the disease. First, we analyzed the phosphatase activity of laforin N163D on complex phosphorylated carbohydrates and observed that it exhibited similar phosphatase activity to wild type. In addition, since the purification of the protein was based on its capacity to bind to amylose, laforin N163D can bind carbohydrate in a similar manner as wild type. The main defect we found with laforin N163D mutant was its reduced ability to interact with recognized laforin partners: laforin N163D showed a decrease interaction with itself, with malin, or with R5/PTG and R6 glycogenic substrates, all recognized partners in laforin function. Crystallographic contacts and analytical ultracentrifugation experiments show that laforin forms a dimer via an extensive hydrophobic patch within the DSP domain, and that mutation of a critical hydrophobic residue within this interface (F321) renders laforin monomeric (Raththagala et al., 2015). However, based on the structure we suspect that N163D alters its ability to interact with many binding partners, which may explain the reduced interaction with itself by yeast two-hybrid assays. It is of note that the N163D mutation is located proximal to the region involved in the interaction with malin [(Lohi et al., 2005), (Zeng et al., 2012)]. Interestingly, other mutations mapping to the DSP domain such as G279C, W287R and F321C have been identified in late-onset forms of Lafora disease [(Jara-Prado et al., 2014), (El Tahry et al., 2015), (Lynch et al., 2016)], although no reports on either phosphatase activity of interaction profile were reported for these mutations. In any case, it would be interesting to assess whether mutations in this area affect malin interaction without affecting phosphatase activity.
Taking all these results together, we conclude that the slow progression of the disease present in this patient could be either due to the specific biochemical properties of laforin N163D or to the presence of alternative genetic modifying factors separate from pathogenicity.
Supplementary Material
Highlights.
-
-
Novel EPM2A mutation (N163D) is present in a slow progression form of Lafora disease
-
-
Gene expression of N163D allele is not affected, but protein levels are undetectable
-
-
Expressed laforin N163D has regular phosphatase activity
-
-
Expressed laforin N163D has a defect in its interaction with binding partners
Acknowledgments
This work was supported by grants from the Spanish Ministry of Economy and Competitiveness SAF2014-54604-C3-1-R and a grant from Generalitat Valenciana (PrometeoII/2014/029); and National Institute of Health grants R01NS070899 and P01NS097197, which established the Lafora Epilepsy Cure Initiative (LECI). We want to thank the family of the Lafora disease patient for their collaboration.
7.- ABBREVIATIONS
- CBM
carbohydrate binding module
- DSP
dual-specificity phosphatase
- LB
Lafora bodies
- LD
Lafora disease
- qPCR
quantitative real-time polymerase chain reaction
Footnotes
CONFLICT OF INTEREST: On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Berkovic SF, Andermann F, Carpenter S, Wolfe LS. Progressive myoclonus epilepsies: specific causes and diagnosis. N Engl J Med. 1986;315:296–305. doi: 10.1056/NEJM198607313150506. [DOI] [PubMed] [Google Scholar]
- Brogna S, Wen J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol. 2009;16:107–113. doi: 10.1038/nsmb.1550. [DOI] [PubMed] [Google Scholar]
- Contreras CJ, Segvich DM, Mahalingan K, Chikwana VM, Kirley TL, Hurley TD, DePaoli-Roach AA, Roach PJ. Incorporation of phosphate into glycogen by glycogen synthase. Arch Biochem Biophys. 2016;597:21–29. doi: 10.1016/j.abb.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJ, Bohlega S, Andermann E, Rouleau GA, Delgado-Escueta AV, Minassian BA, Scherer SW. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- Cheng A, Zhang M, Gentry MS, Worby CA, Dixon JE, Saltiel AR. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 2007;21:2399–2409. doi: 10.1101/gad.1553207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Contreras CJ, Segvich DM, Heiss C, Ishihara M, Azadi P, Roach PJ. Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. J Biol Chem. 2015;290:841–850. doi: 10.1074/jbc.M114.607796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285:25372–25381. doi: 10.1074/jbc.M110.148668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukhande VV, Rogers DM, Roma-Mateo C, Donderis J, Marina A, Taylor AO, Sanz P, Gentry MS. Laforin, a dual specificity phosphatase involved in lafora disease, is present mainly as monomeric form with full phosphatase activity. PLoS One. 2011;6:e24040. doi: 10.1371/journal.pone.0024040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Tahry R, de Tourtchaninoff M, Vrielynck P, Van Rijckevorsel K. Lafora disease: psychiatric manifestations, cognitive decline, and visual hallucinations. Acta Neurol Belg. 2015;115:471–474. doi: 10.1007/s13760-014-0399-3. [DOI] [PubMed] [Google Scholar]
- Emanuelle S, Brewer MK, Meekins DA, Gentry MS. Unique carbohydrate binding platforms employed by the glucan phosphatases. Cell Mol Life Sci. 2016;73:2765–2778. doi: 10.1007/s00018-016-2249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlazzo E, Canafoglia L, Michelucci R, Gambardella A, Gennaro E, Pasini E, Riguzzi P, Plasmati R, Volpi L, Labate A, Gasparini S, Villani F, Casazza M, Viri M, Zara F, Minassian BA, Turnbull J, Serratosa JM, Guerrero-Lopez R, Franceschetti S, Aguglia U. Mild Lafora disease: clinical, neurophysiologic, and genetic findings. Epilepsia. 2014;55:e129–133. doi: 10.1111/epi.12806. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Gambardella A, Canafoglia L, Striano P, Lohi H, Gennaro E, Ianzano L, Veggiotti P, Sofia V, Biondi R, Striano S, Gellera C, Annesi G, Madia F, Civitelli D, Rocca FE, Quattrone A, Avanzini G, Minassian B, Zara F. Clinical and genetic findings in 26 Italian patients with Lafora disease. Epilepsia. 2006;47:640–643. doi: 10.1111/j.1528-1167.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Munz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon HU, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh S, Puri R, Singh S, Mittal S, Dubey D. Recent advances in the molecular basis of Lafora’s progressive myoclonus epilepsy. J Hum Genet. 2006;51:1–8. doi: 10.1007/s10038-005-0321-1. [DOI] [PubMed] [Google Scholar]
- Garcia-Haro L, Garcia-Gimeno MA, Neumann D, Beullens M, Bollen M, Sanz P. The PP1-R6 protein phosphatase holoenzyme is involved in the glucose-induced dephosphorylation and inactivation of AMP-activated protein kinase, a key regulator of insulin secretion, in MIN6 beta cells. FASEB J. 2010;24:5080–5091. doi: 10.1096/fj.10-166306. [DOI] [PubMed] [Google Scholar]
- Gentry MS, Brewer MK, Vander Kooi CW. Structural biology of glucan phosphatases from humans to plants. Curr Opin Struct Biol. 2016;40:62–69. doi: 10.1016/j.sbi.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Dowen RH, 3rd, Worby CA, Mattoo S, Ecker JR, Dixon JE. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J Cell Biol. 2007;178:477–488. doi: 10.1083/jcb.200704094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Guinovart JJ, Minassian BA, Roach PJ, Serratosa JM. Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem. 2018;293:7117–7125. doi: 10.1074/jbc.R117.803064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Abad C, Gómez-Garre P, Gutiérrez-Delicado E, Saygi S, Michelucci R, Tassinari CA, Rodriguez de Cordoba S, Serratosa JM. Lafora disease due to EPM2B mutations. A clinical and genetic study. Neurology. 2005;64:982–986. doi: 10.1212/01.WNL.0000154519.10805.F7. [DOI] [PubMed] [Google Scholar]
- Guerrero R, Vernia S, Sanz R, Abreu-Rodriguez I, Almaraz C, Garcia-Hoyos M, Michelucci R, Tassinari CA, Riguzzi P, Nobile C, Sanz P, Serratosa JM, Gomez-Garre P. A PTG variant contributes to a milder phenotype in Lafora disease. PLoS One. 2011;6:e21294. doi: 10.1371/journal.pone.0021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara-Prado A, Ochoa A, Alonso ME, Lima Villeda GA, Fernandez-Valverde F, Ruano-Calderon L, Vargas-Canas S, Duron RM, Delgado-Escueta AV, Martinez-Juarez IE. Late onset Lafora disease and novel EPM2A mutations: breaking paradigms. Epilepsy Res. 2014;108:1501–1510. doi: 10.1016/j.eplepsyres.2014.08.017. [DOI] [PubMed] [Google Scholar]
- Kecmanovic M, Keckarevic-Markovic M, Keckarevic D, Stevanovic G, Jovic N, Romac S. Genetics of Lafora progressive myoclonic epilepsy: current perspectives. Appl Clin Genet. 2016;9:49–53. doi: 10.2147/TACG.S57890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafora GR, Glueck B. Beitrag zur histogpathologie der myoklonischen epilepsie. Gesamte Neurol Psychiatr. 1911;6:1–14. [Google Scholar]
- Lohi H, Ianzano L, Zhao XC, Chan EM, Turnbull J, Scherer SW, Ackerley CA, Minassian BA. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet. 2005;14:2727–2736. doi: 10.1093/hmg/ddi306. [DOI] [PubMed] [Google Scholar]
- Ludin K, Jiang R, Carlson M. Glucose-regulated interaction of a regulatory subunit of protein phosphatase 1 with the Snf1 protein kinase in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1998;95:6245–6250. doi: 10.1073/pnas.95.11.6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch DS, Wood NW, Houlden H. Late-onset Lafora disease with prominent parkinsonism due to a rare mutation in EPM2A. Neurol Genet. 2016;2:e101. doi: 10.1212/NXG.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins DA, Raththagala M, Auger KD, Turner BD, Santelia D, Kotting O, Gentry MS, Vander Kooi CW. Mechanistic Insights into Glucan Phosphatase Activity against Polyglucan Substrates. J Biol Chem. 2015;290:23361–23370. doi: 10.1074/jbc.M115.658203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian BA, Ianzano L, Meloche M, Andermann E, Rouleau GA, Delgado-Escueta AV, Scherer SW. Mutation spectrum and predicted function of laforin in Lafora’s progressive myoclonus epilepsy. Neurology. 2000;55:341–346. doi: 10.1212/wnl.55.3.341. [DOI] [PubMed] [Google Scholar]
- Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S, Jardim L, Satishchandra P, Andermann E, Snead OC, 3rd, Lopes-Cendes I, Tsui LC, Delgado-Escueta AV, Rouleau GA, Scherer SW. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- Nitschke F, Wang P, Schmieder P, Girard JM, Awrey DE, Wang T, Israelian J, Zhao X, Turnbull J, Heydenreich M, Kleinpeter E, Steup M, Minassian BA. Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab. 2013;17:756–767. doi: 10.1016/j.cmet.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Poyrazoglu HG, Karaca E, Per H, Gumus H, Onay H, Canpolat M, Canoz O, Ozkinay F, Kumandas S. Three patients with lafora disease: different clinical presentations and a novel mutation. J Child Neurol. 2015;30:777–781. doi: 10.1177/0883073814535489. [DOI] [PubMed] [Google Scholar]
- Raththagala M, Brewer MK, Parker MW, Sherwood AR, Wong BK, Hsu S, Bridges TM, Paasch BC, Hellman LM, Husodo S, Meekins DA, Taylor AO, Turner BD, Auger KD, Dukhande VV, Chakravarthy S, Sanz P, Woods VL, Jr, Li S, Vander Kooi CW, Gentry MS. Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Mol Cell. 2015;57:261–272. doi: 10.1016/j.molcel.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ. Glycogen phosphorylation and Lafora disease. Mol Aspects Med. 2015;46:78–84. doi: 10.1016/j.mam.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J. 2012;441:763–787. doi: 10.1042/BJ20111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roma-Mateo C, Aguado C, Garcia-Gimenez JL, Ibanez-Cabellos JS, Seco-Cervera M, Pallardo FV, Knecht E, Sanz P. Increased oxidative stress and impaired antioxidant response in lafora disease. Mol Neurobiol. 2015;51:932–946. doi: 10.1007/s12035-014-8747-0. [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics, a laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor; New York: 1990. [Google Scholar]
- Rubio-Villena C, Garcia-Gimeno MA, Sanz P. Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin-malin complex. Int J Biochem Cell Biol. 2013;45:1479–1488. doi: 10.1016/j.biocel.2013.04.019. [DOI] [PubMed] [Google Scholar]
- Sakai M, Austin J, Witmer F, Trueb L. Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology. 1970;20:160–176. doi: 10.1212/wnl.20.2.160. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin P, Raththagala M, Bridges TM, Husodo S, Gentry MS, Sanz P, Roma-Mateo C. Dimerization of the glucan phosphatase laforin requires the participation of cysteine 329. PLoS One. 2013;8:e69523. doi: 10.1371/journal.pone.0069523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santelia D, Kotting O, Seung D, Schubert M, Thalmann M, Bischof S, Meekins DA, Lutz A, Patron N, Gentry MS, Allain FH, Zeeman SC. The phosphoglucan phosphatase like sex Four2 dephosphorylates starch at the C3-position in Arabidopsis. Plant Cell. 2011;23:4096–4111. doi: 10.1105/tpc.111.092155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serratosa JM, Delgado-Escueta AV, Posada I, Shih S, Drury I, Berciano J, Zabala JA, Antunez MC, Sparkes RS. The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum Mol Genet. 1995;4:1657–1663. doi: 10.1093/hmg/4.9.1657. [DOI] [PubMed] [Google Scholar]
- Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D, Augustijn PB, Tassinari CA, Malafosse RM, Topcu M, Grid D, Dravet C, Berkovic SF, de Cordoba SR. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum Mol Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- Sherwood AR, Johnson MB, Delgado-Escueta AV, Gentry MS. A bioassay for Lafora disease and laforin glucan phosphatase activity. Clin Biochem. 2013a;46:1869–1876. doi: 10.1016/j.clinbiochem.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood AR, Paasch BC, Worby CA, Gentry MS. A malachite green-based assay to assess glucan phosphatase activity. Anal Biochem. 2013b;435:54–56. doi: 10.1016/j.ab.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaz-Fuster MC, Gimeno-Alcaniz JV, Ros S, Fernandez-Sanchez ME, Garcia-Fojeda B, Criado Garcia O, Vilchez D, Dominguez J, Garcia-Rocha M, Sanchez-Piris M, Aguado C, Knecht E, Serratosa J, Guinovart JJ, Sanz P, Rodriguez de Cordoba S. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet. 2008;17:667–678. doi: 10.1093/hmg/ddm339. [DOI] [PubMed] [Google Scholar]
- Striano P, Zara F, Turnbull J, Girard JM, Ackerley CA, Cervasio M, De Rosa G, Del Basso-De Caro ML, Striano S, Minassian BA. Typical progression of myoclonic epilepsy of the Lafora type: a case report. Nat Clin Pract Neurol. 2008;4:106–111. doi: 10.1038/ncpneuro0706. [DOI] [PubMed] [Google Scholar]
- Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, Roach PJ. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283:33816–33825. doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, Azadi P, Hurley TD, DePaoli-Roach AA, Roach PJ. Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab. 2011;13:274–282. doi: 10.1016/j.cmet.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, Depaoli-Roach AA, Roach PJ. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104:19262–19266. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Tiberia E, Striano P, Genton P, Carpenter S, Ackerley CA, Minassian BA. Lafora disease. Epileptic Disord. 2016;18:38–62. doi: 10.1684/epd.2016.0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, Kameka AP, Pencea N, Zhao X, Ackerley CA, Minassian BA. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol. 2010;68:925–933. doi: 10.1002/ana.22156. [DOI] [PubMed] [Google Scholar]
- Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, Serafin A, Canas X, Soriano E, Delgado-Garcia JM, Gruart A, Guinovart JJ. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med. 2011;3:667–681. doi: 10.1002/emmm.201100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Rubio T, Heredia M, Rodriguez de Cordoba S, Sanz P. Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PLoS One. 2009;4:e5907. doi: 10.1371/journal.pone.0005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, Criado-Garcia O, Fernandez-Sanchez E, Medrano-Fernandez I, Dominguez J, Garcia-Rocha M, Soriano E, Rodriguez de Cordoba S, Guinovart JJ. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nature Neurosci. 2007;10:1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- Wang J, Stuckey JA, Wishart MJ, Dixon JE. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J Biol Chem. 2002;277:2377–2380. doi: 10.1074/jbc.C100686200. [DOI] [PubMed] [Google Scholar]
- Wang W, Roach PJ. Glycogen and related polysaccharides inhibit the laforin dual-specificity protein phosphatase. Biochem Biophys Res Commun. 2004;325:726–730. doi: 10.1016/j.bbrc.2004.10.083. [DOI] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, Dixon JE. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281:30412–30418. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, Dixon JE. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG) J Biol Chem. 2008;283:4069–4076. doi: 10.1074/jbc.M708712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Hubbard EJ, Carlson M. A protein kinase substrate identified by the two-hybrid system. Science. 1992;257:680–682. doi: 10.1126/science.1496382. [DOI] [PubMed] [Google Scholar]
- Yu TS, Kofler H, Hausler RE, Hille D, Flugge UI, Zeeman SC, Smith AM, Kossmann J, Lloyd J, Ritte G, Steup M, Lue WL, Chen J, Weber A. The Arabidopsis sex1 mutant is defective in the R1 protein, a general regulator of starch degradation in plants, and not in the chloroplast hexose transporter. Plant Cell. 2001;13:1907–1918. doi: 10.1105/TPC.010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Wang Y, Baba O, Zheng P, Liu Y. Laforin is required for the functional activation of malin in endoplasmic reticulum stress resistance in neuronal cells. FEBS J. 2012;279:2467–2478. doi: 10.1111/j.1742-4658.2012.08627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.