

Abstract
Since my last review on neuroimmune communication afferents in 2008, this area has witnessed substantial growth. At a basic science level, numerous new and exciting phenomena have been described, adding both depth and complexity to the crosstalk between the immune system and the nervous system. At a translational level, accumulating evidence indicates neuroimmune interaction could be a contributing factor for many disease states, as well as an effective physiological mechanism that coordinates the activities of these two systems in healthy individuals or during tissue distress. Furthermore, new evidence suggests neuroimmune interactions are inherently dynamic: varying activities in either the nervous system or the immune system could impact interactions between them. In this review I will attempt to integrate multifarious, and sometimes disparate, findings into a modified conceptual framework that describes the concordance of neuroimmune communication through the cooperative connection between these two systems and the dysfunction that may arise when their inappropriate crosstalk occurs.
Keywords: Cytokines, Neuroimmune communication, Neuroinflammation
1. Crossing the barrier: classical neuroimmune afferents
The founding fathers of the PNIRS, Robert Ader and Nicholas Cohen, laid the corner stones of neuroimmune communication by demonstrating both immuno-suppression and immuno-enhancement can be behaviorally conditioned in experimental animals (Cohen et al., 1994). Their findings showed prior pairings of a sensory event (i.e., taste; the conditioned stimulus) with immunomodulatory stimulations ultimately led to subsequent elicitation of the same immunomodulation by the conditioned stimulus alone. This discovery powerfully suggested that the immune system, like all the other physiological systems, is connected with and regulated by the central nervous system (CNS). Recent studies in this line of research have identified the insular cortex and amygdala as the key neural structures involved in behavioral conditioning of these types of immune modulations and demonstrated behavioral conditioning of immunosuppression can be achieved in humans (Hadamitzky et al., 2013). A corollary question that arises from such intimate connection between these two systems is: what are the afferent pathways by which immune system sends signals to the CNS?
For immunological afferents, most studies thus far have focused on the origin of signaling from innate immune activity because it is an initial response when the host first encounters infection or injury. Common to innate immune activation is the production of inflammatory cytokines including IL-1, IL-6, and TNFα. Direct injection of these cytokines into the brain causes fever, prolonged slow wave sleep, activation of the HPA axis, reduced food and water intake, and decreased locomotor activity, generally mimicking CNS-controlled sickness symptoms during immune challenge. Therefore, afferent pathways that relay immune signals to the brain were initially thought as those by which inflammatory cytokines from the periphery could be conveyed into the brain.
One of the first proposed afferent immune-to-brain pathways was circulating cytokines gained entrance to the CNS through circumventricular organs (CVOs) (Blatteis et al., 1987; Stitt, 1990). These regions possess leaky a blood brain barrier (BBB) and many of them are located near the areas of the brain that are known to react to peripheral immune challenges. For example, the organum vasculosum of the lamina terminalis is situated very close to the preoptic area of the hypothalamus, a region generally believed to control the febrile response. Another is the area postreama which is located atop the nucleus of the solitary tract, a major relay nucleus of the visceral afferents. A technical difficulty in examining the role of the CVOs is procedures used to specifically interrupt the functions of these small brain regions, such as electrolytic lesions, are likely to injure adjacent critical brain parenchyma, thereby producing experimental artifacts. In addition, direct injection of cytokines into the CVOs is likely to introduce cytokines beyond the CVOs into the nearby cerebral ventricles, rendering definitive conclusions about site specificity difficult to ascertain. More importantly, the concept that CVOs are brain sites which conduct passive signal transduction by being simply leaky is probably false. This is because CVOs are structured as small sensory organs of the brain. They are involved in the bi-directional exchange of metabolic information between blood and brain (Cottrell and Ferguson, 2004). Although these structures lack a vascular blood–brain barrier (BBB), they are contained by an ependymal CVO–CSF barrier formed by specialized tanycytes with intercellular tight junctions (Langlet et al., 2013). In this manner, the CVOs are separated from the main CNS proper. Circulating cytokines can indeed enter the CVOs. But further diffusion of cytokines to nearby parenchyma is restricted by the tanycyte limitan (Maness et al., 1998). In addition, the CVOs contain neurons that project to the nucleus of the solitary tract (Cai et al., 1996) and the paraventricular nucleus of the hypothalamus (Kawano and Masuko, 2010). These regions are well known targets of neuroimmune activation. Recent studies showed cytokines inside CVOs can directly act on CVO sensory neurons without the need to engage neurons beyond the CVO proper (Desson and Ferguson, 2003). Furthermore, after peripheral immune challenge, cytokine mRNA expression was found to be highly expressed within 30 min in the CVOs after the challenge (Quan et al., 1998). IL-1β-expressing immune cells within the CVOs have also been found to be in close association with the neuronal elements in the CVO after peripheral LPS challenge (Goehler et al., 2006). Therefore, cytokines produced in situ in the CVOs, rather than imported from circulation, are more likely to mediate neuroimmune communication. Interestingly, a recent study showed high levels of IL-1β and TNFα in circulation activate neurons in the subfornical organ, a CVO involved in sensing angiotensin II, to trigger increased blood pressure, heart rate, and renal sympathetic activity (Wei et al., 2013). This region also contains receptors that sense the acidity of the blood (McGuire et al., 2009) which may be involved in the initiation of panic responses. Therefore, actions of inflammatory cytokines in the CVOs could play a critical role in emergency responses of the host.
A less direct pathway was proposed by Banks and Erickson. By measuring active transport activity, it was demonstrated that many inflammatory cytokines can be transported into the brain across the BBB after animals were challenged with large doses of lipopolysaccharide (LPS), a component of the cell wall of gram negative bacteria (Banks and Erickson, 2010). The transport of circulating inflammatory cytokines can account, at least in part, for some peripheral immune activity-induced sickness behavior (Erickson et al., 2012). A recent development in this line of research was the finding that chronic peripheral LPS treatment can reduce the activity of LRP-1 (the low-density lipoprotein receptor-related protein-1), which functions at the BBB to excrete CNS inflammagens such as amyloid-β into the circulation (Erickson et al., 2012). Therefore, peripheral inflammation may indirectly increase neuroinflammation by contributing to the retention of CNS-generated inflammagens.
The discovery that neural, rather than humoral, pathways are able to transmit inflammatory signals to the brain were first reported by Wan et al. showing that brain c-fos induction after intraperitoneal administration of LPS can be blocked by subdiaphragmatic vagotomy (Wan et al., 1994). Independently, Watkins et al. showed peripheral LPS-induced hyperalgesia can be blocked by vagotomy (Watkins et al., 1994) and Bret-Dibat et al. showed peripheral LPS-induced depression in food motivated behavior was also blocked by vagotomy (Bret-Dibat et al., 1995). Currently, a large body of literature exists demonstrating subdiaphragmatic vagotomy can effectively reduce many sickness symptoms after peripheral LPS challenge. Vagally transmitted immune signals, however, can be masked by the effects induced by the above mentioned humoral pathways. In addition, the vagus nerve does not innervate areas other than visceral organs. Therefore the relevance of this pathway may be particularly important for infections localized to the visceral organs. Indeed, a cleaner demonstration of the vagal afferent was reported by Goehler et al. who showed administration of live bacteria in the gut activated vagal ganglia without causing increased circulating cytokines (Goehler et al., 2007).
Our lab took a different approach to examine the specific neural elements that might be activated by inflammatory cytokines. Many inflammatory cytokines signal through the NF-κB pathway. Activation of NF-κB causes the production of IκBα as an immediate early gene. Assuming CNS inflammatory cytokine activity is a crucial link in the immune-to-brain afferents, one should be able to trace brain IκBα expressing cells to map this link. Surprisingly, we found brain IκBα was induced after peripheral LPS challenge primarily in brain endothelial cells (Quan et al., 1997). This pattern of IκBα expression is similar to the expression of COX-2 which is known to be a downstream cytokine-induced mediator of sickness symptoms. We further demonstrated that blocking endothelial IL-1 receptors or endothelial COX-2 activity reduced peripheral immune challenge-induced neural activation and sickness behaviors (Ching et al., 2007; Quan et al., 2003). Further support for this pathway was provided by studies that showed endothelial production of PGE2, a terminal prostanoid catalyzed by COX-2 is crucial in mediating peripheral immune challenge-induced neural activation and sickness behaviors (Engstrom et al., 2012; Lazarus et al., 2007). In addition, Engblom et al. showed that immune stimulation induced fever required the expression of mPGES-1 in brain endothelial cells (Engblom et al., 2003). mPGES-1 is the final enzyme in the synthesis of PGE2. Thus, the BBB itself appears to be a critical immune-to-brain signal transducer. Interestingly, in this context, COX-2 is shown not to be the exclusive COX involved in the trans-BBB signaling during neuroimmune activation. For example, the early phase of corticosterone secretion induced by peripheral LPS was found to be mediated by the COX-1 dependent PGE2 production (Elander et al., 2009). Again, induced COX-1 expression was found in brain endothelial cells and perivascular monocytes (Garcia-Bueno et al., 2009). Thus, both COX-1 and COX-2 located near the BBB play important roles in transducing immune-to-brain signaling. Recent theoretical extensions of this pathway were added by the elegant work from Rummel et al. They showed IL-6 also plays an important role in mediating immune-to-brain signaling by activating the JAK/STAT3 system. Activated STAT3 was found in brain endothelium and other BBB cell types, and was colocalized with the COX2/mPGES expression (Rummel et al., 2008, 2011). Thus, multiple inflammatory cytokines can act on cells of the BBB to mediate immunological afferent signaling without the need for the presence of these cytokines within the brain.
2. The diverse sources, targets, and contrasting functions of inflammatory cytokines
A nagging question in the classical immune afferent pathway literature is: do cytokines acting on the brain side and those acting on the blood side of the BBB perform the same immune-to-brain signaling function? Induction of inflammatory cytokines inside the brain after peripheral immune challenge has been reported in many studies. The convenient and sensitive qRT-PCR method has allowed numerous studies to show tissue from various brain regions produce inflammatory cytokines in response to peripheral immune challenge. However, this method does not differentiate if these cytokines are originating from brain parenchymal cells, cells trapped in cerebral vasculature, or cells of the BBB. Similarly, detection of proinflammatory cytokine proteins by ELISA does not reveal the source of the cytokine producing cells. An important point here is that the source of inflammatory cytokine production matters and they may determine which side of the BBB cytokines can act on. The difference could engender completely different functional consequences. For example, intracerebroventricular (icv) injection of IL-1β, TNFα, or IFNγ causes leukocyte infiltration into the brain in an IL-1R1 dependent manner (Ching et al., 2005), whereas intravenous injection of IL-1β does not (Proescholdt et al., 2002). In addition, prior peripheral immune challenge even reduces CNS IL-1β induced leukocyte infiltration (Ching et al., 2006). Furthermore, BBB-produced cytokine such as IL-6 is preferentially secreted to the blood side of the BBB (Verma et al., 2006). Therefore, the presence of circulating cytokines, or even the presence of BBB cytokine-producing cells, does not automatically imply the presence of cytokines active on the brain side of the BBB. A definitive demonstration of the presence of cytokine producing cells within the brain parenchyma can be achieved by in situ hybridization histochemistry (ISHH). After septic doses of peripheral LPS, IL-1β (Quan et al., 1998) and TNFα (Nadeau and Rivest, 1999), but not IL-6 (Vallieres and Rivest, 1999) mRNA expression in microglial cells can be found in brain parenchyma 6–10 h after the LPS injection, whereas mRNAs for IL-1β, IL-6, and TNFα expression are induced in cells of the BBB at earlier time points. Consistent with this time course, Danzter et al. demonstrated that peripheral LPS-induced sickness behavior is typically manifested between 2 and 6 h after the LPS injection whereas LPS-induced depressive-like behavior, such as increased immobility in the forced swim test, are initiated 6 h or more after the LPS injection (Dantzer et al., 2008). It is possible that inflammatory cytokines in circulation or those produced by cells of the BBB act primarily through BBB-dependent pathways to induce sickness behavior, which can be an adaptive response to infection at the earlier time points. On the hand, the maladaptive depressive-like behavior is induced primarily by the action of cytokines produced and active on the brain side of the BBB after the LPS injection at later time points.
It is important to point out in this context that inflammatory cytokines inside the CNS also perform neurophysiological functions in the normal brain. An interesting demonstration of this point was the induction of cytokine expression by whisker stimulation. Stimulation of a specific whisker in rats induced TNFα and IL-1β immunoreactivity in the appropriate somatosensory cortical neuronal columns (Churchill et al., 2008; Hallett et al., 2010), but not in the irrelevant neighboring columns. It was proposed that these inflammatory cytokines are involved in synchronizing neuronal activity in the specifically activated cortical columns. Another example of brain cytokine induction unrelated to immunological status is the involvement of cytokines in long term potentiation (LTP), a critical cellular process in learning and memory. The induction of LTP was found to cause the production of IL-1β by hippo-campal cells, which play a role in LTP maintenance (del Rey et al., 2013). Thus, whereas cytokines acting on BBB cells may be more relevant in neuroimmune signaling, cytokines acting on neurons inside BBB may be more involved in mediating or disrupting the neurophysiological roles of the cytokines.
One explanation for the ability of cytokines to carry out such diverse functions in the CNS could be different cytokine activities are mediated by cytokine receptors expressed on different cell types in the brain. Initial cloning of the transcription start sites for the IL-1 receptor (IL-1R1) suggested more than one promoter control the expression of this receptor in the human IL-1R1 gene (Ye et al., 1996). Importantly, different IL-1R1 promoters have been associated with different disease susceptibility in human clinical studies (Bergholdt et al., 1995). Because cell type specific expression of a gene is often controlled by its promoters, the abovementioned evidence suggested that analysis of cytokine receptor promoters could be a path to decipher the cell type specific activities of cytokines. To this end, we thoroughly investigated the existence of multiple IL-1R1 promoters. Using the sensitive CapFishing method in combination with nested PCR, we ultimately found 4 murine and 7 human IL-1R1 promoters (Chen et al., 2009; Li et al., 2010). In neurons, IL-1R1 in the mouse is driven by two different promoters that are not active in the non-neuronal cell types. One expresses the full length IL-1R1 specifically in the dentate gyrus, the other drives neuronal expression of a truncated IL-1R1 which we termed IL-1R3 (Qian et al., 2012). IL-1R3 does not interact with the conventional co-receptor for IL-1, IL-1RAcP, to stimulate NF-κB activation which results in the induction of inflammatory gene expression. Instead, IL-1R3 interacts with neuronal specific IL-1RAcPb to mediate IL-1β-induced increase of potassium currents through fast activation of Akt kinase. It is likely that the neurophysiological functions of IL-1 are mediated by neuronal specific IL-1 receptors and their non-inflammatory signaling pathways. Furthermore, in vitro promoter–reporter analysis shows different IL-1R1 promoters are differentially regulated by glucocorticoids, suggesting responsiveness to IL-1 could be differentially regulated in different cell types by modulating hormones (Chen et al., 2009). The distinct cell type specific actions of IL-1 on various CNS cells can also be demonstrated in cultured CNS cell lines (An et al., 2011). Further substantiation of these cell type specific cytokine actions remains to be provided in an in vivo animal model in which the cytokine receptors can be expressed in specific cell types.
The above-mentioned examples suggest neurophysiological functions of inflammatory cytokines could to be mediated by very low physiological levels of CNS inflammatory cytokines produced in precise neuroanatomical locations. Therefore, they could be disrupted if centrally active cytokine levels exceed the physiological range. Indeed, Goshen et al. (2007) showed that small increases in brain IL-1β improved memory function whereas higher levels of IL-1β impaired it. Consequently, peripheral LPS induced microglial production of inflammatory cytokines inside the BBB could cause neuro- and psycho-pathologies by elevating centrally active cytokines to pathophysiological levels. In this context, high dose systemic LPS is not the only condition the untoward propagation of inflammatory elements from periphery to the brain occurs. For example, D’Mello and Swain showed in a chronic liver inflammation model, monocyte infiltration into the CNS ‘‘transported’’ inflammatory cytokine TNFα into the brain, which in turn triggered fatigue related sickness behavior (D’Mello and Swain, 2013). Furthermore, in a model of repeated social defeat, it was shown recently that defeated animals exhibit heightened anxiety-like behavior that was correlated to infiltration of peripheral inflammatory monocytes into the CNS (Wohleb et al., 2013). Besides monocytes, an interesting recent study reported that transmigration of IL-1β-expressing neutrophils into the brain contributed to peripheral LPS-induced depressive-like behavior 48 h after an intraperitoneal injection of 2.5 mg/kg LPS (Aguliar-Valles et al., 2013). At this time point sickness behavior induced by LPS, which occurred much earlier, was largely recovered. Previous studies showed injection of this dose of LPS typically induces high level IL-1β expression in cells associated with the BBB and in the CVOs between 0.5 and 8 h post injection, but much lower levels of IL-1β in cells within the brain parenchyma thereafter (Quan et al., 1998). Therefore, the depression-inducing effect caused by infiltrating neutrophils may only require small amounts of IL-1β that could act on cells types within the brain parenchyma to propagate late phase depressive-like behavior.
It is still unclear why under certain conditions peripheral inflammation causes neuroinflammation while under other circumstances it does not. Nevertheless, the concept that afferent immune-to-brain signaling causes neuroinflammation is now becoming the foundation for many theories that propose peripheral inflammation as an etiological factor for various psychopathologies. Thus far, this mechanism has been proposed to partially explain the pathogenesis of inflammation-induced major depression (Dantzer et al., 2011), PTSD (Pace and Heim, 2011), schizophrenia and autism (Gibney and Drexhage, 2013), and addiction (Crews et al., 2011). It should be noted, however: (1) correlations between these disease states and peripheral inflammatory cytokines levels are not always convincing, (2) signs of CNS inflammation can be found in the brain of some of the patients, but whether they are the cause or the consequence of the disease is not clear, (3) the correlative increases of inflammatory cytokines in the blood of these patients are usually very small, often magnitudes lower than those detected during systemic infection or sepsis, and (4) neither systemic infection, nor chronic liver inflammation, nor repeated social defeat can accurately recapitulate the pathogenic processes of these diseases. From a teleological standpoint, it seems unreasonable to assume that neuroinflammation should be an obligatory final step in all the neuroimmune afferent pathways, which appears to be a main culprit for the pathogenesis of many psychopathologies related to peripheral inflammation. Indeed, even though current research is largely biased towards investigating the peripheral immune activity-to-neuroinflammation axis in neuroimmune afferent signaling, this type of neuroimmune activation may only represent one end of a spectrum of neuroimmune afferent pathways. Here, a review by Pollmacher et al. (2002) on the levels of cytokines in various disease conditions provides several helpful points. First, many chronic disease conditions do not cause blood cytokine levels to exceed 5 times of the baseline levels, which are often very low (low pg/kg range). Second, acute infection and inflammation could raise blood cytokines to 10–100 times of the basal levels and therapeutic administration of cytokines even further, probably reaching supra-physiological levels. Third, low levels of blood cytokines could affect some of the same neural functions as those affected by the high levels of blood cytokines, but sometimes in opposite direction. Therefore, a thorough understanding the neuroimmune afferents must inspect the entire spectrum of neuroimmune activation, from the ‘‘physiological’’ to the supra-physiological, in order to determine which pathways are relevant to the particular condition under consideration.
3. The spectrum and kinetics of neuroimmune afferent pathways
There is no question that high levels of peripheral inflammagens including LPS (Buttini et al., 1996) and poly I:C (Zhang et al., 2005) can induce microglial activation and neuroinflammation in the CNS that is characterized by increased production of centrally active inflammatory cytokines, increased expression of microglial activation markers, and dramatically altered microglial morphology (Fenn et al., 2013). This process does not create overt neural damage and significant amount of infiltration of leukocytes into the brain of normal animals (Buttini et al., 1996); but it exacerbates neural and behavioral abnormalities in vulnerable conditions such as aging (Godbout et al., 2005) and prion induced chronic CNS degeneration (Cunningham et al., 2009). These sequelae are induced when LPS was administered in a dose range between 100 μg/kg and 10 mg/kg, a condition that causes systemic inflammation in the experimental rodents. However, lower subseptic LPS doses, below 50 μg/kg, only causes proinflammatory cytokine expression in the barrier sites (Quan et al., 1999) and restricted microglial activation in the CVOs (Kloss et al., 2001). Still, these LPS doses are capable of inducing the entire spectrum of sickness behavior including fever, sleep alteration, anorexia (Szentirmai and Krueger, 2013), and activation of the HPA axis (Wang and Dunn, 1999). Therefore, neuroimmune afferent signaling is operational without the induction of the potentially detrimental neuroinflammation, probably via BBB-dependent signaling. This is substantiated by the fact that intravenous injection of a high dose of IL-1β activated cells of the BBB and brain vascular COX-2 expression without inducing microglial and astrocyte activation in the brain parenchyma (Proescholdt et al., 2002).
One can further question whether the BBB-dependent immune-to-brain signaling is an obligatory step in all neuroimmune afferents. The reason this question should be raised is because even subseptic levels of LPS are not usually present in an infected host. Creasey et al. showed in a live Escherichia coliinfection model significant amount of LPS in the blood was only detected when lethal, but not sublethal, levels of E. coli were infused intravenously in baboons (Creasey et al., 1991). Therefore, infection or inflammation that occurs below the level of inducing significant amount of systemic LPS could utilize different neuroimmune afferents. We found in a model of localized inflammation induced by intraperitoneal or intra-airpouch injection of casein, long lasting (more than 7 h) sickness behaviors including fever and reduced locomotor activity can be induced without the induction of inflammatory cytokine and COX-2 mRNA expression in the brain (Zhang et al., 2008). The induction of the sickness behavior was correlated with locally produced inflammatory cytokines and locally produced COX-2. These results are consistent with the observation by Ross et al. who injected LPS into a subcutaneous chamber that was constructed to restrict the injected LPS to the implanted chamber. They found fever induced in this manner can be completely blocked by a locally administered COX inhibitor (Ross et al., 2003). These results suggest that a neuronal route, rather than circulating cytokines, directly projecting from the site of inflammation transmits inflammatory signal to the brain during localized inflammation. The relevant neural pathways must include somato-sensory nerves because vagal sensory pathways do not innervate body parts outside visceral organs. Such neural afferents that relay localized immune information to the brain also raise the issue whether such afferents code for the location of inflammation. We found local inflammation induced in the right leg primarily activated c-fos expression in the left PVN whereas local inflammation induced in the left leg primarily activated c-fos expression in the right PVN. Unilateral sciatic nerve transection abolished PVN c-fos induction if the inflammation was induced in the hindlimb with nerve transection, but had no effect if local inflammation was induced in the hindlimb with intact sciatic nerve. Furthermore, forelimb local inflammation induced c-fos expression in the anterior PVN, whereas hindlimb local inflammation induced c-fos expression in the posterior PVN. These results show localized inflammation activate neuroimmune afferents in a location-specific manner (Belevych et al., 2010). Whether these types of neuroimmune afferents could help coordinate two separate local immune activities in different parts of the body remains a fascinating possibility.
Thus far, neuroimmune afferents relaying peripheral immune signals have been investigated when there is overt neuroimmune activation and/or sickness symptoms. To push the envelope further, we investigated neuroimmune activation and sickness behaviors in animals that received intraperitoneal injections of sub-pyrogenic doses (1–10 μg/kg) of LPS. In this dose range sickness behavior, especially reduced locomotor activity in the open field, can be induced (Tarr et al., 2012; Teeling et al., 2007). We found repeated daily injection of sub-threshold levels of LPS (1 μg/kg) progressively induced both sickness behavior and PVN c-fos expression (Tarr et al., 2012). Therefore, chronic low grade inflammation induced by sub-threshold levels of LPS could precipitate observable sickness behavior over time. This could serve as a more appropriate model to investigate the inflammatory pathogenic theory of psychological disorders including major depression because most patients with these disorders are more likely to have had protracted insidious low grade peripheral inflammation rather than a major systemic infection.
Interestingly the chronic sub-threshold LPS effect is not replicated following live E. coli administration, a more natural immune stimulant. On the contrary, administration of sub-threshold levels of E. coli on a previous day significantly increased the threshold of the host to display sickness behavior upon E. coli challenge on the next day. In addition, PVN c-fos expression was not induced after repeated sub-threshold E. coli challenge on the following days (Chen et al., 2013). This phenomenon led us to define a peripheral inflammation that does not cause overt neuroimmune activation and sickness behavior, yet primes the immune system to provide more resistance to a subsequent inflammatory stimulation, as an ‘‘euflammation’’ and the highest level of inflammagen that causes euflammation at a given time point as an index of maximal euflammatory potential (MEP). Similar to resistance strength training, euflammation appears to be a dynamic phenomenon—prior euflammation may increase MEP when animals receive a subsequent inflammatory challenge, resulting in the induction of escalating MEP over time. We found after a 3-day stepwise escalating euflammatory challenge MEP has increased 50 times from the resting MEP. In addition, increased MEP in previously stimulated mice is associated with reduced IL-1 and IL-6 expression in the injection site (peritoneal cavity) upon subsequent E. coli challenge (Chen et al., 2013). Further investigation of the euflammatory animals revealed that myeloid cells at the site of euflammation induction locus (EIL) show reduced MHCII, TLR4, and CD86 expression but increased phagocytic activity, resembling the classical endotoxin tolerance phenotype. However, in myeloid cells from spleen and blood, the MHCII, TLR4, and CD86 expression levels were increased, suggesting that cells outside EIL are activated rather than deactivated, exhibiting the opposite phenotypes from the endotoxin tolerant cells. These results suggest under euflammatory conditions, gradual and controlled activation of the innate immune system may prevent subsequent induction of sickness behavior and activation of neuroimmune afferents. Indeed, euflammatory animals are resistant to inflammation-induced sickness behavior even if E. coli was administered outside EIL in a subcutaneous airpouch where no endotoxin tolerant myeloid cells were found (our unpublished observation), probably due to higher bactericidal activity of the activated immune cells outside EIL.
Here, three important points should be noted. First, neuroimmune activation patterns caused by infectious bacteria were very different from those induced by free LPS. One plausible explanation for this discrepancy is, besides LPS, bacteria such as E. coli harbor other immune stimulating molecules such as peptidoglycan and bacterial DNA, thereby could elicit immune reactions that may not be inducible by LPS alone. In addition, LPS molecules in E. coli do not exist in the free form; therefore they may be preferentially absorbed by phagocytic cells before they can interact with non-phagocytic cells as free LPS would. Indeed, it has been shown that encapsulation of LPS in liposomes, which are preferentially taken up by phagocytes, can reduce the fever-inducing potential of free LPS by 1000 fold and significantly reduce sickness behavior induced by subsequent free LPS challenge (Petrov et al., 1992). As a result, bacteria as an inflammagen could be much more euflammatory than free LPS. Second, sub-threshold (in the context of neuroimmune activation) infection or inflammation that is quickly resolved is a daily occurrence that often goes unnoticed. Healing of minor wounds, mild sinus inflammation, or mild dermatitis, for example, does not trigger sickness symptoms. These types of inflammation, however, could evoke important physiological host defense mechanisms to maintain homeostasis. Our results on euflammation show a protective inflammation can be induced to prevent sickness symptoms induced by subsequent immune challenges. Third, the euflammatory condition in a host is not maintained by endotoxin tolerance at the site inflammation alone. There are systemic innate immune activations that are inducible by the sub-threshold immune challenge. By defining euflammation as an inflammation that does not result in overt sickness behaviors, the maximal immunomodulatory effect induced by a sub-threshold immune challenge can now be determined under a concrete criterion.
4. Summary and concluding remarks
Fig. 1 summarizes all the neuroimmune afferents discussed in this review. In a physiological homeostatic state, both neural and humoral afferents are in a resting state. This is maintained if localized inflammation can control sub-threshold levels of infection (euflammation). This resting state is not static; rather, animals that received prior euflammatory stimulation could better maintain a quiescent neuroimmune afferent state upon a subsequent inflammatory challenge (scenario 1). However, when local inflammagens exceed a certain threshold, breaching the boundary of homeostatic control, significant production of local inflammatory cytokines ensues. Locally produced inflammatory mediators including cytokines and prostaglandins together with the un-eliminated excess inflammagens activate relevant sensory neurons to relay immune signals to the CNS (scenario 2). In this scenario, the location of immune activity is transmitted to the CNS. Further increases in the intensity of inflammation can cause significant spreading of inflammatory cytokines and bacterial toxins into circulation, resulting in systemic inflammation. Under such conditions, both neural and BBB-dependent neuroimmune afferents can be activated; but this activation may not cause microglial mediated neuroinflammation if the inflammation is below certain level (scenario 3). Finally, strong systemic immune challenge, chronic liver inflammation, sepsis, and repeated social defeat are some of the severe conditions under which not only all neuroimmune afferents are activated, but induction of microglia-mediated neuroinflammation is also induced (scenario 4). In this scenario, high levels of inflammatory cytokines are active on the brain side of the BBB-both acute and chronic disruption of the CNS function might ensue. In addition, infiltration of cytokine expressing monocytes and/or neutrophils into the brain may occur, contributing to the induction of neuroinflammation and consequent behavioral dysfunctions.
Fig. 1.
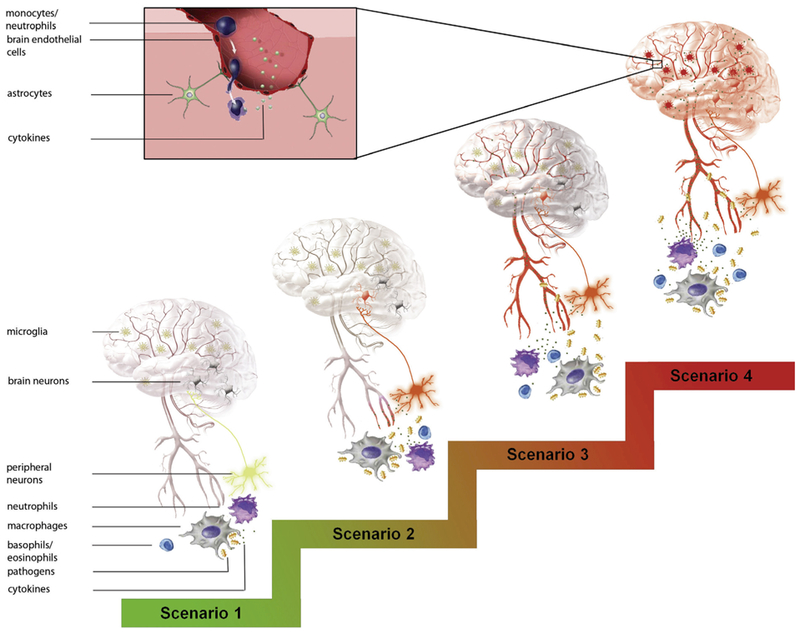
Summary of a spectrum of neuroimmune afferents activated under different conditions. In scenario 1, neuroimmune afferents are kept at quiescent states when sub-threshold levels of inflammagens are controlled by local immune activity. In scenario 2, local excess inflammagens cause the release of significant amounts of local inflammatory mediators to drive the immune sensory afferents which stimulate appropriate neurons in the brain corresponding to the local inflammation. In scenario 3, excess inflammagens and inflammatory mediators enter the circulation. The systemic inflammation signals are transmitted to the brain via humoral routes and their BBB-dependent mechanisms as well as the neural route. Humoral immune-to-brain signaling could be the more dominate afferent pathway in this scenario. In scenario 4, systemic inflammation, chronic liver inflammation, or repeated social defeat stimulates microglia-mediated neuroinflammation in addition to all the neuroimmune afferents. Inset in this scenario show monocyte/neutrophil infiltration contributes to neuroimmune activation. Activated blood vessels, sensory neurons, and microglia are indicated by the red color. Inflammagens are indicated by the yellow symbols. Inflammatory mediators are indicated by the small dots. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The spectrum of activation of distinct neuroimmune afferents may reflect different needs for the recruitment of different CNS responses when the host is faced with different degrees of infectious challenge. Fully controlled sub-threshold infection in scenario 1 may require activation of innate immunity without the induction of fever, reduced locomotor activity, and HPA activation. Some of these centrally mediated responses to infection represent a cost to the host (fever costs additional energy, reduced locomotor activity could mean reduced gathering of food), others reduced the effectiveness of the immune response itself (HPA activation causes the release of glucocorticoids which are immunosuppressive). These responses are avoided in homeostatic maintenance of the neuroimmune quiescence. In scenario 2, localized inflammation activates the CNS primarily via the neural route, allowing location specific feedback regulation by the CNS. Much remains to be understood in this scenario. It is known that neurotransmitters such as norepinephrine and acetylcholine released at the site of inflammation can modulate local immune activity (Matteoli and Boeckxstaens, 2013; Straub et al., 2005). But the physiological functions of these modulations are not entirely clear. In scenario 3, systemic inflammation becomes the dominant concern of the host, induction of CNS mediated immunosuppressive regulations by the HPA axis or the cholinergic anti-inflammatory reflex could be critical to prevent hyper-inflammation induced tissue damage (Rosas-Ballina and Tracey, 2009). The physiological function of neuroinflammation induced in scenario 4 may be related to the protection of the CNS itself. Monocyte infiltration into the brain is known to be important in CNS repair after injury (London et al., 2013). After repeated high dose peripheral LPS injection, activated microglia cells have been found to physically ensheath projection neurons to provide neuroprotection (Chen et al., 2012). In addition, Qin et al. found a single intraperitoneal injection of 5 mg/kg of LPS caused neuronal loss in substantial nigra and elevated brain expression of TNFα 10 months after the injection (Qin et al., 2007). Therefore, neuroinflammation in scenario 4 could be a double edged sword: it may provide certain neuroprotective mechanisms to limit further CNS damage but confer long term consequences related to chronic neurodegeneration. It is also important to point out induction of neuroinflammation by peripheral inflammation may depend upon factors other than inflammation per se. For example, in the repeated social defeat model, neuroinflammation also depended upon the level of stress animals received (Wohleb et al., 2013).
The awareness of a full spectrum of the neuroimmune afferents is important because it will serve as a better guide for investigating how neuroimmune afferent signaling might contribute to psychological disorders. Although the most extensively studied is scenario 4, it may not represent the appropriate conditions under which human psychological disorders such as major depression, PTSD, and autism are initiated. On the other hand, homeostatic control of sub-threshold infection (scenario 1) is an understudied area. The euflammatory mechanism we discovered suggest it is possible to amplify the homeostatic control of the innate immunity by repeated challenges with escalating sub-threshold inflammagens. Whether this mechanism can be translated into clinical prevention or treatment of the more damaging inflammatory responses remains to be explored. Finally, the different scenarios depicted in this review are kinetically connected. Induction of scenario 4 could result in a shifted set point for neuroimmune homeostasis such that the host may have altered sensitivity to sub-threshold immune challenges in the future.
Acknowledgment
This study is supported by NIH funding RO1 AI076926 to NQ. I would like to thank Xioayu Liu for making the illustration in the paper and Dr. Andrew Tarr for his careful reading of the manuscript and many helpful suggestions.
References
- Aguliar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN, 2013. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol. psychiatry. 10.1038/mp.2013.37. Advance online publication. [DOI] [PubMed]
- An Y, Chen Q, Quan N, 2011. Interleukin-1 exerts distinct actions on different cell types of the brain in vitro. J. Inflamm. Res. 2011, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Erickson MA, 2010. The blood–brain barrier and immune function and dysfunction. Neurobiol. Dis. 37, 26–32. [DOI] [PubMed] [Google Scholar]
- Belevych N, Buchanan K, Chen Q, Bailey M, Quan N, 2010. Location-specific activation of the paraventricular nucleus of the hypothalamus by localized inflammation. Brain Behav. Immun. 24, 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergholdt R, Karlsen AE, Johannesen J, Hansen PM, Dinarello CA, Nerup J, Pociot F, 1995. Characterization of polymorphisms of an interleukin 1 receptor type 1 gene (IL1RI) promotor region (P2) and their relation to insulin-dependent diabetes mellitus (IDDM). the Danish Study Group of Diabetes in Childhood. Cytokine 7, 727–733. [DOI] [PubMed] [Google Scholar]
- Blatteis CM, Hales JR, McKinley MJ, Fawcett AA, 1987. Role of the anteroventral third ventricle region in fever in sheep. Can. J. Physiol. Pharmacol. 65, 1255–1260. [DOI] [PubMed] [Google Scholar]
- Bret-Dibat JL, Bluthe RM, Kent S, Kelley KW, Dantzer R, 1995. Lipopolysaccharide and interleukin-1 depress food-motivated behavior in mice by a vagal-mediated mechanism. Brain Behav. Immun. 9, 242–246. [DOI] [PubMed] [Google Scholar]
- Buttini M, Limonta S, Boddeke HW, 1996. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem. Int. 29, 25–35. [DOI] [PubMed] [Google Scholar]
- Cai Y, Hay M, Bishop VS, 1996. Synaptic connections and interactions between area postrema and nucleus tractus solitarius. Brain Res. 724, 121–124. [DOI] [PubMed] [Google Scholar]
- Chen Q, Tarr AJ, Liu X, Wang Y, Reed NS, Demarsh CP, Sheridan JF, Quan N, 2013. Controlled progressive innate immune stimulation regimen prevents the induction of sickness behavior in the open field test. J. Inflamm. Res. 6, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang H, Li Q, An Y, Herkenham M, Lai W, Popovich P, Agarwal S, Quan N, 2009. Three promoters regulate tissue- and cell type-specific expression of murine interleukin-1 receptor type I. J. Biol. Chem. 284, 8703–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD, 2012. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J. Neurosci. 32, 11706–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching S, He L, Lai W, Quan N, 2005. IL-1 type I receptor plays a key role in mediating the recruitment of leukocytes into the central nervous system. Brain Behav. Immun. 19, 127–137. [DOI] [PubMed] [Google Scholar]
- Ching S, Zhang H, Belevych N, He L, Lai W, Pu XA, Jaeger LB, Chen Q, Quan N, 2007. Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J. Neurosci. 27, 10476–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching S, Zhang H, Lai W, Quan N, 2006. Peripheral injection of lipopolysaccharide prevents brain recruitment of leukocytes induced by central injection of interleukin-1. Neuroscience 137, 717–726. [DOI] [PubMed] [Google Scholar]
- Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, Krueger JM, 2008. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience 156, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N, Moynihan JA, Ader R, 1994. Pavlovian conditioning of the immune system. Int. Arch. Allergy Immunol. 105, 101–106. [DOI] [PubMed] [Google Scholar]
- Cottrell GT, Ferguson AV, 2004. Sensory circumventricular organs: central roles in integrated autonomic regulation. Regul. Pept. 117, 11–23. [DOI] [PubMed] [Google Scholar]
- Creasey AA, Stevens P, Kenney J, Allison AC, Warren K, Catlett R, Hinshaw L, Taylor FB Jr., 1991. Endotoxin and cytokine profile in plasma of baboons challenged with lethal and sublethal Escherichia coli. Circ. Shock 33, 84–91. [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L, 2011. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav. Immun. 25 (Suppl. 1), S4–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH, 2009. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 65, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello C, Swain MG, 2013. Liver–brain interactions in inflammatory liver diseases: implications for fatigue and mood disorders. Brain Behav. Immun. 35, 9–20. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW, 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Lawson MA, Kelley KW, 2011. Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology 36, 426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO, 2013. A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav. Immun. 33, 15–23. [DOI] [PubMed] [Google Scholar]
- Desson SE, Ferguson AV, 2003. Interleukin 1beta modulates rat subfornical organ neurons as a result of activation of a non-selective cationic conductance. J. Physiol. 550, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elander L, Engstrom L, Ruud J, Mackerlova L, Jakobsson PJ, Engblom D, Nilsberth C, Blomqvist A, 2009. Inducible prostaglandin E2 synthesis interacts in a temporally supplementary sequence with constitutive prostaglandin-synthesizing enzymes in creating the hypothalamic–pituitary–adrenal axis response to immune challenge. J. Neurosci. 29, 1404–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Saha S, Engstrom L, Westman M, Audoly LP, Jakobsson PJ, Blomqvist A, 2003. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat. Neurosci. 6, 1137–1138. [DOI] [PubMed] [Google Scholar]
- Engstrom L, Ruud J, Eskilsson A, Larsson A, Mackerlova L, Kugelberg U, Qian H, Vasilache AM, Larsson P, Engblom D, Sigvardsson M, Jonsson JI, Blomqvist A, 2012. Lipopolysaccharide-induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology 153, 4849–4861. [DOI] [PubMed] [Google Scholar]
- Erickson MA, Dohi K, Banks WA, 2012. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood–brain barrier. Neuroimmunomodulation 19, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP, 2013. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol. Psychiatry. 10.1016/j.biopsych.2013.10.014. Advance online publication. [DOI] [PMC free article] [PubMed]
- Garcia-Bueno B, Serrats J, Sawchenko PE, 2009. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic–pituitary–adrenal axis activation by inflammatory stimuli. J. Neurosci. 29, 12970–12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney SM, Drexhage HA, 2013. Evidence for a dysregulated immune system in the etiology of psychiatric disorders. J. Neuroimmune Pharmacol. 8, 900–920. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW, 2005. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 19, 1329–1331. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Erisir A, Gaykema RP, 2006. Neural-immune interface in the rat area postrema. Neuroscience 140, 1415–1434. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Lyte M, Gaykema RP, 2007. Infection-induced viscerosensory signals from the gut enhance anxiety: implications for psychoneuroimmunology. Brain Behav. Immun. 21, 721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, Levy-Lahad E, Yirmiya R, 2007. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 32, 1106–1115. [DOI] [PubMed] [Google Scholar]
- Hadamitzky M, Engler H, Schedlowski M, 2013. Learned immunosuppression: extinction, renewal, and the challenge of reconsolidation. J. Neuroimmune Pharmacol. 8, 180–188. [DOI] [PubMed] [Google Scholar]
- Hallett H, Churchill L, Taishi P, De A, Krueger JM, 2010. Whisker stimulation increases expression of nerve growth factor- and interleukin-1beta-immunoreactivity in the rat somatosensory cortex. Brain Res. 1333, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano H, Masuko S, 2010. Region-specific projections from the subfornical organ to the paraventricular hypothalamic nucleus in the rat. Neuroscience 169, 1227–1234. [DOI] [PubMed] [Google Scholar]
- Kloss CU, Bohatschek M, Kreutzberg GW, Raivich G, 2001. Effect of lipopolysaccharide on the morphology and integrin immunoreactivity of ramified microglia in the mouse brain and in cell culture. Exp. Neurol. 168, 32–46. [DOI] [PubMed] [Google Scholar]
- Langlet F, Mullier A, Bouret SG, Prevot V, Dehouck B, 2013. Tanycyte-like cells form a blood–cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. J. Comp. Neurol. 521, 3389–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, Saper CB, 2007. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat. Neurosci. 10, 1131–1133. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhang H, Chen Q, Quan N, 2010. Existence of seven human IL-1R1 promoters. J. Inflamm. Res. 2010, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London A, Cohen M, Schwartz M, 2013. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front. Cell. Neurosci. 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness LM, Kastin AJ, Banks WA, 1998. Relative contributions of a CVO and the microvascular bed to delivery of blood-borne IL-1alpha to the brain. Am. J. Physiol. 275, E207–E212. [DOI] [PubMed] [Google Scholar]
- Matteoli G, Boeckxstaens GE, 2013. The vagal innervation of the gut and immune homeostasis. Gut 62, 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire J, Herman JP, Ghosal S, Eaton K, Sallee FR, Sah R, 2009. Acid-sensing by the T cell death-associated gene 8 (TDAG8) receptor cloned from rat brain. Biochem. Biophys. Res. Commun. 386, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Rivest S, 1999. Regulation of the gene encoding tumor necrosis factor alpha (TNF-alpha) in the rat brain and pituitary in response in different models of systemic immune challenge. J. Neuropathol. Exp. Neurol. 58, 61–77. [DOI] [PubMed] [Google Scholar]
- Pace TW, Heim CM, 2011. A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. Brain Behav. Immun. 25, 6–13. [DOI] [PubMed] [Google Scholar]
- Petrov AB, Semenov BF, Vartanyan YP, Zakirov MM, Torchilin VP, Trubetskoy VS, Koshkina NV, L’Vov VL, Verner IK, Lopyrev IV, et al. , 1992. Toxicity and immunogenicity of Neisseria meningitidis lipopolysaccharide incorporated into liposomes. Infect. Immun. 60, 3897–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollmacher T, Haack M, Schuld A, Reichenberg A, Yirmiya R, 2002. Low levels of circulating inflammatory cytokines – do they affect human brain functions? Brain Behav. Immun. 16, 525–532. [DOI] [PubMed] [Google Scholar]
- Proescholdt MG, Chakravarty S, Foster JA, Foti SB, Briley EM, Herkenham M, 2002. Intracerebroventricular but not intravenous interleukin-1beta induces widespread vascular-mediated leukocyte infiltration and immune signal mRNA expression followed by brain-wide glial activation. Neuroscience 112, 731–749. [DOI] [PubMed] [Google Scholar]
- Qian J, Zhu L, Li Q, Belevych N, Chen Q, Zhao F, Herness S, Quan N, 2012. Interleukin-1R3 mediates interleukin-1-induced potassium current increase through fast activation of Akt kinase. Proc. Natl. Acad. Sci. USA 109, 12189–12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT, 2007. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, He L, Lai W, 2003. Endothelial activation is an intermediate step for peripheral lipopolysaccharide induced activation of paraventricular nucleus. Brain Res. Bull. 59, 447–452. [DOI] [PubMed] [Google Scholar]
- Quan N, Stern EL, Whiteside MB, Herkenham M, 1999. Induction of proinflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J. Neuroimmunol. 93, 72–80. [DOI] [PubMed] [Google Scholar]
- Quan N, Whiteside M, Herkenham M, 1998. Time course and localization patterns of interleukin-1beta messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience 83, 281–293. [DOI] [PubMed] [Google Scholar]
- Quan N, Whiteside M, Kim L, Herkenham M, 1997. Induction of inhibitory factor kappaBalpha mRNA in the central nervous system after peripheral lipopolysaccharide administration: an in situ hybridization histochemistry study in the rat. Proc. Natl. Acad. Sci. USA 94, 10985–10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Tracey KJ, 2009. Cholinergic control of inflammation. J. Intern. Med. 265, 663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross G, Hubschle T, Pehl U, Braun HA, Voigt K, Gerstberger R, Roth J, 2003. Fever induction by localized subcutaneous inflammation in guinea pigs: the role of cytokines and prostaglandins. J. Appl. Physiol. 94, 1395–1402. [DOI] [PubMed] [Google Scholar]
- Rummel C, Inoue W, Sachot C, Poole S, Hubschle T, Luheshi GN, 2008. Selective contribution of interleukin-6 and leptin to brain inflammatory signals induced by systemic LPS injection in mice. J. Comp. Neurol. 511, 373–395. [DOI] [PubMed] [Google Scholar]
- Rummel C, Matsumura K, Luheshi GN, 2011. Circulating IL-6 contributes to peripheral LPS-induced mPGES-1 expression in the rat brain. Brain Res. Bull. 86, 319–325. [DOI] [PubMed] [Google Scholar]
- Stitt JT, 1990. Passage of immunomodulators across the blood–brain barrier. Yale J. Biol. Med. 63, 121–131. [PMC free article] [PubMed] [Google Scholar]
- Straub RH, Stebner K, Harle P, Kees F, Falk W, Scholmerich J, 2005. Key role of the sympathetic microenvironment for the interplay of tumour necrosis factor and interleukin 6 in normal but not in inflamed mouse colon mucosa. Gut 54, 1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentirmai E, Krueger JM, 2013. Sickness behaviour after lipopolysaccharide treatment in ghrelin deficient mice. Brain Behav. Immun. 36, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr AJ, Chen Q, Wang Y, Sheridan JF, Quan N, 2012. Neural and behavioral responses to low-grade inflammation. Behav. Brain Res. 235, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling JL, Felton LM, Deacon RM, Cunningham C, Rawlins JN, Perry VH, 2007. Sub-pyrogenic systemic inflammation impacts on brain and behavior, independent of cytokines. Brain Behav. Immun. 21, 836–850. [DOI] [PubMed] [Google Scholar]
- Vallieres L, Rivest S, 1999. Interleukin-6 is a needed proinflammatory cytokine in the prolonged neural activity and transcriptional activation of corticotropin-releasing factor during endotoxemia. Endocrinology 140, 3890–3903. [DOI] [PubMed] [Google Scholar]
- Verma S, Nakaoke R, Dohgu S, Banks WA, 2006. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav. Immun. 20, 449–455. [DOI] [PubMed] [Google Scholar]
- Wan W, Wetmore L, Sorensen CM, Greenberg AH, Nance DM, 1994. Neural and biochemical mediators of endotoxin and stress-induced c-fos expression in the rat brain. Brain Res. Bull. 34, 7–14. [DOI] [PubMed] [Google Scholar]
- Wang J, Dunn AJ, 1999. The role of interleukin-6 in the activation of the hypothalamo–pituitary–adrenocortical axis and brain indoleamines by endotoxin and interleukin-1 beta. Brain Res. 815, 337–348. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, Smith KP, Maier SF, 1994. Neurocircuitry of illness-induced hyperalgesia. Brain Res. 639, 283–299. [DOI] [PubMed] [Google Scholar]
- Wei SG, Zhang ZH, Beltz TG, Yu Y, Johnson AK, Felder RB, 2013. Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF, 2013. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 33, 13820–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Vannier E, Clark BD, Sims JE, Dinarello CA, 1996. Three distinct promoters direct transcription of different 5′ untranslated regions of the human interleukin 1 type I receptor: a possible mechanism for control of translation. Cytokine 8, 421–429. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ching S, Chen Q, Li Q, An Y, Quan N, 2008. Localized inflammation in peripheral tissue signals the CNS for sickness response in the absence of interleukin-1 and cyclooxygenase-2 in the blood and brain. Neuroscience 157, 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Trautmann K, Schluesener HJ, 2005. Microglia activation in rat spinal cord by systemic injection of TLR3 and TLR7/8 agonists. J. Neuroimmunol. 164, 154–160. [DOI] [PubMed] [Google Scholar]