

Chronic Helicobacter pylori infection can lead to gastric ulcers and gastric cancers. The enzyme urease contributes to the survival of the bacterium in the harsh environment of the stomach by increasing the local pH. In addition to combating acid, H. pylori must survive host-produced reactive oxygen species to persist in the gastric mucosa. We describe a cyclic amino acid-based antioxidant role of urease, whereby oxidized methionine residues can be recycled by methionine sulfoxide reductase to again quench oxidants. This work expands our understanding of the role of an already acknowledged pathogen virulence factor and specifically expands our knowledge of H. pylori survival mechanisms.
KEYWORDS: gastric pathogen, gastritis, methionine, oxidation, oxidative stress, ulcer
ABSTRACT
The well-studied catalytic role of urease, the Ni-dependent conversion of urea into carbon dioxide and ammonia, has been shown to protect Helicobacter pylori against the low pH environment of the stomach lumen. We hypothesized that the abundantly expressed urease protein can play another noncatalytic role in combating oxidative stress via Met residue-mediated quenching of harmful oxidants. Three catalytically inactive urease mutant strains were constructed by single substitutions of Ni binding residues. The mutant versions synthesize normal levels of urease, and the altered versions retained all methionine residues. The three site-directed urease mutants were able to better withstand a hypochlorous acid (HOCl) challenge than a ΔureAB deletion strain. The capacity of purified urease to protect whole cells via oxidant quenching was assessed by adding urease enzyme to nongrowing HOCl-exposed cells. No wild-type cells were recovered with oxidant alone, whereas urease addition significantly aided viability. These results suggest that urease can protect H. pylori against oxidative damage and that the protective ability is distinct from the well-characterized catalytic role. To determine the capability of methionine sulfoxide reductase (Msr) to reduce oxidized Met residues in urease, purified H. pylori urease was exposed to HOCl and a previously described Msr peptide repair mixture was added. Of the 25 methionine residues in urease, 11 were subject to both oxidation and to Msr-mediated repair, as identified by mass spectrometry (MS) analysis; therefore, the oxidant-quenchable Met pool comprising urease can be recycled by the Msr repair system. Noncatalytic urease appears to play an important role in oxidant protection.
IMPORTANCE Chronic Helicobacter pylori infection can lead to gastric ulcers and gastric cancers. The enzyme urease contributes to the survival of the bacterium in the harsh environment of the stomach by increasing the local pH. In addition to combating acid, H. pylori must survive host-produced reactive oxygen species to persist in the gastric mucosa. We describe a cyclic amino acid-based antioxidant role of urease, whereby oxidized methionine residues can be recycled by methionine sulfoxide reductase to again quench oxidants. This work expands our understanding of the role of an already acknowledged pathogen virulence factor and specifically expands our knowledge of H. pylori survival mechanisms.
INTRODUCTION
Persistent Helicobacter pylori infections can cause chronic gastritis, peptic ulcer disease, and gastric cancer (1–4). Decades of immune response to the infection can lead to chronic inflammation and tissue damage (5, 6). H. pylori must first survive the harsh conditions of stomach gastric acid and then a prolonged host immune response after its colonization of the gastric mucosa (5). To combat the low pH of the stomach lumen, H. pylori expresses urease, which catalytically converts urea into ammonia and carbon dioxide, the former enabling H. pylori to resist the acidic gastric environment (7–9). Urease has been shown to be the most highly expressed protein in H. pylori, making up to 10% of the total protein content of the gastric pathogen (10). The enzyme consists of two structural proteins, UreA and UreB, with 12 subunits each that form a 1.1-MDa dodecamer (11). The UreAB heterodimer contains 25 methionine (Met) residues, 3% of the total amino acid content (12). Enzyme activity requires the addition of two nickel atoms per UreB monomer, and such maturation is facilitated by the urease accessory proteins UreEFGH and the hydrogenase maturation proteins HypAB (10, 13–15).
Although the catalytic activity of urease is required for initial colonization, (10, 16–18) several studies also indicate that non-acid-neutralizing roles may exist for urease. For instance, urease has been shown to be required for persistence in the mouse gastric mucosa, where the pH approaches neutrality (16, 19) and a urease-negative strain was unable to colonize a pH neutral piglet stomach (20). In addition, using a tetracycline-inducible system to turn on or off urease expression, Debowski et al. were able to show that urease-expressing bacteria were selected over time in a murine infection model, indicating that urease is required for chronic infection (16). Additionally, H. pylori urease has been shown to play multiple roles in modulating the host immune response. Indeed, it was shown to decrease opsonization (21), stimulate the chemotaxis of neutrophils and monocytes (22), induce apoptosis in gastric epithelial cells after binding to class II major histocompatibility complex (MHC) receptors (23), and induce proinflammatory cytokines (24). Recently, urease has been linked to the formation of gastric carcinoma through an ability to promote angiogenesis (25).
After reaching the gastric epithelium, H. pylori triggers responses by the host innate immune cells, which respond to the infection by generating reactive oxygen species (ROS) such as superoxide anion (O2−), hydrogen peroxide (H2O2), hydroxyl radicals (˙OH), and hypochlorous acid (HOCl) (10, 26, 27). Indeed, the exposure of gastric cells (28) or phagocytes (29) to H. pylori increases host cell ROS production. Patients with H. pylori infections have been shown to have larger amounts of ROS in their gastric mucosa (30). ROS can damage protein, DNA, and lipids (31). With regard to proteins, the amino acids most susceptible to oxidation are Met and cysteine (Cys) due to their sulfur-containing ligands (32).
H. pylori has many mechanisms to protect itself from, as well as repair damage caused by, oxidative stress. For instance, catalase and superoxide dismutase act to convert H2O2 and O2− into less harmful products. Catalase was recently shown to protect H. pylori against oxidative damage via an oxidant-quenching mechanism of its Met residues (33). The reaction involves methionine sulfoxide reductase (Msr) to reduce Met-SO to Met (34, 35). Msr reduces oxidized methionines of damaged proteins and has been shown to restore function to the damaged protein (36, 37). Cross-linking and direct repair assays showed that H. pylori Msr has at least five repair target proteins (34, 38, 39). These include AhpC, UreG, GroEL, catalase, and a site-specific recombinase (SSR), but urease was not observed as a repair target in those studies. In the present study, we describe the evidence for a noncatalytic role for urease, which involves Met-S/Met-SO recycling. This role aids in protecting the pathogen against oxidation-mediated cell death.
(A preliminary account of this work was presented at the American Society for Microbiology Microbe, New Orleans, LA, 1 to 5 June 2017 [40].)
RESULTS
Construction of catalytically inactive urease.
Strains that synthesize inactive urease (herein referred to as apo-urease) were constructed to determine if the catalytically inactive enzyme is sufficient to quench oxidants. A sucrose-kanamycin selection-counterselection was used to make markerless single-amino-acid substitutions in the UreB subunit of urease. The three single substitutions (His136Ala, His138Ala, and Lys219Ala) were constructed to replace nickel binding residues known to be required for enzyme activity (41). As expected, the three site-directed strains (ureBH136A, ureBH138A, and ureBK219A strains) lacked urease activity compared to the wild-type (WT) strain 43504 (Table 1). Next, we sought to verify that the lack of urease activity was not linked to a decrease or loss of urease, e.g., that the mutant strains were still making apo-urease. Since urease is the most abundant protein in H. pylori, both UreA (27 kDa) and UreB (62 kDa) structural subunits can readily be visualized on Coomassie-stained SDS-PAGE gels (Fig. 1A). UreAB levels in the three site-directed mutants were found to be similar or slightly less than WT levels (Fig. 1A). This was confirmed by immunoblot, using anti-UreA antiserum (Fig. 1B). No UreA-specific band was seen in the ΔureAB deletion strain, our negative control. These results indicate that apo-urease is still synthesized in each of the three site-directed mutants.
TABLE 1.
Urease activity of H. pylori wild-type, urease deletion, and site-directed mutant strains
| Strain | Urease activity (μmol/min/mg)a |
|---|---|
| 43504 (wild type) | 4.65 ± 0.96a |
| ΔureAB strain | 0.06 ± 0.01 |
| ureBH136A strain | 0.07 ± 0.02 |
| ureBH138A strain | 0.07 ± 0.01 |
| ureBK219A strain | 0.08 ± 0.01 |
Urease activity expressed in micromoles of NH3 produced per minute per milligram of total protein. Results shown are the means ± SDs from 3 to 8 independent experiments with assays performed in triplicate.
FIG 1.
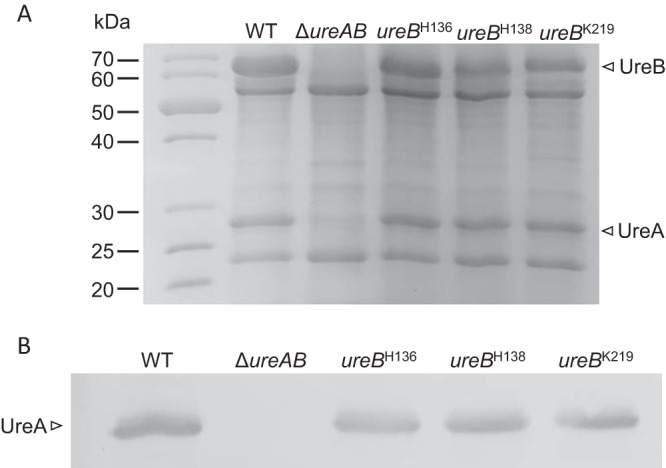
Urease in cell-free and whole-cell extracts of WT and ureAB mutants. (A) SDS-PAGE (12.5%) analysis of cell-free extract. (B) Immunoblot analysis. Whole-cell extracts were separated via 12.5% SDS-PAGE, transferred to a nitrocellulose membrane, and blotted with anti-UreA antibodies. Strains are indicated above each lane. UreA and UreB are indicated.
Catalytically inactive urease confers resistance to HOCl.
The three catalytically inactive strains, as well as the WT (positive control) and the ΔureAB deletion strain (negative control), were exposed to HOCl to determine their ability to quench an oxidant and survive. The cells were incubated with HOCl for 1 h, diluted, and the CFU were counted (Fig. 2). The catalytically inactive mutants were as resistant as the WT and significantly (P < 0.01) less sensitive to HOCl oxidative damage than the urease gene deletion strain (Fig. 2). This suggests that the presence of urease, independent of its catalytic activity, can play a role in preventing cell death due to oxidative damage.
FIG 2.
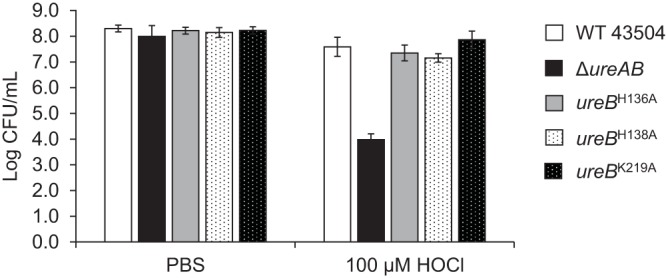
Apo-urease confers resistance to HOCl. Cells grown on BA plates with 10 μM NiCl2 were incubated with PBS or with PBS supplemented with 100 μM HOCl for 1 h at 37°C, diluted, and plated. CFU were counted after 72 h at 37°C. Error bars indicate standard deviations from 4 to 10 independent experiments.
Both holo-urease and apo-urease protect against HOCl-mediated killing.
To determine if holo-urease (purified from WT strain 43504) and apo-urease (purified from either strain ureBH138A or strain ureBK219A) were able to protect against HOCl-mediated oxidative damage, each purified urease was preincubated with the oxidant for 15 min and the mixture was added to whole nongrowing WT or ΔureAB cells. Cell survival was then monitored and compared to that with a HOCl-only treatment (no protein) (Fig. 3). While no cells were recovered from the HOCl treatment alone (detection limit, 102 CFU/ml), when HOCl was preincubated with purified urease (both holo and apo), the cell recovery levels reached those of cells without HOCl treatment (Fig. 3). This suggests that purified urease itself can quench HOCl, irrespective of urease activity. Interestingly, the ΔureAB strain, shown to be more sensitive than the WT to HOCl challenge (Fig. 2), was almost fully protected against HOCl that had been preincubated with purified urease (Fig. 3). An additional protein, UreE, with a low Met content (1%) was shown to not protect against the HOCl challenge, suggesting that Met content might contribute to the protective effect.
FIG 3.
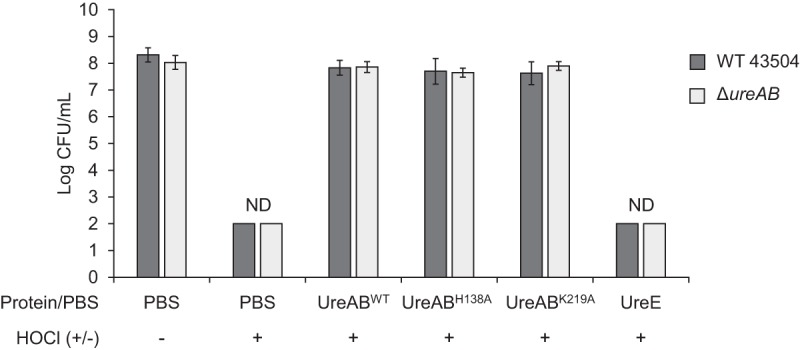
Holo- and apo-urease proteins protect against HOCl. Wild-type and ΔureAB 43504 cells were incubated for 1 h in PBS, PBS with HOCl, or PBS with HOCl that had been previously incubated for 15 min at 37°C with either purified UreABWT, UreABH138A, UreABK219A, or UreE as indicated below each column. Final protein concentration was 0.25 μM, and final HOCl concentration was 200 μM in each reaction. Error bars indicate standard deviations from 3 to 9 independent experiments. ND, no CFU could be detected (detection limit, 102 CFU/ml).
MS/MS identification of oxidized and reduced methionine residues in UreAB.
Urease is abundantly expressed, is found outside cells as well as in the cytosol, and contains many Met residues (25 of 807 residues, accounting for 3% of the total residues) (10, 42, 43). To identify which Met residues of urease (UreAB) are oxidized and repaired, we used liquid chromatography-tandem mass spectrometry (LC-MS/MS). The urease holoenzyme was first oxidized with HOCl, and then a sample of oxidized urease was subsequently incubated with an Msr repair mixture. Oxidized, oxidized and repaired, and untreated samples were analyzed by LC-MS/MS. Of the 25 methionine residues in urease, 19 were detected by LC-MS/MS (Fig. 4). UreA Met1 and UreB Met1, Met104, Met182, Met191, and Met478 were either not detected or could not be quantified reliably. An examination of the protein sequence shows that four of these methionines reside in peptides containing multiple basic residues that were digested by trypsin resulting in small hydrophilic fragments, making their detection by LC-MS/MS highly unlikely. A fifth methionine-containing peptide was not included in the data analysis, as the signal-to-noise ratio was far too low for reliable quantitation. Most of the detected Met residues in the unoxidized samples had low levels of oxidation (less than 0.15 average oxidation per peptide) except UreA Met71, Met77, and Met86 and UreB Met262. HOCl-treated Met residues UreA Met44, Met71, Met77, and Met86 and UreB Met12, Met262, Met315, Met317, Met319, Met353, and Met366 had greater than 0.8 average oxidation per peptide. UreA Met12 and UreB Met448 showed approximately 0.5 average oxidation per peptide, and all other residues showed less than 0.5 average oxidation per peptide. After HOCl-oxidized urease was treated with an Msr repair mixture, most Met residues were reduced to levels similar to that observed for the unoxidized sample, with the exception of UreB Met12, Met262, Met353, and Met366. Most of the digested peptides contained a single Met residue, the exceptions being UreA peptide 63 to 92 and UreB peptide 289 to 326, which contain three Met residues each (Met71, Met77, and Met86 and Met315, Met317, and Met319, respectively). Likewise, UreB peptide 339 to 368 contains two Met residues (Met353 and Met366). For peptides with more than one Met residue, the oxidation of each Met cannot be accurately quantified by collision-induced dissociation (CID) MS/MS (44, 45). In summary, the Msr repair mixture reduced 11 of the oxidized Met residues to close to the baseline non-oxidant-treated levels. This suggests that oxidized urease can be reduced by Msr and thus probably recycles the oxidant-quenchable pool of urease.
FIG 4.

LC-MS/MS analysis of oxidized and Msr-repaired urease. Purified urease was incubated with a 60-fold molar excess of HOCl for 15 min. Excess HOCl was quenched with 15 mM Met and removed via dialysis. After dialysis, oxidized urease was incubated with purified Msr, Trx1, TrxR, NADPH, and DTT for 2 h at 37°C. Urease samples were digested by trypsin, and oxidation levels of methionine residues were quantified by LC-MS/MS. The oxidation level for the untreated sample was below the limit of detection for UreA Met58. White bars, untreated; black bars, HOCl treated; gray bars, treated with HOCl plus Msr repair mixture. Error bars indicate standard deviations from 2 independent experiments with 3 replicates each.
DISCUSSION
Previous studies have focused on the catalytic by-products of urea hydrolysis by urease (NH3 and CO2) and their contribution to pathogen virulence. The ammonia produced by the enzyme supplements nitrogen metabolism, promotes alkalinization, and contributes to damage of host cell tissue (46). Additionally, carbon dioxide has been shown to protect the bacterium against highly toxic peroxynitrite during H. pylori infection (47). Now, there is growing evidence that urease plays noncatalytic roles in H. pylori infection. Indeed, urease is required for persistence in the mouse gastric epithelium, where the pH approaches neutrality (16, 19). Urease has been shown to play roles in modulating the immune response to infection (21–24), and ureA was found to be upregulated in response to oxidative stress (48).
We demonstrate that urease has a previously undescribed role as an antioxidant, thus adding to the growing number of noncatalytic roles that urease can play. Indeed, some of the observations cited above (e.g., urease requirement for persistence even at neutrality) may be due to an antioxidant role. This role depends at least in part on the primary sequence of the protein (e.g., Met residues). Although gastric acid exposure is an impressive obstacle to overcome, H. pylori also survives a barrage of host-produced ROS, including HOCl, which can reach levels as high as 5 mM at inflamed sites (49). H. pylori survives this via multiple mechanisms of detoxification and repair, enabling the infection to oftentimes persist throughout the life span of the host. Nickel-deficient mutants that lacked catalytic activity were as resistant as the WT to HOCl-mediated oxidative stress. In contrast, a urease deletion mutant was much more sensitive than the WT to HOCl-mediated oxidative stress. Thus, catalytic activity is not required for combating the oxidant. Rather, we propose that the Met residues of urease can quench oxidants, forming Met-SO in the process. Catalase, another abundant protein, was similarly shown to quench oxidants to aid bacterial survival, and the mechanism was independent of catalytic activity (33). A similar antioxidant mechanism of relying on Met residues was assigned to pure E. coli glutamine synthetase; however, the system was not shown to confer oxidative stress resistance to cells (50).
We predict that Met-SO residues of urease could be reduced by Msr and then used again to quench oxidants. Indeed, Msr was previously shown to reduce and repair several H. pylori proteins such as AhpC, UreG, GroEL, catalase, and site-specific recombinase (SSR) (34, 38, 39). However, it was previously unknown whether Msr could reduce urease Met residues. Through mass spectrometry analysis, we found that of the 25 Met residues in urease, 11 were shown to be susceptible to oxidation and were reduced (i.e., repaired) by Msr. Thus, urease can be added to the list of Msr repair targets. Upon PDBePISA analysis of surface accessibility, using the X-ray crystal structure of H. pylori urease (Protein Data Bank code 1E9Z), 9 of the 11 Met residues that were oxidized and then subsequently reduced are surface exposed (11, 51). This is consistent with previous reports of surface methionine residues being subject to oxidation and subsequent repair by Msr (26, 38). H. pylori Msr deletion mutants showed lowered urease activity when exposed to oxidative stress (21% O2) (38). This was attributed to the inability of the strain to repair UreG, a urease accessory protein required for urease activity; however, we can now hypothesize the lowered activity could also be due to the lack of reduction of oxidized Met residues of the structural proteins of urease (UreAB).
An Msr deletion mutant has been shown to be more sensitive to HOCl challenge than its parent strain (SS1) (39). Here, we show a high HOCl sensitivity of a strain lacking one of the described repair targets of Msr, namely, urease. Conclusions based on comparing the phenotypes of the ΔureAB strain to that of the Δmsr strain (39) are not possible due to the use of different parent strains. Comparisons are further complicated by the fact that Msr has multiple repair targets besides urease.
While urease lacks a described secretion pathway in H. pylori, it has been found outside the cell, possibly due to cell lysis (42, 52). We show that purified urease added exogenously and whole cells expressing urease quench HOCl and survive HOCl challenge, respectively. Regardless of location, urease activity is not required for the described oxidant quenching effect. Seemingly, both extracellular and intracellular ureases likely act to quench oxidants. However, intracellular urease may play a larger role in protecting against oxidants because of its Met turnover—Msr is unlikely to be active extracellularly. Although this study focused on the role of Met residues in urease oxidant quenching, the Cys residues of urease might also contribute to the antioxidant ability we describe. While Cys residues are very sensitive to oxidation, the UreAB heterodimer only contains 4 Cys residues compared to 25 Met residues. Oxidized Cys residues could be reduced enzymatically through the thioredoxin/thioredoxin reductase (Trx/TrxR) system (32). H. pylori contains two thioredoxins (Trx1 and Trx2), which have been shown to play a role in preventing macromolecule damage, and one TrxR; however, it is not known if the Trx system can repair oxidized Cys residues in H. pylori (53, 54).
Future site-directed mutagenesis studies of individual Met residues in urease could elucidate the role of individual residues in the quenching. Also, the use of the mouse model to gauge colonization by the catalytically inactive strains is likely to be a promising approach to assess in vivo function, although the acid barrier would have to be minimized to observe colonization by urease-activity-negative strains. Catalytically inactive urease may contribute to the persistence of H. pylori infections of the host gastric epithelium, where the pathogen is exposed to multiple oxidants (30).
Although the role of urease in H. pylori pathogenesis has been extensively studied, urease is also a virulence factor for several pathogens, including Proteus mirabilis and Klebsiella pneumoniae (55). During P. mirabilis infection of the urinary tract, urease is required for mineral precipitation to form urinary stones, which impede the elimination of the infection (56). During mice gastrointestinal tract infections, urease-negative strains of K. pneumoniae were outcompeted by WT strains, and so it was suggested that the ammonia produced by urease serves as a supplementary nitrogen source (57). K. pneumoniae and P. mirabilis must also defend against HOCl produced by neutrophils as a part of the innate immune response to infection (56, 58). These species also contain Msr, enabling the possibility that a similar method for urease methionine oxidation and repair by Msr would be found in additional pathogens. Additionally, other highly expressed proteins that are Met rich might play a similar antioxidant role. Indeed, catalase, a Met-rich and abundant H. pylori protein, has been shown to have similar noncatalytic antioxidant properties (33).
Urease and Msr can be found in plants, fungi, archaea, and bacteria (59, 60). Throughout the three domains of life, urease is increasingly seen as a “moonlighting protein” with a variety of noncatalytic roles, including fungicidal, insecticidal, and proinflammatory effects (61). For H. pylori, an antioxidant mechanism based on urease Met residue recycling is expected to explain at least part of the in vivo requirement for this important enzyme.
MATERIALS AND METHODS
Growth conditions.
Escherichia coli cells were cultured at 37°C aerobically in Luria-Bertani (LB) broth or on agar LB plates with 100 μg/ml ampicillin and 30 μg/ml chloramphenicol as needed. H. pylori cells (ATCC 43504) were routinely grown at 37°C on brucella agar plates supplemented with 10% defibrinated sheep blood (BA) under microaerophilic conditions (4% O2, 5% CO2, and 91% N2). As needed, H. pylori was grown on BA supplemented with 5 or 10 μM NiCl2, 5% sucrose, 20 μg/ml kanamycin, or 30 μg/ml chloramphenicol. For urease protein purification, H. pylori cells were grown on BA plates supplemented 5 μM NiCl2.
Strain construction.
The E. coli and H. pylori strains and plasmids used in this study are listed in Table 2. Site-directed mutants were constructed using a kanamycin-sucrose selection-counterselection system described previously (33, 39, 62). Briefly, a 1,400-bp DNA fragment containing a partial ureB gene was amplified by PCR using primers UreBF1 and UreBR1 (Table 3) from the chromosomal DNA of H. pylori 26695 (12) and cloned into pBluescript KS(+) to obtain pKS-ureB. The Kanr-sacB-PflaA (KSF) cassette (62) was inserted into ureB resulting in pKS-ureB::KSF, which was then naturally transformed into WT strain 43504, thereby replacing parent ureB with ureB::KSF. The resulting strain (ΔureB::KSF strain) was found to be urease negative and kanamycin resistant. Site-directed mutations were introduced to pKS-ureB by overlapping PCR using internal primers UreB:H136AF, UreB:H136AR, UreB:H138AF, UreB:H138AR, UreB:K219AF, and UreB:K219AR and external primers UreBF1 and UreBR1 (Table 3). The resulting plasmids (pKS-ureBH136A, pKS-ureBH138A, and pKS-ureBK219A) were introduced into the ΔureB::KSF strain by natural transformation. UreB site-directed mutants were selected on BA plates containing 5% sucrose. The resulting strains were sucrose resistant and urease negative. The ΔureAB strain was constructed as described previously (15). All plasmids and PCR products were sequenced at the Georgia Genomics Facility, University of Georgia, Athens, GA, and compared with DNA sequences from strain 26695 (12) to ensure that no error had been introduced following PCR amplification, as well as to verify the presence of engineered site-directed mutations within ureB.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| E. coli TOP10 | Cloning strain | Invitrogen |
| H. pylori | ||
| 43504 | Parental strain | ATCCb |
| ΔureB mutant | ΔureB::KSF Kanr Sucs | This study |
| ΔureAB mutant | ΔureAB::cat Cmr | 15 |
| ureBH136A strain | Codon for His136 replaced by Ala | This study |
| ureBH138A strain | Codon for His138 replaced by Ala | This study |
| ureBK219A strain | Codon for Lys219 replaced by Ala | This study |
| Plasmids | ||
| pBluescript KS(+) | Cloning vector; Apr | Stratagene |
| pKS-ureB | ureB cloned into the KpnI-SacI site of pBluescript KS(+) | This study |
| pKS-ureB::KSF | KSF cassette cloned into the BamHI site of pKS-ureB | This study |
| pKS-ureBH136A | pKS-ureB with His136 replaced by Ala | This study |
| pKS-ureBH138A | pKS-ureB with His138 replaced by Ala | This study |
| pKS-ureBK219A | pKS-ureB with Lys219 replaced by Ala | This study |
Cm, chloramphenicol; Ap, ampicillin; Kan, kanamycin; Suc, sucrose; KSF, Kanr-sacB-PflaA (62).
American Type Culture Collection, Manassas, VA.
TABLE 3.
Primers used in this paper
| Name | Sequence (5′→3′)a | Restriction site |
|---|---|---|
| UreBF1 | GCTTCGGTACCCCTGAGAGAAGGCATGAG | KpnI |
| UreBR1 | GCATCGAGCTCCCGGCAACACTTGTCT | SacI |
| UreB:H136AF | GTGGTATTGACACAGCTATCCACTTCATTTCAC | |
| UreB:H136AR | GTGAAATGAAGTGGATAGCTGTGTCAATACCAC | |
| UreB:H138AF | ATTGACACACACATCGCCTTCATTTCACCCC | |
| UreB:H138AR | GGGGTGAAATGAAGGCCATGTGTGTGTCAAT | |
| UreB:K219AF | GGTGCGATTGGCTTTGCAATCCACGAAGACTGG | |
| UreB:K219AR | CCAGTCTTCGTGGATTGCAAAGCCAATCGCACC |
Primers were purchased from Integrated DNA Technology, Coralville, IA.
Urease assays.
Whole cells grown on BA plates under microaerophilic conditions at 37°C were resuspended in 50 mM HEPES (pH 7.5), lysed by sonication (Heat Systems Ultrasonics sonicator: 30 s at 4-W output power and 50% duty cycle), and centrifuged at 21,100 × g for 5 min. The cell-free supernatant was assayed for urease activity following the phenol-hypochlorite (Weatherburn) method (63). Protein concentration was determined using the bicinchoninic acid (BCA) kit (Thermo Scientific Pierce).
SDS-PAGE and immunoblotting.
Whole cells grown on BA plates supplemented with 10 μM NiCl2 were resuspended in phosphate-buffered saline (PBS) and treated as above to obtain cell-free extracts (CFE). Total protein from CFE (0.4 μg protein per lane) was loaded onto 12.5% SDS-PAGE gels and stained with Coomassie brilliant blue G-250 or transferred to a nitrocellulose membrane for immunoblotting as previously described with modifications (33, 38). Briefly, the nitrocellulose membrane was blocked with 3% gelatin in Tris-buffered saline (TBS; 50 mM Tris [pH 7.6] and 150 mM NaCl [pH 7.6]). After blocking, the membrane was incubated with anti-UreA antibody (rabbit polyclonal; Santa Cruz Biotechnology) diluted 1:2,000 in TBS with 0.1% Tween 20 (TTBS) containing 1% gelatin for 1 h. The membrane was then washed with TTBS and incubated with goat anti-rabbit IgG conjugated to alkaline phosphatase (Bio-Rad) diluted 1:2,000 in TTBS with 1% gelatin. After 1 h of incubation, the blot was developed with Nitro Blue Tetrazolium–5-bromo-4-chloro-3-indolyl phosphate in 10 mM Tris-HCl (pH 9.5) and 150 mM NaCl.
Whole-cell HOCl tolerance assay.
Whole-cell HOCl tolerance assays were performed as previously described (33) with the following modification. Briefly, cells grown on BA plates with 10 μM NiCl2 were resuspended in PBS to an optical density at 600 nm (OD600) of approximately 1.1 and then mixed with either PBS or PBS with NaOCl (100 μM final concentration, 10 to 15% available chlorine [referred to as HOCl]) for 1 h at 37°C. The HOCl-challenged cells were diluted in PBS and plated on BA plates, and the CFU were counted after 72 h of incubation at 37°C under microaerophilic conditions.
Purified protein HOCl tolerance assay.
HOCl tolerance assays with purified proteins were performed as previously described (33). Briefly, wild-type and ΔureAB 43504 cells (OD600 of 1.3) were incubated for 1 h in PBS, PBS with 200 μM HOCl, or PBS with HOCl that had been previously incubated with 0.25 μM UreAB or UreE for 15 min at 37°C. After the PBS or HOCl challenge, the cells were plated on BA plates and the CFU were counted after 72 h of incubation at 37°C under microaerophilic conditions.
Protein purification.
H. pylori 43504 cells were grown on BA plates supplemented with 5 μM NiCl2 under microaerophilic conditions (4% O2, 5% CO2, 91% N2) at 37°C and broken by passage through a French pressure cell at 18,000 lb/in2 or by cell disruption at 20,000 lb/in2 (Constant Systems One Shot). Cell debris was removed by centrifugation at 4,500 × g. Urease was purified from the resulting supernatant (cell-free extract) using fast protein liquid chromatography (AKTA; GE Healthcare). Cell-free extract was loaded onto a Q-Sepharose HiTrap column that had been equilibrated with 50 mM NaPO4 (pH 7.2) and 25 mM NaCl (buffer A). The protein was eluted with a linear gradient of 0.025 to 1 M NaCl in buffer A. Urease-containing fractions, as determined by the phenol red urease assay (64), were concentrated with Amicon Ultra-4 devices with a 10-kDa molecular mass cutoff (Merck Millipore). The concentrated urease was further purified with a HiLoad 16/60 Superdex 75 column in buffer A with 0.35 M NaCl. Urease-containing fractions were identified and concentrated as above, and the final protein concentration was determined using the BCA kit (Thermo Scientific Pierce). UreAB proteins were purified to near homogeneity (>90%), as determined by SDS-PAGE. UreE was purified as previously described (65).
Urease oxidation and Msr repair.
Urease oxidation and subsequent Msr repair were performed as previously described with modifications (26). Briefly, purified urease (10 μM) was incubated with a 60-fold molar excess of HOCl for 15 min at room temperature in the dark. HOCl was quenched with excess Met (15 mM final) for 30 min on ice. Excess Met and HOCl were removed via dialysis (Spectra/Por; molecular weight cutoff [MWCO], 12,000 to 14,000) in 50 mM sodium phosphate buffer. Oxidized and quenched urease was incubated with 6.2 μM Msr, 5 μM Trx1, 0.5 μM TrxR, 400 μM NADPH, and 100 μM dithiothreitol (DTT) for 2 h at 37°C. The reaction was stopped by incubating at 95°C for 5 min.
Sample preparation and mass spectrometry analysis.
Concentrated ammonium bicarbonate buffer was added to the protein to adjust to a final buffer concentration of 50 mM (pH 8.0). The samples were denatured by heating at 95°C for 20 min in the presence of 10 mM DTT. After cooling the sample, 20 mM iodoacetamide was added and the sample was kept at room temperature in dark for 45 min. Next, 20 mM DTT was added to quench the excess iodoacetamide and the mixture was incubated for 20 min at room temperature. Finally, the samples were digested with sequencing-grade trypsin at a 1:20 ratio of protease to protein by incubating at 37°C overnight with rotation. The samples, after trypsin digestion, were acidified with 0.1% formic acid and spun briefly, and the supernatant was transferred to sample vials. An Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) coupled to an Ultimate 3000 high-performance liquid chromatography (HPLC) system was used for the nano-LC-MS/MS analysis. The samples were first loaded onto a trap cartridge containing C18 stationary phase to desalt the samples. After 4 min, the samples were back-eluted from the trapping cartridge onto a nano PepMap column (75-μm inner diameter, 150-mm length, 3-μm particle size). Mobile phase A consisted of water with 0.1% formic acid, and mobile phase B consisted of acetonitrile with 0.1% formic acid. The peptide separation was achieved with a linear binary gradient from 0 to 40% phase B for 25 min at 0.3 μl/min. The peptides were fragmented by collision-induced dissociation, and the normalized collision energy was set to 35%. The MS scan range was m/z 150 to 2,000, and the top five peaks were selected in precursor scan for the data-dependent CID fragmentation in each cycle. The data files were searched against the protein sequences of UreA and UreB of H. pylori strain 26695. The peak intensities of the unoxidized peptides and their corresponding methionine oxidation products observed in LC-MS were used to calculate the average oxidation events per peptide in the sample. The ion intensity of the oxidized peptides was multiplied by the number of oxidation events required for the mass shift (e.g., one oxidation on methionine for +16, two events for +32) and then divided by the sum of the ion intensities of all unoxidized and oxidized peptide masses as represented by the equation below.
In this equation, P represents the average oxidation level of the peptide and I represents the peak intensities of oxidized and methionine unoxidized peptides.
ACKNOWLEDGMENTS
We thank Amelia Carlie for technical assistance with data presentation.
This work was supported by the University of Georgia Foundation and the National Institute of General Medical Sciences Research Resource for Integrated Glycotechnology (P41GM103390 to S.K.M. and J.S.S.).
REFERENCES
- 1.Blaser M. 1995. The role of Helicobacter pylori in gastritis and its progression to peptic ulcer disease. Aliment Pharmacol Ther 9:27–30. doi: 10.1111/j.1365-2036.1995.tb00780.x. [DOI] [PubMed] [Google Scholar]
- 2.Sipponen P, Hyvärinen H, Seppälä K, Blaser M. 1998. Pathogenesis of the transformation from gastritis to malignancy. Aliment Pharmacol Ther 12:61–71. doi: 10.1111/j.1365-2036.1998.00005.x. [DOI] [PubMed] [Google Scholar]
- 3.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. 1991. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 4.Karnes W, Samloff I, Siurala M, Kekki M, Sipponen P, Kim S, Walsh J. 1991. Positive serum antibody and negative tissue staining for Helicobacter pylori in subjects with atrophic body gastritis. Gastroenterology 101:167–174. doi: 10.1016/0016-5085(91)90474-Y. [DOI] [PubMed] [Google Scholar]
- 5.Peek RM, Fiske C, Wilson KT. 2010. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol Rev 90:831–858. doi: 10.1152/physrev.00039.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Augusto AC, Miguel F, Mendonça S, Pedrazzoli J, Gurgueira SA. 2007. Oxidative stress expression status associated to Helicobacter pylori virulence in gastric diseases. Clin Biochem 40:615–622. doi: 10.1016/j.clinbiochem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Scott DR, Marcus EA, Wen Y, Singh S, Feng J, Sachs G. 2010. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with UreI, a channel for urea and its metabolites, CO2, NH3, and NH4+, is necessary for acid survival of Helicobacter pylori. J Bacteriol 192:94–103. doi: 10.1128/JB.00848-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunn BE, Campbell GP, Perez-Perez GI, Blaser MJ. 1990. Purification and characterization of urease from Helicobacter pylori. J Biol Chem 265:9464–9469. [PubMed] [Google Scholar]
- 9.Scott DR, Marcus EA, Weeks DL, Sachs G. 2002. Mechanisms of acid resistance due to the urease system of Helicobacter pylori. Gastroenterology 123:187–195. doi: 10.1053/gast.2002.34218. [DOI] [PubMed] [Google Scholar]
- 10.Bauerfeind P, Garner R, Dunn B, Mobley H. 1997. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut 40:25–30. doi: 10.1136/gut.40.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha N-C, Oh S-T, Sung JY, Cha KA, Mann HL, Oh B-H. 2001. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol 8:505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 12.Tomb J-F, White O, Kerlavage AR, Clayton RA. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 13.Mobley H. 1996. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment Pharmacol Ther 10:57–64. doi: 10.1046/j.1365-2036.1996.22164006.x. [DOI] [PubMed] [Google Scholar]
- 14.Mehta N, Benoit S, Maier RJ. 2003. Roles of conserved nucleotide-binding domains in accessory proteins, HypB and UreG, in the maturation of nickel-enzymes required for efficient Helicobacter pylori colonization. Microb Pathog 35:229–234. doi: 10.1016/S0882-4010(03)00151-7. [DOI] [PubMed] [Google Scholar]
- 15.Benoit SL, Mehta N, Weinberg MV, Maier C, Maier RJ. 2007. Interaction between the Helicobacter pylori accessory proteins HypA and UreE is needed for urease maturation. Microbiology 153:1474–1482. doi: 10.1099/mic.0.2006/003228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Debowski AW, Walton SM, Chua EG, Tay AC, Liao T, Lamichhane B, Himbeck R, Stubbs KA, Marshall BJ, Fulurija A, Benghezal M. 2017. Helicobacter pylori gene silencing in vivo demonstrates urease is essential for chronic infection. PLoS Pathog 13:e1006464. doi: 10.1371/journal.ppat.1006464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kavermann H, Burns BP, Angermüller K, Odenbreit S, Fischer W, Melchers K, Haas R. 2003. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med 197:813–822. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eaton KA, Brooks C, Morgan D, Krakowka S. 1991. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun 59:2470–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling H-O, Josenhans C, Suerbaum S. 2004. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci U S A 101:5024–5029. doi: 10.1073/pnas.0308386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eaton KA, Krakowka S. 1994. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect Immun 62:3604–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rokita E, Makristathis A, Presterl E, Rotter ML, Hirschl AM. 1998. Helicobacter pylori urease significantly reduces opsonization by human complement. J Infect Dis 178:1521–1525. doi: 10.1086/314459. [DOI] [PubMed] [Google Scholar]
- 22.Mai U, Perez-Perez G, Allen J, Wahl S, Blaser M, Smith P. 1992. Surface proteins from Helicobacter pylori exhibit chemotactic activity for human leukocytes and are present in gastric mucosa. J Exp Med 175:517–525. doi: 10.1084/jem.175.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan X, Gunasena H, Cheng Z, Espejo R, Crowe SE, Ernst PB, Reyes VE. 2000. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J Immunol 165:1918–1924. doi: 10.4049/jimmunol.165.4.1918. [DOI] [PubMed] [Google Scholar]
- 24.Harris P, Mobley H, Perez-Perez G, Blaser M, Smith P. 1996. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokine production. Gastroenterology 111:419–425. doi: 10.1053/gast.1996.v111.pm8690207. [DOI] [PubMed] [Google Scholar]
- 25.Olivera-Severo D, Uberti AF, Marques MS, Pinto MT, Gomez-Lazaro M, Figueiredo C, Leite M, Carlini CR. 2017. A new role for Helicobacter pylori urease: contributions to angiogenesis. Front Microbiol 8:1883. doi: 10.3389/fmicb.2017.01883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahawar M, Tran V, Sharp JS, Maier RJ. 2011. Synergistic roles of Helicobacter pylori methionine sulfoxide reductase and GroEL in repairing oxidant-damaged catalase. J Biol Chem 286:19159–19169. doi: 10.1074/jbc.M111.223677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G, Alamuri P, Maier RJ. 2006. The diverse antioxidant systems of Helicobacter pylori. Mol Microbiol 61:847–860. doi: 10.1111/j.1365-2958.2006.05302.x. [DOI] [PubMed] [Google Scholar]
- 28.Bagchi D, Bhaitacharya G, Stohs S. 1996. Production of reactive oxygen species by gastric cells in association with Helicobacter pylori. Free Radic Res 24:439–450. doi: 10.3109/10715769609088043. [DOI] [PubMed] [Google Scholar]
- 29.Ramarao N, Gray-Owen SD, Meyer TF. 2000. Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol Microbiol 38:103–113. doi: 10.1046/j.1365-2958.2000.02114.x. [DOI] [PubMed] [Google Scholar]
- 30.Davies G, Simmonds N, Stevens T, Sheaff M, Banatvala N, Laurenson I, Blake D, Rampton D. 1994. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35:179–185. doi: 10.1136/gut.35.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stadtman ER, Levine RL. 2000. Protein oxidation. Ann N Y Acad Sci 899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 32.Ezraty B, Gennaris A, Barras F, Collet J-F. 2017. Oxidative stress, protein damage and repair in bacteria. Nat Rev Microbiol 15:385–396. doi: 10.1038/nrmicro.2017.26. [DOI] [PubMed] [Google Scholar]
- 33.Benoit SL, Maier RJ. 2016. Helicobacter catalase devoid of catalytic activity protects the bacterium against oxidative stress. J Biol Chem 291:23366–23373. doi: 10.1074/jbc.M116.747881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alamuri P, Maier RJ. 2006. Methionine sulfoxide reductase in Helicobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J Bacteriol 188:5839–5850. doi: 10.1128/JB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez Porqué P, Baldesten A, Reichard P. 1970. The involvement of the thioredoxin system in the reduction of methionine sulfoxide and sulfate. J Biol Chem 245:2371–2374. [PubMed] [Google Scholar]
- 36.Cao G, pil Lee K, van der Wijst J, de Graaf M, van der Kemp A, Bindels RJ, Hoenderop JG. 2010. Methionine sulfoxide reductase B1 (MsrB1) recovers TRPM6 channel activity during oxidative stress. J Biol Chem 285:26081–26087. doi: 10.1074/jbc.M110.103655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khor HK, Fisher MT, Schöneich C. 2004. Potential role of methionine sulfoxide in the inactivation of the chaperone GroEL by hypochlorous acid (HOCl) and peroxynitrite (ONOO−). J Biol Chem 279:19486–19493. doi: 10.1074/jbc.M310045200. [DOI] [PubMed] [Google Scholar]
- 38.Kuhns LG, Mahawar M, Sharp JS, Benoit S, Maier RJ. 2013. Role of Helicobacter pylori methionine sulfoxide reductase in urease maturation. Biochem J 450:141–148. doi: 10.1042/BJ20121434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benoit SL, Bayyareddy K, Mahawar M, Sharp JS, Maier RJ. 2013. Alkyl hydroperoxide reductase repair by Helicobacter pylori methionine sulfoxide reductase. J Bacteriol 195:5396–5401. doi: 10.1128/JB.01001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmalstig AA, Misra SK, Sharp JS, Maier RJ. 2017. A non-catalytic oxidant-combating role for Helicobacter pylori urease, poster 826. Abstr ASM Microbe, New Orleans, LA, 3 June 2017. [Google Scholar]
- 41.Mobley HLT, Mendz GL, Hazell SL (ed). 2001. Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC. [PubMed] [Google Scholar]
- 42.Phadnis SH, Parlow MH, Levy M, Ilver D, Caulkins CM, Connors JB, Dunn BE. 1996. Surface localization of Helicobacter pylori urease and a heat shock protein homolog requires bacterial autolysis. Infect Immun 64:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunn BE, Vakil NB, Schneider BG, Miller MM, Zitzer JB, Peutz T, Phadnis SH. 1997. Localization of Helicobacter pylori urease and heat shock protein in human gastric biopsies. Infect Immun 65:1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srikanth R, Wilson J, Bridgewater JD, Numbers JR, Lim J, Olbris MR, Kettani A, Vachet RW. 2007. Improved sequencing of oxidized cysteine and methionine containing peptides using electron transfer dissociation. J Am Soc Mass Spectrom 18:1499–1506. doi: 10.1016/j.jasms.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Li Z, Xie B, Sharp JS. 2013. Improved identification and relative quantification of sites of peptide and protein oxidation for hydroxyl radical footprinting. J Am Soc Mass Spectrom 24:1767–1776. doi: 10.1007/s13361-013-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smoot DT, Mobley H, Chippendale GR, Lewison J, Resau J. 1990. Helicobacter pylori urease activity is toxic to human gastric epithelial cells. Infect Immun 58:1992–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuwahara H, Miyamoto Y, Akaike T, Kubota T, Sawa T, Okamoto S, Maeda H. 2000. Helicobacter pylori urease suppresses bactericidal activity of peroxynitrite via carbon dioxide production. Infect Immun 68:4378–4383. doi: 10.1128/IAI.68.8.4378-4383.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CH, Chiou SH. 2011. Proteomic analysis of upregulated proteins in Helicobacter pylori under oxidative stress induced by hydrogen peroxide. Kaohsiung J Med Sci 27:544–553. doi: 10.1016/j.kjms.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss SJ. 1989. Tissue destruction by neutrophils. N Engl J Med 320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 50.Levine RL, Mosoni L, Berlett BS, Stadtman ER. 1996. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci U S A 93:15036–15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 52.Marcus EA, Scott DR. 2001. Cell lysis is responsible for the appearance of extracellular urease in Helicobacter pylori. Helicobacter 6:93–99. doi: 10.1046/j.1523-5378.2001.00014.x. [DOI] [PubMed] [Google Scholar]
- 53.Lu J, Holmgren A. 2014. The thioredoxin antioxidant system. Free Radic Biol Med 66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 54.Kuhns LG, Wang G, Maier RJ. 2015. Comparative roles of the two Helicobacter pylori thioredoxins in preventing macromolecule damage. Infect Immun 83:2935–2943. doi: 10.1128/IAI.00232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mora D, Arioli S. 2014. Microbial urease in health and disease. PLoS Pathog 10:e1004472. doi: 10.1371/journal.ppat.1004472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaffer JN, Norsworthy AN, Sun T-T, Pearson MM. 2016. Proteus mirabilis fimbriae-and urease-dependent clusters assemble in an extracellular niche to initiate bladder stone formation. Proc Natl Acad Sci U S A 113:4494–4499. doi: 10.1073/pnas.1601720113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maroncle N, Rich C, Forestier C. 2006. The role of Klebsiella pneumoniae urease in intestinal colonization and resistance to gastrointestinal stress. Res Microbiol 157:184–193. doi: 10.1016/j.resmic.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Hirche TO, Gaut JP, Heinecke JW, Belaaouaj A. 2005. Myeloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating neutrophil elastase: effects on host defense. J Immunol 174:1557–1565. doi: 10.4049/jimmunol.174.3.1557. [DOI] [PubMed] [Google Scholar]
- 59.Ligabue-Braun R, Andreis FC, Verli H, Carlini CR. 2013. 3-to-1: unraveling structural transitions in ureases. Naturwissenschaften 100:459–467. doi: 10.1007/s00114-013-1045-2. [DOI] [PubMed] [Google Scholar]
- 60.Cabreiro F, Picot CR, Friguet B, Petropoulos I. 2006. Methionine sulfoxide reductases. Ann N Y Acad Sci 1067:37–44. doi: 10.1196/annals.1354.006. [DOI] [PubMed] [Google Scholar]
- 61.Carlini CR, Ligabue-Braun R. 2016. Ureases as multifunctional toxic proteins: a review. Toxicon 110:90–109. doi: 10.1016/j.toxicon.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 62.Copass M, Grandi G, Rappuoli R. 1997. Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect Immun 65:1949–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weatherburn M. 1967. Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 39:971–974. doi: 10.1021/ac60252a045. [DOI] [Google Scholar]
- 64.Mobley H, Cortesia MJ, Rosenthal L, Jones B. 1988. Characterization of urease from Campylobacter pylori. J Clin Microbiol 26:831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benoit S, Maier RJ. 2003. Dependence of Helicobacter pylori urease activity on the nickel-sequestering ability of the UreE accessory protein. J Bacteriol 185:4787–4795. doi: 10.1128/JB.185.16.4787-4795.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]