

Abstract
Professional antigen presenting cell (APCs) are important modulators of acute graft-versus-host disease (GVHD). Although dendritic cells (DCs) are most potent APC subset, other myeloid cells, especially macrophages (MFs) and neutrophils have recently been shown to play a role in the severity of GVHD. However, the critical molecular mechanisms that determine the functions of myeloid cells in GVHD are unclear. Signal transducer and activator of transcription 3 (STAT3) is a master transcription factor that plays a crucial role in regulating immunity. But its role in MF biology and in acute GVHD remains unknown. To determine the role of myeloid cell specific expression of STAT3 on the severity of acute GVHD, we utilized myeloid cell specific STAT3 deficient LysM-Cre/STAT3fl/− animals as recipients and donors in well-characterized experimental models of acute GVHD. We found that reduced expression of STAT3 in myeloid cells from the hosts, but not the donors, increased inflammation, donor T cell activation and exacerbated GVHD. Our data demonstrate that STAT3 in host myeloid cells, such as MFs, dampens acute GVHD.
Keywords: bone marrow transplantation, graft-versus-host disease, STAT-3, myeloid cells
INTRODUCTION
Graft-versus-host disease (GVHD) is a major life threatening complications of allogeneic hematopoietic cell transplantation (allo-HCT). Approximately, 35–50% of patients suffer from acute GVHD despite prophylaxis with calcineurin inhibitors, which suppress the main effector cell of GVHD pathogenesis, allogeneic T cells1, 2. Therefore, new GVHD prophylaxis and treatment strategies are needed.
The biology of GVHD is complex. Antigen presenting cells (APCs), derived from both donor and host, play important roles in the development of acute GVHD by producing pro-inflammatory cytokines, such as TNF-alpha (TNF-α) and IL-6, as well as by directly stimulating donor T cells3–9. The complex role of dendritic cells (DCs), a potent subset of APCs, in GVHD pathogenesis is being increasingly appreciated9. But the effect of macrophages (MFs), another type of APC which infiltrate GVHD target organs10, 11 on GVHD is less well-understood. In support of a role for MFs influencing GVHD, acute GVHD was reduced when macrophage recruitment to GVHD target organs was inhibited by CYM-5442, a sphingosine 1-phosphate 1(S1P1) receptor agonist12, and the anti-GVHD properties of corticosteroids are likely due in part to inhibition of MF functions13. However, these studies did not distinguish the role of donor versus recipient-derived MFs on acute GVHD, which is important because both host and donor MFs are present early post allo-HCT due to the resistance of host MFs to transplant conditioning regimens14. In an effort to clarify this, Hashimoto et al, showed that deleting host MFs increases donor T cell expansion and exacerbates GVHD while colony stimulating factor (CSF)-1 expanded host MFs and reduced acute GVHD15. In contrast to host-derived MFs, CSF-1-dependent donor MFs exacerbate chronic GVHD16. However, it is unclear exactly how host versus donor MFs modulate GVHD, what macrophage molecular and signaling pathways are required for GVHD modulation, or how MFs affect allogeneic T cell responses.
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family and plays a central role in regulating innate and adaptive immune responses17, 18. STAT3 signaling in T cells exacerbates both acute and chronic GVHD potentially due to its essential role for donor T cell activation and inflammatory TH17 cell differentiation19–26. In addition, STAT3 signaling exacerbates GVHD by reducing the stability of naïve regulatory T (Treg) cells and by limiting the expansion of induced donor Treg cells27. In contrast, absence of STAT3 in donor T cells ameliorates chronic GVHD by reducing follicular helper T (Tfh) cells and increasing T follicular regulatory (Tfr) cells28.
STAT3 activation is required for their immune regulatory functions and survival of MFs29, 30. Interestingly, IL-10 secretion by MFs driven by STAT3 signaling is crucial for inhibition of inflammatory responses31, 32, whereas deficiency of STAT3 in MFs increases pro-inflammatory cytokine production in response to lipopolysaccharide (LPS) and helps create a TH1 promoting milieu33. In DCs, STAT3 signaling is crucial for both DC activation and induction of immune tolerance34, 35. Increased STAT3 expression in donor derived plasmacytoid DCs (pDCs) reduces acute GVHD36. STAT3 acetylation negatively regulates DC function by promoting indoleamine 2,3-dioxygenase (IDO) expression and ameliorates acute GVHD37–40. Taken together, these studies suggest that STAT3 signaling influences inflammation in a cell intrinsic and presumably context dependent manner.
To determine whether STAT3 contributes to the role of myeloid cells such as MFs in regulation of GVHD, we tested the influence of STAT3-deficient myeloid lineage cells generated from LysM-Cre/STAT3fl/− animals in experimental models of acute GVHD33. We found that the absence of STAT3 in host- derived but not donor-derived myeloid cells (i.e. MFs) exacerbates GVHD. These data suggest that STAT3 signaling in host myeloid cells such as MFs and PMNs mitigates the severity of acute GVHD.
RESULTS
LysM-Cre/STAT3fl/− animals showed reduced expression of STAT3 in macrophages (MF)
Host macrophages (MFs) regulate the severity of acute GVHD9, 15. STAT3 is a master regulator of MF dependent innate immune responses. But the role of STAT3 in professional APCs, specifically in MFs, in regulation of their allo-stimulatory functions is not known. To evaluate the role of STAT3 in MFs on allogeneic immune responses, we generated STAT3 deficient MFs by crossing B6 STAT3 hemizygous mice in which one STAT3 allele was floxed and the other was already ablated (STAT3fl/−), with B6 mice expressing Cre recombinase under the control of the myeloid lineage (MFs and granulocytes) specific promoter, lysosome M (LysM-Cre)41, 42. We next confirmed that both mRNA and protein expression of STAT3 in peritoneal MFs from LysM-Cre/STAT3fl/− animals were decreased relative to wild type LysM-Cre/STAT3flox/flox (WT) animals (Figure 1A)33. STAT3 expression in bone marrow derived DCs (BMDCs) from LysM-Cre/STAT3fl/− animals was not reduced suggesting that, of myeloid-derived APCs, STAT3 expression was exclusively decreased in MFs from LysM-Cre/STAT3fl/− animals33. The MFs from both naïve WT and LysM-Cre/STAT3fl/− animals showed similar expression of the co-stimulatory molecules, such as CD40, CD80, PD-L1, and I-Ab (Figure 1B–C).
Figure 1. LysM-Cre/STAT3fl/− aanimals showed reduced expression of STAT3 in macrophages (MF).
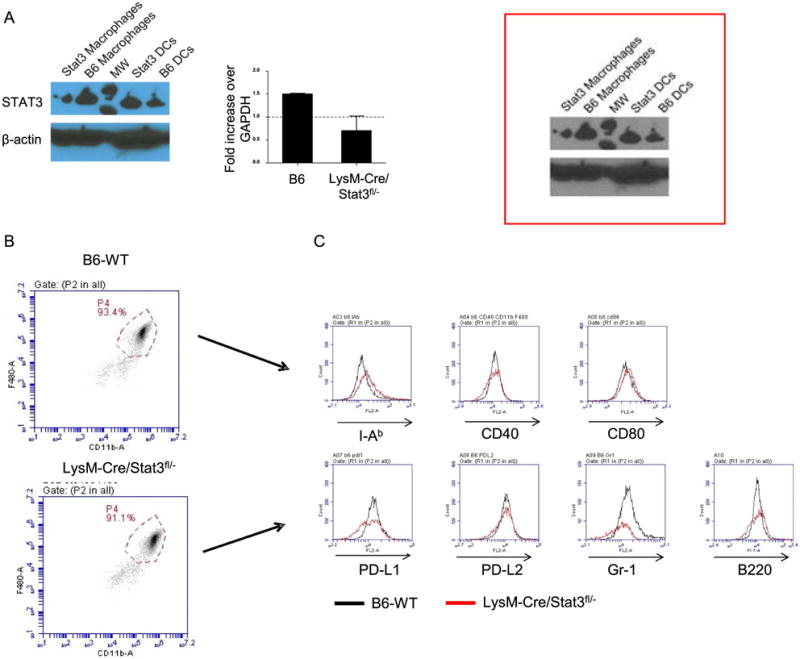
(A)Western blot showing total STAT3 (top) and β-actin (middle) for B6 macrophages (MFs), LysM-Cre/STAT3fl/− MFs, B6-WT dendritic cells (DCs) and LysM-Cre/STAT3fl/− DCs. (bottom) Relative increase in RNA expression of STAT3 in both B6-WT and LysM-Cre/STAT3fl/− MFs by qRT-PCR. (B)Representative dot-plots and gates of peritoneal CD11b+F4/80+ cells from B6-WT or LysM-Cre/STAT3fl/− animals are shown. (C) Representative expression levels of MHC class II (I-Ab), CD40, CD80, PD-L1, PD-L2, Gr-1 and B220 on CD11b+F4/80+ peritoneal MFs from B6-WT or LysM-Cre/STAT3fl/− animals are shown. All error bars show the mean ± SEM.
Reduced STAT3 expression in host myeloid APCs amplifies the severity of acute GVHD
Next, we determined whether reduced expression of STAT3 in host myeloid cells, specifically MFs but not in DCs, will modulate GVHD severity. To accomplish this, we utilized the well-established major histocompatibility complex (MHC) mismatched acute GVHD model, BALB/c into B6. Both B6-WT and LysM-Cre/STAT3fl/− animals were lethally irradiated and transplanted with bone marrow (BM) and splenic T cells from either syngeneic B6-WT or allogeneic BALB/c donors. Compared to allogeneic B6-WT animals, the allogeneic LysM-Cre/STAT3fl/− animals demonstrated significantly greater mortality (p=0.02) and increased clinical severity of GVHD (Figure 2A–B).
Figure 2. Reduced STAT3 expression in host myeloid APCs amplifies the severity of acute GVHD.
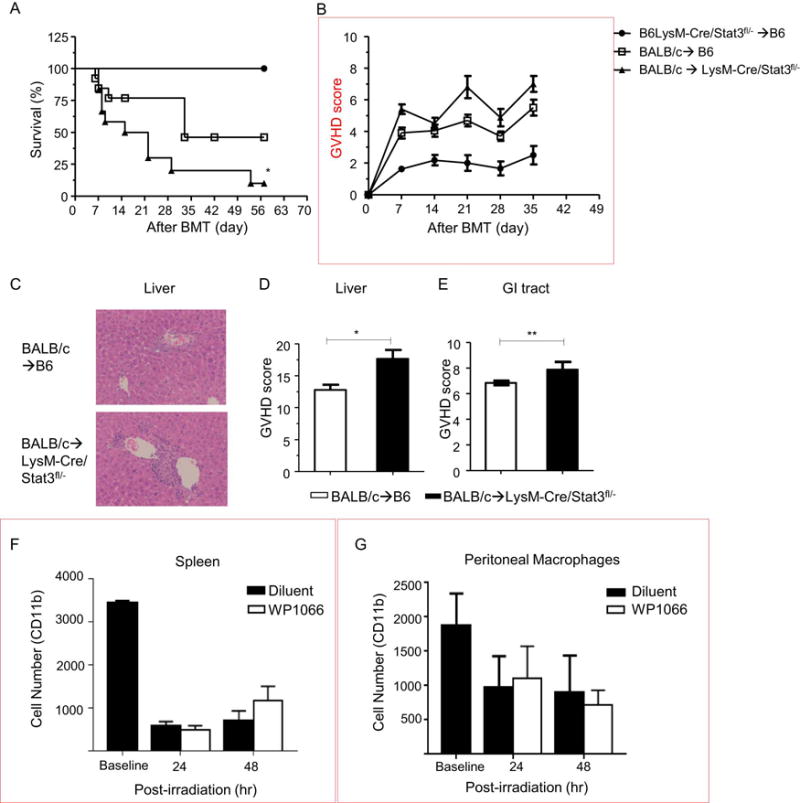
Host B6-WT and B6-LysM/Cre STAT3fl/− animals were lethally irradiated (11 Gy) on day −1 and infused with 2 × 106 CD90.2+ T cells along with 5 × 106 bone marrow (BM) cells from either syngeneic B6 or allogeneic BALB/c animals on day 0. (A) Survival (n=12–16 per group). Data are combined from three experiments with similar results. (B) GVHD clinical score. (C) Representative hematoxylin and eosin (H&E) stained images of liver on day 14 after allo-BMT are shown. (D and E) Histopathological GVHD scores from liver (D) and gastrointestinal (GI) tract (E) on day 14 after allo-BMT (n=12 per group, pooled from two experiments). Balb/c-WT mice were irradiated (8 Gy) and then immediately i.p. injected with 20 mg/Kg WP1066 STAT3 inhibitor or diluent. At 24 and 48 hours after injections mice were euthanized and whole spleens (F) and peritoneal (G) macrophages and were harvested (n=3 per group). Cells were analyzed for CD11b+ cells within the myeloid gate. All error bars show the mean ± SEM. **p<0.01, *p<0.05.
The increase in mortality and GVHD severity was associated with more severe histopathological damage of GVHD target organs, liver (Figure 2C–D) and gastrointestinal (GI) tract (Figure 2 E) on day 14 post-BMT compared with B6-WT allogeneic animals. Next, in order to determine whether the impact on GVHD is due to differences in the survival between the STAT3 deficient WT macrophages after conditioning and BMT, we analyzed MF survival following similar conditioning protocol as above. To this end, wild-type Balb/c animals were lethally irradiated and then treated with either 20mg/kg of WP1066 (a STAT3 inhibitor)56 or diluent immediately after conditioning and analyzed the MF recovery and survival from both spleen and peritoneal cavity. This dose has been shown to inhibit STAT3 in vivo56. Peritoneal and splenic CD11b+ MFs were then analyzed at 24 and 48 hours post irradiation. MF counts in non-conditioned animals were used as a baseline. No significant differences in MF survival were observed at either time points in MFs regardless of STAT3 inhibition (Figure 2 G–H). This approach suggests, but is not definitive, that STAT3 deficiency in MF does not alter their survivability after conditioning.
To rule out strain, model, and major MHC mismatch dependent artifacts, we utilized a multiple minor histocompatibility antigens (miHAs) –mismatched model of GVHD, C3H.sw into B6. Similar to the major MHC mismatched model, serum levels of IFN-γ (Supplementary Figure 1a) and IL-17A (Supplemental Figure 1b) were increased in the allogeneic LysM-Cre/STAT3fl/− recipient animals, and their livers and GI tracts exhibited greater GVHD specific histopathological damage (Supplemental Figure 1c-e). These data demonstrate that STAT3 signaling in host myeloid APCs regulates the severity of GVHD in a strain independent manner.
Deficiency of STAT3 in host MFs enhances donor T cell expansion after allogeneic BMT
We next explored the putative mechanisms for enhanced acute GVHD when STAT3 is deficient in host APCs of myeloid origin. In light of the role of STAT3 in attenuating pro-inflammatory responses by the MFs33, we determined whether the absence of STAT3 in host myeloid APCs also enhanced T cell expansion and inflammation at day +14 after BMT, which is in the time range of the peak donor T cell response in this model. Consistent with this notion, compared to the WT recipients, the enhanced mortality of the allogeneic LysM-Cre/STAT3fl/− recipient animals was associated with increased donor CD4+ and CD8+ T cell expansion (Figure 3A) and increased serum levels of T cell derived pro-inflammatory cytokines, IFN-γ (Figure 3B) and IL-17A (Figure 3C) on day +14 after BMT. Furthermore, the expansion of CD4+Foxp3+ Treg was not changed, but the effector T cells/regulatory T cell ratio (TEff)/Treg) was significantly increased in allogeneic LysM-Cre/STAT3fl/− compared to WT animals (Figure 3D–E). These data show that STAT3 signaling in host myeloid cells inhibits murine allogeneic T cell proliferation in vivo and reduced acute GVHD.
Figure 3. Deficiency of STAT3 in host MFs enhances donor T cell expansion after allogeneic BMT.
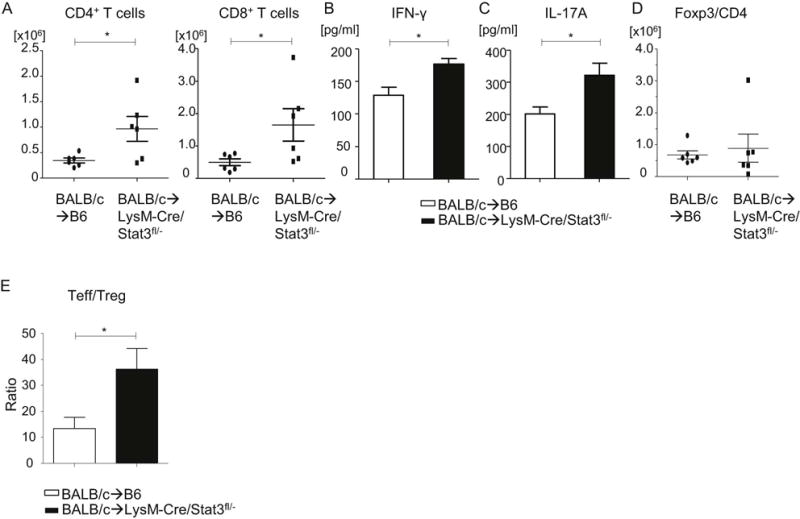
Host B6-WT and B6-LysM/Cre STAT3fl/− animals were lethally irradiated (11 Gy) on day −1 and infused with 2 × 106 CD90.2+ T cells along with 5 × 106 bone marrow (BM) cells from either syngeneic B6 or allogeneic BALB/c animals on day 0. (A) To evaluate donor T cell (H-2kd+CD4+ aor H-2kd+CD8+) expansion, spleen cells from B6-WT or B6 LysM/Cre STAT3fl/− animals were harvested on day 14 after allogeneic bone marrow transplantation (allo-BMT), stained, and analyzed by flow cytometry (n=6 per group, pooled from two experiments). (B and C) Serum was collected from recipients on day 14 and IFN-γ (B) and IL-17A (C) levels were determined by ELISA. (n=6 per group, pooled from two experiments). (D and E) Donor CD4+Foxp3+ regulatory T cell (Treg) expansion (D) and the ratio of TEff cells (CD4+FoxP3− and CD8+FoxP3−) to Treg cells (E) on day 14 after allo-BMT are shown. All error bars show the mean ± SEM. *p<0.05.
STAT3 deficient macrophages show enhanced stimulation of allogeneic T cells in vitro
Amongst the myeloid derived cells, MFs are bona fide APCs. Because in vivo STAT3 deficiency in host myeloid cells showed enhanced in vivo expansion of allogeneic T cells, we therefore reasoned that the absence of STAT3 in MFs (and not the other myeloid derived cells namely neutrophils) might be the main driver of donor T cell expansion and hence the cause of amplified GVHD. To determine the cell intrinsic effect of STAT3 deficiency in MF, we examined whether reduced expression of STAT3 in MFs from LysM-Cre/STAT3fl/− animals affects their ability to stimulate allogeneic T cell response in vitro. To this end, peritoneal MFs were used as stimulators for co-cultured splenic T cells from either syngeneic B6 or allogeneic BALB/c animals in mixed lymphocyte reaction (MLR). We found that MFs from LysM-Cre/STAT3fl/− animals enhanced allogeneic T cell proliferation and production of IL-2, IFN-γ, and IL-17A but not IL-4 and IL-10 compared to WT MFs (Figure 4A–F). Importantly, BMDCs from LysM-Cre/Stat3fl/− animals showed equivalent allogeneic T cell responses to WT DCs in vitro (Supplemental Figure 2). These data show that STAT3 signaling in MFs inhibits allogeneic T cell responses and possibly TH1 or TH17 differentiation in vitro and suggest that the in vivo effects are from deficiency in host MFs.
Figure 4. STAT3 deficient macrophages show enhanced stimulation of allogeneic T cells in vitro.
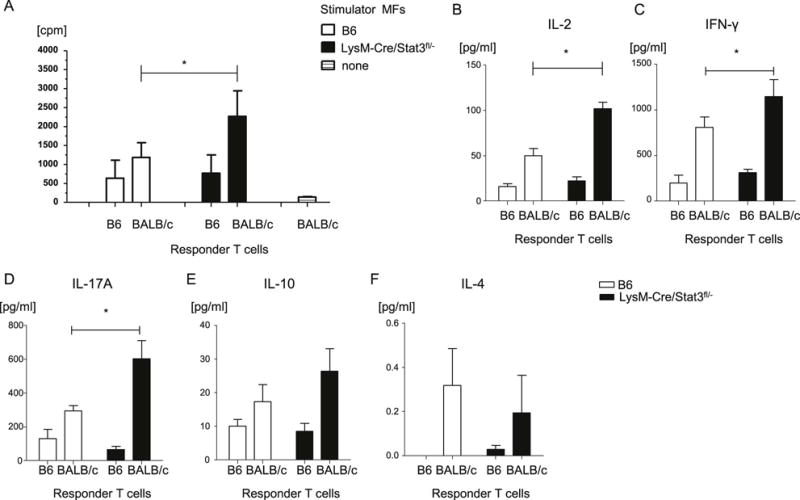
(A) Peritoneal MFs from B6-WT and LysM-Cre STAT3fl/− animals were used as stimulators in an MLR with T cells from either syngeneic B6 or allogeneic BALB/c animals and analyzed for T-cell proliferation via 3H-thymidine incorporation at 72 h. (B-F) Supernatants from MLR cultures were collected at 72 h and analyzed for IL-2 (B), IFN-γ (C), IL-17A (D), IL-10 (E), and IL-4 (F) by ELISA. The data are representative of three independent experiments. Error bars show the mean ± SEM. * p<0.05.
STAT3 deficient macrophages exhibit enhanced innate immune responses
Allo-HCT conditioning causes tissue damage which results in the generation of damage- and pathogen-associated molecular patterns (DAMPs and PAMPs, respectively), such as LPS. DAMPs and PAMPs activate GVHD-promoting inflammation via pattern recognition receptor signaling, particularly in APCs43–46. Therefore, we next determined whether STAT3 expression alters APC responses to LPS. Consistent with previous observations, STAT3 deficient MFs showed enhanced production of IL-1β, IL-6 and TNF-α, and decreased production of IL-10 relative to WT MFs (Supplemental Figure 3, a-d) when stimulated with LPS (1μg/ml) for 16 hours33. In contrast, STAT3 deficient DCs produced similar levels of these cytokines compared to WT DCs upon LPS stimulation (Supplemental Figure 4 a-d). These data demonstrate increased LPS-stimulated innate immune responses in STAT3 deficient MFs which may contribute to their ability to aggravate acute GVHD.
STAT3 deficiency in donor myeloid cells is dispensable for acute GVHD severity
Donor APCs also contribute to GVH responses8, 47–49; therefore, we explored whether murine GVHD was affected by STAT3 signaling in donor myeloid cells. To test this, we utilized the well-established major MHC mismatched B6 into BALB/c acute GVHD model. Recipient BALB/c animals were lethally irradiated and transplanted with splenic CD90.2+T cells from B6-WT animals and with T cell depleted BM (TCD-BM) from either B6-WT or LysM-Cre/STAT3fl/− animals. Survival, GVHD clinical score, serum pro-inflammatory cytokine levels, and donor T cell expansion except for IL-17A at day 14 after allo-BMT were similar between BALB/c animals that received either allogeneic WT or STAT3-deficient TCD-BM (Figure 5 A–J). These data indicate that STAT3 expression in donor myeloid cells is dispensable for regulation of the severity of acute GVHD.
Figure 5. STAT3 deficiency in donor myeloid cells is dispensable for acute GVHD severity.
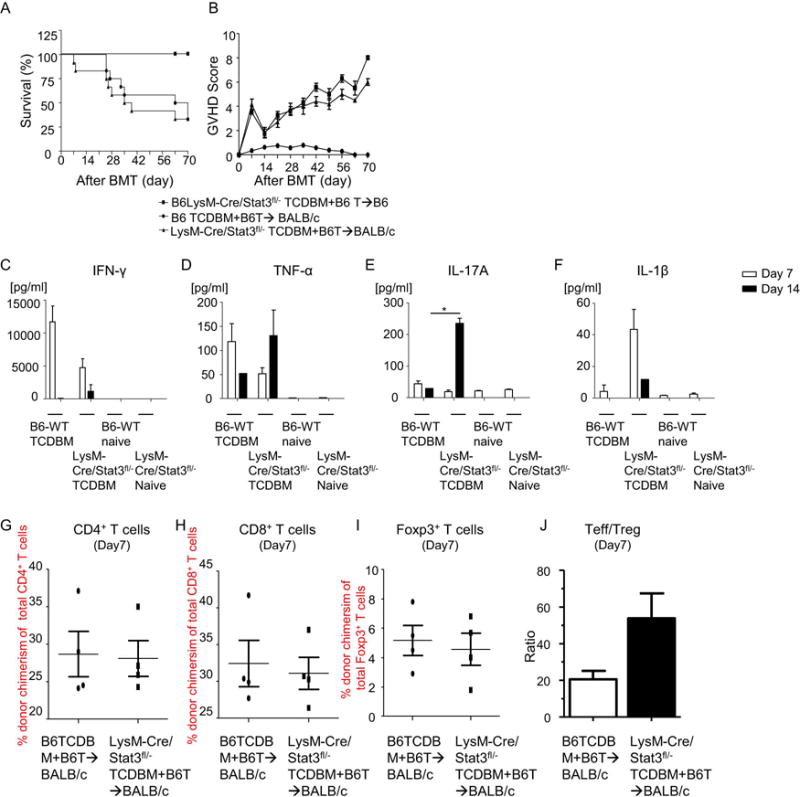
syngeneic controls were irradiated (8 Gy) on day −1 and infused with 5 × 106 TCD-BM cells along with 1 × 106 CD90.2+ T cells from B6-LysM-Cre/STAT3fl/− animals on day 0 (■, n=12). Allogeneic host BALB/c animals were irradiated as above and infused with B6-WT 2×106 CD90.2+ T cells along with 5×106 TCD-BM from either WT-B6 (•, n=12) or B6-LysM/Cre STAT3fl/− animals (▲, n=12). The recipients were monitored for (A) Survival and (B) GVHD clinical scores on a weekly basis. Data are combined from three experiments with similar results. (C–F) Serum was collected from recipients on day 7 and day14 after allo-BMT and cytokine levels were determined by ELISA for IFN-γ (C), TNF-α (D), IL-17A (E), and IL-1β (F). *p<0.05. (G-I) Spleen cells from B6-WT or B6 LysM/Cre STAT3fl/− animals were harvested on day 7 after allo-BMT, stained, and analyzed by flow cytometry. Donor T cell (H-2kb+CD4+ a(G) or H-2kb+CD8+ (H)) expansion, donor CD4+Foxp3+ Tregcell expansion (I), and the ratio of TEff cells (CD4+FoxP3− and CD8+FoxP3−) to Treg cells (J) on day 7 after allo-BMT are shown (n=4 per group, pooled from two experiments). Error bars show the mean ± SEM.
DISCUSSION
Previous reports suggested that host myeloid derived APCs such as MFs and neutrophils aggravate acute GVHD9. For example, host MFs survive BMT conditioning, modulate donor T cell functions14, infiltrate GVHD target organs10, 11, and inhibiting their ability to migrate to GVHD target organs ameliorates GVHD12. In addition, dexamethasone ameliorates GVHD by reducing production of pro-inflammatory cytokines from host derived MFs13, while decreasing the number of host MFs enhances donor T cell expansion and aggravates acute GVHD15. However, the molecular signaling mechanisms required for host myeloid derived APCs such as MF-dependent aggravation of acute GVHD are incompletely characterized. Herein we addressed this important question. We found that STAT3 limits the ability of host myeloid cells to enhance GVHD presumably by limiting host macrophage and neutrophil mediated inflammatory innate immune responses and their intrinsic ability to stimulate allogeneic T cell proliferation.
Importantly, our data show that STAT3 expression in host MFs does not completely block GVHD. This is likely because other APCs, including DCs or neutrophils may be sufficient for induction of murine GVHD. In contrast, STAT3 in donor MFs has no effect on murine acute GVHD. These data suggest that host MFs, much like DCs, are critical APCs for regulating the severity of GVHD while not being mandatory for the induction of GVH responses when all other APCs are intact9.
Thus our results are consistent with and expand on previous reports that host MFs, when expanded by administrating CSF-1, can decrease GVHD severity and mortality by modulating donor T cell function15. Our data points to STAT3 as being a critical molecule that regulates the ability of host MFs in mitigating GVHD responses. Future studies will address the downstream targets of STAT3 that are critical for negatively regulating host MFs after allogeneic BMT.
STAT3 expression in myeloid cells is required for modulating inflammatory responses to PAMPs in vivo33, 34. Therefore, we confirmed that STAT3 deficiency in MFs enhances their production of inflammatory cytokines when stimulate with LPS. In addition, we show that recipient animals deficient in STAT3 in their myeloid lineage showed aggravated GVHD associated with increased allogeneic T cell proliferation. Remarkably, we couldn’t find any significant aggravation of GVHD when using STAT3 deficient donor myeloid cells. These data suggest that STAT3 expression in host MFs may play an important role in regulating allogeneic immune responses possibly by limiting inflammatory host macrophage innate immune responses and creating a milieu that is less advantageous for allogeneic T cell proliferation. However, our in vitro data suggest that MF intrinsic deficiency amplified their ability to stimulate allogeneic T cell proliferation and inflammatory cytokine secretion. These data, when taken in light of the lack of impact of STAT3 deficiency in donor MFs on GVHD, point to the notion that the alteration of the allo-stimulatory capacity of MFs might be a critical component for the aggravation of GVHD. The reasons for enhanced allo-stimulatory effects by STAT3 deficient macrophages remains to be explored. It may be secondary to changes in expression of PDL1 and/or other co-inhibitory markers or due to impact on other immune-regulatory pathways such as IL-10 in the macrophages. Furthermore, although the deficiency of STAT3 in host myeloid cells aggravated GVHD across multiple models, the magnitude of GVHD aggravation was greater in the major MHC mismatched (BALB/c into B6) model than in the MHC matched multiple miHAs mismatched (C3H.sw into B6) model of acute GVHD, indicating that the anti-GVHD effect of STAT3 in host myeloid cells, especially MFs may be dependent on the strength of allogenic stimulation. Overall, our data suggest that activating STAT3 in host myeloid cells, decreases allogeneic immune responses. Further efforts will focus on determining the molecular signaling events and the specific effects in MF versus those in neutrophils that are critical for this phenomenon.
We wish to point out certain caveats that are germane to our study. First, the LysM-Cre/STAT3fl/− are not complete knock-outs but are merely deficient (not absent) of STAT3 due to leakiness of the LyM Cre driver. Second, the deficiency of STAT3 in the LysM-Cre/STAT3fl/− animals is not limited to MFs, but also includes other myeloid derived cells, specifically neutrophils. Importantly, neutrophils also drive allogeneic T cell responses50, therefore, our in vivo results are due the net effect of STAT3 deficiency in all host myeloid cells and we cannot formally rule out a role for STAT3-expressing neutrophils mitigating GVHD in the LysM-Cre/STAT3fl/− recipient animals.
We and others previously demonstrated that STAT3 negatively regulates DCs39, 40, 51. Interestingly, the HDAC inhibitor, vorinostat, which is currently being investigated in clinical trials, reduces GVHD by modulating DC functions via enhancing STAT3 acetylation thereby promoting the transcription of IDO, which is critical for the amelioration of acute GVHD by HDAC inhibitors37–40, 52, 53. Therefore, the activation and acetylation of STAT3 in host MFs, in addition to DCs, may be required for the anti-GVHD activity of HDAC inhibitors and suggests the intriguing possibility that HDAC inhibitors may skew host MFs to an anti-inflammatory phenotype after allo-HCT.
In conclusion, STAT3 expression in host, but not donor myeloid cells, regulates experimental acute GVHD severity and suggest that STAT3 inhibition in these cells could serve as a potential target for development of novel approaches to mitigate clinical GVHD.
MATERIALS and METHODS
Mice
Female BALB/c (H-2d, CD45.2+) and C57BL/6 (B6, H-2b+, CD45.2+) mice were purchased from Charles River Laboratories (Wilmington, MA). B6Ly5.2 (H-2b+, CD45.1+) mice were purchased from National Cancer Institute-Frederick (Frederick, MD). C3H.SW (H-2b+, CD45.2+) mice were purchased from Jackson Laboratory (Bar Harbor, ME). B6-background Stat3fl/+ animals were provided by Dr. Akira (Osaka University) and have been described previously54. All animals were cared for according to regulations reviewed and approved by the University Committee on Use and Care of Animals of the University of Michigan, based on University Laboratory Animal Medicine guidelines.
STAT3 genotyping
Stat3fl/+ females and Stat3fl/+ males were crossbred and genotyped to obtained Stat3fl/− as previously described33. The mice were inbred and maintained by the University of Michigan Breeding Colony.
Bone marrow derived dendritic cells (DCs)
To obtain bone marrow derived DCs (BMDCs), bone marrow (BM) cells from B6-WT or LysM-Cre/Stat3fl/− animals were cultured with murine recombinant GM-CSF (20 ng/ml; PeproTech, Rocky Mill, NJ) for 7 days and harvested as described previously. BMDCs were isolated by using CD11c (N418) MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) on an autoMACS (Miltenyi Biotec).
STAT3 detection
Peritoneal macrophages (MFs, 1 × 106), purified BMDCs (1 × 106), and whole splenic cells (1 × 106) from naïve B6 or LysM-Cre/Stat3fl/− animals were washed with PBS and lysed with RIPA buffer. Cell lysates were subjected to western blot analysis using the following antibodies: STAT3 (Abcam), β-actin-HRP (Abcam), and Cre-lox (Imegenix) per the manufacturer’s instructions.
STAT3 inhibitor study
WT Balb/c animals were irradiated (137Cs source) with 800 cGy total body irradiation (TBI). Immediately after irradiation, mice were treated via intraperitoneal injection with a single dose (20 mg/Kg) of WP1066 (Sigma Aldrich, St. Louis, MO) STAT3 inhibitor dissolved in DMSO/polyethylene glycol (PEG) 300 (1:4, v/v) (REF). Control mice were treated with only diluent. At 24 and 48 hours post injection mice were euthanized and peritoneal (20 ml wash) and whole splenic CD11b+ cells were analyzed. Cells were analyzed on an Attune NxT Acoustic Focusing Flow Cytometer (Life Technologies, Carlsbad, CA).
Quantitative real-time PCR
Total RNA was obtained by phenol/chloroform separation. Gapdh (forward: 5′-GACATGCCGCCTGGAGAAAC-3′; reverse 5′-AGCCCAGGATGCCCTTTAGT-3′) was used as the control house-keeping gene. The primers for Stat3fl/fl detection were: forward 5′-GGGGTGAGAGTTACCGTGAA-3′ and reverse 5′-CACACACACACAAGCCATCA-3′. Real-time PCR was performed with a SYBR green PCR mix (ABI biosystems, Waltham, MA) in a Realplex Eppendorf Real-Time PCR instrument (Eppendorf, Hamburg, Germany).
FACS analysis
Flow cytometry was performed as previously described. Briefly, to analyze phenotypes of peritoneal MFs, isolated cells were suspended in FACS wash buffer (2% BSA in PBS) and stained with conjugated monoclonal antibodies (FITC-, PE-, or APC). Anti-mouse monoclonal antibodies (mAb) for CD11b (M1/70), F4/80(BM8), I-Ab (AF6-120.1), CD40 (3/23), CD80 (16-10A1), Gr-1 (RB6-8C5), PD-L2 (TY25), and B220 (RA3-6B2) were purchased from Biolegend (San Diego, CA). PE-conjugated anti-PD-L1 mAb (M1H5) was purchased from eBioscience (San Diego, CA). After staining, cells were washed with FACS wash buffer and fixed with FACS Lysing Solution (BD Biosciences). Samples were analyzed on a C6 cytometer (BD bioscience). To analyze donor T cell expansion after allo-BMT, isolated spleen cells were processed as above and stained with CD4 (RM4-4), CD8a (53-6.7), CD90.2 (53-2.1), CD229.1 (30C7), CD45.1 (A20), and CD45.2 (104) mAbs purchased from Biolegend. For intracellular staining of Foxp3, cells were washed with permeabilization buffer (eBioscience) and stained with PE-conjugated anti-Foxp3 mAb (FJK-16s; eBioscience) for 30 min at 4°C. The cells were then washed with FACS wash buffer and analyzed on a C6 cytometer (BD bioscience).
Bone marrow transplantation
Host animals were irradiated (137Cs source) with 800-1100 cGy total body irradiation (TBI) on day -1 before allo-BMT. Donor BM cells were harvested from the femur and tibia. Where indicated, T cells were magnetically depleted from the BM (TCD-BM) using mouse CD90.2-microbeads and MACS™ LS columns (Miltenyi Biotec). Splenic T cells were magnetically isolated by using mouse CD90.2-microbeads and MACS™ LS columns. T cells purity was checked by flow cytometry and adjusted accordingly. Syngeneic or allogeneic BM (either whole or TCD-BM) and T cells were infused through the tail vein. Host mice were housed in sterilized micro-isolator cages and maintained on acidified water (pH<3) for three weeks after allo-BMT, as described previously55. Survival was monitored daily, and clinical GVHD was assessed weekly. All animal studies were performed per the Institutional Animal Care and Use Committee guidelines of the University of Michigan.
Mixed lymphocyte reaction (MLR)
Splenic T cells from B6- or BALB/c-WT animals (magnetically separated by MACS using CD90.2 microbeads) were used as responders and B6-WT or B6- LysM-Cre/Stat3fl/− derived BMDCs or peritoneal MFs were used as stimulators. 1×105 T cells and irradiated (20Gy) 2.5×103 BMDCs or 1×105 MFs were co-cultured on 96-well U-bottom plates for 72 hours. The incorporation of 3H-thymidine (1μCi/well) by proliferating T cells during the final 16 hours of co-culture was measured by a Betaplate reader (Wallad, Turku, Finland).
ELISA
IFN-γ, TNF-α, IL-6, Il-17A, and IL-10 were measured in culture supernatants or mouse serum by ELISA with specific anti-mouse mAbs for capture and detection. The appropriate standards were purchased from BD Systems (TNF-α and IL-6) or BD OptEIA (IFN-γ, IL-17A and IL-10). Assays were performed according to the manufacturer’s protocol and read at 450 nm using a microplate reader (Molecular Devices).
Histology
Formalin-preserved livers and GI tracts were embedded in paraffin, cut into 5mm-thick sections and stained with hematoxylin and eosin (H&E) for histological examination. Slides were scored in a blind fashion by Dr. C Liu using a semi-quantitative scoring system that assess the abnormalities known to be associated with GVHD.
Statistical analysis
Student’s t-test was used for in vitro data, and the Wilcoxon rank test was used to analyze survival data. p < 0.05 was considered statistically significant.
Supplementary Material
Supplemental Figure 1. Reduced STAT3 expression in host macrophages exacerbates acute GVHD in a minor antigens mismatched BMT model.
Host B6-WT and LysM/Cre STAT3fl/− animals were irradiated (11 Gy) on day −1 and transplanted with 5 × 106 BM cells along with 1×106 CD90.2+ T cells from either syngeneic B6 or allogeneic C3H.sw donors. Serum was collected from recipients on day 14 after allo-BMT and cytokine levels were determined by ELISA for IFN-γ (a) and IL-17A (b). (c) Representative hematoxylin and eosin (H&E) stained images of liver on day 14 after allo-BMT are shown. (d and e) Histopathological GVHD scores of liver (d) and gastrointestinal (GI) tract (e) on day 14 after allo-BMT are shown. (n=6 per group, pooled from two experiments). **p<0.01, *p<0.05. Error bars show the mean ± SEM.
Supplemental Figure 2. Reduced expression of STAT3 in dendritic cells doesn’t alter allogeneic T cell proliferative capacity in vitro.
(a)Bone marrow derived dendritic cells (BMDCs) from B6-WT or LysM/Cre STAT3fl/− animals were used as stimulators in an MLR by co-culturing with T cells from either B6 (syngeneic) or BALB/c (allogeneic) animals and analyzing T-cell proliferation via 3H-thymidine incorporation at 72 hours. (b–e) Supernatants from the above MLRs were collected at 72 hours and analyzed for IL-2 (b), IL-4 (c), IL-10 (d) and IFN-γ (d) by ELISA. Data are combined from three independent experiments. Error bars show the mean ± SEM.
Supplemental Figure 3. STAT3 deficient macrophages exhibit enhanced innate immune responses
(A-D) Peritoneal MFs were isolated as described in materials and methods. Cells were (1×106 cells/ml) plated and stimulated with LPS (1μg/ml) or diluent for 16 h at 37°C. Supernatants from cultured wells were harvested and examined for IL-1β (A), IL-6 (B), TNF-α (C), and IL-10 (D) by ELISA. Experiments were performed in triplicate. ****p<0.0001, *p<0.05. Error bars show the mean ± SEM.
Supplemental Figure 4. Reduced expression of STAT3 in dendritic cells doesn’t alter their LPS induced cytokine production.
BMDCs were isolated as described in materials and methods. DCs were (1×106 cells/mL) were plated and stimulated with LPS (1μg/ml) or diluent for 16 hours at 37°C. Supernatants were analyzed for IL-1β (a), IL-6 (b), TNF-α (c), and IL-10 (d) by ELISA. Experiments were performed in triplicate. Error bars show the mean ± SEM.
Highlights.
STAT3 negatives regulates macrophage responses
Expression of STAT3 in host derived myeloid cells such as macrophages and neutrophils restrains allo-immune responses and reduces GVHD severity
Expression of STAT3 in donor derived myeloid cells is dispensable for GVHD severity
Acknowledgments
This work was supported by National Institutes of Health grants HL090775, CA173878, HL128046 and CA203542 (PR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions:
Evelyn C. Nieves: designed and performed experiments, analyzed the data and wrote the paper
Tomomi Toubai: performed experiments, analyzed the data and wrote the paper
Daniel C. Peltier: performed experiments, wrote the manuscript
Katherine Oravecz-Wilson: performed experiments
Chen Liu: performed histopathological analysis
Hiroya Tamaki: performed experiments
Yaping Sun: performed experiments
Pavan Reddy: designed experiments, analyzed the data and wrote the paper
Conflict of interest statement: The authors have no conflict of interest
References
- 1.Jagasia M, Arora M, Flowers ME, Chao NJ, McCarthy PL, Cutler CS, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119(1):296–307. doi: 10.1182/blood-2011-06-364265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SE, Cho BS, Kim JH, Yoon JH, Shin SH, Yahng SA, et al. Risk and prognostic factors for acute GVHD based on NIH consensus criteria. Bone Marrow Transplant. 2013;48(4):587–592. doi: 10.1038/bmt.2012.187. [DOI] [PubMed] [Google Scholar]
- 3.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285(5426):412–415. doi: 10.1126/science.285.5426.412. e-pub ahead of print 1999/07/20; doi: 7674 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8(6):575–581. doi: 10.1038/nm0602-575. e-pub ahead of print 2002/06/04; doi: 10.1038/nm0602-575 nm0602-575 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med. 2005;11(11):1244–1249. doi: 10.1038/nm1309. [DOI] [PubMed] [Google Scholar]
- 6.Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J, Jr, et al. Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102(10):1882–1891. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duffner UA, Maeda Y, Cooke KR, Reddy P, Ordemann R, Liu C, et al. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. J Immunol. 2004;172(12):7393–7398. doi: 10.4049/jimmunol.172.12.7393. [DOI] [PubMed] [Google Scholar]
- 8.Merad M, Hoffmann P, Ranheim E, Slaymaker S, Manz MG, Lira SA, et al. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat Med. 2004;10(5):510–517. doi: 10.1038/nm1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koyama M, Hill GR. Alloantigen presentation and graft-versus-host disease: fuel for the fire. Blood. 2016;127(24):2963–2970. doi: 10.1182/blood-2016-02-697250. [DOI] [PubMed] [Google Scholar]
- 10.Nishiwaki S, Terakura S, Ito M, Goto T, Seto A, Watanabe K, et al. Impact of macrophage infiltration of skin lesions on survival after allogeneic stem cell transplantation: a clue to refractory graft-versus-host disease. Blood. 2009;114(14):3113–3116. doi: 10.1182/blood-2009-03-209635. [DOI] [PubMed] [Google Scholar]
- 11.Terakura S, Martin PJ, Shulman HM, Storer BE. Cutaneous macrophage infiltration in acute GvHD. Bone Marrow Transplant. 2015;50(8):1135–1137. doi: 10.1038/bmt.2015.114. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Q, Ma S, Lin D, Mei Y, Gong H, Lei L, et al. The S1P1 receptor-selective agonist CYM-5442 reduces the severity of acute GVHD by inhibiting macrophage recruitment. Cell Mol Immunol. 2015;12(6):681–691. doi: 10.1038/cmi.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishiwaki S, Nakayama T, Murata M, Nishida T, Terakura S, Saito S, et al. Dexamethasone palmitate ameliorates macrophages-rich graft-versus-host disease by inhibiting macrophage functions. PLoS One. 2014;9(5):e96252. doi: 10.1371/journal.pone.0096252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haniffa M, Ginhoux F, Wang XN, Bigley V, Abel M, Dimmick I, et al. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med. 2009;206(2):371–385. doi: 10.1084/jem.20081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, Leboeuf M, et al. Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med. 2011;208(5):1069–1082. doi: 10.1084/jem.20101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, Olver SD, et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest. 2014;124(10):4266–4280. doi: 10.1172/JCI75935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane A, Deenick EK, Ma CS, Cook MC, Uzel G, Tangye SG. STAT3 is a central regulator of lymphocyte differentiation and function. Curr Opin Immunol. 2014;28:49–57. doi: 10.1016/j.coi.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016;31:1–15. doi: 10.1016/j.cytogfr.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103(21):8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beadling C, Guschin D, Witthuhn BA, Ziemiecki A, Ihle JN, Kerr IM, et al. Activation of JAK kinases and STAT proteins by interleukin-2 and interferon alpha, but not the T cell antigen receptor, in human T lymphocytes. EMBO J. 1994;13(23):5605–5615. doi: 10.1002/j.1460-2075.1994.tb06898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2(4):331–339. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, et al. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181(5):1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Betts BC, Sagatys EM, Veerapathran A, Lloyd MC, Beato F, Lawrence HR, et al. CD4+ T cell STAT3 phosphorylation precedes acute GVHD, and subsequent Th17 tissue invasion correlates with GVHD severity and therapeutic response. J Leukoc Biol. 2015;97(4):807–819. doi: 10.1189/jlb.5A1114-532RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu SX, Alpdogan O, Lin J, Balderas R, Campos-Gonzalez R, Wang ×, et al. STAT-3 and ERK 1/2 phosphorylation are critical for T-cell alloactivation and graft-versus-host disease. Blood. 2008;112(13):5254–5258. doi: 10.1182/blood-2008-03-147322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radojcic V, Pletneva MA, Yen HR, Ivcevic S, Panoskaltsis-Mortari A, Gilliam AC, et al. STAT3 signaling in CD4+ T cells is critical for the pathogenesis of chronic sclerodermatous graft-versus-host disease in a murine model. J Immunol. 2010;184(2):764–774. doi: 10.4049/jimmunol.0903006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu F, Zhong XM, Qiao J, Liu Q, Sun HY, Chen W, et al. Cytotoxic T Lymphocyte Antigen-4 Down-Regulates T Helper 1 Cells by Increasing Expression of Signal Transducer and Activator of Transcription 3 in Acute Graft-versus-Host Disease. Biol Blood Marrow Transplant. 2016;22(2):212–219. doi: 10.1016/j.bbmt.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, et al. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity. 2012;37(2):209–222. doi: 10.1016/j.immuni.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood. 2016;127(17):2144–2154. doi: 10.1182/blood-2015-10-678706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Ma Y, Cole SM, Zander C, Chen KH, Karras J, et al. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood. 2003;102(1):344–352. doi: 10.1182/blood-2002-11-3396. [DOI] [PubMed] [Google Scholar]
- 31.Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119(13):e110–119. doi: 10.1182/blood-2011-09-381483. [DOI] [PubMed] [Google Scholar]
- 32.Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem. 1999;274(23):16513–16521. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10(1):39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 34.Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, et al. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19(3):425–436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- 35.Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344(6181):310–313. doi: 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- 36.Capitini CM, Nasholm NM, Chien CD, Larabee SM, Qin H, Song YK, et al. Absence of STAT1 in donor-derived plasmacytoid dendritic cells results in increased STAT3 and attenuates murine GVHD. Blood. 2014;124(12):1976–1986. doi: 10.1182/blood-2013-05-500876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101(11):3921–3926. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008;118(7):2562–2573. doi: 10.1172/JCI34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Y, Chin YE, Weisiger E, Malter C, Tawara I, Toubai T, et al. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. 2009;182(10):5899–5903. doi: 10.4049/jimmunol.0804388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Y, Iyer M, McEachin R, Zhao M, Wu YM, Cao X, et al. Genome-Wide STAT3 Binding Analysis after Histone Deacetylase Inhibition Reveals Novel Target Genes in Dendritic Cells. J Innate Immun. 2016 doi: 10.1159/000450681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonifer C, Bosch FX, Faust N, Schuhmann A, Sippel AE. Evolution of gene regulation as revealed by differential regulation of the chicken lysozyme transgene and the endogenous mouse lysozyme gene in mouse macrophages. Eur J Biochem. 1994;226(1):227–235. doi: 10.1111/j.1432-1033.1994.tb20045.x. [DOI] [PubMed] [Google Scholar]
- 42.Cross M, Mangelsdorf I, Wedel A, Renkawitz R. Mouse lysozyme M gene: isolation, characterization, and expression studies. Proc Natl Acad Sci U S A. 1988;85(17):6232–6236. doi: 10.1073/pnas.85.17.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90(8):3204–3213. [PubMed] [Google Scholar]
- 44.Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, et al. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest. 2001;107(12):1581–1589. doi: 10.1172/JCI12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. 2010;16(12):1434–1438. doi: 10.1038/nm.2242. [DOI] [PubMed] [Google Scholar]
- 46.Toubai T, Hou G, Mathewson N, Liu C, Wang Y, Oravecz-Wilson K, et al. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood. 2014;123(22):3512–3523. doi: 10.1182/blood-2013-12-545335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Das R, Chen X, Komorowski R, Hessner MJ, Drobyski WR. Interleukin-23 secretion by donor antigen-presenting cells is critical for organ-specific pathology in graft-versus-host disease. Blood. 2009;113(10):2352–2362. doi: 10.1182/blood-2008-08-175448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koyama M, Cheong M, Markey KA, Gartlan KH, Kuns RD, Locke KR, et al. Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. J Exp Med. 2015;212(8):1303–1321. doi: 10.1084/jem.20150329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Markey KA, Banovic T, Kuns RD, Olver SD, Don AL, Raffelt NC, et al. Conventional dendritic cells are the critical donor APC presenting alloantigen after experimental bone marrow transplantation. Blood. 2009;113(22):5644–5649. doi: 10.1182/blood-2008-12-191833. [DOI] [PubMed] [Google Scholar]
- 50.Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med. 2014;20(6):648–654. doi: 10.1038/nm.3517. [DOI] [PubMed] [Google Scholar]
- 51.Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19(6):903–912. doi: 10.1016/s1074-7613(03)00332-7. [DOI] [PubMed] [Google Scholar]
- 52.Choi SW, Braun T, Chang L, Ferrara JL, Pawarode A, Magenau JM, et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 2014;15(1):87–95. doi: 10.1016/S1470-2045(13)70512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi SW, Gatza E, Hou G, Sun Y, Whitfield J, Song Y, et al. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood. 2015;125(5):815–819. doi: 10.1182/blood-2014-10-605238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161(9):4652–4660. [PubMed] [Google Scholar]
- 55.Toubai T, Sun Y, Tawara I, Friedman A, Liu C, Evers R, et al. Ikaros-Notch axis in host hematopoietic cells regulates experimental graft-versus-host disease. Blood. 2011;118(1):192–204. doi: 10.1182/blood-2010-12-324616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J, Yuan X, Liu Y, Zhang K, Wang J, Zhang H, Liu F. Delayed administration of WP1066, an STAT3 inhibitor, ameliorates radiation-induced lung injury in mice. Lung. 2016;194(1):67–74. doi: 10.1007/s00408-015-9821-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Reduced STAT3 expression in host macrophages exacerbates acute GVHD in a minor antigens mismatched BMT model.
Host B6-WT and LysM/Cre STAT3fl/− animals were irradiated (11 Gy) on day −1 and transplanted with 5 × 106 BM cells along with 1×106 CD90.2+ T cells from either syngeneic B6 or allogeneic C3H.sw donors. Serum was collected from recipients on day 14 after allo-BMT and cytokine levels were determined by ELISA for IFN-γ (a) and IL-17A (b). (c) Representative hematoxylin and eosin (H&E) stained images of liver on day 14 after allo-BMT are shown. (d and e) Histopathological GVHD scores of liver (d) and gastrointestinal (GI) tract (e) on day 14 after allo-BMT are shown. (n=6 per group, pooled from two experiments). **p<0.01, *p<0.05. Error bars show the mean ± SEM.
Supplemental Figure 2. Reduced expression of STAT3 in dendritic cells doesn’t alter allogeneic T cell proliferative capacity in vitro.
(a)Bone marrow derived dendritic cells (BMDCs) from B6-WT or LysM/Cre STAT3fl/− animals were used as stimulators in an MLR by co-culturing with T cells from either B6 (syngeneic) or BALB/c (allogeneic) animals and analyzing T-cell proliferation via 3H-thymidine incorporation at 72 hours. (b–e) Supernatants from the above MLRs were collected at 72 hours and analyzed for IL-2 (b), IL-4 (c), IL-10 (d) and IFN-γ (d) by ELISA. Data are combined from three independent experiments. Error bars show the mean ± SEM.
Supplemental Figure 3. STAT3 deficient macrophages exhibit enhanced innate immune responses
(A-D) Peritoneal MFs were isolated as described in materials and methods. Cells were (1×106 cells/ml) plated and stimulated with LPS (1μg/ml) or diluent for 16 h at 37°C. Supernatants from cultured wells were harvested and examined for IL-1β (A), IL-6 (B), TNF-α (C), and IL-10 (D) by ELISA. Experiments were performed in triplicate. ****p<0.0001, *p<0.05. Error bars show the mean ± SEM.
Supplemental Figure 4. Reduced expression of STAT3 in dendritic cells doesn’t alter their LPS induced cytokine production.
BMDCs were isolated as described in materials and methods. DCs were (1×106 cells/mL) were plated and stimulated with LPS (1μg/ml) or diluent for 16 hours at 37°C. Supernatants were analyzed for IL-1β (a), IL-6 (b), TNF-α (c), and IL-10 (d) by ELISA. Experiments were performed in triplicate. Error bars show the mean ± SEM.