

Summary
Extracellular proTGF-β is covalently linked to “milieu” molecules in the matrix or on cell surfaces and is latent until TGF-β is released by integrins. Here we show that LRRC33 on the surface of microglia functions as a milieu molecule and enables highly localized, integrin αVβ8-dependent TGF-β activation. Lrrc33−/− mice lack CNS vascular abnormalities associated with deficiency in TGF-β activating integrins, but have microglia with a reactive phenotype and after 2 months develop ascending paraparesis with loss of myelinated axons and death by 5 months. Whole bone marrow transplantation results in selective repopulation of Lrrc33−/− brains with WT microglia and halts disease progression. The phenotypes of WT and Lrrc33−/− microglia in the same brain suggest that there is little spreading of TGF-β activated from one microglial cell to neighboring microglia. Our results suggest that interactions between integrin-bearing cells and cells bearing milieu molecule-associated TGF-β provide localized and selective activation of TGF-β.
Keywords: LRRC33, TGF-β, microglia, milieu molecules, Integrins
Introduction
The three TGF-βs present in mammals have important roles throughout the body in development and homeostasis, including in the CNS. TGF-β1 organizes responses to neurodegeneration and is constitutively expressed in microglia into adulthood (Kiefer et al., 1995). Tgfb1−/− mice die from uncontrolled lymphocyte proliferation and autoimmunity (Robertson and Rifkin, 2016). Longer-lived II2-Tgfb1; Tgfb1−/− mice develop neurological defects and marked microglia alterations (Butovsky et al., 2014).
The TGF-β prodomain (~250 amino acid residues) and growth factor domain (GF, ~110 amino acid residues) are derived from a single gene. At the earliest stage of proTGFβ glycoprotein biosynthesis, in the endoplasmic reticulum (ER), “milieu molecules” non-covalently associate with and disulfide link to the TGF-β prodomain. In the Golgi, a pro-protein convertase cleaves between the TGF-β prodomain and GF domains; however, after secretion the GF maintains tight, noncovalent association with the prodomain and is latent. Previously identified milieu molecules are latent TGF-β binding proteins (LTBPs) and glycoprotein-A repetitions predominant protein (GARP, leucine-rich repeat-containing protein 32, LRRC32). Latent TGF-β complexes with LTBPs 1, 3, and 4 are stored in the extracellular matrix (ECM). Complexes with GARP are stored on the surface of endothelium, platelets, and induced T regulatory cells (Robertson and Rifkin, 2016). Two integrins, αVβ6 and αVβ8, bind with high affinity to an RGD motif in the prodomains of TGF-βs 1 and 3. Binding is not sufficient for activation; application of force by the cytoskeleton to the integrin that is resisted by a milieu molecule linked to the prodomain appears to be required to distort the prodomain, release the GF from the prodomain, and hence activate TGF-β. Thus stimulation of cells by TGF-β may be localized to the site of TGF-β activation. The importance of integrins in activation of TGF-β1 is emphasized by mutation of the prodomain RGD sequence to RGE, which phenocopies TGF-β1-deficiency (Robertson and Rifkin, 2016)
Localized release of TGF-β may explain why it can mediate such a wide range of functions in vertebrates, many of which appear contradictory to one another. Here, we show that molecules that covalently associate with proTGFβ provide unique milieus for activation such that TGF-β can have highly localized effects within tissues. We identify LRRC33 as a milieu molecule that is uniquely associated with, and required for, integrin-dependent activation of TGF-β1 in macrophages and their relatives in the nervous system, microglia. Multiple approaches define highly specific association between LRRC33 and proTGF-β1 Although no previous studies on Lrrc33−/− (Nrros−/−) mice have suggested a role in TGF-β1 activation, reported phenotypes (Noubade et al., 2014; Su et al., 2014) are downstream of TGF-β1 dysregulation. Lrrc33−/− mice appear normal up to 2 months of age, but then develop ascending paraparesis that is characterized by demyelination, loss of axons, loss of neurons in the somatomotor cortex and spinal cord, and death by 5 months. Transplantation with WT whole bone marrow arrests disease progression and WT microglia selectively repopulate the CNS. Transcriptional profiling of Lrrc33−/− microglia demonstrates defective TGF-β1 signaling and concordance with the profile from Il2-Tgfb1; Tgfb1−/− mice (Butovsky et al., 2014). Lrrc−/− mice have the neurological but not the vascular defects found in mice deficient in TGF-β1 and integrin αVβ8 and suggest that neural-glial and vascular milieus for TGF-β1 activation are separate in the CNS.
Results
Homology and tissue expression of LRRC33 and GARP.
LRRC33 is highly homologous with GARP (34% amino acid sequence identity) and only distantly related to other LRR super-family proteins (Fig. 1A, B). Identity between LRRC33 and GARP is comparable to that seen with proteins that associate with related ligands, such as toll-like receptors TLR7 and TLR8 (Fig 1B). LRRC33 and GARP each contain a signal sequence, an ectodomain containing 23 LRRs, a transmembrane domain, and a cytoplasmic domain (Fig. 1A). Cysteine residues 192 and 331 in GARP disulfide link to a Cys in each proTGF-β monomer to form a 1:2 complex (Wang et al., 2012). Fascinatingly, these Cys residues are conserved in LRRC33 (Fig. 1A, asterisks), and in none of the other LRR family proteins shown in Fig. 1B.
Figure 1. LRRC33 homology to GARP and tissue-specific expression.
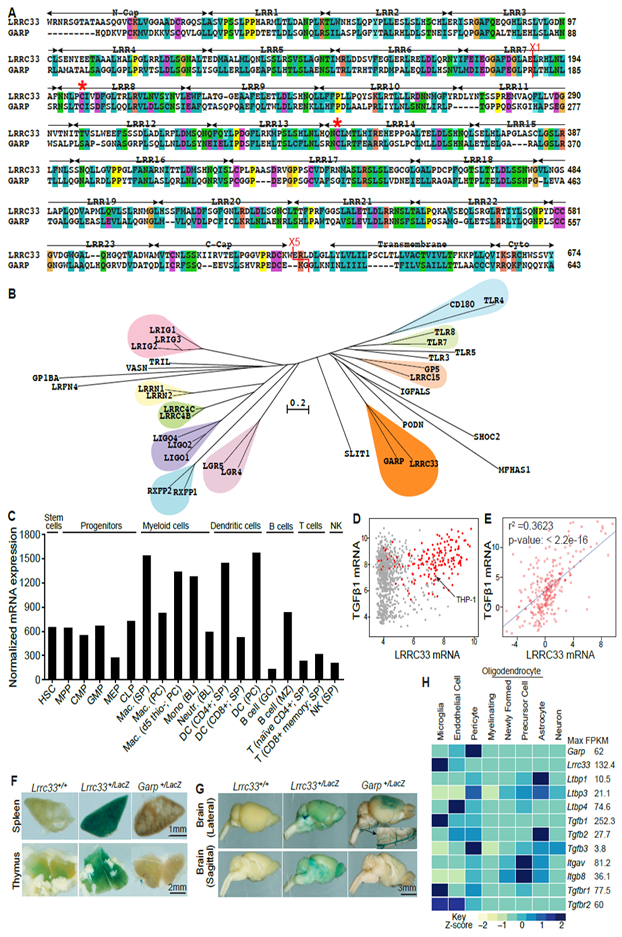
(A) Sequence alignment. Red asterisks (*) mark cysteines that disulfide link to proTGF-β1 (Wang et al., 2012). X1 and X5 mark GARP/LRRC33 chimaera exchange positions. (B) Phylogram of closest LRR-superfamily relatives of LRRC33. Trees were calculated with the NJ method on ectodomains aligned with MAFFT (G-INS-i, gap insertion and extension penalties of 3 and 1, respectively). (C) LRRC33 mRNA expression in murine hematopoietic cells from the ImmGen microarray database. (D) LRRC33 and TGF-β1 mRNA expression in human cancer cell lines in the Cancer Cell Line Encyclopedia; red dots: haematopoietic cell lines. (E) LRRC33 and TGF-β1 mRNA levels positively correlate in normal human tissue, datasets from BioGPS. (F, G) X-gal staining showing LacZ expression in 4-month-old WT, Lrrc33+/− and Garp+/− heterozygous mice. (H) Mouse brain RNAseq data (Zhang, 2014); relative gene expression is shown among 8 cell types isolated from the CNS with FPKM (Fragments Per Kilobase of transcript per Million mapped reads) value shown for the highest expressing cell type.
Lrrc33 expression is largely limited to cells of hematopoietic origin. Among normal and tumor cell lines, Lrrc33 expression is highest in myeloid lineage cells including macrophages and dendritic cells, is also high in B cells, and is generally low in T cells and NK cells (Fig. 1C, D). Among normal human tissues, LRRC33 and TGF-β1 mRNA expression correlates (Fig. 1E). X-gal staining of organs from heterozygotes with a LacZ reporter showed that Lrrc33 was expressed strongly in spleen and at lower levels in thymus (Fig. 1F). In contrast, little Lrrc33 was expressed in liver, kidney, heart, lung, and skin. In the brain, Lrrc33 was widely and diffusely expressed (Fig. 1G). In contrast, Garp was localized within the frontal cerebral cortex (Fig. 1G). RNAseq data on 8 cell populations of validated purity from the brain (Zhang, 2014) showed that Lrrc33 is highly expressed in microglia but less in other CNS cell types, in resemblance to TGF-β1 (Fig. 1H and Supplemental Table 1). In contrast, Garp is highest on pericytes and endothelial cells (Fig. 1H), in agreement with its presence in blood vessels (Fig. 1G, inset).
ProTGF-β1 associates with LRRC33 on the cell surface
Immunoprecipitation (IP) and Western blotting (WB) showed highly specific association between LRRC33 and proTGF-β1. IP followed by WB of transfectants showed that proTGF-β1, GARP, and LRRC33 could each be detected in cell lysates when TGF-β1 and milieu molecules were expressed individually or together (Fig. 2A). Furthermore, Flag-tagged milieu molecules were found to co-associate with proTGF-β1 when the IP was done either with the milieu molecule (first panel) or proTGF-β1 (third panel). Moreover, IP of supernatants from the same transfectants showed that secretion of proTGF-β1 into the supernatant (Fig. 2B, lane 3) was prevented by co-expression with LRRC33 (Fig. 2B, lane 6) or GARP (Fig. 2B, lane 4) (Wang et al., 2012). Thus, LRRC33 associates with proTGF-β1 and stores it in a cell-associated form, whereas in absence of a milieu molecule, proTGF-β1 is secreted.
Figure 2. LRRC33 association with proTGF-β1 and TGF-β1 activation.
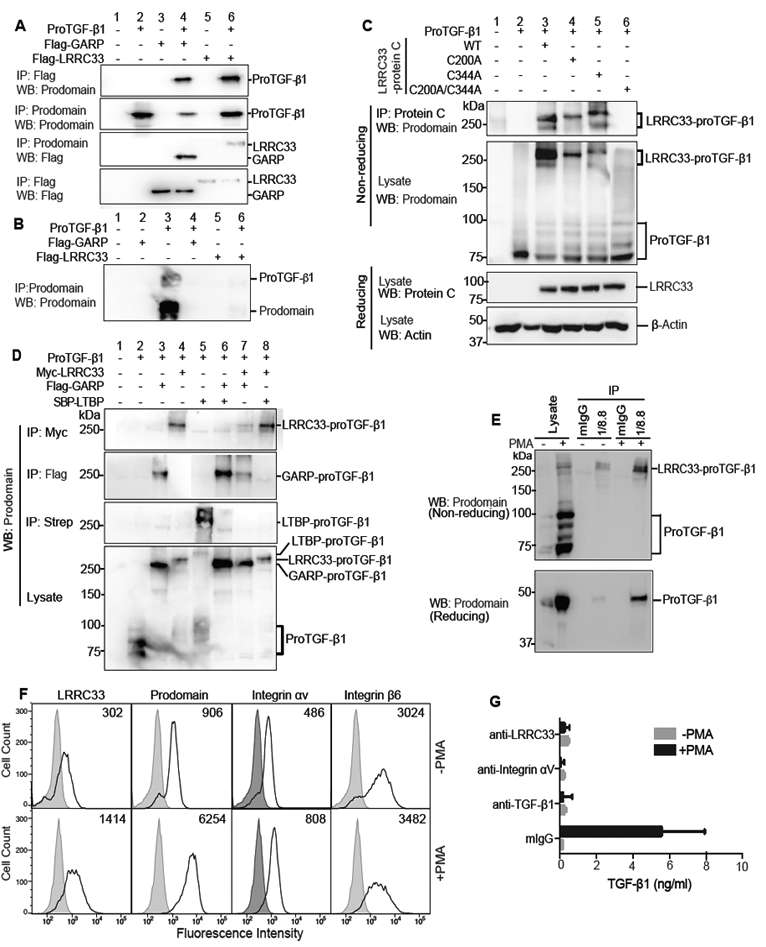
(A and B) Lysates of 293T cells transfected with indicated constructs (A) or culture supernatants (B) were immunoprecipitated (IP) and subjected to reducing SDS 10% PAGE and blotted (WB) as indicated. (C) Disulfide linkage. 293T cells transfected with indicated constructs were subjected to IP, 7.5% non-reducing or 10% reducing SDS-PAGE, and WB as indicated. (D) LRRC33 outcompetes LTBP for proTGF-β1 293T transfectant lysates were IP, subjected to non-reducing SDS 7.5% PAGE, and WB as indicated. (E) LRRC33-proTGF-β1 complex in THP-1 cells. THP-1 cells were treated with or without PMA (80 nM, 24 h) and cell lysates were IP with 1/8.8 to LRRC33 or mouse IgG control, reducing and non-reducing SDS 7.5% PAGE, and WB as indicated. (F) Flow cytometry. THP-1 cells treated with or without PMA were stained with anti-LRRC33 (1/8.8), anti-prodomain (TW4–2F8), anti-integrin αV (17E6) or anti-integrin β6 (7.1G10) and subjected to FACS. Numbers in histograms show specific mean fluorescence intensity. (G) Blockade of active TGF-β1 release. THP-1 cells treated with or without PMA were incubated with antibody 1/8.8 to LRRC33, 17E6 to αV integrin, or MAB240 to TGF-β1 and cocultured with TMLC to measure TGF-β activation. Data represent mean ± SEM of quadruplicate samples.
Non-reducing SDS-PAGE showed that LRRC33 was disulfide linked to proTGF-β1. Whereas proTGF-β1 migrated at 75 to 100 kDa in non-reducing SDS-PAGE (Fig. 2C, lane 2), WT LRRC33 co-expression shifted the mass of proTGF-β1 immunoreactive material to 270–290 kDa (Fig. 2C, lane 3). C200 and C344 in LRRC33 are homologous to the cysteines in GARP that disulfide link to proTGF-β1 (Fig. 1A). The LRRC33 C200A and C344A single mutants and C200A/C344A double mutant were expressed at similar levels as WT LRRC33 in 293T cells (Fig. 2C, third panel). Single cysteine mutations did not abolish disulfide formation, but slowed migration in SDS-PAGE (Fig. 2C, upper two panels, lanes 4 and 5 compared to 3), as expected from greater elongation in SDS after removal of one disulfide cross-link (Wang et al., 2012). In contrast, the C200A/C344A double mutant failed to disulfide crosslink to proTGF-β (Fig. 2C, panels 1 and 2, lane 6). Thus, LRRC33 disulfide links to proTGF-β1 and uses cysteines homologous to those in GARP to do so (Wang et al., 2012).
The specificity of association between LRRC33 and proTGF-β1 was further tested by competition with LTBP and GARP, which are highly validated partners of proTGF-β1 (Robertson and Rifkin, 2016). Impressively, LRRC33 outcompeted LTBP for proTGF-β1 (Fig. 2D, lane 8), despite ability of LTBP to covalently link to proTGF-β1 in absence of LRRC33 (Fig. 2D, lane 5). GARP and LRRC33 competed equally with one another for proTGF-β1 (Fig. 2D, lane 7), and as previously described (Wang et al., 2012) GARP outcompeted LTBP (Fig. 2D, lane 6). Thus, LRRC33 completely outcompetes the highly specific association of LTBP with proTGF-β1.
Myeloid cells and cell lines express TGF-β1 (Khalil et al., 1989; Taipale, 1994) but a corresponding milieu molecule has not been defined. The myelomonocytic cell line THP-1 expresses Lrrc33 and Tgfb1 mRNA (Fig. 1D) and phorbol myristyl acetate (PMA) increases Tgfb1 mRNA (Taipale, 1994). We made mAb 1/8.8 specific to human LRRC33 (Supplemental Fig. 1). IP of THP-1 cell lysates with LRRC33 mAb and WB with anti-TGF-β1 prodomain demonstrated a PMA-stimulated increase in the LRRC33—proTGF-β1 non-reduced complex at 290 kDa and an LRRC33-associated reduced proTGF-β1 monomer at 48 kDa (Fig. 2E). Importantly, LRRC33 and proTGF-β1 were present on the cell surface and their expression was increased by PMA (Fig. 2F). In contrast, expression of the αV and β6 subunits of αVβ6 integrin, which can activate TGF-β1 (Robertson and Rifkin, 2016), were little affected by PMA stimulation (Fig 2F).
We next determined whether LRRC33 and αV integrins participated in TGF-β1 activation in THP-1 cells. PMA stimulation greatly increased TGF-β1 activation (Fig 2G). Activation was inhibited by 1/8.8 antibody to LRRC33 and 17E6 antibody to integrin αV (Mitjans et al., 1995) and specificity was demonstrated via inhibition by antibody to TGF-β1 (Fig 2G). Thus, LRRC33 specifically associates with and is disulfide-linked to proTGF-β, is co-expressed with proTGF-β1 on the cell surface, and plays a role in TGF-β activation.
Deficiency of Lrrc33 in mice results in ascending paraparesis.
Mice with heterozygous deficiency of Lrrc33 (Supplemental Fig. 2A) were fertile and healthy. Homozygous Lrrc33−/− mice showed increased susceptibility to respiratory tract infection, were sometimes runted (Fig. 3A, B) and had a yield at weaning of ~20% compared to the Mendelian expectation of 25%, but otherwise appeared normal.
Figure 3. Phenotype and neuropathology of Lrrc33−/− mice.
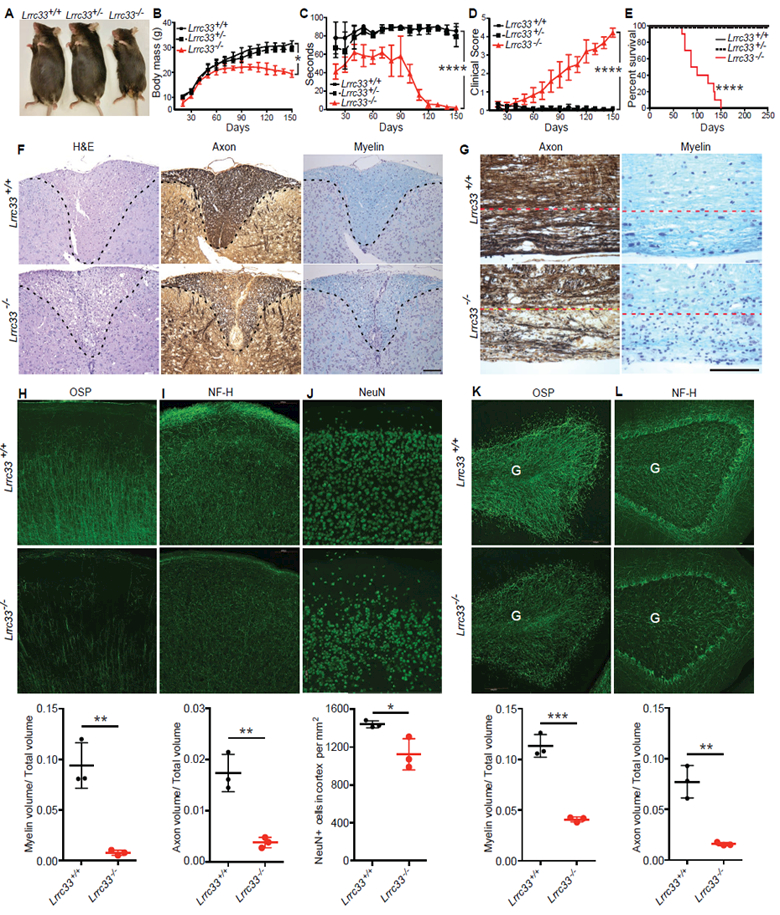
(A-E) Appearance at 4 months (A), body mass (B), rotarod performance (C), clinical score (D), and Kaplan-Meier survival curve (E) of Lrrc33+/+, Lrrc33+/− and Lrrc33−/− mice. Data are mean ± SEM. P values for last time point (*, <0.05; ****, <0.0001) are from unpaired Student’s t-test (B-D) and log-rank test (E). (F) Demyelination and axon degeneration at 4.5 month in spinal cord dorsal column (area above dashed line). Left panel: H&E stain shows accumulation of foamy macrophages and cholesterol clefts (sign of myelin breakdown) in Lrrc33−/− mice. Middle panel: Bielschowsky’s silver stain. Right panel: Luxol fast blue stain. Scale bar: 1 mm. (G) Demyelination and axon degeneration at 4.5 month in corticospinal tract of brain stem (below dashed line). Left: Bielschowsky’s silver stain. Right: Luxol fast blue stain. Scale bar: 1 mm. (H-L) Fluorescent immunostaining (upper two rows) with quantitation (lower row) of 40 μm sagittal sections at 4 months of somatomotor cortex region M1 (H-J) and cerebellum (K-L). M (molecular) and II/III mark layers of the cerebral cortex. G marks granule cell layer and dashed lines demarcate the Purkinje cell layer in cerebellum. Scale bars: 100 μm. Quantitation in lower row shows mean ± SEM for three mice from measurements averaged over 2–3 sections per mouse with 1–2 images per section. *: P<0.05; **: P<0.01; ***: P<0.001, unpaired t-test.
By 2 months of age, Lrrc33−/− mice began to display neurological symptoms including defects in motor control and strength, an increase in locomotor activity and exploration, and declining ability to maintain themselves on a rotating cylinder (Fig. 3C, Supplemental Fig. 3A). Lrrc33−/− mice developed ascending paraparesis with progressive loss of hind limb grasp, hind limb coordination, bladder control, and ability to right themselves. Eventually, quadriplegia required euthanasia (Fig. 3D). Lack of bladder control correlated with palpable, highly distended bladders and a failure of reflex urination upon euthanasia (Supplemental Fig. 3B). All Lrrc33−/− mice required euthanasia or died by 150 days (Fig. 3E).
Progressive paraparesis was associated with loss of myelin and axons in the spinal cord and brainstem. Spinal cord sections of 4–5 month-old Lrrc33−/− mice stained with hematoxylineosin (H&E) showed specific loss of cellularity in the dorsal column paired with appearance of lipid-laden (foamy) macrophages (Fig. 3F). Bielschowsky silver and Luxol fast blue stains revealed depletion of axons and myelin, respectively, in the same region (Fig. 3F). Axons and myelin were also depleted in the corticospinal tract of the brain stem (Fig. 3G), through which motor neuron axons pass from the cerebral cortex to the spinal cord. Although TGF-β1-deficiency results in uncontrolled autoimmunity, no autoimmune-like infiltration of the CNS by mononuclear cells or T cells was observed by H&E staining and anti-CD3 immunohistochemistry, respectively. All neonates deficient in the integrin αV or β8 subunits have grossly visible intracerebral hemorrhage, which is also seen in excised brains (Mobley et al., 2009). No such hemorrhage was ever seen in Lrrc33−/− neonates.
Immunofluorescent staining demonstrated localized loss of axons, oligodendrocytes and neurons in brain regions including the somatomotor M1 region of the cerebral cortex (Lein et al., 2007) (Fig. 3H-J) and cerebellum (Fig. 3K-L) in 4-month-old Lrrc3−/− mice. Oligodendrocyte-specific protein (OSP)-positive myelin tracts were well organized and linear in the II/III layer of the somatomotor M1 region in WT mice but severely and significantly depleted in Lrrc33−/− mice (Fig. 3H). Neurofilament-heavy (NF-H)-positive axons were also significantly depleted in the M and II/III layers in Lrrc33−/− mice (Fig. 3I). NeuN+ neurons were also significantly depleted, although not to the same quantitative extent as axons, in the II/III layer of the M1 region (Fig. 3J) with some mice showing striking patches of neuron loss (Supplemental Fig. 4A). Localized decreases in myelin and axon density were also significant in the granule cell layer of the cerebellum (Fig. 3K,L). Losses were much smaller in the somatosensory cortex, with significance reached only for myelin volume (Supplemental Fig. 4C). Losses of neurons, myelin and axons in Lrrc33−/− mice provide a cellular mechanistic basis for impaired motor coordination and ascending paraparesis.
Abnormalities in microglia, macrophages, and TGF-β1 in Lrrc33−/− mice
Staining for Lrrc33 expression with anti-LacZ in the cerebral cortex of Lrrc33+/LacZ mice and co-staining with anti-Iba1 to mark microglia (Butovsky et al., 2014) showed that Lrrc33 was expressed in microglia (Fig. 4A). Lrrc33+/LacZ cells had the highly ramified morphology of microglia. Thus both RNA-Seq (Fig. 1H) and fluorescent microscopy showed that Lrrc33 is specifically expressed in microglia within the brain.
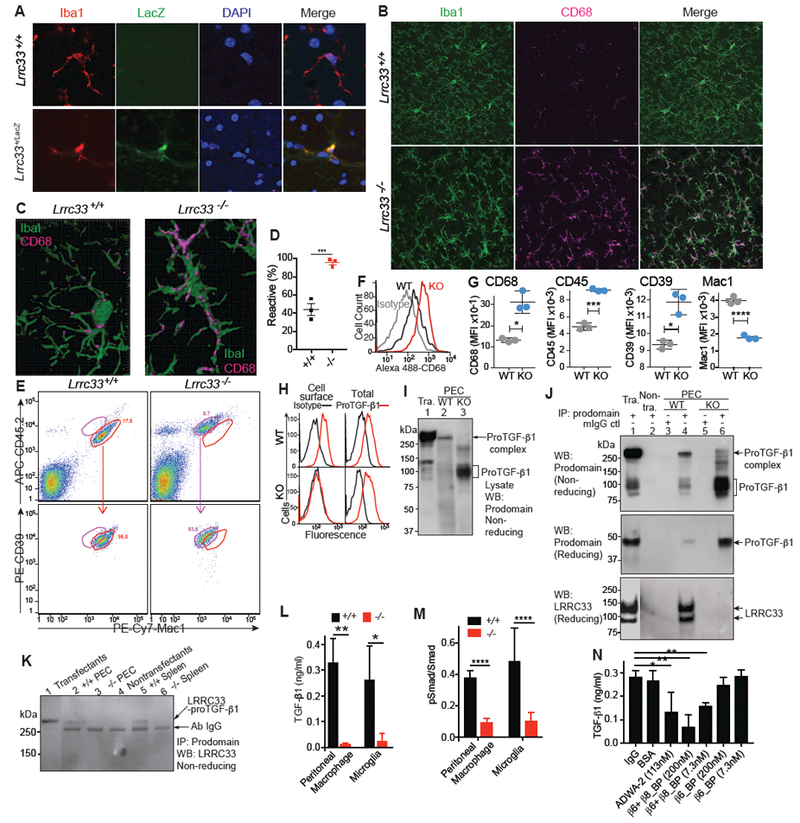
Figure 4. Alterations in microglia, macrophages, and TGF-β complex formation and activation in Lrrc33−/− mice.
(A) Immunostaining of 8 μm brain sections for LacZ and Iba1. (B and C) Immunostaining of 40 μm brain sections for Iba1 and CD68 (B) and 3D reconstruction (C). (D) Quantification of reactive microglia cells in somatomotor cortex region M1, mean ± SEM for n=3 mice; ***P=0.008, two-tailed t-test). (E-G) Immunophenotype of CD45low Mac1high CD39high microglia from day 21 mice and quantification of expression. (E) Representative gating comparing gates for WT (red) and KO (violet) microglia. (F-G). Quantitation with representative histograms for CD68 (F) and mean fluorescence intensity (G, MFI, mean ± SEM, n = 3 mice). *: p=0.012 to 0.02; ***: p=0.006; ****: p=0.0006. (H) Staining of F4/80+ Mac1+ PEC with TGF-β prodomain (TW7–16B4) or control antibodies with or without permeabilization. One representative of 4 mice. (I-K) High MW proTGF-β1 complexes in WT and not Lrrc33−/− cells or transfected (Tra.) or non-transfected (Non-tra.) cells. (I) Anti-proTGF-β1 WB of PEC (106/lane) or 33-G-X5 - proTGF-β1 L1.2 transfectant (105/lane) lysates. (J,K) Lysates from WT and Lrrc33−/− PEC and spleen cells, LRRC33 proTGF-β1 co-transfectants or untransfected cells, with or without IP with prodomain antibody TW7–16B4 or mouse IgG coupled to Sepharose, were subjected to non-reducing or reducing SDS 7.5% PAGE and WB with anti-denatured mouse LRRC33 (Noubade et al., 2014) or anti-proTGF-β1 Ratio of transfectant:native cell equivalents was 1:10. (L) TGF-β activation. PEC from adult WT or Lrrc33−/− mice or 1:1 cocultures of WT astrocytes with WT or Lrrc33−/− microglia were assayed for TGF-β production; N=3 mice, mean ± SEM; **P<0.01, *P<0.05 (unpaired Student’s t-test). (M) pSMAD. PEC and microglia from WT or Lrrc33−/− mice were assayed for fluorescence intensity with SMAD2 and phospho-SMAD2/3 antibodies by In-cell-western. Mean ± SEM; N=3 mice (3 replicates each). ****P<0.0001, unpaired Student’s t-test. (N) Integrin dependence of TGF-β activation. WT astrocyte and microglia co-cultures were assayed for TGF-β production using reporter cells in the presence of indicated inhibitors. Mean ± SEM, N=3 mice (3 replicates each); **P<0.01, *P<0.05 (unpaired Student’s t-test).
Loss of Lrrc33 affected microglia morphology and immunophenotype. CD68 (macrosialin), a marker of reactive microglia, was markedly upregulated in Lrrc33−/− Iba1+ microglia (Fig. 4B). While WT microglia had a typical ramified morphology, Lrrc33−/− microglia had an altered, activated morphology with fewer cellular processes that were thicker, less ramified, and did not extend as far into their cellular neighborhoods (Fig. 4B&C, Movies 1&2). By CD68 immunoreactivity and morphology (Schafer et al., 2012), Lrrc33−/− microglia were significantly more reactive-like than WT microglia (Fig. 4D). We further characterized microglia immunophenotype by flow cytometry by gating on CD45low, Mac1+, and CD39high cells to exclude CD39neg/low monocytes/macrophages (Butovsky et al., 2014). Because CD45 and other markers are up-regulated in immune-reactive microglia during viral infection (Sedgwick et al., 1991) we compared CD45, Mac1, and CD39 as well as CD68 for expression on WT and Lrrc3−/− microglia. Lrrc33−/− microglia from multiple mice had statistically significantly increased CD45 and CD68 expression (Fig. 4F and G), consistent with the reactive-like phenotype seen in tissue sections with immunohistochemistry. Interestingly, compared to WT, Lrrc33−/− microglia also showed significantly increased expression of CD39 and decreased expression of Mac1 (Fig.4G).
Loss of LRRC33 also affected TGF-β1 expression. As predicted by dependence on one another for LRRC33 and proTGF-β1 surface expression on transfectants (Supplemental Fig. 1), WT, but not Lrrc33−/−, peritoneal exudate cell (PEC) macrophages expressed proTGF-p1 on the cell surface (Fig 4H). In contrast, staining of permeabilized cells showed that intracellular proTGF-β1 is present in both WT and Lrrc33−/− macrophages. Thus, LRRC33 is required for expression of proTGF-β1 on the surface of macrophages.
Western blotting of WT PEC lysates showed a high molecular weight proTGF-β1 complex of the same size as in cells co-transfected with LRRC33 and proTGF-β1 (Fig.4I, lanes 1 and 2). In contrast, Lrrc33−/− PEC lacked a corresponding high molecular weight proTGF-β1 complex, and instead expressed aberrantly large amounts of uncomplexed proTGF-β1 (Fig. 4I, lane 3). Similarly, IP of PEC with anti-proTGF-β1 followed by WB with anti-proTGF-β1 showed the high MW complex in WT PEC and lack of the high MW complex and large amounts of aberrant proTGF-β1 in Lrrc33−/− PEC (Fig. 4J, upper two panels). Moreover, IP with anti-proTGF-β1 followed by WB with an antibody specific for denatured mouse LRRC33 (Noubade et al., 2014) showed LRRC33 associated with proTGF-β1 in WT but not Lrrc33−/− PEC (Fig. 4J, lower panel). LRRC33 is detected as two bands of 120 and 90 kDa in WT and not Lrrc33−/− lysates (Fig. 4J, lower panel). Only the 120 kDa LRRC33 band was present on the cell surface (Supplemental Fig. 5B,C), suggesting that the 120 kDa and 90 kDa bands represent mature LRRC33 with complex N-glycans and immature LRRC33 with high mannose glycans, respectively. After IP from PEC and spleen lysates with anti-proTGF-β1, WB with anti-denatured LRRC33 detected a non-reduced, ~290 kDa LRRC33-proTGF-β1 complex from WT but not Lrrc33−/− cells (Fig. 4K). Thus, LRRC33 and proTGF-β1 specifically associate and disulfide link to one another in macrophages and spleen cells from WT and not Lrrc33−/− mice, and proTGF-β1 is aberrant in Lrrc33−/− macrophages.
We then tested the requirement of LRRC33 and integrins for TGF-β1 activation. Production of active TGF-β1 by WT or Lrrc33−/− (KO) macrophages or 1:1 mixtures of WT astrocytes with WT or Lrrc33−/− microglia was greatly decreased with Lrrc33−/− compared to WT macrophages or microglia (Fig. 4L). SMAD 2/3 phosphorylation was also significantly decreased in Lrrc33−/− compared to WT microglia (Fig. 4M), demonstrating canonical TGF-β signaling. Astrocytes express αVβ8 and not αVβ6 (Zhang, 2014) (Fig. 1H). We used αVβ6 and αVβ8 inhibitors to test the integrin dependence of TGF-β1 activation in astrocyte: microglia cocultures. Blocking antibody ADWA-2 to αVβ8 and a protein that blocks ligand binding to both αVβ6 and αVβ8 (β6+β8_Bp) but not a blocking protein selective for αVβ6(β6_BP) significantly inhibited TGF-β activation in astrocyte cocultures with WT microglia (Fig. 4N). Thus, activation of TGF-β was dependent on both LRRC33 and αVβ8.
Overall, the results show that LRRC33 specifically associates with and disulfide links to proTGF-β1 in WT myeloid cells, that proTGF-β1 is intracellular and aberrant in Lrrc33−/− macrophages, that LRRC33 is required for expression of proTGF-β1 on the surface of macrophages, that LRRC33 is required for activation of TGF-β and phosphorylation of SMAD2/3 in macrophages and astrocyte:microglia cocultures, and that activation of TGF-β is dependent on integrin αVβ8 and not αVβ6 in astrocyte:microglia cocultures. These results provide mechanistic insight into the aberrant, reactive phenotype of microglia in the CNS of Lrrc33−/− mice.
Microglia repopulation and arrest of disease progression by bone marrow transplantation of Lrrc33−/− mice.
Whole bone marrow (wBM) transplantation has been effective in rescuing microglia-related CNS pathologies (Wirenfeldt et al., 2011). We utilized the CD45.1 and CD45.2 alleles in congenic mice to distinguish donor and recipient BM-derived cells. Whole BM cells (8 ×106) freshly harvested from 10-week-old mice were intravenously transplanted into lethally irradiated recipients in all four possible Lrrc33−/− and Lrrc33+/+ combinations. Mice were neurologically scored as in Fig. 3D and sacrificed 5 months post-transplantation to determine donor cell contribution to the host BM and CNS. Independent transplantation experiments gave similar results (Fig. 5 and Supplemental Fig. 6).
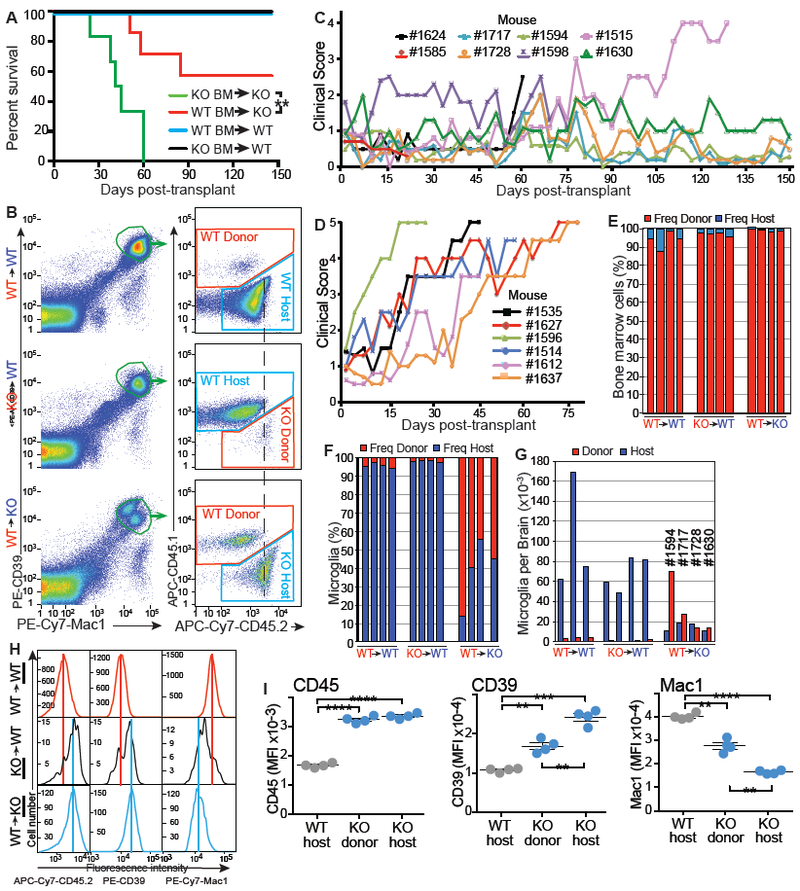
Figure 5. Whole bone marrow transplantation rescues neurological defects.
(A) Kaplan-Meier survival curve, **: P<0.01 (Mantel–Cox) test. (B) FACS analysis of microglia chimerism in representative mice 5 months post BM transplantation. Vertical dashed line illustrates greater intensity of CD45.2 in KO than WT microglia. (C-D) Clinical scores for WT→KO (C) and KO→KO (D) recipients. (E-G) Bone marrow (E) and microglia chimerism (F,G). Bars represent individual mice. (H-I) Microglia immunophenotype. Cells were gated as in (B). Immunophenotypes of host or donor cells (underlined to left in H) are shown for representative mice (H) or for four mice (symbols with mean and SEM in I). Colored lines in H compare peak intensities of microglia in upper and lower panels to the middle panel. MFI: Mean fluorescence intensity. **: p=0.0011 to 0.0037; ***: p=0.0004; ****: p<0.0001; unpaired t-test with Welch’s correction.
Lrrc33−/− mice were 12 weeks old at transplantation and already showing clinical symptoms; nonetheless, WT wBM rescued 60% of Lrrc33−/− mice from disease progression and death (Fig. 5A). Lrrc33−/− recipients of WT wBM showed either no progression or slowed progression of neurological symptoms after transplantation, while Lrrc33−/− recipients of Lrrc33−/− wBM showed neurological disease progression and age at death similar to naive Lrrc33−/− mice (Fig. 5C, D). WT mice transplanted with WT or Lrrc33−/− wBM showed no neurological signs or deaths (Fig. 5A and Supplemental Fig 6).
Lrrc33−/− mice transplanted with WT wBM were 8 months old at sacrifice, having survived well beyond the 5-month lifespan of naive Lrrc33−/− mice. Bone marrow chimerism averaging over 90% showed successful transplantation (Fig. 5E). Microglia were assessed for chimerism by flow cytometry as CD45.1low or CD45.2low, Mac1+, and CD39high cells (Fig. 5B, left). Including CD39high as a sort criterion excluded donor-derived CD39neg/low monocytes/macrophages that might have also engrafted into recipient mouse brains. Microglia chimerism was expressed as % of total (Fig. 5F) or microglia recovered per brain (Fig 5G). There was little chimerism in WT recipients transplanted with either wild type wBM (4.1±1.4%) or Lrrc33−/− wBM (2.4±0.65%). In marked contrast, Lrrc33−/− mice transplanted with wild type wBM showed significantly higher levels of donor-derived microglia (68±16%, p=0.007; Fig. 5F) and increased total number of microglia (Fig. 5G).
Microglia are the only known cell of hematopoietic origin with an important functional role in CNS physiology. To test whether spreading of TGF-β from WT microglia had converted Lrrc33−/− microglia to a phenotype indistinguishable from WT microglia, we examined microglia immunophenotype of host Lrrc33−/− microglia that coexisted with similar numbers of donor WT microglia and donor Lrrc33−/− microglia that coexisted with a large excess of host WT microglia. Compared to WT host microglia, CD45.2 was significantly increased on donor and host Lrrc33−/− microglia (Fig. 5B, dashed line, and Fig. 5H and I), as in microglia in naive Lrrc33−/− mice (Fig. 4E and G). Moreover, Mac1 was significantly decreased and CD39 was significantly increased on donor and host Lrrc33−/− microglia (Fig. 5I) as found in untransplanted mice (Fig. 4G). Interestingly, for all three markers, donor Lrrc33−/− microglia, which were outnumbered 40-fold by host WT microglia, showed a minor subpopulation with the fluorescence intensity of WT microglia and a major subpopulation with intensity similar to host Lrrc33−/− microglia but slightly shifted toward WT intensity (Fig. 5H and Supplemental Fig. 6K). Differences between donor and host Lrrc33−/− microglia reached significance for CD39 and Mac1 (Fig. 5I). Thus, when similar numbers of WT and Lrrc33−/− microglia are present in Lrrc33−/− recipients, the defect in Lrrc33−/− microglia is cell-autonomous, suggesting that activation of TGF-β from proTGF-β1-LRRC33 complexes displayed by WT microglia is so localized that there is little spreading of the WT immunophenotype to Lrrc33−/− microglia. In contrast, when 40-fold more WT than Lrrc33−/− microglia are present in WT recipients, there is limited but significant spreading of the WT immunophenotype to Lrrc33−/− microglia.
Molecular signature of Lrrc33−/− microglia.
To gain broader and independent insight into how loss of Lrrc33 affects microglia, we compared transcriptional profiles of microglia isolated from Lrrc33+/+ and Lrrc33−/− mice. CD45low, Mac1+, CD39high, Ter119- microglia sorted from 3 Lrrc33+/+ and 3 Lrrc33−/− 3-week old mice were analyzed using the Affymetrix Murine Exon 1.0 ST platform (Supplemental Fig. 7). Significant (p<0.05 after correction for multiple comparisons) up or down regulation of mRNA expression of >2.8-fold in Lrrc33−/− microglia was found for 158 and 190 genes, respectively (Fig. 6A; Supplemental Fig. 7A). Both wild type and Lrrc33−/− cell populations expressed markers previously found to be selective microglia-specific markers including CD39 (Butovsky et al., 2014). Previously, 354 genes were found to be selectively expressed in microglia compared to macrophages, and 62 of these were found to differ in expression more than 5-fold between WT and Il2-Tgfb1; Tgfb1−/− microglia (Butovsky et al., 2014). Up or down regulation of these 62 genes was completely concordant between Il2-Tgfb1; Tgfb1−/− microglia and Lrrc33−/− microglia (Fig. 6B; concordance bar). This concordance strongly suggests that Lrrc33 is required for Tgfb1 signaling in microglia and further suggests that LRRC33 is the only milieu molecule required for TGF-β1 function in microglia.
Figure 6. Effect of Lrrc33 deficiency on transcriptional phenotype of microglia.
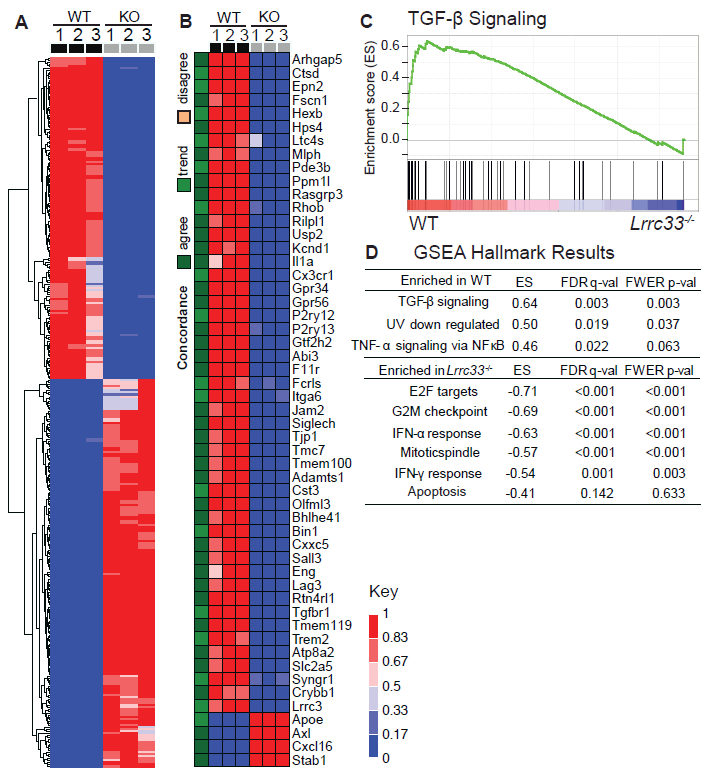
(A) Microarray data on microglia isolated from brains of 3 WT and 3 KO animals at 3 weeks. Genes are shown with at least a 2.8-fold change and a p value adjusted for multiple comparisons <0.05. Scale shows quotient of difference between sample intensity and row minimum intensity over the row range. (B) Comparison to genes identified as differentially regulated between WT and II2-Tgfb1;Tgfb1−/− microglia (Butovsky et al., 2014). Green-orange scale: agree, change in same direction in our dataset with fold change >2.8 and p-value<0.05; trend, change in same direction in our dataset with fold change <2.8; disagree, opposite direction of change in two datasets. Red-blue scale is same as in (A). (C) GSEA of TGF-β signaling in WT microglia. (D) GSEA results showing the most significant Hallmark differences (Subramanian, 2005). ES, enrichment scores.
Gene Set Enrichment Analysis (GSEA) examines sets of genes that function together in key biological processes and show coordinate differential expression (Subramanian, 2005). Quite strikingly, among 50 biological pathways, “TGF-β signaling” was the most statistically enriched pathway in wild type microglia compared to Lrrc33−/− microglia (Fig. 6C, D, Supplemental Fig. 7C, D), providing independent evidence that LRRC33 is required for TGF-β signaling in microglia. GSEA further revealed significant negative ES scores (enrichment in Lrrc33−/− relative to wild type microglia) for “Interferon-α response” and “Interferon-γ response” pathways (Fig. 6D). Enrichment of these pathways suggests activation of Lrrc33−/− microglia. Lrrc33−/− microglia were also significantly enriched for “E2F targets,” “G2M checkpoint,” and “Mitotic spindle” (Fig. 6D), which are all associated with cell cycling. GSEA thus provided independent, hypothesis-free support for the requirement of LRRC33 for TGF-β signaling in microglia and for the reactive phenotype of Lrrc33−/− microglia.
Discussion
Restriction of LRRC33, pleiotropy of TGF-β, and prior reports.
We have established that LRRC33 is a milieu molecule that associates highly specifically with proTGF-β1 and is required for activation of latent TGF-β in macrophages and microglia. LRRC33 functions analogously to LTBPs 1, 3, and 4 and GARP (LRRC32), which are expressed by other cell types (Robertson and Rifkin, 2016). We propose the term milieu molecule for all such partners of proTGF-β. Macrophages have long been known to be important biological sources of TGF-β (Khalil et al., 1989); however, despite investigations (Taipale, 1994) a corresponding milieu molecule was not defined until now.
Previous publications on LRRC33 have proposed other functions. The hypothesis for a relationship to TLRs emerged from common content of LRRs (Liu et al., 2013); however, sequence identity between LRRC33 and TLRs is too low at 15–18% to suggest a functional relationship, in contrast to the 34% identity of LRRC33 with GARP (Fig. 1B). Sequence relationship between GARP and LRRC33 has previously been pointed out and they have been given similar gene names: Lrrc32 (Garp) and Lrrc33 (GARP-like 1; Garpl1) (Dolan et al., 2007). More recently, Lrrc33 was proposed to be renamed Nrros by one group (Noubade et al., 2014). The hypothesis that LRRC33 negatively regulated reactive oxygen species arose from the observation that combined treatment of macrophages with interferon-γ (IFN-γ) and lipopolysaccharide (LPS) increased reactive oxygen species and decreased Lrrc33 expression. However, many other genes were similarly down-regulated, the oxidase component Cybb was already known to be positively regulated by these agents and sufficed to explain the effect, and contrary to the hypothesis, no difference was seen between Lrrc33−/− and WT macrophages in ROS generation either with or without LPS+ IFN-γ treatment (Noubade et al., 2014).
Two previous publications reported no neurological phenotype in Lrrc33−/− mice (Noubade et al., 2014; Su et al., 2014); hence, we were initially apprehensive of the reception of the findings described here. However, one of these groups recently reported a neurological phenotype similar to that described here, and abnormalities in Lrrc33−/− microglia; however, this group did not link the defect to TGF-β signaling and was unsuccessful in halting disease progression with BM transplantation (Wong et al., 2017). Strikingly, the neurological phenotype was not reversed in Lrrc33−/−;Cybb−/− mice that lacked the ability to generate reactive oxygen species. This finding strongly suggests that Lrrc33 does not function in the ROS pathway, in agreement with the lack of effect on “Reactive oxygen species” in GSEA (Supplemental Fig. 7C), and supports retaining the name Lrrc33 rather than renaming to Nrros.
As TGF-β is pleiotropic, multiple downstream effects of LRRC33, which is required for TGF-β activation from macrophages and microglia, are to be expected. We found that expression of 348 genes was significantly altered in Lrrc33−/− microglia. Weak, 1.5 to 2-fold effects on ROS in certain assays with Lrrc33−/− macrophages (Noubade et al., 2014) can be explained by inhibition by TGF-β of ROS generation by macrophages (Tsunawaki et al., 1988). Findings on TLR signaling (Su et al., 2014) can be explained by down-regulation by TGF-β of innate immune signaling (Flavell et al., 2010) including IFN-γ signaling in microglia (Zhou et al., 2015) in agreement with our finding with GSEA of enhanced IFN-α and IFN-γ signaling in Lrrc33−/− microglia.
Molecular and cellular function of LRRC33.
Our study demonstrated that association between LRRC33 and TGF-β1 is exquisitely specific and is required for cell surface expression of the LRRC33 -- TGF-β1 complex and TGF-β signaling. Co-IP demonstrated association between proTGF-β1 and LRRC33 in human and mouse myeloid cells and in transfectants that expressed mouse and human proteins. Moreover, a covalent, disulfide-linked, high MW LRRC33 -- proTGF-β1 complex was demonstrated in all of these cell types. Specificity was further demonstrated because only two of 15 LRRC33 ectodomain cysteines were required for covalent linkage to proTGF-β1 and these corresponded to the two in GARP required for disulfide linkage to proTGF-β1. As additional proofs of specificity, LRRC33, like GARP, prevented secretion of aberrant TGF-β1 and out-competed LTBP in formation of covalent complexes with proTGF-β1. In yet further proofs of specificity, LRRC33 and proTGFβ associated with one another on the surface of myeloid cells, co-expression with proTGFβ was required for surface expression of LRRC33, and Lrrc33−/− macrophages lacked the high MW LRRC33 -- proTGF-β1 complex and surface expression of proTGF-β1. Moreover, Lrrc33−/− macrophages and microglia were profoundly deficient in TGF-β signaling. Association was further demonstrated genetically and transcriptionally by the identical phenotypes of LRRC33 and proTGF-β1-deficient mice, their identical microglia gene expression signatures, and identification by GSEA of TGF-β signaling as the most down-regulated pathway in Lrrc33−/− microglia.
Ascending paraparesis with multiple aberrant behaviors began to appear in Lrrc33−/− mice by 2 months with inevitable progression to death by 5 months. As early as 3 weeks, microglia in Lrrc33−/− mice are strongly perturbed as shown by transcriptional profiles, surface marker expression, and morphology in brain sections. However, H&E-staining revealed no overall brain development abnormalities at this age, consistent with results in Tgfb1−/− mice and the stronger expression of TGF-β2 and TGF-β3 than TGF-β1 in early development (Butovsky et al., 2014; Kiefer et al., 1995). We found little spinal cord pathology in 3 month-old Lrrc33−/− mice (Supplemental Fig. 4E-F). Another group examined spinal cords at 15 weeks and reported no loss of axons or myelination (Wong et al., 2017). We found that ascending paraparesis was associated with localized loss of myelin and axons in the cerebellum and within the M1 motor region cortex at 4 months, and with profound loss of myelin and axons in the corticospinal tract and spinal cord at 4.5 months. These regions are associated with motor function. Myelination occurs largely in the first two months after birth in mice. The relatively late appearance of symptoms in Lrrc33−/− mice may relate to important trophic and homeostatic functions of microglia in the CNS, including in phagocytosis, synapse pruning, and interactions with neurons, astrocytes, and myelin-producing oligodendrocytes (Bilimoria and Stevens, 2015; Wirenfeldt et al., 2011).
Lrrc33−/− Iba1+ microglia had a reactive phenotype evidenced by less ramified morphology, CD45 and CD68 up-regulation, and transcriptional up-regulation of pathways for INF-α and INF-γ signaling and cell cycling. Lrrc33−/− microglia also showed increased CD39 and decreased Mac1 expression. CD4 and CD45 are both increased in reactive microglia (Sedgwick et al., 1991); microarrays showed a 19-fold increase in CD4 in Lrrc33−/− microglia (arrow, Supplemental Fig. 7A). Measurement of TGF-β in microglia astrocyte co-cultures, GSEA, and phospho-SMAD staining showed that Lrrc33−/− microglia were deficient in TGF-β activation and signaling. Because TGF-β down-regulates microglia reactivity (Liu et al., 2016; Zhou et al., 2015), the reactive phenotype is likely a direct consequence of the lack of the LRRC33-proTGF-β1 complex required for integrin and force-dependent TGF-β activation (Robertson and Rifkin, 2016).
Remarkably, transplantation with WT wBM rescued a large portion of Lrrc33−/− mice from disease progression and extended survival to 8 months of age when animals were sacrificed for analysis. Survival of transplanted Lrrc33−/− mice receiving WT wBM was associated with appearance of much greater numbers of microglia in the CNS of donor origin (68±16%) than in WT mice receiving WT or Lrrc33−/− wBM (4.1±1.4% and 2.4±0.65%, respectively). Lrrc33 is specifically expressed in microglia within the CNS, and microglia are the only known hematopoietic-derived cell type with an important role in regulating CNS physiology. These facts, together with the reactive phenotype of Lrrc33−/− microglia and selective repopulation of transplanted Lrrc33−/− mice with WT microglia, suggest that microglia are the cells responsible both for the neuropathology in Lrrc33−/− mice and clinical improvement after transplantation. After transplantation, host Lrrc33−/− microglia in the presence of similar numbers of WT microglia retained the reactive immunophenotype defined with CD45, CD39, and Mac1. These results suggest that neuropathology in Lrrc33−/− mice may result not from injury by reactive microglia, but from an absence of the TGF-β-dependent trophic and homeostatic effects of microglia described two paragraphs above. Donor Lrrc33−/− microglia in WT mice did not induce neuropathology; however, their numbers were much smaller than host WT microglia and they showed a partial shift toward a WT immunophenotype.
Activation of TGF-β in cellular milieus.
Milieu molecules can function on multiple levels to provide highly selective and localized activation of TGF-β. They enable TGF-β to be recognized on the plasma membrane of specific cells (LRRC33 and GARP) or within extracellular matrices (LTBPs) or on different cell types, e.g. LRRC33 on microglia and macrophages or GARP on endothelium and pericytes. Furthermore, because milieus differ in expression of adhesion and chemoattractant molecules, they will attract distinctive cells that bear TGF-β activating integrins. When the integrin-bearing, TGF-β-activating cell is distinct from the cell that secretes proTGF-β into the matrix or displays proTGF-β on the cell surface, TGF-β activation requires two distinct cell types (Fig. 7). This requirement enables great cellular selectivity in TGF-β activation in much the same way that the three-step model for leukocyte emigration from the blood stream enables selectivity. Of further importance, milieu molecules enable localization of TGF-β activation to the interface between an integrin-bearing activating cell and another cell or matrix bearing the proTGF-β-milieu molecule complex. In contrast, many other members of the TGF-β family such as bone morphogenetic proteins diffuse over long distances to establish morphogenetic gradients (Robertson and Rifkin, 2016).
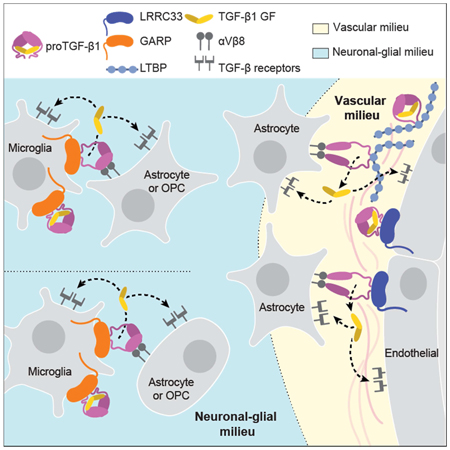
Figure 7. The milieu model.
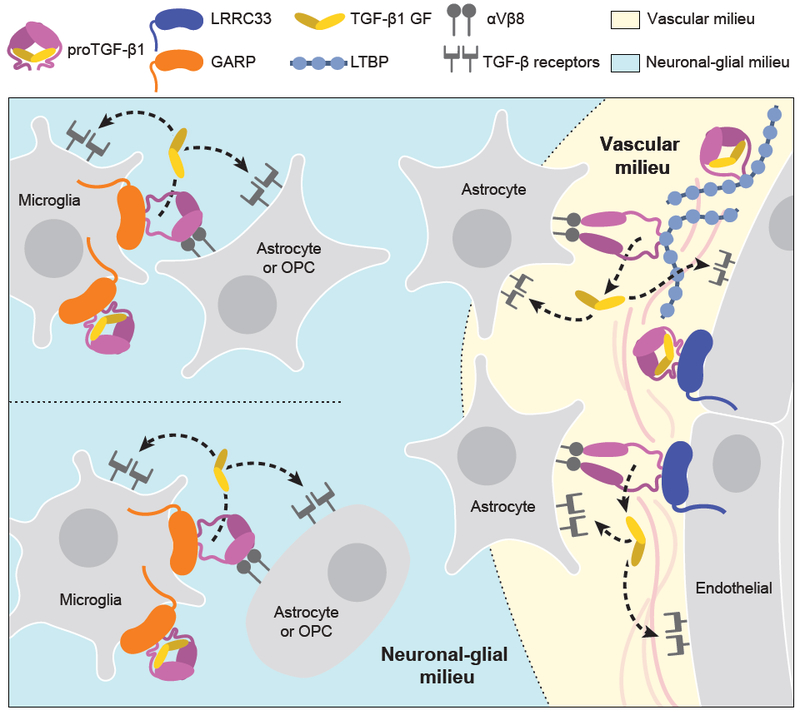
In vivo evidence summarized in Discussion suggests that TGF-β is activated in separate neuronal-glial and vascular milieus in the CNS, with little or no diffusion of TGF-β between milieus (dotted lines). Furthermore, there is little spreading between microglia of TGF-β activated within the neuronal-glial milieu (dotted lines between microglia).
The close similarity in CNS phenotypes and microglia transcriptional profiles in Tgfb1 and Lrrc33-deficient mice strongly suggests that LRRC33 is the only milieu molecule important for TGF-β1 activation in microglia. Il2-Tgfb1; Tgfb1−/− mice develop motor abnormalities at 3–4 months, show rotarod deficits, and paralysis steadily progresses until death by 6 months (Butovsky et al., 2014). Furthermore, all 62 up and down-regulated genes reported in Il2-Tgfb1; Tgfb1−/− microglia were concordantly regulated in Lrrc33−/− microglia.
Despite characterization of the phenotypes in mice of knockouts of milieu molecules including LTBPs 1 and 3, GARP, and each of the three TGF-βs (Robertson and Rifkin, 2016; Wu et al., 2017), the clear correspondence between the neurological phenotypes of Tgfb1 and Lrrc33 deficiency reported here, and comparisons to the phenotypes of knockouts of TGF-β-activating integrins described in the following paragraphs, provide the first compelling evidence for the existence of distinctive milieus that can make TGF-β activation highly localized within tissues (Fig. 7). Association of LTBPs and GARP with multiple proTGFβs, and some redundancy among milieu molecules, may contribute to the difficulty of teasing out the importance of specific milieu molecules for each of the functions of the three TGF-β isoforms (Robertson and Rifkin, 2016; Wu et al., 2017). Sorting out milieu molecule functions in white blood cell lineages is easier, because these cells express TGF-β1 and not TGF-β2 or β3, and LRRC33 is expressed selectively in these lineages.
Integrating current and previous results in mice with deficiencies in TGF-β activation with transcriptomics data enables us to propose a model for milieu-dependent TGF-β1 signaling in the CNS (Fig. 7). RNAseq of CNS cell populations of validated purity (Zhang, 2014) shows that proTGF-β1 and LRRC33 are each most highly expressed within microglia and that microglia express much less GARP or LTBPs than other cells (Fig. 1H), in agreement with our conclusion that LRRC33 is the sole milieu molecule required for TGF-β1 activation in microglia. Among genes encoding TGF-β-activating integrins, none of the profiled cells express the Itgb6 gene for the integrin β6 subunit, whereas Itgav and Itgb8, which encode the subunits of integrin αvβ8, are well expressed on oligodendrocyte precursor cells, newly formed oligodendrocytes, and astrocytes, but not on microglia (Fig. 1H). These transcriptomic results are in excellent agreement with our observation that TGF-β activation in microglia astrocyte co-cultures is dependent on integrin αVβ8 and not αVβ6.
Mice mutant for Itgav and Itgb8 exhibit neurological defects similar to those of Lrrc33−/− mice, as well as CNS hemorrhagic defects not shared with Lrrc33−/− mice, supporting separate neuronal-glial and vascular milieus for TGF-β activation in the CNS (Fig. 7). Itgb8−/− mice develop neurological phenotypes including ascending paraparesis and ataxia that progress until death by 5 months (Aluwihare et al., 2009; Mobley et al., 2009). Mice conditionally deficient in Itgav in neural cells (glia and neurons but not microglia) similarly develop defects in hind limb coordination by 2–3 months, progress to severe paraparesis, and show both glial and axonal degeneration in the spinal cord (McCarty et al., 2005). The results on Itgav, Itgb8, Lrrc33, and Tgfb1-deficient mice together with the transcriptional profiles of CNS cell types (Fig. 1H) and our results showing Lrrc33-dependent and integrin αVβ8-dependent TGF-β activation in microglia astrocyte co-cultures provide strong support for the hypothesis that interactions in the milieu between integrin αVβ8-bearing neural cells and proTGF-β1-LRRC33-microglia are required for maintenance of the integrity of myelinated axons (Fig. 7).
CNS vascular defects in mice mutant for Itgav and Itgb8 suggest an additional, Lrrc33-independent milieu for TGF-β activation. Itgav−/− and Itgb8−/− mice show brain-specific hemorrhage and vascular abnormalities at birth (Aluwihare et al., 2009; McCarty et al., 2005; Mobley et al., 2009). In contrast, cerebral hemorrhage was never grossly evident in Lrrc33−/− neonates or their excised brains or brain sections. Conditional Itgav−/− mutations show that CNS vascular development requires αV integrin in neural cells but not in endothelial cells. Astrocytes express the subunits for integrin αVβ8 (Fig. 1H). Astrocytes, pericytes, and endothelium express all three TGF-β isoforms together with LTBP1 and LTBP3, while pericytes and endothelium additionally express GARP (Fig. 1H).
Both microglia and blood capillaries are widely distributed throughout the brain and are typically within a cell diameter or so of one another. Nonetheless, the severe effects of Lrrc33 deficiency on microglia and neurologic function with an absence of hemorrhage in the CNS suggest that the microglia-neural cell milieu and the astrocyte-pericyte-endothelium vascular milieu are functionally distinct milieus for TGF-β activation in the CNS. The distinct immunophenotypes of co-existing Lrrc33−/− host and WT donor microglia also suggest that TGF-β activation is highly localized within the microglia-neural cell milieu. Only when WT host microglia were in large excess over Lrrc33−/− donor microglia was slight correction of the Lrrc33−/− immunophenotype seen. These results suggest that there is little spreading of TGF-β activated within microglial-neural interfaces to neighboring microglia.
Overall, the results suggest that when αVβ8-bearing cells activate TGF-β1, it is released into a highly localized milieu where αVβ8-bearing cells interface with microglia bearing proTGF-β1-LRRC33 complexes (Fig. 7). Once activated, TGF-β1 can bind to its receptors on either microglia (autocrine signaling) or other glial cells or neurons (paracrine signaling). Thus, our findings on a previously uncharacterized TGF-β milieu molecule support the hypothesis that TGF-β activation is highly localized in vivo. The association of milieu molecules with different in vivo functions of TGF-β opens up new approaches for therapy with antibodies or other antagonists that target proTGFβ associated with specific milieu molecules.
Star Methods
CONTACT FOR REAGENT AND RESOURCE SHARING:
For further information and reagents, the Lead Contact is Timothy A. Springer (springer_lab@crystal.harvard.edu).
EXPERIMENTAL MODEL
Mice.
All animal experiments were approved by the Institutional Animal Care and Use Committees (IACUC) at Harvard Medical School and Boston Children’s Hospital. C57BL/6N embryonic stem (ES) cells with the Lrrc33 gene inactivated by homologous recombination (Supplemental Fig. 2A) from UC Davis Knockout Mouse Project (KOMP) Repository (Project ID CSD40790) were injected into C57BL/6N blastocysts. Heterozygous C57BL/6N Lrrc33+/− mice were fertile with no abnormalities and were used to breed Lrrc33−/− mice. Progeny mice were intercrossed at least six generations before use in experiments. Genotyping used two sets of PCR primers (Supplemental Fig. 2B), primer 7: 5’-GAA CCC AGG ACA TCT GGA AA-3’, primer 8: 5’-TGA GTG ACA GCA TCC TGG AG-3’, and primer 9: 5’-GCG CAA CGC AAT TAA TGA TA-3’.
C57BL/6N embryonic stem (ES) cells with the Garp/Lrrc32 gene deleted was obtained from the UC Davis KOMP Repository (Project ID VG18567) and injected into C57BL/6N blastocysts. A LacZ reporter replaced the protein coding region of the Garp gene. ES cell-derived mice were genotyped by a PCR-based protocol provided by KOMP (Supplemental Fig. 2C,D). Heterozygous mice were fertile but we obtained no evidence of Garp−/− viable or perinatally dead neonates.
METHOD DETAILS
Reagents
DNA constructs.
Human GARP, proTGF-β1 and LTBP1 constructs for 293 cell transfection were described (Wang et al., 2012). Human LRRC33 and mouse proTGF-β1 cDNAs were from Origene (Rockville, MD) and mouse LRRC33 cDNA from GE Dharmacon (Lafayette, CO). Human and mouse LRRC33 were subcloned into pLEXm_v1 vector, which is derived from the pLEXm vector (Aricescu et al., 2006) and contains a protein secretion signal peptide generated from Hidden Markov model (Barash et al., 2002), followed by Flag tag. LRRC33 constructs with N-terminal Flag tag and secretion peptide were further subcloned into pcDNA3.1(+) vector (Invitrogen). Human LRRC33 was also subcloned into ET1 vector (Mi et al., 2008), which contains C-terminal protein C tag. LRRC33 cysteine mutations were generated in ET1 vector using QuickChange (Stratagene). LRRC33 and GARP chimeras were constructed by overlap PCR and cloned into pEF1-puro (Dong et al., 2017) or pcDNA3.1(+) vectors. The 33-G-X1 and 33-G-X5 chimeras contain LRRC33 N-terminal amino acids 1–188 and 1–625, respectively; fusion boundaries are shown in Fig. 1. Human and mouse proTGF-β1 expression vectors were generated by subcloning into pEF1-puro vector without tags.
Antibodies and inhibitors.
Primary antibodies included anti-FLAG (mouse clone M2 and rabbit clone Sig1–25, Sigma-Aldrich), APC-anti-FLAG (clone L5, Biolegend), anti-Myc (clone 9E10, Thermo Fisher), anti-protein C (clone HPC-4, Roche), mouse anti-human LAP1 (27232; R&D Systems), unconjugated and biotinylated goat anti-LAP1 (BAF246; R&D Systems), Pacific-blue-anti-mouse TGF-β1 prodomain (clone TW7–16B4, Biolegend), anti-mouse CD68 (clone FA-11, Abcam), anti-mouse Iba1 (Wako), anti-mouse OSP (Abcam), anti-mouse NF-H (Abcam), anti-mouse NeuN (clone EPR12763, Abcam), PE-F4/80 (clone BM8, Biolegend), PE-Cy7-Mac-1 (clone M1/70, Biolegend), horseradish peroxidase (HRP), and anti-β-actin (C4, Santa Cruz). Unconjugated anti-mouse TGF-β1 prodomain (TW7–16B4) and anti-mouse and human TGF-β1 (TW4–2F8) (Oida and Weiner, 2010)) were generously provided by Dr. Howard Weiner. Hamster monoclonal antibody to denatured mouse LRRC33/NRROS (Noubade et al., 2014) was provided by Genentech Inc. Secondary antibodies included APC or Alexa 488 or 546-labeled goat anti-mouse, anti-rat or anti-rabbit IgG (Invitrogen), HRP conjugated sheep anti-mouse IgG, goat anti-rabbit IgG and rabbit anti-goat IgG (GE Healthcare), and HRP-goat anti-hamster IgG (Jackson Laboratory). HRP-streptavidin was from GE Healthcare. 17E6 Mab to αV integrin (Mitjans et al., 1995) was from E. Merck, Darmstadt, Germany. MAB240 to TGF-β1 was from R&D Systems. 7.1G10 to integrin β6 (Weinreb et al., 2004) was from Biogen, Cambridge, MA. Binding proteins to αVβ6 or αVβ8 (β6+β8_BP and β8_BP) and β8 antibody ADWA-2 (Sheppard, 2014) were measured for affinity using K562 transfectants overexpressing human integrin αVβ6 or αVβ8. Cells were incubated with ADWA-2 or biotinylated binding proteins at a range of concentrations, followed by incubation with fluorophore labeled secondary antibody or streptavidin, and subjected to fluorescence flow cytometry. ADWA-2 is αVβ8-specific and binds with a Kd of 0.6±0.1 nM. The Kd of β6+β8_ΒΡ is 1.7±0.2 nM with αVβ6 and 7.3±1.4 nM with αVβ8. The Kd of β6_ΒΡ is 0.11±0.09 nM with αVβ6 and 580±40 nM with αVβ8.
LRRC33 monoclonal antibody (mAb).
LRRC33 mAb was generated by immunizing Lrrc33−/− mice with L1.2 transfectants that stably expressed the 33-G-X1 chimera, which contains the N-terminal 188 amino acids of LRRC33. Mice were immunized once intraperitoneally with 107 cells and boosted intravenously with transfectants 3 days before fusion. Splenocytes from two immunized mice were fused with P3X63Ag8.653 myeloma cells (Springer, 1980). Hybridoma supernatants were screened by immunofluorescence flow cytometry. An LRRC33 positive hybridoma was identified that stained 33-G-X1 chimera but not GARP transfectants (Supplemental Fig 1A and 1B). Clone 1/8.8 (IgG3, lambda) recognizes human LRRC33 in permeabilized 293T transfectants (not shown) and on the surface of human myeloid THP-1 cells (Fig 2F), immunoprecipitates human LRRC33 complexed with proTGF-β1 from cell lysates of co-transfectants, and does not crossreact with mouse LRRC33 complexes (Supplemental Fig 1C).
1/8.8 VH and VL sequences were used to make a recombinant mouse IgG1 antibody, 1/8.8R, which was expressed in Expi293F cells. 1/8.8R exhibited the same specificity in Western blotting and immunoprecipitation as 1/8.8; however, 1/8.8 was more potent in blocking TGF-β activation, suggesting a role for the IgG3 subclass in functional assays.
Mouse experiments
X-Gal staining.
Lrrc33+/lacZ, Garp+/lacZ and WT 3-month old mice were euthanized and transcardially perfused with ice cold PBS. Organs were removed, fixed with 2% paraformaldehyde, stained overnight at 37°C with 1 mg/ml X-gal (Sigma B4252), 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, in PBS pH 7.4, and washed 5 times with PBS.
Scoring neurological symptoms.
A 5-point clinical score was used for Lrrc33 knockout mice with 1 point each for loss of hind limb grasp, loss of bladder control, loss of coordination leading to hind limb paraplegia, inability to walk upright, and full quadriplegia (requiring euthanasia). SHIRPA tests (Rogers et al., 1997) were conducted by the Neurodevelopmental Behavioral Core, Boston Children’s Hospital.
Whole bone marrow transplantation.
Congenic mouse transplant recipients (10-week-old CD45.1+ C57BL/6N; 12-week-old CD45.2+ Lrrc33+/+ and Lrrc33−/−) were lethally irradiated (950 rad) and tail-vein injected with 8×106 fresh whole bone marrow cells from Lrrc33+/+ and Lrrc33−/− donor mice. Transplant recipients were given water containing 30 mg/L neomycin sulfate, 30 mg/L kanamycin sulfate and 50 mg/L gentamycin sulfate for 4 weeks.
Cell isolation, sorting, transfection, TGF-β assay, and microarrays
Isolation, sorting, and culture of mouse cells.
For peritoneal exudate cell (PEC) macrophages, mice were injected with 1 ml of 4% thioglycollate medium. After 4 days, PEC were collected by lavage using 10 ml PBS. Cells were used directly for WB or IP, or allowed to adhere for 24 hours in 6 well plates to isolate macrophages.
Cell suspensions enriched in microglia were obtained from brains of 21-day-old, transcardially perfused mice as described (Lee and Tansey, 2013) using 330 units of papain (Worthington) per brain. Cells that sedimented through a cushion of 37% percoll were then analyzed or purified further. For 8-month-old mice, brain cell suspensions were prepared by Dounce homogenization instead of papain digestion, and microglia enriched using an otherwise identical procedure. For purification by sorting for RNA preparation or analysis by flow cytometry, microglia were labeled with antibodies to CD45 (APC-CD45.2, clone 30-F11; or APC-Cy7-CD45.1, clone A20, Biolegend), Mac1 (PE-Cy7-Mac1, clone M1/70, Biolegend), CD39 (PE-CD39, clone 5F2, Biolegend), and CD68 (Alexa488-CD68, FA-11, Biolegend). In some experiments (with CD68 and for isolation of mRNA), APC-Cy7-Ter-119 (Biolegend) was also used. In flow cytometry, the first gates used were scatter (side versus forward scatter), singlets (peak intensity versus area under the curve of forward scatter), and live cells (propidium iodide).
RNAseq data shown in Fig. 1H is from (Zhang, 2014) and quantitative values for the genes used in Fig. 1H are shown in Supplemental dataset 1.
Microarray analysis.
Euthanized mice were perfused to minimize possible contamination with monocytes, macrophages, and other blood cells. FACS-sorted microglia (CD45low CD11b+ CD39+ Ter119- propidium iodide-) were isolated from 3-week old mice as described in the previous paragraph. Total RNA was isolated using Trizol (Invitrogen) according to the manufacturer’s protocol. RNA preps with sufficient quality and integrity as verified using a Bioanalyzer 2100 (Agilent Technologies) were hybridized with Affymetrix Mouse Exon 1.0 ST arrays at the Molecular Biology Core Facilities of the Dana-Farber Cancer Institute. Arrays were processed using the BioConductor oligo package (Carvalho and Irizarry, 2010) and normalized using RMA (Irizarry et al., 2003) to provide expression summaries at the exon level. Quality was assessed with the Bioconductor array QualityMetrics package. Affymetrix annotation files were used to collapse probe sets to the ‘core’ set of annotations and summarize the expression data at the gene level, which was used as input for differential expression analysis (Supplemental Dataset 2). Significantly different genes were identified using limma (Smyth et al., 2005). GSEA (Subramanian, 2005) was carried out on the full expression data matrix with the “hallmark (H)” gene sets database, which represents 50 specific well-defined biological states or processes.
Microglia and astrocytes.
Microglia were isolated from 3-week old mice as described in the previous section. Microglia were purified by immuno-panning (Zhang, 2014) with a polyclonal CD45 antibody (catalog #AF114; R&D systems). Prior to co-culture, microglia were cultured for 5 days with no change of medium at 37 °C, 5% CO2 in 6-well poly-D-lysine-coated plates in Neurobasal medium with B-27 Supplement (Thermo Fisher), 5 μg/ml insulin, 2 mM L-glutamine, 1 mM sodium pyruvate, 5 μg/ml N-acetyl cysteine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 μM thyroxine, 100 μg/ml transferrin, 100 μg/ml BSA, 16 μg/ml putrescine, 60 ng/ml progesterone, 40 ng/ml sodium selenite (Sigma) and 10 ng/ml mouse recombinant, carrier-free colony stimulating factor (R&D Systems) (serum-free microglia culture medium).
Astrocytes were isolated and cultured from the cerebrum as described (McCarthy and de Vellis, 1980). For microglia and astrocyte co-culture, microglia and astrocytes (2×104 cells of each/well) were added in 200 μl to 48-well poly-D-lysine-coated plates and cultured in serum-free microglia culture medium containing 10 ng/ml heparin-binding EGF-like growth factor at 37°C in 5% CO2 for 7 days.
Culture and transfection of cell lines
293T cells were maintained in DMEM medium with 10% FBS, transfected with the indicated plasmid(s) using Lipofectamine 2000 (Life Technologies) according to manufacturer’s instructions, and analyzed 48 hours later. THP-1 and L1.2 cells were maintained in RPMI-1640 medium with 10% FBS. L1.2 cells (107) were transfected with 20 μg linearized plasmid DNA by electroporation at 250 V, 960 μF (Gene Pulser™, Bio-RAD). After 48 hours, 2 μg/ml puromycin or 500 μg/ml G418 was added for selection. Transfectants were further subcloned by limiting dilution, and clones with surface expression confirmed by flow cytometry using antibodies to Flag tag and proTGF-β1 were expanded and used for experiments.
TGF-β activation assay
TGF-β was assayed using transformed mink lung cells (TMLC) transfected with luciferase under the control of a TGF-β-activated promoter (Abe et al., 1994). To peritoneal macrophages (2×104 in 200 μl DMEM, 0.1% BSA in 48-well tissue culture plates incubated in 10% CO2) or to microglia and astrocyte co-cultures were added 1× 104 TMLC cells. For standard curves, TMLC cells were also added to recombinant mature human TGF-β (#T7039, Sigma) diluted from 100 ng/ml to 0.05 ng/ml in the same medium. After 24 hours, cell lysates were assayed for luciferase (Promega).
Immunostaining, immunofluorescence, immunoprecipitation (IP), and Western blotting (WB)
Flow cytometry
Cells were stained and analyzed as described previously (Butovsky et al., 2014; Wang et al., 2009; Wang et al., 2012). In brief, cells were incubated with primary antibodies or directly conjugated antibodies in PBS with 2% FBS for 30–60 min on ice. Cells were washed, and for indirect immunofluorescence labeled with conjugated secondary antibodies for 30 min on ice, washed twice with the same buffer, and subjected to flow cytometry with a FACScanto (BD Biosciences) and analyzed with Flowjo software.
Immunofluorescent staining of mouse tissue sections
Brains were harvested from mice after transcardial perfusion with PBS followed by 4% paraformaldehyde. Sagittal cryo-sections of the brain (8 or 40 pm) were treated for 1 h with permeabilization and blocking solution (PBS, 5% BSA, 0.05% Triton X-100 (Sigma-Aldrich)). Primary and secondary antibodies were diluted in PBS containing 5% BSA and 0.03% Triton X-100. Sections were incubated with the primary antibody overnight at 4 °C, washed with PBS, and incubated with the secondary antibody for 1 h at room temperature (20–22°C) while protected from light (Schafer et al., 2012). Sections were then washed with PBS and coverslipped in ProLong Gold antifade reagent with DAPI (Invitrogen) and imaged using Olympus Fluoview FV1000 confocal laser scanning microscope. Brain regions were defined according to the Allen Mouse Brain Atlas (Lein et al., 2007).
Quantitation was with Image J software. The same threshold was applied to all images for each type of antibody to outline cells. To measure volumes, the “measure stack” plugin was used to get areas in each stack. Areas in each stack were summed and multiplied by the stack step size to obtain volumes. To count cell number, “automated counting of single color images” was used in Image J.
Microglial activation state and morphology
Activation states were quantified as described (Schafer et al., 2012) in 3 week old mice. Immunofluorescence was on 40 μm cryosections with antibodies against Iba1 (1:400, Wako, Cat. # 019–19741) and CD68 (clone FA-11, 1:500, Abcam). For each brain section, two 20× fields of view were analyzed on a spinning disk confocal using 1.22 μm thickness z-stacks. Activation states were determined using maximum intensity projections. Microglia were scored from 0 (low) to 5 (high activation) based on branching of microglia (Iba1 staining) and lysosomal content (CD68 positive) (Schafer et al., 2012). Branching was scored as 0, 1, 2, or 3, respectively, if Iba1 staining revealed >15 thin processes with multiple branches, 5–15 thick processes with branches, 1–5 thick processes with few branches, or no clear processes. CD68 staining was scored as 0 (no/scarce expression), 1 (punctate expression), or 2 (aggregated expression or punctate expression throughout the cell). These two scores were summed to give a final score of microglial activation. Microglia with an activation state of 3 or higher were defined as reactive.
For 3D reconstruction, Imaris software (Bitplane) was used to create 3D volume surface renderings of each z-stack. To visualize the volume of the CD68+ component, CD68 fluorescence that was not enclosed within the Iba1+ microglia cell surface was subtracted from the image using the mask function.
In cell Western assays
PECs were cultured in poly-D-lysine coated 96-well plates (1×105/well) at 37 °C in 5% CO2 for 2 hours. Microglia were cultured in poly-D-lysine coated 96-well plates (1×104/well) as described above. Cells were fixed with 4% paraformaldehyde, treated with blocking solution (PBS, 5% BSA with 0.03% Triton X-100) at room temperature for 1 hour, and then stained with anti-Smad2 (D43B4, Cell Signaling Technology; 1:200 dilution) or anti-pSmad2/3 (D27F4, Cell Signaling Technology; 1:200 dilution) overnight at 4 °C, followed by 3 washes and staining with secondary antibodies IRDye 800CW goat-rabbit (925–3321, LI-COR, 1:15000 dilution) or IRDye 680 donkey anti-rabbit (925–68073, LI-COR, 1:15000 dilution), respectively, at room temperature for 1 hour. Plates were washed 3 times with PBS and 3 times with ddH2O, then dried and imaged using LI-COR Odyssey Clx infrared imaging system and analyzed with Image Studio software.
Immunoprecipitation and Western blot
Cells were lysed at ~107/ml for 293 transfectants and ~108/ml for other cell types in lysis buffer (1% NP-40 or 1% Triton X-100, 50 mM Tris pH 7.4, 150 mM NaCl, 2 mM N-ethylmaleimide (NEM), 1 mM EDTA and protease inhibitor cocktail (Roche), and centrifuged at 15,000 rpm at 4oC for 10 min. IP using soluble antibodies and protein G-Sepharose beads was as described (Wang et al., 2012). For IP using antibody-coupled Sepharose 4B beads, cell lysates (300–400 μl) were incubated with 10 μl of packed beads (3 mg Ab/ml packed beads) overnight at 4oC with rotation. For WB, cell lysates or washed beads from IP were heated at 95°C in SDS sa mple buffer containing 5 mM NEM or 5% β-mercaptoethanol for non-reducing and reducing SDS-polyacrylamide gel electrophoresis (PAGE), respectively. Proteins were transferred to PVDF membrane using Trans-Blot Turbo System (Bio-RAD), probed with specific antibodies, and detected with HRP-conjugated secondary antibodies and Amersham ECL detection reagents (GE Healthcare). Biotinylated primary antibodies were detected with HRP-streptavidin. Blots were imaged using LAS4000 luminescence imager (Fujifilm).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis.
GraphPad Prism 6 software (La Jolla, CA) was used for statistical tests including unpaired Student’s t-test and log-rank (Mantel–Cox) test and data are presented as mean ± SEM. No statistical methods were used to pre-determine sample sizes. However, our sample sizes are similar to those in previous publications (Zhang, 2014). Data collection and analysis were performed blinded to conditions in all experiments. A p-value of less than 0.05 was considered as significant.
DATA AVAILABILITY
The probe (exon level) and gene level microarray data for microglia freshly isolated from WT and Lrrc33−/− mice have been deposited at GEO as accession number GSE112604. Supplemental Figure Legends
Supplementary Material
Related to Fig. 2. (A, B) mAb specificity and co-expression requirements. 293T cells were transfected with the indicated Flag-tagged constructs with or without proTGF-β1, stained with the indicated antibodies using indirect immunofluorescence and subjected to flow cytometry. Inset numbers show mean fluorescence intensity. (C) mAb clone 1/8.8 does not cross react with mouse LRRC33. 293T cells were co-transfected with human proTGF-β1 and human or mouse Flag-tagged LRRC33. Cell lysates, or immunoprecipitates (IP) with mAb clone 1/8.8 from lysates, were subjected to 10% reducing SDS-PAGE and Western blot (WB) as indicated.
Movie 1. Related to Fig. 3D. Reconstructions of WT (Movie 1) and Lrrc33−/− (Movie 2) microglia from the M1 motor cortex region of 21-day-old mice. Microglia are stained with antibodies to Iba1 (green fluorescence) and CD68 (magenta fluorescence) as described in the Fig. 4C legend and the movies correspond to the projection view shown in Fig. 4C. Grids in the movies are 5 μm.
Movie 2. Related to Fig. 3D. Reconstructions of WT (Movie 1) and Lrrc33−/− (Movie 2) microglia from the M1 motor cortex region of 21-day-old mice. Microglia are stained with antibodies to Iba1 (green fluorescence) and CD68 (magenta fluorescence) as described in the Fig. 4C legend and the movies correspond to the projection view shown in Fig. 4C. Grids in the movies are 5 μm.
Supplemental dataset 1. Related to Fig. 1. Excel spreadsheet containing the FPKM values for genes shown in Fig. 1H and additionally Itgb6. Data is from {Zhang, 2014 #24481}.
Supplemental dataset 2. Related to Fig. 6. Excel spreadsheet containing microarray data for all genes analyzed in WT AND KO microglia from 3 animals each.
Related to Fig. 3. (A and B) Lrrc33 knockout. (A) construct. (B) Genotyping results showing the WT band (PCR product using primers 7 and 8) and knockout (KO) band (PCR product using primers 7 and 9). (C and D) Garp knockout. (C) construct. (D) Genotyping results showing the WT band (PCR product using primers TUF and TUR) and knockout (KO) band(PCR product using primers LacInf and LacInR). VG18567
Related to Fig. 3. (A) SHIRPA tests showing score for each animal (Black dots: WT mice, red dots: Lrrc33−/− mice). Coat appearance: 0 = normal, 1 = abnormal; Limb grasping: 0 =present, 1 = absent; Tail elevation: 0 = dragging, 1 = horizontal extension, 2 = elevated/straub tail; Trunk curl: 0 = present, 1 = absent; Morphology: 0 = normal, 1 = abnormal; Defecation: 0 = present, 1 = absent; Gait: 0 = fluid movement, 1 = lack of fluidity; Body position: 0 = active, 1 = inactive, 2 = excessive activity; Locomotor activity: total # of squares entered in 30s. Statistical analysis for Grip strength, Locomotor activity, and Open field test used unpaired Student’s t-test, n=5 mice, bars show mean ± SEM. Statistical analysis for Coat appearance, Limb grasping, Tail elevation, Trunk curl, Morphology, Defecation, Gait and Body position used the Z score for 2 population proportions; n=5 mice. *: P<0.05, **: P<0.01, ***: P<0.001, ****: P<0.0001. (B) Lrrc33−/− mice have enlarged bladders compared to WT mice (red arrows).
Related to Figure 3. (A) Variation among micein neuron number revealed by NeuN staining in somatomotor cortex region M1. Sections shown are from 3 different WT and Lrrc33−/− 4 monthold mice, and were stained with NeuN antibody as described in Fig. 3 legend. (B-D) Somatosensory cortex region is relatively normal in 4-month old Lrrc33−/− mice. Immunostaining with antibodiesto (B) NeuN (neuron marker), (C) OSP (myelin marker) and (D) NF-H (axon marker) in somatosensory cortex. Quantitation in lower row shows mean ± SEM (n=3, *P <0.05, unpaired Student’s t-test) for three mice from measurements averaged over 2–3 sections per mouse with 1–2 images per section. Although some significant differences arepresent, the differences and their significance are lesser than in the M1 region (Fig. 3). A and B: M(molecular) and II/III layers of the cerebral cortex. Scale bars: 100 μm. (E) Apparent lack of demyelination and axon degeneration at 3 month in spinal cord dorsal column (area above dashed line). Left panel: H…E stain. Middle panel: Bielschowsky's silver stain. Right panel: Luxol fast blue stain. Scale bar: 1 mm. (F) Lack of obvious demyelination and axon degeneration at 3 month in corticospinal tract of brain stem (below dashed line). Left: Bielschowsky's silver stain. Right: Luxol fast blue stain. Scale bar: 1 mm.
Related to main text Fig. 4. (A) Expression of Flag-tagged mouse LRRC33-GARP chimera 33-G-X5 and proTGF-β1 in L1.2 stable transfectants. Surface expression was detected by flow cytometry with APC-anti-Flag and Pacific blue-anti-TGF-β1prodomain (clone TW7–16B4) or identically conjugated mouse IgG as control (grey curves). Inset numbers show mean fluorescence intensity. (B) IP studies on the LRRC33-GARP chimera 33-G-X5 suggest24 processing of the chimera to a higher molecular weight glycoform when it associates with proTGF-β1. Lysatesof L1.2 transfectants co-expressing mouse proTGF-β and Flag-tagged mouse LRRC33 chimera 33-G-X5 were immunoprecipitated (IP) with mouse proTGF-β1 prodomain antibody TW7–16B4 or control mouse IgG. Immunoprecipitates or lysates were subjected to non-reducing or reducing SDS 7.5% PAGE and Western blotting (WB) with anti-Flag (rabbit clone SIGI-25) or the hamster antibody to denatured mouse LRRC33 {Noubade, 2014 #22620}. Both anti-Flag and anti-denatured LRRC33 reacted with the LRRC33-proTGF-β1 complex (~320 kDa) under non-reducing conditions (top panels) and LRRC33 bands of ~120 kDa and ~90 kDa under reducing conditions. The anti-prodomain precipitate contains a much larger proportion of the ~120 kDa material than in the whole lysate. Since glycoproteins increase in molecular weight during carbohydrate processing, the results suggest that association with proTGF-β is required for LRRC33 chimera transit from the endoplasmic reticulum to the Golgi and carbohydrate processing. The LRRC33 polypeptide has a mass of 74,294 Da and 10 predicted N-glycosylation sites, consistent with a putative intracellular high mannose form of 90 kDa and substantial increase in size to ~120 kDa upon conversion to complex carbohydrates. (C) The ~120 kD glycoform of LRRC33 chimera 33-G-X5 localizes to the cell surface. Prodomainantibody TW7–16B4 (20ug/ml) or control IgG was incubated with L1.2 transfectants for 2 hrs at 4oC to bind cellsurface proTGF-β1. After 3 washes to remove unbound antibody, cells were lysed. Lysates were incubated with protein G beads to pull down antibody-proTGF-β1 complexes, and after removal of beads, supernatants were subjected to two further rounds of incubation with protein G beads to completely deplete pre-formed antibody-proTGF-β1 complexes. (No bands from the 2nd and 3rd pull-down were observed in WB, not shown). To isolate intracellular proTGF-β1 complexes, the supernatant from the third round was immunoprecipitated with prodomain antibody TW7–16B4 or control mouse IgG coupled to Sepharose. As a control, lysates from the same number of cells were alsoimmunoprecipitated in parallel. Immunoprecipitates of cell surface proTGF-β1 complexes, intracellular proTGF-β1 complexes, or total lysates (left, middle, and right panels, respectively) were heated in sample buffer and subjected to reducing SDS-PAGE and WB with Flag antibody. The results show that the cell surface LRRC33 chimera 33-G-X5 complexedwith proTGF-β1 is almost entirely of the high molecular weight form, its intracellular counterpart is largely of the low molecular weight form, and complexes with proTGF-β in total lysate contain similar amounts of the two forms. (D) and (E) are identical to the experiments on IP and WB of LRRC33 complexes with proTGF-β from macrophages and spleen cells shown in main text Figure 4J,K, but include the full molecular weight range resolved in SDS-PAGE.
Related to main text Fig. 5. (A-D) An independent experiment of wBM transplantation into lethallyirradiated WT recipients (10 weeks). (E-H) An independent experiment of wBM transplantation into lethally irradiated Lrrc33−/− recipients (10 weeks). (B…F)Kaplan-Meier survival curve of transplanted mice, **: P<0.01 (Mantel—Cox) test. (I-J): Clinical scores for transplanted WT recipients from the experiment shown in main text Fig. 5. (C, D, G, H, I, J) Clinical scores. See Fig. 5 legend for other details. (K) The immunophenotypes of resident KO microglia and WT donor derived microglia in chimeric brain from the experiment shown in main text Fig.5H and I. Total cells and mouse # are shown in upper right. Microglia shown in Fig. 5H are from mice #1638, #4, and #1630 and appear in the rightmost column in each group. This column was chosen because it contains the WT recipient with the largest number of KO donor microglia. See Fig. 5 legend for other details.
Related to main text Fig. 6. (A) Details of the heat map of Lrrc33−/− microglia signature in 3 WT and 3 KO animals shown in Fig. 6A with gene names. CD4 expression (red arrow) is described in main text. (B) Unsupervised Global Hierarchical Clustering shows the WT and Lrrc33 knockout (KO) samples fall into 2 separate groups. (C) Enrichment score curves for the GSEA results shown in main text Fig. 6D. Also included are results for the “Apoptosis” gene set, which nearly reached significance, and for the “Reactive oxygen species” gene set, which showed no difference between WT and KO. (D)Results for all of the genes included in the “TGF-beta signaling” gene set.
Highlights:
Expression and activation of TGF-β in myeloid cells require association with LRRC33
Lrrc33−/− mice develop paraparesis, neurodegeneration, and have reactive microglia
Bone marrow transplantation halts disease progression and restores microglia
TGF-β activation is highly localized within distinctive cellular milieus in the CNS
The cell surface milieu molecule LRRC33 selectively modulates downstream signaling outcomes and through direct association with the TGF-β ligand in specific cell types.
Acknowledgements.
We thank Dvora Ghitza and Klaus Rajewsky for production of chimeric mice, Ed Lamperti for early work, Allison Bialas for advice, Nick Andrews and Sophie Griswold of the Neurodevelopmental Behavioral Core, Boston Children’s Hospital, the Molecular Biology Core at the Dana-Farber Cancer Institute, Ana Zguro for help with animal irradiation and tail vein injections, Christina Welsh for help with microglia isolation, and Margaret Nielsen for illustrations. Supported by NIH grant AR067288, the Harvard NeuroDiscovery Center, and the Harvard Stem Cell Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions. Y.Q., B.S.G., and D.A.R. designed and conducted experiments and wrote the paper. W.M., R.W., A.J., D.S., and C.F. conducted experiments. M.M. and R.T.B analyzed data. B.S. and D.J.R supervised experiments and wrote the paper. B.S., D.J.R, C.L. and T.A.S. designed, supervised and wrote the paper.
Declaration of interests disclosure: Y.Q., A.J., C.L., and T.A.S. are inventors on a patent on LRRC33 that may be licensed. T.A.S. owns stock in Scholar Rock.
References
(yellow highlight denotes referenced in Star Methods)
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, and Rifkin DB (1994). An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem 216, 276–284. [DOI] [PubMed] [Google Scholar]
- Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, and Munger JS (2009). Mice that lack activity of αVβ6- and αVβ8-integrins reproduce the abnormalities of TGFβ1- and TGFβ3-null mice. J Cell Sci 122, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, Lu W, and Jones EY (2006). A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr 62, 1243–1250. [DOI] [PubMed] [Google Scholar]
- Barash S, Wang W, and Shi Y (2002). Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem Biophys Res Commun 294, 835–842. [DOI] [PubMed] [Google Scholar]
- Bilimoria PM, and Stevens B (2015). Microglia function during brain development: New insights from animal models. Brain Res 1617, 7–17. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. (2014). Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho BS, and Irizarry RA (2010). A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan J, Walshe K, Alsbury S, Hokamp K, O’Keeffe S, Okafuji T, Miller SF, Tear G, and Mitchell KJ (2007). The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BCM Genomics 8, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, and Springer TA (2017). Force interacts with macromolecular structure in activation of TGF-β. Nature 542, 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell RA, Sanjabi S, Wrzesinski SH, and Licona-Limón P (2010). The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 10, 554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, and Speed TP (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. [DOI] [PubMed] [Google Scholar]
- Khalil N, Bereznay O, Sporn M, and Greenberg AH (1989). Macrophage production of transforming growth factor β and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 170, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer R, Streit WJ, Toyka KV, Kreutzberg GW, and Hartung HP (1995). Transforming growth factor-β1: a lesion-associated cytokine of the nervous system. Int J Dev Neurosci 13, 331–339. [DOI] [PubMed] [Google Scholar]
- Lee JK, and Tansey MG (2013). Microglia isolation from adult mouse brain. Methods Mol Biol 1041, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang Z, Chai L, Che Y, Min S, and Yang R (2013). Identification and characterization of a unique leucine-rich repeat protein (LRRC33) that inhibits Toll-like receptor-mediated NF-κB activation. Biochem Biophys Res Commun 434, 28–34. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen H, Huang Y, Qiu Y, and Peng Y (2016). Transforming growth factory-β1 acts via TβR-I on microglia to protect against MPP(+)-induced dopaminergic neuronal loss. Brain Behav Immun 51, 131–143. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, and de Vellis J (1980). Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85, 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, and Hynes RO (2005). Selective ablation of αv integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132, 165–176. [DOI] [PubMed] [Google Scholar]
- Mi LZ, Grey MJ, Nishida N, Walz T, Lu C, and Springer TA (2008). Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry 47, 10314–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitjans F, Sander D, Adan J, Sutter A, Martinez JM, Jaggle CS, Moyano JM, Kreysch HG, Piulats J, and Goodman SL (1995). An anti-α v-integrin antibody that blocks integrin function inhibits the development of a human melanoma in nude mice. J Cell Sci 108 (Pt 8), 2825–2838. [DOI] [PubMed] [Google Scholar]
- Mobley AK, Tchaicha JH, Shin J, Hossain MG, and McCarty JH (2009). β8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci 122, 1842–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez P.a., Ding J, Peng I, Sebrell A, Caplazi P, et al. (2014). NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 509, 235–239. [DOI] [PubMed] [Google Scholar]
- Oida T, and Weiner HL (2010). TGF-β induces surface LAP expression on murine CD4 T cells independent of Foxp3 induction. PLoS One 5, e15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson IB, and Rifkin DB (2016). Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, and Martin JE (1997). Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8, 711–713. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, and Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, and ter Meulen V (1991). Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A 88, 7438–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DA, Amha; Henderson Neil Cowan (2014). Methods and Compositions for Treating and Preventing Disease Associated with αVβ8 Integrin, W.I.P. Organization, ed. (USA: The Regents of the University of California; ). [Google Scholar]
- Smyth GK, Michaud J, and Scott HS (2005). Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21, 2067–2075. [DOI] [PubMed] [Google Scholar]
- Springer TA (1980). Cell-surface differentiation in the mouse. Characterization of “jumping” and “lineage” antigens using xenogeneic rat monoclonal antibodies In Monoclonal antibodies, Kennett RH, McKearn TJ, and Bechtol KB, eds. (New York: Plenum Press; ), pp. 185–217. [Google Scholar]
- Su X, Mei S, Liang X, Wang S, Liu J, Zhang Y, Bao Y, Chen Y, Che Y, Chunhua Zhao R, et al. (2014). Epigenetically modulated LRRC33 acts as a negative physiological regulator for multiple Toll-like receptors. J Leukoc Biol 95, 1–10. [DOI] [PubMed] [Google Scholar]
- Subramanian AT, P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES; Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale JM, S; Hurme M; Keski-Oja J (1994). Induction of transforming growth factor β1 and its receptor expression during myeloid leukemia cell differentiation. Cell Growth Differ 5, 1309–1319. [PubMed] [Google Scholar]
- Tsunawaki S, Sporn m., Ding a., and Nathan C (1988). Deactivation of macrophages by transforming growth factor-β. Nature 334, 260–264. [DOI] [PubMed] [Google Scholar]
- Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, and Unutmaz D (2009). Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 106, 13439–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhu J, Dong X, Shi M, Lu C, and Springer TA (2012). GARP regulates the bioavailability and activation of TGF-β. Mol Biol Cell 23, 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb PH, Simon KJ, Rayhorn P, Yang WJ, Leone DR, Dolinski BM, Pearse BR, Yokota Y, Kawakatsu H, Atakilit A, et al. (2004). Function-blocking integrin αVβ6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem 279, 17875–17887. [DOI] [PubMed] [Google Scholar]
- Wirenfeldt M, Babcock AA, and Vinters HV (2011). Microglia - insights into immune system structure, function, and reactivity in the central nervous system. Histol Histopathol 26, 519–530. [DOI] [PubMed] [Google Scholar]
- Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, Friedman BA, Pappu R, Scearce-Levie K, and Ouyang W (2017). Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol 18, 633–641. [DOI] [PubMed] [Google Scholar]
- Wu BX, Li A, Lei L, Kaneko S, Wallace C, Li X, and Li Z (2017). Glycoprotein A repetitions predominant (GARP) positively regulates transforming growth factor (TGF) β3 and is essential for mouse palatogenesis. J Biol Chem 292, 18091–18097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YC, K; Sloan SA; Bennett ML; Scholze AR; O’Keeffe S; Phatnani HP; Guarnieri P; Caneda C; Ruderisch N; Deng S; Liddelow SA; Zhang C; Daneman R; Maniatis T; Barres BA; Wu J (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zöller T, Krieglstein K, and Spittau B (2015). TGFβ1 inhibits IFNγ-mediated microglia activation and protects mDA neurons from IFNγ-driven neurotoxicity. J Neurochem 134, 125–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Fig. 2. (A, B) mAb specificity and co-expression requirements. 293T cells were transfected with the indicated Flag-tagged constructs with or without proTGF-β1, stained with the indicated antibodies using indirect immunofluorescence and subjected to flow cytometry. Inset numbers show mean fluorescence intensity. (C) mAb clone 1/8.8 does not cross react with mouse LRRC33. 293T cells were co-transfected with human proTGF-β1 and human or mouse Flag-tagged LRRC33. Cell lysates, or immunoprecipitates (IP) with mAb clone 1/8.8 from lysates, were subjected to 10% reducing SDS-PAGE and Western blot (WB) as indicated.
Movie 1. Related to Fig. 3D. Reconstructions of WT (Movie 1) and Lrrc33−/− (Movie 2) microglia from the M1 motor cortex region of 21-day-old mice. Microglia are stained with antibodies to Iba1 (green fluorescence) and CD68 (magenta fluorescence) as described in the Fig. 4C legend and the movies correspond to the projection view shown in Fig. 4C. Grids in the movies are 5 μm.
Movie 2. Related to Fig. 3D. Reconstructions of WT (Movie 1) and Lrrc33−/− (Movie 2) microglia from the M1 motor cortex region of 21-day-old mice. Microglia are stained with antibodies to Iba1 (green fluorescence) and CD68 (magenta fluorescence) as described in the Fig. 4C legend and the movies correspond to the projection view shown in Fig. 4C. Grids in the movies are 5 μm.
Supplemental dataset 1. Related to Fig. 1. Excel spreadsheet containing the FPKM values for genes shown in Fig. 1H and additionally Itgb6. Data is from {Zhang, 2014 #24481}.
Supplemental dataset 2. Related to Fig. 6. Excel spreadsheet containing microarray data for all genes analyzed in WT AND KO microglia from 3 animals each.
Related to Fig. 3. (A and B) Lrrc33 knockout. (A) construct. (B) Genotyping results showing the WT band (PCR product using primers 7 and 8) and knockout (KO) band (PCR product using primers 7 and 9). (C and D) Garp knockout. (C) construct. (D) Genotyping results showing the WT band (PCR product using primers TUF and TUR) and knockout (KO) band(PCR product using primers LacInf and LacInR). VG18567
Related to Fig. 3. (A) SHIRPA tests showing score for each animal (Black dots: WT mice, red dots: Lrrc33−/− mice). Coat appearance: 0 = normal, 1 = abnormal; Limb grasping: 0 =present, 1 = absent; Tail elevation: 0 = dragging, 1 = horizontal extension, 2 = elevated/straub tail; Trunk curl: 0 = present, 1 = absent; Morphology: 0 = normal, 1 = abnormal; Defecation: 0 = present, 1 = absent; Gait: 0 = fluid movement, 1 = lack of fluidity; Body position: 0 = active, 1 = inactive, 2 = excessive activity; Locomotor activity: total # of squares entered in 30s. Statistical analysis for Grip strength, Locomotor activity, and Open field test used unpaired Student’s t-test, n=5 mice, bars show mean ± SEM. Statistical analysis for Coat appearance, Limb grasping, Tail elevation, Trunk curl, Morphology, Defecation, Gait and Body position used the Z score for 2 population proportions; n=5 mice. *: P<0.05, **: P<0.01, ***: P<0.001, ****: P<0.0001. (B) Lrrc33−/− mice have enlarged bladders compared to WT mice (red arrows).
Related to Figure 3. (A) Variation among micein neuron number revealed by NeuN staining in somatomotor cortex region M1. Sections shown are from 3 different WT and Lrrc33−/− 4 monthold mice, and were stained with NeuN antibody as described in Fig. 3 legend. (B-D) Somatosensory cortex region is relatively normal in 4-month old Lrrc33−/− mice. Immunostaining with antibodiesto (B) NeuN (neuron marker), (C) OSP (myelin marker) and (D) NF-H (axon marker) in somatosensory cortex. Quantitation in lower row shows mean ± SEM (n=3, *P <0.05, unpaired Student’s t-test) for three mice from measurements averaged over 2–3 sections per mouse with 1–2 images per section. Although some significant differences arepresent, the differences and their significance are lesser than in the M1 region (Fig. 3). A and B: M(molecular) and II/III layers of the cerebral cortex. Scale bars: 100 μm. (E) Apparent lack of demyelination and axon degeneration at 3 month in spinal cord dorsal column (area above dashed line). Left panel: H…E stain. Middle panel: Bielschowsky's silver stain. Right panel: Luxol fast blue stain. Scale bar: 1 mm. (F) Lack of obvious demyelination and axon degeneration at 3 month in corticospinal tract of brain stem (below dashed line). Left: Bielschowsky's silver stain. Right: Luxol fast blue stain. Scale bar: 1 mm.
Related to main text Fig. 4. (A) Expression of Flag-tagged mouse LRRC33-GARP chimera 33-G-X5 and proTGF-β1 in L1.2 stable transfectants. Surface expression was detected by flow cytometry with APC-anti-Flag and Pacific blue-anti-TGF-β1prodomain (clone TW7–16B4) or identically conjugated mouse IgG as control (grey curves). Inset numbers show mean fluorescence intensity. (B) IP studies on the LRRC33-GARP chimera 33-G-X5 suggest24 processing of the chimera to a higher molecular weight glycoform when it associates with proTGF-β1. Lysatesof L1.2 transfectants co-expressing mouse proTGF-β and Flag-tagged mouse LRRC33 chimera 33-G-X5 were immunoprecipitated (IP) with mouse proTGF-β1 prodomain antibody TW7–16B4 or control mouse IgG. Immunoprecipitates or lysates were subjected to non-reducing or reducing SDS 7.5% PAGE and Western blotting (WB) with anti-Flag (rabbit clone SIGI-25) or the hamster antibody to denatured mouse LRRC33 {Noubade, 2014 #22620}. Both anti-Flag and anti-denatured LRRC33 reacted with the LRRC33-proTGF-β1 complex (~320 kDa) under non-reducing conditions (top panels) and LRRC33 bands of ~120 kDa and ~90 kDa under reducing conditions. The anti-prodomain precipitate contains a much larger proportion of the ~120 kDa material than in the whole lysate. Since glycoproteins increase in molecular weight during carbohydrate processing, the results suggest that association with proTGF-β is required for LRRC33 chimera transit from the endoplasmic reticulum to the Golgi and carbohydrate processing. The LRRC33 polypeptide has a mass of 74,294 Da and 10 predicted N-glycosylation sites, consistent with a putative intracellular high mannose form of 90 kDa and substantial increase in size to ~120 kDa upon conversion to complex carbohydrates. (C) The ~120 kD glycoform of LRRC33 chimera 33-G-X5 localizes to the cell surface. Prodomainantibody TW7–16B4 (20ug/ml) or control IgG was incubated with L1.2 transfectants for 2 hrs at 4oC to bind cellsurface proTGF-β1. After 3 washes to remove unbound antibody, cells were lysed. Lysates were incubated with protein G beads to pull down antibody-proTGF-β1 complexes, and after removal of beads, supernatants were subjected to two further rounds of incubation with protein G beads to completely deplete pre-formed antibody-proTGF-β1 complexes. (No bands from the 2nd and 3rd pull-down were observed in WB, not shown). To isolate intracellular proTGF-β1 complexes, the supernatant from the third round was immunoprecipitated with prodomain antibody TW7–16B4 or control mouse IgG coupled to Sepharose. As a control, lysates from the same number of cells were alsoimmunoprecipitated in parallel. Immunoprecipitates of cell surface proTGF-β1 complexes, intracellular proTGF-β1 complexes, or total lysates (left, middle, and right panels, respectively) were heated in sample buffer and subjected to reducing SDS-PAGE and WB with Flag antibody. The results show that the cell surface LRRC33 chimera 33-G-X5 complexedwith proTGF-β1 is almost entirely of the high molecular weight form, its intracellular counterpart is largely of the low molecular weight form, and complexes with proTGF-β in total lysate contain similar amounts of the two forms. (D) and (E) are identical to the experiments on IP and WB of LRRC33 complexes with proTGF-β from macrophages and spleen cells shown in main text Figure 4J,K, but include the full molecular weight range resolved in SDS-PAGE.
Related to main text Fig. 5. (A-D) An independent experiment of wBM transplantation into lethallyirradiated WT recipients (10 weeks). (E-H) An independent experiment of wBM transplantation into lethally irradiated Lrrc33−/− recipients (10 weeks). (B…F)Kaplan-Meier survival curve of transplanted mice, **: P<0.01 (Mantel—Cox) test. (I-J): Clinical scores for transplanted WT recipients from the experiment shown in main text Fig. 5. (C, D, G, H, I, J) Clinical scores. See Fig. 5 legend for other details. (K) The immunophenotypes of resident KO microglia and WT donor derived microglia in chimeric brain from the experiment shown in main text Fig.5H and I. Total cells and mouse # are shown in upper right. Microglia shown in Fig. 5H are from mice #1638, #4, and #1630 and appear in the rightmost column in each group. This column was chosen because it contains the WT recipient with the largest number of KO donor microglia. See Fig. 5 legend for other details.
Related to main text Fig. 6. (A) Details of the heat map of Lrrc33−/− microglia signature in 3 WT and 3 KO animals shown in Fig. 6A with gene names. CD4 expression (red arrow) is described in main text. (B) Unsupervised Global Hierarchical Clustering shows the WT and Lrrc33 knockout (KO) samples fall into 2 separate groups. (C) Enrichment score curves for the GSEA results shown in main text Fig. 6D. Also included are results for the “Apoptosis” gene set, which nearly reached significance, and for the “Reactive oxygen species” gene set, which showed no difference between WT and KO. (D)Results for all of the genes included in the “TGF-beta signaling” gene set.
Data Availability Statement
The probe (exon level) and gene level microarray data for microglia freshly isolated from WT and Lrrc33−/− mice have been deposited at GEO as accession number GSE112604. Supplemental Figure Legends