

Abstract
Identifying sickle cell disease patients at high risk of complications could lead to personalized treatment and better prognosis but despite many advances prediction of the clinical course of these patients remains elusive. We propose a system-type approach to discover profiles of multiple, common biomarkers that correlate with morbidity and mortality in sickle cell disease. We used cluster analysis to discover 17 signatures of 17 common circulating biomarkers in 2320 participants of the Cooperative Study of Sickle Cell Disease, and evaluated the association of these signatures with risk for stroke, pain, leg ulceration, acute chest syndrome, avascular necrosis, seizure, death, and trend of fetal hemoglobin and hemolysis using longitudinally collected data. The analysis shows that some of the signatures are associated with reduced risk for complications, while others are associated with increased risk for complications. We also show that these signatures repeat in two more contemporary studies of sickle cell disease and correlate with recently discovered biomarkers of pulmonary vascular disease. With replication and further study, these biomarker signatures could become an important and affordable precision medicine tool to aid treatment and management of the disease.
Keywords: sickle cell disease, disease severity, molecular signature, clustering, fetal hemoglobin, hemolytic anemia
Introduction
Sickle cell disease is caused by mutations in the β-hemoglobin gene (HBB) and its epidemiology, pathophysiology and clinical complications have been recently reviewed1. The two most common genotypes of this disease are homozygosity for the HBB glu6val mutation (HbS; rs334) called sickle cell anemia (HbSS), and compound heterozygosity for HbS and HbC (glu6lys) mutations, called sickle cell–HbC (HbSC) disease. Both of these genotypes, and the less common sickle-β thalassemias, have extremely variable phenotypes2. In each genotype some individuals have mild disease that in the extreme can be clinically unapparent while others can have most of the known severe complications. These include pulmonary hypertension, priapism, stroke, leg ulceration, acute painful episodes, acute chest syndrome, avascular necrosis of bone and severe hemolytic anemia3. Identifying patients at high risk of complications could lead to better treatment and prognosis and substantial work has focused on discovering those genetic and non-genetic risk factors of sickle cell disease that could be used for risk prediction4.
The greatest progress has been made toward characterizing genetic variants that explain variability of fetal hemoglobin (HbF), which is a major marker and modulator of disease severity, and the effects of concurrent α thalassemia on subphenotypes of disease5-14. Additional genetic variants linked to higher risk for complications of the disease have been found through genetic association studies but they often explain only a small part of the disease variability15-23. We showed that an ensemble of genetic risk scores can predict with substantial precision HbF in patients with HbSS24, but the value of HbF level to predict disease complications is limited due to the variable distribution of erythrocyte HbF concentrations among patients with similar total HbF25. Despite these efforts, predicting patients at higher risk for complications using only genetic data remains difficult.
Between a genetic variant and a physiologic outcome, or between genotype and phenotype, are proteins and cells whose levels and biologic effects can be captured by laboratory measures and could be more powerful than genetic data to predict morbidity and mortality. The review by Rees and Gibson in 201126, reported more than 100 blood and urine biomarkers that have been correlated to at least one of the complications of sickle cell disease. However, as the authors of the review noted, it is still unclear whether this large collection of biomarkers adds any value to simple biomarkers such as hemoglobin concentration because often the discovery of predictive biomarkers is done “one-biomarker-at-a-time” and the relative added values of sets of biomarkers is not always established.
Scores reflecting disease severity have been proposed to stratify patients by risk of complications and typically they include a combination of laboratory data and patient medical history. Using data from the Cooperative Study of Sickle Cell Disease (CSSCD), we generated a model of disease severity that integrated some biomarkers with complications of the disease to define an overall measure of disease severity that can predict death within five years27. This model, independently validated in part28, is useful to understand the relationships among clinical and laboratory measures of disease expression, although it is not designed to predict complications of the disease29. Recent work by Desai et al30 discovered signatures based on a combination of blood mononuclear cell gene expression profiles and medical history of sickle cell disease patients to predict mortality. Despite these advances, prediction of the clinical course of patients with sickle cell disease remains elusive. We propose here a system-type approach to discover profiles of multiple, common biomarkers that correlate with morbidity and mortality in sickle cell disease.
Materials and Methods
Study populations
The CSSCD was a 10-year, multi-center, longitudinal study of sickle cell disease that has been extensively described31,32. The study began recruiting patients in 1979 from 23 centers resulting in over 3,600 patients in four age groups: newborns, children, adolescents, and adults. The Pulmonary Hypertension and the Hypoxic Response in Sickle Cell Disease (PUSH) and Sickle Cell Disease with Sildenafil Therapy (Walk-PHaSST) trials were used as more contemporary cohorts to replicate the findings33,34. All subjects provided informed consent approved by the field centers IRB. More detailed information about the studies is in supplement material.
Statistical Analysis
Biomarker selection and data preparation
In the CSSCD, 39 biomarkers were recorded at the baseline exam, including four subsequently shown to estimate the severity of the intravascular component of hemolytic anemia, along with other laboratory data35,36. Biomarkers that were dichotomous, homogeneous, and had more than 25% missing data were ignored. One marker was selected from groups of highly correlated biomarkers based on clinical interpretation (correlation coefficient > 0.5). This selection removed 22 biomarkers and left 17 biomarkers for analysis (Supplement Table 1). Outliers defined as values that differ from the mean by more than six standard deviation were removed and biomarkers were transformed using a log or a cubic root transformation as needed. Regression-based imputation with age and sex was used to create complete profiles for each subject and biomarkers were standardized within sex and age groups (0-4, 5-12, 13-19, 20-29, 30+).
Hierarchical Clustering
Hierarchical clustering with complete linkage and Euclidean distance was used to group CSSCD participants based on the 17 standardized biomarkers (Supplement Figure 1). To determine the number of significant clusters, we used the resampling procedure introduced in37 (Supplement Figure 1). Biomarker signatures were defined by the median biomarker values in each cluster, and interquartile range, and are summarized in Supplement Table 1. Clusters are visually displayed using side-by-side boxplots of the 17 biomarkers (Figure 1 and Supplement Figure 2) and color-coded based on biomarker function (magenta = liver function, green=kidney function, red= anemia).
Figure 1. Most common biomarker signatures discovered in CSSCD.
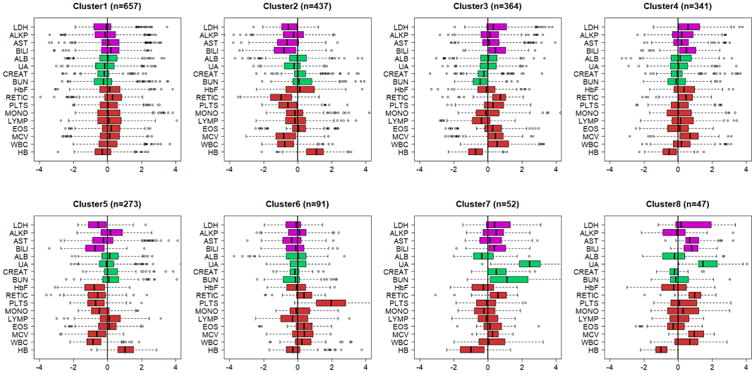
The side-by-side boxplots show the patterns of 17 biomarkers in the 8 clusters with > 40 subjects. Biomarkers are age and sex standardized and color coded by function: magenta=liver function; green= kidney function; red=anemia. All 17 clusters are displayed in Supplement Figure 2. (LDH-lactic dehydrogenase; ALKP-alkaline phosphatase; AST-aspartate aminotransferase; BILI-total bilirubin; ALB-serum albumen; URA-uric acid; CREAT-creatinine; BUN-Blood urea nitrogen; HbF-fetal hemoglobin level; RETIC-reticulocyte count; PLTS-platelets; MON-monocyte count; LYMP-lymphocyte count; EOS-eosinophil count; MCV-mean corpuscular volume; WBC white blood cell count; HB-hemoglobin level).
Annotation with disease complication risk
We estimated that a sample size of 40 subjects provides 80% power to detect odds ratios of at least three for common complications. Therefore, clusters with at least 40 people were correlated with HBB genotype (HbSS, HBSC, HbSS-α thalassemia and β thalassemia, or another variant), morbidity (stroke, seizure, painful episodes, acute chest syndrome (ACS), avascular necrosis of bone (AVN) and leg ulceration), mortality, and trends of HbF and of a score of hemolytic anemia in prospectively collected data. In all analyses, the cluster with the majority of standardized biomarkers that were symmetrically distributed around zero was set as the reference cluster (Cluster 1). Relative risk for mortality, AVN, ACS, leg ulceration, stroke, and seizure of the various clusters compared to the referent clusters were estimated using Cox Proportional Hazards model controlling for age at enrollment, sex, and hemoglobin genotype. Since dates of seizures were not recorded, time to the first follow-up visit after a seizure was used in the survival analysis. The proportional hazard assumption was tested using Schoenfeld residuals38. To assess the effect of cluster membership on risk for painful episodes, three different outcomes were analyzed. The first outcome was the severity of a painful episode when subjects reported to a hospital for an acute episode. Severity was self-assessed as mild, moderate, severe. Since patients reported multiple acute episodes with hospitalization over the course of the study, we estimated the odds of having a severe painful episode compared to a mild or moderate painful episode using logistic regression estimated with generalized estimating equations (GEE). The second outcome was the number of painful episodes reported at each routine follow-up visit that were analyzed using logistic regression with GEE. The third outcome was the time to the first acute episode (reporting to the hospital with an episode) that was analyzed using a Cox Proportional Hazards model. All models controlled for baseline age, sex, and HBB genotype.
Annotation with longitudinal trend of HbF and hemolysis
The association between biomarker signatures and longitudinal trend of HbF was assessed using a linear regression model that included cluster membership, age, sex and HBB genotype. GEE was used to estimate the regression parameters from repeated measurements. To estimate the association between intravascular hemolysis and biomarker signatures, we used principal component analysis (PCA) of AST, LDH, reticulocyte and bilirubin collected at enrollment and at the first follow up visit, to derive a score of hemolysis as in39. The first principal components from each analysis were analyzed in a regression model that included age, sex, HBB genotype and cluster membership using GEE. In neither analysis the interaction between cluster membership and time reached statistical significance suggesting that signatures associated with different clusters affect levels of hemolysis and HbF but not their change over time.
Replication in PUSH and Walk-PHaSST Studies
PUSH40 and Walk-PHaSST41 were used for replication and validation. The PUSH data contained 16 out of the 17 biomarkers (missing uric acid), and the Walk-PHaSST data contained 13 out of the 17 biomarkers (missing uric acid, eosinophils, lymphocytes and monocytes). We used the Bayesian classification method described in42 to compute the most likely cluster membership for each subject in PUSH and Walk-PhaSST. This method essentially computed the probability that each subject was in one of the 17 clusters discovered in the CSSCD using the subject biomarker data, and allocated the subject to the most likely cluster. Subjects in the two studies were stratified using the most likely cluster assignment, and the distribution of the biomarkers in the various clusters are displayed using side-by-side boxplots (Figures 3, 4 and Supplement Figures 3, 4). We correlated the clusters predicted in PUSH and Walk-PHaSST with HBB genotypes, history of hydroxyurea treatment, tricuspid regurgitant jet velocity (TRV) and additional biomarkers including nt-proBNP measured at enrollment. We also analyzed the association between biomarker signatures and mortality data in Walk-PHaSST (Table 3). In these two data sets, the referent clusters were chosen based on the majority of standardized biomarkers that were symmetrically distributed around zero.
Figure 3. Predicted signatures in PUSH.
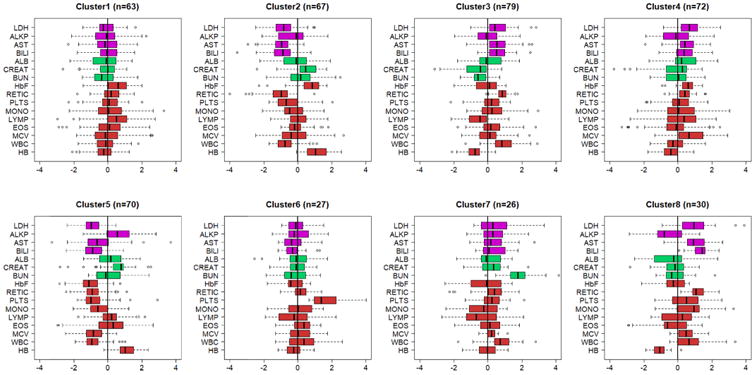
Cluster membership of each PUSH subject was predicted using a Bayesian classifier and the side-by-side boxplots display the patterns of 16 biomarkers stratified by predicted clusters. Biomarkers are age and sex standardized and color coded by function: magenta=liver function; green= kidney function; red=anemia. Additional clusters are displayed in Supplement Figure 3. (LDH-lactic dehydrogenase; ALKP-alkaline phosphatase; AST-aspartate aminotransferase; BILI-total bilirubin; ALB-serum albumen; URA-uric acid; CREAT-creatinine; HbF-fetal hemoglobin level; RETIC-reticulocyte count; PLTS-platelets; MON-monocyte count; LYMP-lymphocyte count; EOS-eosinophil count; MCV-mean corpuscular volume; WBC white blood cell count; HB-hemoglobin level).
Figure 4. Predicted signatures in Walk-PHaSST.
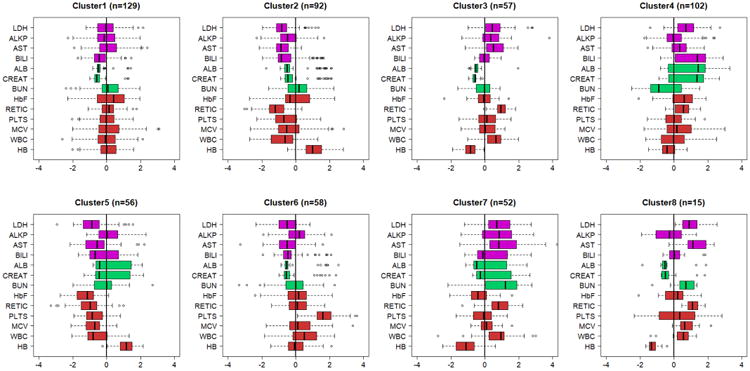
Cluster membership of each Walk-PHaSST subject was predicted using a Bayesian classifier and the side-by-side boxplots display the patterns of 15 biomarkers stratified by predicted clusters. Biomarkers are age and sex standardized and color coded by function: magenta=liver function; green= kidney function; red=anemia. Additional clusters are displayed in Supplement Figure 4. (LDH-lactic dehydrogenase; ALKP-alkaline phosphatase; AST-aspartate aminotransferase; BILI-total bilirubin; ALB-serum albumen; URA-uric acid; CREAT-creatinine; HbF-fetal hemoglobin level; RETIC-reticulocyte count; PLTS-platelets; EOS-eosinophil count; MCV-mean corpuscular volume; WBC white blood cell count; HB-hemoglobin level).
Table 3. Replication analysis in the PUSH and Walk-PHaSST studies.
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Cluster 7 | Cluster 8 | Other | |
|---|---|---|---|---|---|---|---|---|---|
| PUSH | |||||||||
| Sample Size | 63 | 67 | 79 | 72 | 70 | 27 | 26 | 30 | 11 |
| Age (years, mean (sd)) | 13.25(4.98) | 12.22(5.82) | 11.08(5.69) | 11.28(5.3) | 10.69(4.61) | 13.04(4.79) | 13.12(4.93) | 12.6(4.53) | 14.82(4.42) |
| Sex = Male (%) | 52.4% | 49.3% | 46.8% | 45.8% | 51.4% | 66.7% | 53.8% | 60.0% | 72.7% |
| Hemoglobin Genotype (%) | |||||||||
| HbSS | 53(88.3%) | 29(46.0%) | 61(91.0%) | 65(100.0%) | 8(12.1%) | 21(84.0%) | 21(91.3%) | 25(96.2%) | 10(90.9%) |
| HbSC | 3(5.0%) | 30(47.6%) | 0(0.0%) | 0(0.0%) | 45(68.2%) | 2(8.0%) | 0(0.0%) | 0(0.0%) | 0(0.0%) |
| Other1 | 4(0%) | 4(0%) | 6(0%) | 0(0%) | 13(0%) | 2(0%) | 2(0%) | 1(0%) | 1(0%) |
| Hydroxyurea (%) | |||||||||
| Yes | 27(42.9%) | 28(41.8%) | 24(30.4%) | 41(56.9%) | 8(11.4%) | 10(37.0%) | 4(15.4%) | 18(60.0%) | 2(18.2%) |
| No | 35(55.6%) | 39(58.2%) | 54(68.4%) | 29(40.3%) | 61(87.1%) | 17(63.0%) | 22(84.6%) | 11(36.7%) | 8(72.7%) |
| Unknown | 1(1.6%) | 0(0.0%) | 1(1.3%) | 2(2.8%) | 1(1.4%) | 0(0.0%) | 0(0.0%) | 1(3.3%) | 1(9.1%) |
| Walk-Phasst | |||||||||
| Sample Size | 129 | 92 | 57 | 102 | 56 | 58 | 52 | 15 | 8 |
| Age | |||||||||
| (years, mean (sd)) | 33.48(13.02) | 37.34(12.51) | 34.61(13.55) | 38.25(11.67) | 37.65(14.12 | ) 36.2(12.58) | 39.2(11.75) | 43.27(16.16) | 34.29(17.22) |
| Sex = Male (%) | 51.9% | 41.3% | 47.4% | 48.0% | 39.3% | 43.1% | 46.2% | 33.3% | 75.0% |
| Hemoglobin Genotype (%) | |||||||||
| HbSS | 109(84%) | 29(32%) | 51(89%) | 95(93%) | 11(20%) | 47(81%) | 49(94%) | 12(80%) | 4(50%) |
| HbSC | 13(10%) | 44(48%) | 3(5%) | 3(3%) | 40(71%) | 6(10%) | 2(4%) | 1(7%) | 2(25%) |
| Other2 | 7(0%) | 19(0%) | 3(0%) | 4(0%) | 5(0%) | 5(0%) | 1(0%) | 2(0%) | 2(0%) |
| Hydroxyurea (%) | |||||||||
| Current | 61(47%) | 33(36%) | 16(28%) | 38(37%) | 5(9%) | 28(48%) | 14(27%) | 8(53%) | 0(0%) |
| No/Past | 68(53%) | 59(64%) | 41(72%) | 64(63%) | 51(91%) | 30(52%) | 38(73%) | 7(47%) | 8(100%) |
| Mortality1 HR (p) | 1 | 0.55(0.67) | No events | 0.39(0.26) | 0.56(0.67) | 0.47(0.048) | 2.3(0.16) | No events |
Other genotypes include HbS/β+ Thal, HbS/β0 Thal, and unspecified
Other genotypes include HbS/β+ Thal, HbS/β0 Thal, HbS/pThal, and unspecified
All analyses were conducted in R3.3.
Results
Table 1 summarizes characteristics of 2320 CSSCD subjects included in the analysis, after exclusion of participants who did not have HbF values (564), or were missing their entire blood panel at enrollment (n=596). The mean age of all participants was 15.30 years (standard deviation [sd]: 12.15) and the subjects ranged from newborns to age 63. The mean follow-up time was 6.44 years (sd: 2.32) and 1217 (52.2%) were men.
Table 1. Demographic characteristics of sickle cell disease patients by cluster.
| Overall | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Cluster 7 | Cluster 8 | |
|---|---|---|---|---|---|---|---|---|---|
| n | 2320 | 657 | 437 | 364 | 341 | 273 | 91 | 52 | 47 |
| Age (years, mean (sd)) | 15.30 (12.15) | 14.97 (11.30) | 15.46 (13.13) | 14.91 (11.61) | 16.26 (11.72) | 15.04 (12.89) | 12.69 (10.78) | 18.98 (14.57) | 15.40 (12.34) |
| Follow-up years (mean (sd))1 | 6.44 (2.32) | 6.49 (2.27) | 6.50 (2.78) | 6.17 (2.09) | 6.44 (2.10) | 6.45 (2.42) | 6.63 (1.86) | 6.16 (1.92) | 6.56 (2.12) |
| Sex = Male (%) | 1217 (52.5) | 327 (49.8) | 229 (52.4) | 188 (51.6) | 208 (61.0) | 144 (52.7) | 41 (45.1) | 26 (50.0) | 28 (59.6) |
| Fetal hemoglobin (%, mean(sd))2 | 8.47 (11.89) | 8.76 (10.83) | 11.56 (15.97) | 7.23 (9.42) | 10.04 (13.81) | 3.99 (5.42) | 7.42 (9.62) | 6.20 (7.44) | 4.94 (3.72) |
| Hemolytic Score3 | 0 | 0.31 | -1.38 | 0.86 | 0.84 | -1.31 | 0.48 | 0.72 | 1.28 |
| Hemoglobin (g/dL) | 9.23 (1.76) | 8.81 (1.34) | 10.88 (1.53) | 8.05 (1.11) | 8.35 (1.05) | 10.97 (1.24) | 9.04 (1.82) | 7.73 (1.45) | 7.49 (0.94) |
Time from baseline until death or date of last entry in any study dataset
Baseline fetal hemoglobin
Hemolytic score at baseline is the first principal component from principal component analysis of AST, reticulocyte counts, LDH and hemoglobin 40. A positive score denotes more hemolysis.
Discovery of Biomarker Signatures
A total of 17 biomarkers were included in the clustering analysis resulting in the dendrogram in Supplement Figure 1. We used a resampling procedure to detect 17 statistically significant clusters of subjects with similar biomarker profiles and a significance level of 0.00537. Supplement Table 1 shows summary statistics of biomarkers in the 17 clusters and Table 1 shows summary age, HbF level, and the hemolytic score based on principal component analysis in the eight clusters with at least 40 subjects. The complete description of patient characteristics by clusters is in Supplement Table 2. Note that the mean age of participants through Clusters 1-6, and 8 are very similar. Sensitivity analysis was conducted to test the selection of 17 biomarkers by testing the difference between each standardized biomarker in each cluster and the referent cluster. All differences were statistically significant implying that the clusters were efficiently capturing the varying biomarker profiles and all biomarkers were necessary.
Figure 1 displays side-by-side boxplots of the biomarkers in the eight largest clusters that included at least 40 people. The largest cluster, Cluster 1 with 657 subjects, was characterized by all biomarkers having a distribution centered at zero, and was used as referent group in all other analyses. In all of the other clusters, the biomarkers displayed characteristic patterns of departures from the average distribution, and the differences were statistically significant. For example, the levels of biomarkers of liver function in patients assigned to Cluster 2 appear to be below the values of age and sex matched referent participants (left of zero), while the patterns of markers of anemia (lower than average reticulocyte counts, white blood cell counts and higher than average hemoglobin) suggest that patients in Cluster 2 have less hemolysis than patients in Cluster 1. Cluster 3 shows almost the opposite situation, with higher than average biomarkers of liver function, higher markers of anemia and worse kidney function, suggesting that patients in this cluster have more hemolysis than patients in Cluster 1. Patients in Cluster 4 show a pattern of biomarkers of liver function that is similar to that of patients in Cluster 3, but less difference from the average distribution of markers of anemia, and increased HbF. Cluster 5 shows a pattern of liver and anemia biomarkers similar to Cluster 2, suggesting that these patients have less hemolysis than patients in Cluster 1 but, in addition, patients in Cluster 5 have substantially lower than average HbF compared to patients in Cluster 2. Patients in Clusters 6 have fairly average biomarkers with the exception of substantially elevated platelet levels (median standardized value =2). Patients in Clusters 7 and 8 are characterized by substantially elevated biomarkers of kidney function (median standardized uric acid >2). Additional clusters are small (13 or less patients) and the distribution of the biomarkers are visualized in Supplement Figure 2. We next investigated whether these different signatures of biomarkers have clinical relevance and can be used to identify sickle cell disease patients at risk for certain complications of the disease.
Annotation with disease complications
Table 2 shows the prevalence of HbSS, HbSC and HbSS -α-thalassemia in the most common clusters. HbSS was most prevalent in Clusters 1, 3, 4, 6, 7 and 8, while Clusters 2 and 5 were enriched for patients with HbSC disease. The prevalence of HbSS-α-thalassemia was highest in Cluster 1. The differences were statistically significant (p-value < 2 E-16). We also correlated cluster membership with HBB haplotypes that we recently inferred in CSSCD patients using a more sensitive algorithm,43 but the analysis did not detect any significant association between cluster membership and haplotype.
Table 2. Annotation of clusters with disease outcomes.
| Cluster 1 (n = 657) | Cluster 2 (n = 437) | Cluster 3 (n = 364) | Cluster 4 (n = 341) | Cluster 5 (n = 273) | Cluster 6 (n = 91) | Cluster 7 (n = 52) | Cluster 8 (n = 47) | |
|---|---|---|---|---|---|---|---|---|
| Hemoglobin Genotype (%) | ||||||||
| HbSS | 396 (60.6) | 68 (15.6) | 279 (76.6) | 270 (79.2) | 22 (8.1) | 56 (61.5) | 37 (71.2) | 45 (95.7) |
| HbSC | 42 (6.4) | 205 (46.9) | 3 (0.8) | 5 (1.5) | 194 (71.1) | 13 (14.3) | 4 (7.7) | 0 (0.0) |
| HbSS-α thalassemia | 167 (25.5) | 50 (11.4) | 62 (17.0) | 62 (18.2) | 18 (6.6) | 17 (18.7) | 11 (21.2) | 1 (2.1) |
| HbS-β+ thalassemia | 49 (7.5) | 114 (26.1) | 20 (5.5) | 4 (1.2) | 39 (14.3) | 5 (5.5) | 0 (0.0) | 1 (2.1) |
| Mortality1 HR (p) | Reference | 0.57 (0.17) | 1.24 (0.37) | 0.51 (0.03) | 1.73 (0.18) | 1.42 (0.39) | 1.08 (0.86) | 0.88 (0.81) |
| Stroke2 HR (p) | Reference | 0.26 (0.082) | 1.62 (0.15) | 0.66 (0.35) | 0.91 (0.88) | 0.74 (0.68) | 1.37 (0.67) | 0.60 (0.62) |
| Seizure1 HR (p) | Reference | 1.30 (0.46) | 1.04 (0.91) | 0.45 (0.06) | 0.58 (0.35) | 2.27 (0.042) | 2.21 (0.11) | 2.36 (0.082) |
| Leg Ulcer1 HR (p) | Reference | 0.57 (0.18) | 1.50 (0.067) | 1.00 (0.98) | 0.51 (0.30) | 1.55 (0.29) | 1.14 (0.75) | 1.35 (0.50) |
| Acute Chest Syndrom1 HR (p) | Reference | 0.75 (0.04)8 | 1.06 (0.61) | 0.90 (0.38) | 0.96 (0.83) | 1.53 (0.01) | 1.06 (0.81) | 1.06 (0.81) |
| Avascular Necrosis1 HR (p) | Reference | 1.08 (0.68) | 0.84 (0.34) | 0.67 (0.033) | 1.31 (0.24) | 2.12 (0.0029) | 0.58 (0.17) | 0.49 (0.17) |
| Time Until First Painful Episode1 HR (p) | Reference | 0.77 (0.026) | 0.87 (0.18) | 0.94 (0.51) | 1.13 (0.39) | 1.00 (0.99) | 0.50 (0.0095) | 0.78 (0.30) |
| Having a Painful Episode OR3,4 (p) | Reference | 0.68 (0.00046) | 0.97 (0.76) | 0.97 (0.74) | 0.87 (0.27) | 0.96 (0.78) | 1.09 (0.69) | 1.04 (0.87) |
| Having a Severe Painful Episode OR5 (p) | Reference | 0.82 (0.27) | 0.76 (0.074) | 0.60 (0.00048) | 0.96 (0.86) | 1.05 (0.83) | 1.74 (0.20) | 1.23 (0.56) |
| Fetal Hemoglobin Trajectory 6 (p) | 9.5% | 1.54 (0.00) | 0.83 (0.004) | 1.20 (0.006) | 0.98 (0.80) | 0.82 (0.05) | 0.91 (0.58) | 0.48 (0.00) |
| Hemolytic anemia7 (p) | Reference | -0.37 (0.00) | 0.18 (0.03) | 0.17 (0.07) | -0.25 (0.01) | 0.22 (0.07) | 0.25 (0.16) | 0.53 (0.07) |
Hazard ratios and p-values obtained from Cox Proportional Regression controlling for baseline age, sex, and hemoglobin genotype
HR adjusted for baseline age, sex and whether patients are HbSS or not
Odds ratios and p-values obtained from logistic regression controlling for baseline age, sex, and hemoglobin genotype
Model adjusted for HbSS or other genotype.
Odds ratios and p-values obtained from longitudinal data using GEE and logistic regression controlling for baseline age, sex, and hemoglobin genotype
Regression coefficients estimated from longitudinal data using GEE and an additive cluster effect. The value in Cluster 1 is the average HbF at age 8, and the values in the other clusters are fold changes compared to Cluster 1.
Regression coefficients estimated from longitudinal data using GEE and an additive cluster effect. Cluster effects are changes in hemolytic score compared to Cluster 1.Positive score = more hemolysis.
The levels of significance were not corrected for multiple testing and a p-value < 0.006 would be required for maintaining statistical significance after Bonferroni correction.
The clusters were annotated with hazard of mortality, ACS, AVN, leg ulceration, stroke, seizure, and acute painful episodes. Overall, 132 subjects included in this analysis died, 56 had at least one stroke, 91 had at least one seizure, 142 had leg ulceration, 700 had at least one ACS episode, 314 had AVN, and 942 had at least one acute painful episode in the longitudinal follow up (Supplemental Table 3). To determine the odds of having a severe acute painful episode (as opposed to a mild episode), we identified a total of 3717 hospital visits for an acute painful event in 923 subjects and, among these, 2282 hospital visits of 639 subjects were severe acute events. Furthermore, to assess the odds of having at least one painful episode between routine visits, we identified 10,207 routine visits for 2257 subjects, with 4324 visits of 1632 subjects who reported at least having one painful episode since the last visit. Table 2 and Figure 2 summarize the results of the analysis of morbidity and mortality in the eight largest clusters. After controlling for hemoglobin genotype, age and sex, the biomarker signature of Cluster 2 was protective against having at least one painful episode in between visits when compared to Cluster 1 (Odds Ratio [OR]: 0.68, p-value:0.0005), and ACS (HR: 0.75, p-value: 0.04). There was suggestive protection against mortality and stroke although the association did not reach statistical significance (Hazard Ratio [HR]: 0.57, p-value: 0.17; HR=0.26, p-value=0.08 respectively). Cluster 3 signature was borderline protective against having a severe painful episode (HR: 0.76, p-value:0.07) and was associated with a suggested increased risk for leg ulceration (HR: 1.5, p-value=0.067), and for stroke and mortality, although these effects did not reach statistical significance. The signature of Cluster 4 was protective against mortality (HR: 0.51, p-value: 0.03), AVN (HR: 0.67, p-value: 0.03), seizure (HR: 0.45, p-value: 0.06), and against having a severe acute painful episode (OR: 0.60, p-value: 0.0005). Participants in Cluster 6 had an increased hazard of seizure (HR: 2.27, p-value: 0.042), ACS (HR: 1.53, p-value: 0.01), and AVN (HR: 2.12, p-value: 0.003). Finally, patients in Cluster 7 had a decreased hazard of reporting to the hospital with an acute painful episode (HR: 0.50, p-value: 0.0095).
Figure 2.
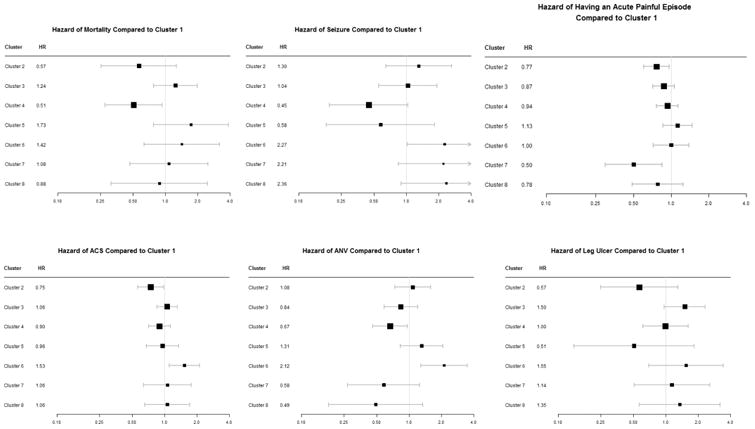
Forest plots of hazard ratios for various complications of sickle cell disease comparing clusters 2 through 8 to cluster 1. Intervals that include 1 show non-significant results.
In the analysis of longitudinal measurements of HbF, 1433 subjects had at least two measurements and over 100 subjects had at least seven measurements. Regression modeling showed that the level of HbF decreased on average 0.96 times for every 1-year increase in age and females had 0.83 times the HbF levels of males. Several clusters were associated with higher or lower HbF, compared to the referent cluster in which we estimated an average 9.5% HbF at age eight years, but cluster membership was not associated with different rate of HbF change with increasing age. Consistent with reduced risk for mortality, stroke and painful episodes, Cluster 2 and 4 were associated with increased HbF levels compared to Cluster 1, while HbF was lower in all other clusters.
The result from the regression analysis of the hemolytic score suggests a strong longitudinal correlation (p<0.0001). We did not find any significant effect of the biomarker signatures on this longitudinal correlation. However, signatures were strongly associated with different levels of hemolysis compared to the referent signature of Cluster 1. Cluster 2 and 5 were significantly associated with lower hemolytic score compared to Cluster 1, while Clusters 3, 4, 6, and 8 were associated with higher hemolytic score, and the signatures of different clusters distinguished between different degrees of intravascular hemolysis.
Replication of the signatures in PUSH and Walk-PHaSST studies
We used a Bayesian classification rule to predict the cluster membership of patients in PUSH and Walk-PHaSST and used the most likely predicted signature to assign each patient to one of the 17 clusters. Only 12 of the 17 signatures were detected in PUSH, and 13 of the 17 signatures were detected in Walk-PHaSST. Figures 3 and 4 display side-by-side boxplots of the biomarker distributions in clusters 1-8 in the two studies that reproduce very closely the signatures in the discovery set (CSSCD). Additional signatures are in Supplement Figures 3 and 4. The HBB genotype distribution in the clusters inferred in PUSH and Walk-PHaSST were similar to the distribution in the clusters discovered in CSSCD (Table 3), for most clusters. Specifically, there was no significant difference in the distribution of HbSS in clusters 3, 5, 7 between CSSCD and PUSH studies, and also there was no significant difference in the distribution of HbSS in clusters 5 and 8 between CSSCD and Walk-PHaSST studies. Interestingly, Cluster 2 in PUSH included a large proportion of HbSS patients compared to Cluster 2 in CSSCD (62% compared to 16%). In the absence of follow up data for major complications of the disease, we correlated the signatures in PUSH and Walk-PHaSSt with history of hydroxyurea treatment and noted a correlation between the “protective effect” of Cluster 2 signature and a predominant portion of subjects with a history of hydroxyurea treatment. Clusters 5 in both PUSH and Walk-PHaSST included a large portion of HbSC disease patients, as in the primary analysis in Table 2, and very small portion of hydroxyurea treated patients. We also examined the association between risk for death and biomarker signatures using the approximate 2-year follow up in Walk-PHaSSt44. Only Clusters 1 and 7 had more than five events, and Cluster 7 was associated with an increased risk for mortality as in the CSSCD, although the association did not reach statistical significance (HR: 2.3, p=0.17).
We then correlated the biomarker signatures with two biomarkers of pulmonary vasculopathy in PUSH and Walk-PHaSST. Figure 5 shows that TRV and nt-proBNP of patients in Clusters 2 and 5 tend to be lower than Cluster 1, while patients in Clusters 3 and 7 have elevated values of both TRV and nt-proBNP. The association between cluster membership and TRV and BNP was statistically significant in both studies (p-value from ANOVA < 10-6).
Figure 5. Distributions of TRV and nt-proBNP in the clusters predicted in PUSH and Walk-PHaSST.
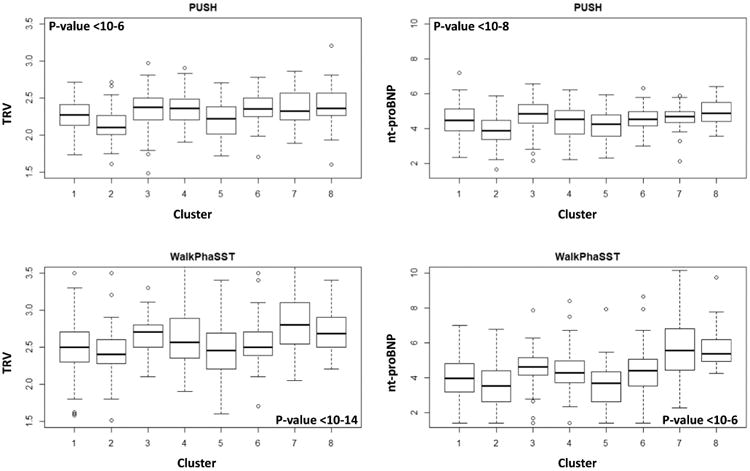
The distributions of TRV and nt-proBNP among clusters were significantly different (p-value from ANOVA, age-adjusted, shown in each panel).
Discussion
We have identified signatures of 17 circulating biomarkers that stratify sickle cell disease patients in groups with different risk for disease complications and mortality. The biomarker signatures were discovered using data from the CSSCD but we replicated the patterns in two independent, contemporary cohorts of sickle cell disease, the PUSH and Walk-PHaSST trials. The former was a study of pulmonary vascular disease focused on children while the latter study of adults was also focused on pulmonary disease. Patients in the contemporary cohorts were sometimes treated with hydroxyurea, which can alter the pathophysiology of disease. The CSSCD was an observational study done in the pre-hydroxyurea era.
The advantage of using many biomarkers concurrently is to be able to simultaneously detect risk for a variety of disease complications, rather than focusing on one complication at a time, and to increase the specificity of risk prediction. For example, the patterns of markers of anemia in Clusters 3 and 7 are very similar and are consistent with more anemic patients, but those patterns combined with different patterns of kidney disease predict different severity. Patients in Cluster 3 have a higher risk for leg ulcers but a lower risk for a severe painful episode than patients in Cluster 7. Similarly, while the lower risk for mortality and stroke in Clusters 2 and 4 is consistent with the higher than average HbF compared to Cluster 1, the differences in risk for AVN and pain in Clusters 2 and 4 would not be detectable with HbF alone. Our analyses with incident events in the CSSCD showed that indeed different biomarker signatures can predict risk for stroke, seizure, acute painful episodes, AVN, ACS, leg ulceration, and ultimately mortality. The analyses also showed that the biomarker signatures can predict trends of HbF and intravascular hemolysis, both of which are associated with different constellations of common complications 14,45. The correlation between signatures and contemporary markers of hemolysis and pulmonary vasculopathy suggests that these signatures could also predict increased risk for these complications although we do not have data to independently verify this hypothesis. Unfortunately we did not have incident events in PUSH or Walk-Phasst to replicate the association of the biomarker signatures with complications we estimated in the CSSCD.
Convincing evidence, accrued by dozens of investigators studying thousands of patients, showed that the complications of sickle cell disease are affected by both sickle vasoocclusion and intravascular hemolysis, and that each is associated with a spectrum of subphenotypes14. Each contributes to the pathophysiology of disease but are often difficult to disentangle in individuals. Our analysis illustrates the complexity of this phenotypic dichotomization when readily available biomarkers are used to group patients and allows this pathophysiologic and mechanistically simplistic separation to be clinically more applicable. For example, while Cluster 2 contained patients with less intravascular hemolysis, these individuals also had a reduction in pain and ACS in addition to the trend to reduced mortality and stroke. Similarly, in Cluster 7 the expected increased mortality, stroke and seizure risk, and worsening of renal function associated with a high level of hemolysis was accompanied by an increase in some measures of acute pain.
Our analysis included all participants from the CSSCD with biomarker data, regardless of the age at enrollment or complications of the disease. Other studies to discover predictors of sickle cell disease complications focused on particular age groups, for example young children 46, and it remains to be seen if the sensitivity and specificity of the biomarker signatures we found are comparable to other scoring systems that target particular complications. Compared to the disease severity score that we introduced27, an important feature of the biomarker signatures is to rely exclusively on biomarkers to predict risk for disease complications. Inclusion of medical history of patients in the signatures could improve the prediction for the risk of death, but make the signature less useful to identify patients at risk for complications, independently of death.
Desai and colleagues have shown that there are two signatures based on gene expression profiles of peripheral blood mononuclear cell that together with other clinical markers of severity such as TRV, white blood cell count, history of ACS, and hemoglobin levels significantly differentiate survival in three small longitudinal studies of sickle cell disease30. Our results are consistent with the general hypothesis that biomarker signatures should be able to identify patients with different clinical presentations and ultimately different risks for death, but promote the specific hypothesis that these data alone should be able to capture signatures of disease complications before their clinical expression.
Another advantage of these signatures is that they rely on commonly available laboratory data, so that their translation into a clinical tool should be relatively easy. However, more extensive validation of these results is necessary before these signatures can be used in clinical practice in time for preventive therapy, or for patient stratification by risk of complications for enrollment in clinical trials.
In addition to the suggestive utility in clinical practice, our analysis discovered one biomarker signature shared predominantly by HbSC disease patients, and a small portion of HbSS patients in the CSSCD (Cluster 2). This cluster was associated with a higher HbF level contributed by the patients with HbSS (mean HbF = 22.7%), but a lower prevalence of HbSS-α thalassemia compared to Cluster 1. There was also a reduced risk for mortality, stroke, chronic pain, and less intravascular hemolysis compared to Cluster 1, all features of HbSC disease. Consistently with the well-established therapeutic effects of hydroxyurea, the same biomarker signature in PUSH was shared by a majority of HbSS patients treated with hydroxyurea. We conjecture that untreated HbSS patients in Cluster 2 with this signature might be enriched for protective genetic variants in addition to HbF, or could be exposed to other non-genetic factors that protects them from complications of HbSS. Additional analysis that is beyond the scope of this work is needed to test this hypothesis.
A limitation of our analysis is the use of CSSCD cases, a cohort of patients from the pre-hydroxyurea era where the contemporary treatment paradigms that might impact morbidity and mortality, in addition to hydroxyurea, were unavailable. Furthermore, some clusters that represent uncommon patterns are small and we had limited statistical power to evaluate their diagnostic and prognostic value. Nevertheless, although we replicated the patterns of biomarkers in more contemporary cohorts, replication in larger studies with longitudinal follow up is needed to establish the clinical utility of these signatures. It will also be important to replicate this work in studies from other locales, particularly Africa, the Middle East, India and Brazil where the health burden of sickle cell disease is high and growing1. With replication and further study, biomarker signatures could become an important and affordable precision medicine tool to aid treatment and management of the disease. They can also be useful in the selection for, and analysis of, the increasing number of clinical trials in sickle cell disease where the studied agents are targeted to different facets of pathophysiology.
Supplementary Material
Key points.
Prediction of the clinical course of patients with sickle cell disease remains elusive.
We discovered signatures of 17 common circulating biomarkers that correlate with morbidity and mortality in sickle cell disease.
We replicate the signatures in two independent studies of sickle cell disease and show that they correlate with recently discovered biomarkers of pulmonary vasculopathy.
With replication and further study, these biomarker signatures could become an important and affordable precision medicine tool to aid treatment and management of the disease.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (NIGMS) Interdisciplinary Training Grant for Biostatisticians (T32 GM74905), and the National Heart, Lung and Blood Institute (R21HL128871, 1R01HL125886).
Footnotes
Authorship contribution. PS and MHS designed the study, planned analyses, wrote the manuscript. SVN, MD and PS conducted statistical analyses and wrote the manuscript. VG, MG, SMN and SN provided data for validation and edited the manuscript.
Disclosure of Conflicts of Interest. Dr. Gladwin is a co-inventor of pending patent applications and planned patents directed to the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning, which have recently been licensed by Globin Solutions, Inc. Dr. Gladwin is a shareholder, advisor and director in Globin Solutions, Inc. Additionally, and unrelated to CO poisoning, Dr. Gladwin is a co-inventor on patents directed to the use of nitrite salts in cardiovascular diseases, which have been licensed by United Therapeutics and Hope Pharmaceuticals, and is a co-investigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguate as a treatment for patients with SCD.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017;376(16):1561–1573. doi: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Cambridge University Press; 2009. [Google Scholar]
- 3.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364(9442):1343–1360. doi: 10.1016/S0140-6736(04)17192-4. [DOI] [PubMed] [Google Scholar]
- 4.Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87(8):795–803. doi: 10.1002/ajh.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinberg MH, Rosenstock W, Coleman MB, et al. Effects of thalassemia and microcytosis on the hematologic and vasoocclusive severity of sickle cell anemia. Blood. 1984;63(6):1353–1360. [PubMed] [Google Scholar]
- 6.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105(5):1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solovieff N, Milton JN, Hartley SW, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood. 2010;115(9):1815–1822. doi: 10.1182/blood-2009-08-239517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342(6155):253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bae HT, Baldwin CT, Sebastiani P, et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood. 2012;120(9):1961–1962. doi: 10.1182/blood-2012-06-432849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masuda T, Wang X, Maeda M, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351(6270):285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrell JJ, Sherva RM, Chen ZY, et al. A 3-bp deletion in the HBS1L-MYB intergenic region on chromosome 6q23 is associated with HbF expression. Blood. 2011;117(18):4935–4945. doi: 10.1182/blood-2010-11-317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaikho EM, Habara AH, Alsultan A, et al. Variants of ZBTB7A (LRF) and its beta-globin gene cluster binding motifs in sickle cell anemia. Blood Cells Mol Dis. 2016;59:49–51. doi: 10.1016/j.bcmd.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinberg MH, Embury SH. Alpha-thalassemia in blacks: genetic and clinical aspects and interactions with the sickle hemoglobin gene. Blood. 1986;68(5):985–990. [PubMed] [Google Scholar]
- 14.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–760. doi: 10.1172/JCI89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bean CJ, Boulet SL, Yang G, et al. Acute chest syndrome is associated with single nucleotide polymorphism-defined beta globin cluster haplotype in children with sickle cell anaemia. British journal of haematology. 2013;163(2):268–276. doi: 10.1111/bjh.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatnagar P, Barron-Casella E, Bean CJ, et al. Genome-Wide Meta-Analysis of Systolic Blood Pressure in Children with Sickle Cell Disease. PLoS ONE. 2013;8(9):e74193. doi: 10.1371/journal.pone.0074193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhatnagar P, Purvis S, Barron-Casella E, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J Hum Genet. 2011;56(4):316–323. doi: 10.1038/jhg.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681–6684. doi: 10.1182/blood-2011-01-332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galarneau G, Coady S, Garrett ME, et al. Gene-centric association study of acute chest syndrome and painful crisis in sickle cell disease patients. Blood. 2013;122(3):434–442. doi: 10.1182/blood-2013-01-478776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin PJ, Sebastiani P, Edward H, et al. The Genetics of Hemoglobin A Regulation in Sickle Cell Anemia. Am J Hematol. 2014 doi: 10.1002/ajh.23811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milton JN, Sebastiani P, Solovieff N, et al. A genome-wide association study of total bilirubin and cholelithiasis risk in sickle cell anemia. PLoS One. 2012;7(4):e34741. doi: 10.1371/journal.pone.0034741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sebastiani P, Solovieff N, Hartley SW, et al. Genetic modifiers of the severity of sickle cell anemia identified through a genome-wide association study. Am J Hematol. 2010;85(1):29–35. doi: 10.1002/ajh.21572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaturvedi S, Bhatnagar P, Bean CJ, et al. Genome-wide association study to identify variants associated with acute severe vaso-occlusive pain in sickle cell anemia. Blood. 2017;130(5):686–688. doi: 10.1182/blood-2017-02-769661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milton JN, Gordeuk VR, Taylor JGt, Gladwin MT, Steinberg MH, Sebastiani P. Prediction of fetal hemoglobin in sickle cell anemia using an ensemble of genetic risk prediction models. Circ Cardiovasc Genet. 2014;7(2):110–115. doi: 10.1161/CIRCGENETICS.113.000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 26.Rees DC, Gibson JS. Biomarkers in sickle cell disease. Br J Haematol. 2012;156(4):433–445. doi: 10.1111/j.1365-2141.2011.08961.x. [DOI] [PubMed] [Google Scholar]
- 27.Sebastiani P, Nolan VG, Baldwin CT, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110(7):2727–2735. doi: 10.1182/blood-2007-04-084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belini Junior E, Silva DG, Torres Lde S, Okumura JV, Lobo CL, Bonini-Domingos CR. Severity of Brazilian sickle cell disease patients: severity scores and feasibility of the Bayesian network model use. Blood Cells Mol Dis. 2015;54(4):321–327. doi: 10.1016/j.bcmd.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 29.Coelho A, Dias A, Morais A, Nunes B, Faustino P, Lavinha J. Sickle cell disease severity scoring: a yet unsolved problem. European Journal of Haematology. 2012;89(6):501–502. doi: 10.1111/ejh.12011. [DOI] [PubMed] [Google Scholar]
- 30.Desai AA, Lei Z, Bahroos N, et al. Association of circulating transcriptomic profiles with mortality in sickle cell disease. Blood. 2017;129(22):3009–3016. doi: 10.1182/blood-2016-11-752279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaston M, Rosse WF. The cooperative study of sickle cell disease: review of study design and objectives. Am J Pediatr Hematol Oncol. 1982;4(2):197–201. [PubMed] [Google Scholar]
- 32.Gaston M, Smith J, Gallagher D, et al. Recruitment in the Cooperative Study of Sickle Cell Disease (CSSCD) Control Clin Trials. 1987;8(4 Suppl):131S–140S. doi: 10.1016/0197-2456(87)90016-x. [DOI] [PubMed] [Google Scholar]
- 33.Gordeuk VR, Minniti CP, Nouraie M, et al. Elevated tricuspid regurgitation velocity and decline in exercise capacity over 22 months of follow up in children and adolescents with sickle cell anemia. Haematologica. 2011;96(1):33–40. doi: 10.3324/haematol.2010.030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minniti CP, Sable C, Campbell A, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica. 2009;94(3):340–347. doi: 10.3324/haematol.13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordeuk VR, Campbell A, Rana S, et al. Relationship of erythropoietin, fetal hemoglobin, and hydroxyurea treatment to tricuspid regurgitation velocity in children with sickle cell disease. Blood. 2009;114(21):4639–4644. doi: 10.1182/blood-2009-04-218040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milton JN, Rooks H, Drasar E, et al. Genetic determinants of haemolysis in sickle cell anaemia. Br J Haematol. 2013;161(2):270–278. doi: 10.1111/bjh.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sebastiani P, Perls TT. Detection of Significant Groups in Hierarchical Clustering by Resampling. Front Genet. 2016;7(144) doi: 10.3389/fgene.2016.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrika. 1982;69:239–241. [Google Scholar]
- 39.Campbell A, Minniti CP, Nouraie M, et al. Prospective evaluation of haemoglobin oxygen saturation at rest and after exercise in paediatric sickle cell disease patients. Br J Haematol. 2009;147(3):352–359. doi: 10.1111/j.1365-2141.2009.07854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul R, Minniti CP, Nouraie M, et al. Clinical correlates of acute pulmonary events in children and adolescents with sickle cell disease. Eur J Haematol. 2013;91(1):62–68. doi: 10.1111/ejh.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sachdev V, Kato GJ, Gibbs JS, et al. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation. 2011;124(13):1452–1460. doi: 10.1161/CIRCULATIONAHA.111.032920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebastiani P, Thyagarajan B, Sun F, et al. Biomarker signatures of aging. Aging Cell. 2017;16(2):329–338. doi: 10.1111/acel.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaikho EM, Farrell JJ, Alsultan A, et al. A phased SNP-based classification of sickle cell anemia HBB haplotypes. BMC Genomics. 2017;18(1):608. doi: 10.1186/s12864-017-4013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gladwin MT, Barst RJ, Gibbs JS, et al. Risk factors for death in 632 patients with sickle cell disease in the United States and United Kingdom. PLoS One. 2014;9(7):e99489. doi: 10.1371/journal.pone.0099489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller ST, Sleeper LA, Pegelow CH, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342(2):83–89. doi: 10.1056/NEJM200001133420203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.