

Abstract
Background
Crassulacean acid metabolism (CAM) enhances plant water-use efficiency through an inverse day/night pattern of stomatal closure/opening that facilitates nocturnal CO2 uptake. CAM has evolved independently in over 35 plant lineages, accounting for ~ 6% of all higher plants. Agave species are highly heat- and drought-tolerant, and have been domesticated as model CAM crops for beverage, fiber, and biofuel production in semi-arid and arid regions. However, the genomic basis of evolutionary innovation of CAM in genus Agave is largely unknown.
Results
Using an approach that integrated genomics, gene co-expression networks, comparative genomics and protein structure analyses, we investigated the molecular evolution of CAM as exemplified in Agave. Comparative genomics analyses among C3, C4 and CAM species revealed that core metabolic components required for CAM have ancient genomic origins traceable to non-vascular plants while regulatory proteins required for diel re-programming of metabolism have a more recent origin shared among C3, C4 and CAM species. We showed that accelerated evolution of key functional domains in proteins responsible for primary metabolism and signaling, together with a diel re-programming of the transcription of genes involved in carbon fixation, carbohydrate processing, redox homeostasis, and circadian control is required for the evolution of CAM in Agave. Furthermore, we highlighted the potential candidates contributing to the adaptation of CAM functional modules.
Conclusions
This work provides evidence of adaptive evolution of CAM related pathways. We showed that the core metabolic components required for CAM are shared by non-vascular plants, but regulatory proteins involved in re-reprogramming of carbon fixation and metabolite transportation appeared more recently. We propose that the accelerated evolution of key proteins together with a diel re-programming of gene expression were required for CAM evolution from C3 ancestors in Agave.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4964-7) contains supplementary material, which is available to authorized users.
Keywords: Crassulacean acid metabolism, Photosynthesis, Comparative genomics, Transcriptome, Positive selection, Circadian rhythm
Background
Among the three modes of photosynthesis in higher plants, the C3 pathway is the most ancient and common, occurring in approximately 90% of higher plant species. C4 and CAM photosynthesis, which account for approximately 3 and 6% of higher plant species, respectively, are evolutionarily derived from C3 photosynthesis and are believed to have arisen in response to selective pressures imposed by global reductions in atmospheric CO2 concentration (C4) and water limitation (CAM) [1, 2]. Both C4 and CAM plants capture CO2 via an initial carboxylation reaction catalyzed outside the chloroplast by phosphoenolpyruvate carboxylase (PEPC), which then subsequently delivers the captured CO2 at increased concentration to ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) in the chloroplast. While C4 operates via a spatial separation of carboxylases in different cell types, CAM operates via a temporal day/night separation of RuBisCO and PEPC with net CO2 uptake shifted predominantly to the night. Nocturnal CO2 uptake is accompanied by an inverse (compared to C3 and C4) day/night pattern of stomatal closure/opening in CAM that results in improved water-use efficiency (i.e., CO2 fixed per molecule of H2O lost) that is six-fold higher than C3 plants and 3-fold higher than C4 plants under comparable conditions [3].
Circadian regulation of gene expression has been implicated as a core component in the diel re-programming of metabolism that distinguishes CAM from C3 and C4 photosynthesis [4, 5]. For instance, the nocturnal activation of PEPC via phosphorylation in CAM plants is catalysed by a dedicated PEPC kinase (PPCK), the transcript abundance of which is regulated by the circadian clock [5, 6]. A number of clock genes which were examined in the facultative CAM species Mesembryanthemum crystallinum indicated both conserved and divergent functions of genes encoding components of the core oscillator and the clock output pathways in this species [7]. In Opuntia ficus-indica (a constitutive CAM species), a transcriptomics study identified several genes implicated in the CAM biochemical pathway and in the circadian clock that displayed a unique 12-h periodicity [8] different from C3 species like Arabidopsis. Other work has suggested that the rhythmic expression of genes required for CAM is a consequence of diel changes in CAM-defining metabolites, such as malic acid [3]. Together, these studies suggest that modifications to the circadian clock, including both input and output pathways, might be critical to CAM evolution. Still, how the circadian clock was integrated within signalling and core biochemical components of CAM during the evolution of this pathway remains unestablished. Genome-wide gene expression profiling in model species has greatly facilitated our understanding of gene regulatory networks that are relevant to circadian regulation [9]. In this work, we develop a genome-wide approach to investigate the evolution of the core metabolic and regulatory elements of CAM via cross-species comparisons.
CAM and C4 photosynthesis are thought to have evolved from C3 ancestors multiple times in response to limitations in CO2 and water. It has been proposed that a propensity for frequent mutation or selection of certain regulatory genes during the early stages of evolution underpinned adaptations to environmental stress conditions [1]. Several studies in C4 have uncovered positive selection of key components of photosynthesis [10, 11], despite the conserved nature of many of these genes. For example, enzymatic kinetics and structure modelling of positively-selected residues of RuBisCO subunits in Flaveria indicated that these key amino acid substitutions were relevant to the functional diversification of C4 [11]. Although little has been characterized in CAM plants, the systematic analysis of positive selection in genes implicated in the circadian clock, photosynthesis, and CAM biochemistry could provide insight to unlock the mechanism(s) underlying CAM evolution.
Agave species are constitutive CAM species and many are important economic crops for beverage and fiber production [12, 13]. The water conserving properties of CAM have also highlighted the value of Agave as potential dedicated bioenergy feedstocks in semi-arid regions [14, 15]. Hence, a fundamental understanding of regulatory pathways underlying CAM in Agave is critical for efforts directed at engineering this pathway into C3 crops to improve water-use efficiency [13, 16]. All of the enzymes required for C4 and CAM appear to be homologs of ancestral forms found in C3 species [17], yet the specific genomic origins and genetic regulation of diel reprogramming of metabolism that distinguishes CAM from C3 and C4 photosynthesis are largely unknown [18]. In this study, we investigated the molecular evolution of CAM in Agave using an approach that integrated gene co-expression networks, comparative genomics, and protein structure analysis. Our findings demonstrate that 1) core metabolic components required for CAM have ancient genomic origins traceable to non-vascular plants, 2) regulatory proteins required for diel re-programming of metabolism have a more recent origin shared among C3, C4, and CAM species and 3) accelerated evolution of key proteins together with a diel re-programming of gene expression were required for the evolution of CAM in Agave.
Results
CAM physiology and identification of co-expression modules relevant to CAM
Using the quantitative gene expression data obtained from the RNA-Seq analysis of 15 tissues, including mature leaf (sampled at 8 time-points over a diel cycle), young leaf (3 time-points), root, meristem, rhizome, and stem in A. americana cultivar ‘Marginata’ [14], we created a gene co-expression network that was partitioned into 16 co-expression modules, with each displaying distinctive diel patterns (Fig. 1). Gene ontology (GO) enrichment analysis identified biological processes over-represented (p < 0.05) in each of these co-expression modules (Additional file 1: Figure S1). We characterized the GO enrichment results together with expression profiles to identify potential modules for CAM. For example, module ‘M11’, containing 1509 transcripts, was significantly (p < 0.05) associated with CAM-defining nocturnal net CO2 uptake (9 pm – 6 am) (Fig. 1a), and was over-represented by biological processes relevant to stomatal movement, carboxylation, and signal transduction (Table 1). Module M11 contains some key genes involved in CAM metabolism, including PHOSPHOENOLPYRUVATE CARBOXYLASE KINASE 1 (PPCK1), which regulates the temporal activation of nocturnal CO2 uptake. Since genes within module M11 are relevant to nocturnal carboxylation and stomatal movement, we propose that this module provides a molecular signature for temporal reprogramming of metabolism underpinning CAM.
Fig. 1.
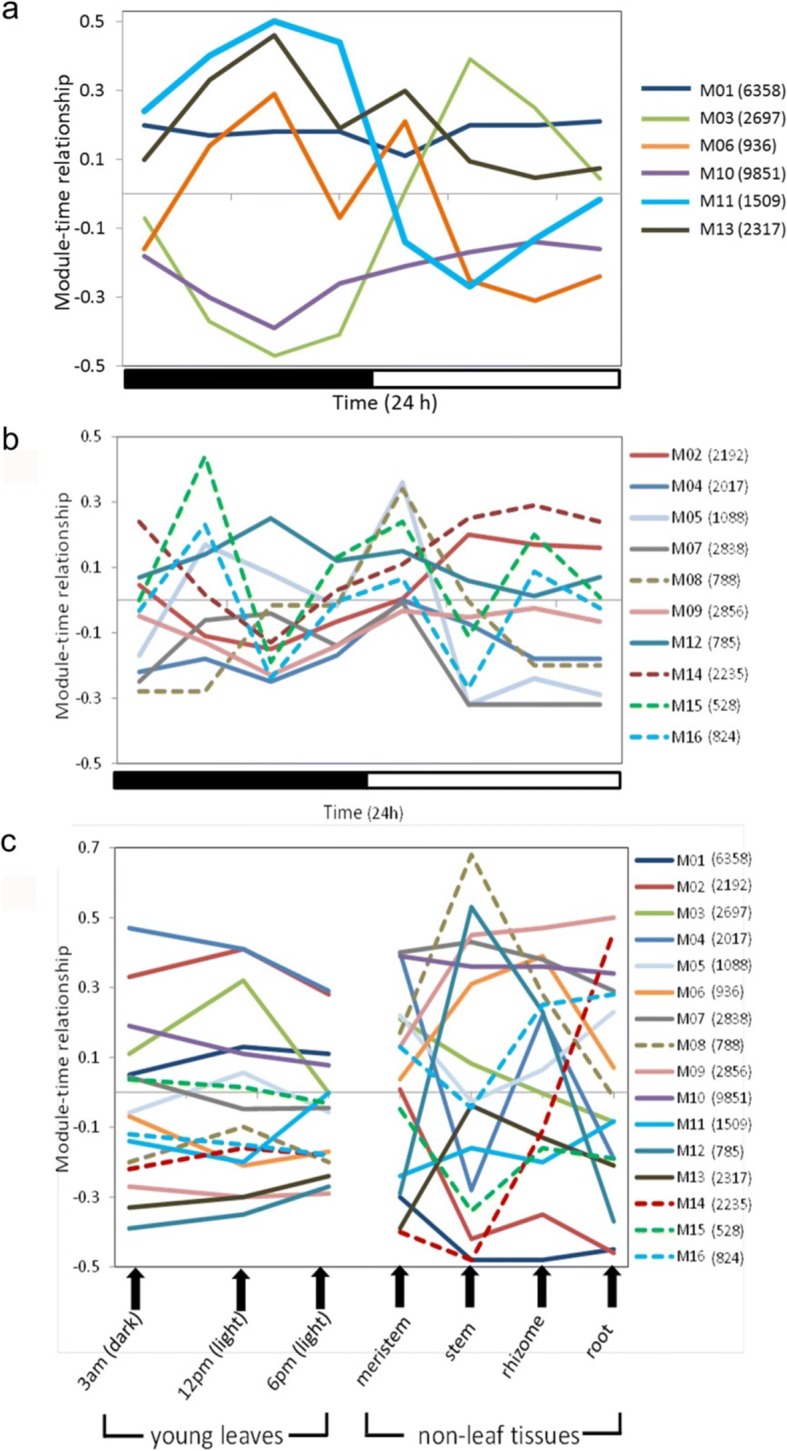
Temporal expression of CAM and gene co-expression modules in Agave americana. a Diel expression pattern of selected co-expression modules in mature leaf, as identified from network analysis of RNA-Seq data with relevance to CAM physiology. The black and white bars indicate nighttime and daytime, respectively. b Diel expression pattern of other modules in mature leaf with distinct profiles. c Expression pattern in young leaves sampled at 3 time points and non-leaf tissues sampled at one time point (9 am). The number in the parentheses is the number of transcripts in each individual module
Table 1.
Biological processes over-represented (p < 0.02) in the co-expression module M11
| GO ID | GO Term | Corrected P-Value |
|---|---|---|
| GO:0009738 | Abscisic acid-activated signaling pathway | 4.2E-08 |
| GO:0016311 | Dephosphorylation | 6.2E-08 |
| GO:0071215 | Cellular response to abscisic acid stimulus | 9.0E-08 |
| GO:0009737 | Response to abscisic acid | 9.3E-08 |
| GO:0097306 | Cellular response to alcohol | 5.1E-07 |
| GO:0071396 | Cellular response to lipid | 6.6E-07 |
| GO:0006470 | Protein dephosphorylation | 7.0E-07 |
| GO:0097305 | Response to alcohol | 7.8E-07 |
| GO:0009611 | Response to wounding | 4.6E-06 |
| GO:0009745 | Sucrose mediated signaling | 4.8E-04 |
| GO:0009753 | Response to jasmonic acid | 6.3E-04 |
| GO:0019722 | Calcium-mediated signaling | 1.7E-03 |
| GO:0009788 | Negative regulation of abscisic acid-activated signaling pathway | 1.8E-03 |
| GO:1901420 | Negative regulation of response to alcohol | 1.8E-03 |
| GO:0009694 | Jasmonic acid metabolic process | 2.5E-03 |
| GO:1901419 | Regulation of response to alcohol | 2.6E-03 |
| GO:0009787 | Regulation of abscisic acid-activated signaling pathway | 2.6E-03 |
| GO:0009968 | Negative regulation of signal transduction | 2.7E-03 |
| GO:0010648 | Negative regulation of cell communication | 2.7E-03 |
| GO:0023057 | Negative regulation of signaling | 2.7E-03 |
| GO:0019856 | Pyrimidine nucleobase biosynthetic process | 2.7E-03 |
| GO:0010224 | Response to UV-B | 5.0E-03 |
| GO:0019932 | Second-messenger-mediated signaling | 6.0E-03 |
| GO:0010243 | Response to organonitrogen compound | 6.1E-03 |
| GO:0006835 | Dicarboxylic acid transport | 6.3E-03 |
| GO:0009875 | Pollen-pistil interaction | 6.3E-03 |
| GO:0010200 | Response to chitin | 6.5E-03 |
| GO:0009695 | Jasmonic acid biosynthetic process | 7.0E-03 |
| GO:0006206 | Pyrimidine nucleobase metabolic process | 7.4E-03 |
| GO:0006984 | ER-nucleus signaling pathway | 8.1E-03 |
| GO:0071324 | Cellular response to disaccharide stimulus | 8.3E-03 |
| GO:0071329 | Cellular response to sucrose stimulus | 8.3E-03 |
| GO:0042538 | Hyperosmotic salinity response | 9.6E-03 |
| GO:0030968 | Endoplasmic reticulum unfolded protein response | 1.1E-02 |
| GO:0015743 | Malate transport | 1.2E-02 |
| GO:0034620 | Cellular response to unfolded protein | 1.3E-02 |
| GO:0035967 | Cellular response to topologically incorrect protein | 1.3E-02 |
| GO:0010118 | Stomatal movement | 1.3E-02 |
| GO:0006986 | Response to unfolded protein | 1.3E-02 |
| GO:0048544 | Recognition of pollen | 1.5E-02 |
| GO:0008037 | Cell recognition | 1.6E-02 |
| GO:0009827 | Plant-type cell wall modification | 1.8E-02 |
| GO:0015740 | C4-dicarboxylate transport | 1.9E-02 |
Orthologous gene groups among CAM, C3 and C4 species
To understand the evolutionary origins and possible shared trajectories of module M11 and other CAM components between different photosynthetic lineages, we performed comparative genomics analysis of 15 plant species, including CAM, C3, C4, and non-vascular plant (NVP) species (Fig. 2a). Specifically, we identified ortholog clades through OrthoMCL analysis, such as clade NVP:C3:CAM:C4, which contains ortholog groups shared by NVP, C3, CAM, and C4 species, and clade C3:CAM:C4, which contains ortholog groups shared only by C3, CAM, and C4 species (Fig. 2b). The genes in the Agave (CAM) species were distributed mainly in three ortholog clades: NVP:C3:CAM:C4, C3:CAM:C4, and CAM-only, whereas the genes found in C4 species were distributed mainly in four ortholog clades: NVP:C3:CAM:C4, C3:CAM:C4, C3:C4, and C4-only (Additional file 2: Table S1), indicating that C4 evolution has one additional major genomic event, as represented by clade C3:C4, relative to CAM evolution.
Fig. 2.

Comparative analysis of protein sequences among CAM and non-CAM plant species. a Plant species used in comparative genomics analysis. b Ortholog groups in 15 plant species as identified by OrthoMCL. Number of ortholog groups is listed in each of the ortholog clades. c Percent of ortholog clade were predicted to be transcription factors in Agave americana. “a” and “b” indicate that the transcription factors are over-represented (p < 0.05) and under-represented (p < 0.05), respectively. d Percent of ortholog clade undergoing positive selection (i.e., nonsynonymous to synonymous substitution ratio (Ka/Ks) > 1, as calculated from Agave-Arabidopsis gene pairs with a sliding window of 50 amino acids). “*” indicates that the ortholog clade was over-represented (p < 0.0001) by Agave genes with Ka/Ks ratio greater than 1. Clade NVP:C3:CAM:C4 is shared by NVP, C3, CAM, and C4; NVP:C3:CAM shared only by NVP, C3, and CAM; C3:CAM:C4 shared only by C3, CAM, and C4; C3:CAM shared only by C3 and CAM; and CAM-only is specific to CAM species
Gene ontology enrichment analyses revealed that A. americana genes in clade NVP:C3:CAM:C4 were over-represented by biological processes relevant to primary metabolic processes (Additional file 3: Table S2), while those in clade C3:CAM:C4 were over-represented by regulatory processes (Additional file 4: Table S3). Notably, the core enzymes in C4 and CAM pathways belong to clade NVP:C3:CAM:C4, whereas the majority of the regulatory proteins belong to clade C3:CAM:C4 (Additional file 5: Table S4). Furthermore, genes in the M11 module were over-represented in the ortholog clade C3:CAM:C4 (Additional file 6: Table S5). Transcription factors were also over-represented (p < 0.05) in clade C3:CAM:C4, but under-represented (p < 0.05) in both NVP:C3:CAM:C4, and CAM-only clades (Fig. 2c). These results indicate that CAM evolution in Agave required genes that are shared across C3, C4 and CAM lineages to act as regulatory agents, whereas the core metabolic CAM machinery predates the C3-CAM-C4 divergence and is shared by NVP, C3, CAM, and C4 lineages.
Since the three CAM species (A. americana, A. deserti and A. tequilana) in this research are closely-related, the CAM-only ortholog groups may contains two types of CAM-specific genes: 1) specific to the Agave lineage and 2) conserved CAM-specific genes shared between Agave and other CAM lineages. To identify the conserved CAM-specific genes shared between Agave and other CAM lineages, the Agave genes in the CAM-only ortholog groups were compared with the protein tribes from the same 15 plant species as those used for ortholog group analysis, which were constructed by using TribeMCL [19]. In general, the protein tribes are equivalent to gene families, with each tribe containing multiple ortholog groups. The A. americana genes in both the CAM-only ortholog groups and CAM-only tribes were then compared with an extended list of CAM and non-CAM species using BLASTp, resulting in the identification of 13 A. americana genes that have homologs in other three independent CAM lineages (i.e., Kalanchoë fedtschenkoi, Ananas comosus, and Phalaenopsis equestris) but not in 21 non-CAM species (Additional file 7: Table S6). Some of these CAM-specific genes displayed variable diel expression patterns (Additional file 8: Figure S2).
Positive selection in CAM evolution
To study protein sequences evolution, we analyzed the non-synonymous to synonymous substitution ratio (Ka/Ks) of orthologous gene pairs between A. americana and three non-CAM species, including two C3 species (Arabidopsis thaliana and Oryza sativa) and one C4 species (Zea mays). A Ka/Ks ratio greater than 1 indicates positive selection or an acceleration of protein evolution [20, 21]. Ka/Ks analysis identified a set of 160 Agave genes that had protein sequence regions with Ka/Ks ratio greater than one, as compared with orthologous genes in the three non-CAM species (Additional file 9: Table S7), indicating that these genes experienced accelerated amino acid substitutions during the divergence between CAM and non-CAM species. The functionally annotated genes in this set included genes involved in circadian clock, starch and sugar metabolism, and decarboxylation (Additional file 10: Table S8). Importantly, we identified 94 Agave genes that had protein sequence regions with Ka/Ks ratio greater than 1, as compared with orthologous genes in the two C3 species, but not the orthologs in the C4 plant (Additional file 11: Table S9). The functionally annotated genes in this 94-gene set include PPCK1 (Fig. 3a, c, and e), a core regulator of nocturnal carboxylation, and PsI-D2 (Additional file 11: Table S9), which encodes a component of photosystem I essential for photosynthesis [22] and a chloroplast beta-amylase (CT-BMY).
Fig. 3.

Positive selection region in phosphoenolpyruvate carboxylase kinase (PPCK1). a Ka/Ks profile of Agave americana (Aa) versus Arabidopsis thaliana (At); b superimposed structures in Aa and At, with the positive selection region highlighted; (c) Ka/Ks profile of Aa versus Oryza sativa (Os); d superimposed structures in Aa and Os, with the positive selection region highlighted; e Ka/Ks profile of Aa versus Zea mays (Zm); f superimposed structures in Aa and Zm. An ATP substrate that may bind to the Aa PPCK1 is marked by an arrow. The proteins are colored in grey for Aa, blue for At, green for Os and red for Zm. g A snapshot of PPCK1 (Aam048341) structure model revealing the positive selected sites. The PPCK1 model bound with an ATP substrate (blue surface) is after a 1-us MD simulation. K42 (codon 124, at the C-end of β3) and N73 (codon219, at the N-end of β4) are located at the two strands connecting to the αC helix, the orientation of which is known to be involved in the activation of the kinase
To further consolidate the results of positive selection analysis based on Ka/Ks ratio, codon-based site analysis was performed to identify specific amino acid sites under positive selection. Out of the 94 genes that were revealed to experience positive selection by Ka/Ks ratio analysis (Additional file 11: Table S9), 64 genes were shown to carry at least one positively selected site, with posterior probabilities > 80% (Additional file 12: Table S10). We found 2 sites in PPCK1 (K42, N73) and 1 site in CT-BMY (E24) were supported by positive selection analysis based on both random effects likelihood (REL) and Fast, Unconstrained Bayesian AppRoximation (FUBAR) models (Table 2). Our 3-D protein structural modeling revealed that the positive-selection regions (sites) occur in important functional domains. The regions with Ka/Ks ratio > 1 of PPCK1 were located in the N-terminal domains responsible for ATP-binding (Fig. 3b and d), and K42 and N73 were likely involved in the activation of kinase (Fig. 3g). The positively selected regions of CT-BMY are responsible for substrate binding (Additional file 13: Figure S3). The clade C3:CAM:C4 was over-represented (p < 0.0001) by Agave genes with Ka/Ks ratio > 1 in the Agave-Arabidopsis gene pair comparison (Fig. 2d). Together, these results indicate that accelerated amino acid substitution has played a key role in the modification of proteins required for the light and carbon processing reactions of photosynthesis, as well as regulatory and signaling pathways in Agave.
Table 2.
The positively selected sites of PPCK1 and CT-BMY under models from HYPHY (REL, FUBAR). The sites were listed as positively selected sites if they had a posterior probability greater than 80%
| Codon(FUBAR) | α | β | β-α | Posterior Prob β > α | Emp. Bayes Factor | PSRF | Neff | |
| PPCK1 | 124* | 0.81 | 2.82 | 2.01 | 0.83 | 10.81 | 1.00 | 1292.65 |
| 219* | 0.81 | 2.71 | 1.90 | 0.82 | 10.27 | 1.00 | 1327.29 | |
| CT-BMY | 73* | 0.60 | 1.93 | 1.34 | 0.83 | 15.97 | 1.01 | 491.79 |
| Codon(REL) | E[dS] | E[dN] | Normalized E[dN-dS] | Posterior Probability | Bayes Factor | |||
| PPCK1 | 124* | 0.96 | 3.43 | 2.47 | 0.95 | 307.87 | ||
| 219* | 0.97 | 3.41 | 2.44 | 0.94 | 268.09 | |||
| 267 | 1.07 | 3.31 | 2.24 | 0.90 | 155.82 | |||
| 273 | 1.06 | 3.32 | 2.25 | 0.90 | 165.39 | |||
| 275 | 1.09 | 3.14 | 2.05 | 0.85 | 97.75 | |||
| 276 | 1.07 | 2.85 | 1.78 | 0.77 | 58.35 | |||
| CT-BMY | 73* | 0.94 | 0.85 | −0.0961 | 0.75 | 52.6 |
The * sites indicated they were identified by both models
Diel re-programming of gene expression between CAM and C3
To further examine the molecular basis of the diel re-programming of metabolism that underpins CAM, we performed a comparative analysis of time-course expression data between A. americana (CAM) and Arabidopsis thaliana (C3). We identified two clusters of Agave genes that exhibited shifts in day/night patterns of abundance relative to the corresponding orthologous genes in Arabidopsis. One cluster, containing 22 genes, showed a morning-to-night shift with alternative peak expression at night and morning between Agave and Arabidopsis, respectively (Fig. 4a). This gene set was over-represented (p < 0.05) by co-expression modules M01 and M11 (Table 3), with M11as the aforementioned molecular marker for CAM-associated nocturnal gene expression. Among the 22 genes showing the morning-to-night shift, 8 encode proteins with unknown function; the 14 annotated genes have functions related to circadian clock, photosynthetic electron transport, malate transport, stomatal movement, and redox homeostasis (Additional file 14: Table S11). The other cluster, containing 20 genes, showed an afternoon-to-night shift, with alternate peak expression during late night and afternoon in Agave and Arabidopsis, respectively (Fig. 4b). This gene set was over-represented by co-expression modules M06 (p < 0.05) and M13 (p < 0.01) (Table 3), both of which showed positive association with gene expression in mature leaves around midnight (Fig. 1b), suggesting that the genes in these two modules are involved in CAM related processes during the night. Among the 20 genes showing the afternoon-to-night shift, 7 encode proteins with unknown function; 13 have known functions related to signaling, sugar metabolism and light processing (Additional file 15: Table S12). In concern of multiple copies of Agave and Arabidopsis genes in an ortholog group, we assessed each ortholog group containing abovementioned genes in Fig. 4, and found 23 ortholog groups containing two genes, with a one-to-one relationship between Agave and Arabidopsis. We evaluated the phylogeny and diel expression pattern for ortholog groups with a total of more than two Agave and Arabidopsis genes, and found that the expression patterns of Agave genes in comparison with their orthologs in Arabidopsis displayed differential day-night patterns (Additional file 16: Figure S4 and Additional file 17: Figure S5), suggesting a functional diversification of gene family members between C3 and CAM plants.
Fig. 4.

Diel shift in gene expression pattern between Agave americana and Arabidopsis thaliana. a Morning-to-night shift with peak expression during morning in Arabidopsis and during night in Agave. b Afternoon-to-night shift with peak expression during afternoon in Arabidopsis and during late night in Agave. See gene annotation in Additional file 14: Table S11 and Additional file 15: Table S12
Table 3.
Distribution of gene co-expression modules in the gene sets with positive selection and diel shift in gene expression pattern, respectively, in Agave americana. Gene set 1a includes Agave genes with Ka/Ks ratio greater than one in the Agave-Arabidopsis, Agave-Oryza and Agave-Zea orthologous gene pairs. Gene set 1b includes Agave genes with Ka/Ks ratio greater than one in Agave-Arabidopsis and Agave-Oryza but not Agave-Zea pairs. Gene sets 2a and 2b includes Agave genes with morning-to-night shift and afternoon-to-night shift, respectively, in expression pattern as compared with the orthologous genes in Arabidopsis. The numbers represent the observed and expected (in parentheses) number of genes
| Co-expression module | Gene set 1a | Gene set 1b | Gene set 2a | Gene set 2b |
|---|---|---|---|---|
| M01 | 14 (27) | 17 (16) | 10 (4)* | 5 (4) |
| M02 | 13 (7)* | 4 (4) | 2 (1) | 0 (1) |
| M03 | 16 (7) ** | 10 (4)* | 0 (1) | 0 (1) |
| M04 | 1 (5) | 0 (3) | 0 (1) | 0 (1) |
| M05 | 0 (4) | 0 (2) | 1 (1) | 0 (1) |
| M06 | 1 (4) | 1 (2) | 0 (1) | 3 (1)* |
| M07 | 5 (10) | 1 (6) | 1 (1) | 2 (1) |
| M08 | 2 (2) | 0 (1) | 0 (0) | 0 (0) |
| M09 | 1 (9) | 2 (5) | 0 (1) | 1 (1) |
| M10 | 49 (28)** | 23 (16) | 0 (4) | 0 (4) |
| M11 | 5 (5) | 2 (3) | 4 (1)* | 0 (1) |
| M12 | 0 (3) | 0 (2) | 0 (0) | 0 (0) |
| M13 | 3 (11) | 2 (6) | 3 (2) | 10 (1)** |
| M14 | 2 (8) | 2 (5) | 0 (1) | 0 (1) |
| M15 | 0 (2) | 1 (1) | 0 (0) | 0 (0) |
| M16 | 1 (2) | 1 (1) | 1 (0) | 0 (0) |
| Non-module | 47 (25)** | 28 (15)* | 0 (4) | 0 (3) |
| Total | 160 | 94 | 22 | 21 |
*Overrepresentation (FDR adjusted p-value< 0.05, cumulative Poisson distribution) of co-expression modules in each category. **Overrepresentation (FDR adjusted p-value< 0.01, cumulative Poisson distribution) of co-expression modules in each category
Comparison of circadian clock pathway between CAM and C3
We compared the diel expression pattern of genes implicated in signal input to the clock (e.g., PHOT2, Phototropin-2), clock oscillation (e.g., CCA1, Circadian Clock Associated 1; TOC1, TIMING OF CAB EXPRESSION1), and regulatory output from the clock (e.g., RVE1, REVEILLE 1) between Agave (CAM) and Arabidopsis (C3). All known genes implicated in input to the clock and the central clock oscillator showed similar diel expression patterns between Agave and Arabidopsis. However, RVE1 exhibited patterns of peak transcript abundance that were substantially out of phase between CAM and C3, with the peak expression of RVE1 occurring at midnight in Agave and morning in Arabidopsis (Fig. 5 and Fig. 6a; Additional file 18: Table S13). These comparative analyses support the concept that the multiple independent evolutionary origins of CAM exploited an existing C3 multi-gene loop oscillator similar to that in Arabidopsis thaliana, and diel re-programming of metabolism was achieved via changes to genes like RVE1 that link metabolic output to the clock.
Fig. 5.
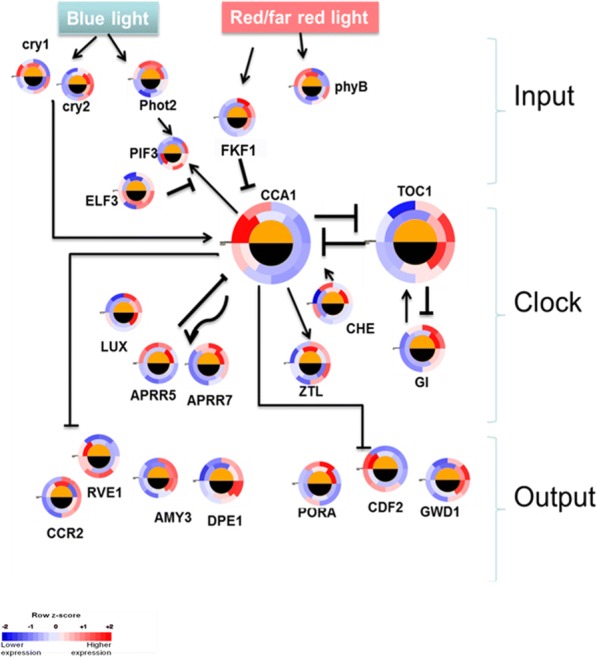
Diel gene expression pattern of circadian system genes in Agave americana and Arabidopsis thaliana. In the circular heatmaps, the outer and inner rings represent Agave americana and Arabidopsis thaliana, respectively. The black and white half-circles inside the circular heatmaps indicate night-time and day-time, respectively. Full gene names are listed in Additional file 18: Table S13
Fig. 6.
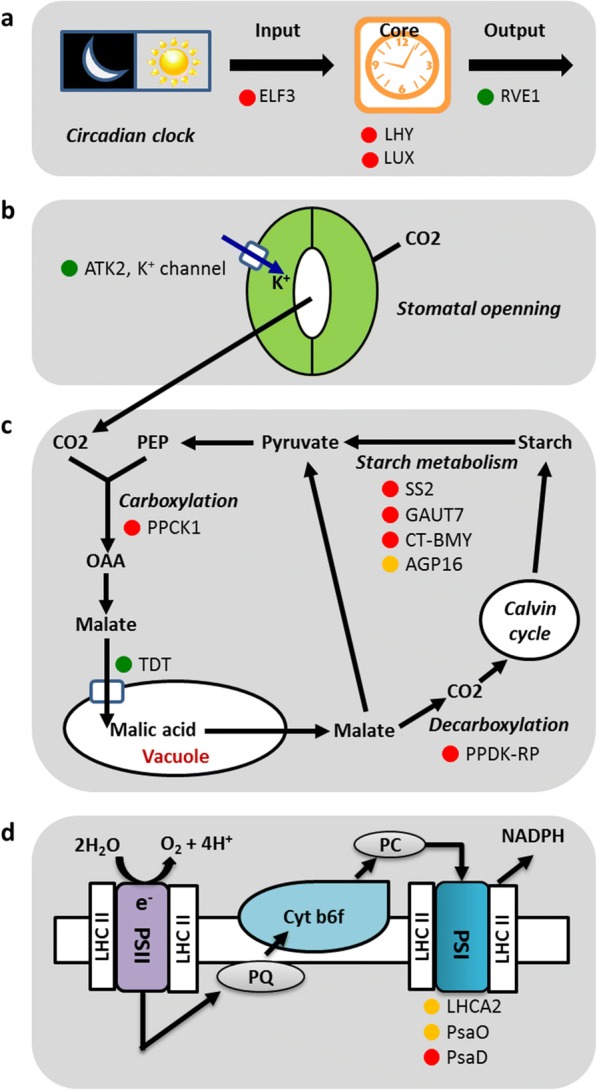
Functionally annotated Agave genes showing positive selection and diel rewiring of expression pattern relative to C3 plants. a Genes involved in circadian clock. A gene involved in stomatal opening. c Genes involved in carboxylation, malate transport, decarboxylation, and starch/sugar metabolism. d Genes involved in photosynthetic electron transport chain. Red circles indicate positive selection. Green circles indicate morning-to-night shift in peak gene expression. Yellow circles indicate afternoon-to-night shift in peak gene expression. AGP16, Arabinogalactan protein 16; AKT2, Arabidopsis Shaker family K+ channels 2/3; CT-BMY, Chloroplast Beta-Amylase; ELF3, Early Flowering 3; GAUT7, Galacturonosyltransferase 7; LHY, Late Elongated Hypocotyl; LUX, Phytoclock 1; PPCK1, Phosphoenlpyruvate Carboxylase Kinase 1; PPDK-RP, Pyruvate orthophosphate dikinase regulatory protein; SS2, Starch Synthase 2; TDT, Tonoplast Dicarboxylate Transporter
Discussion
Regulatory genes are critical for C3-to-CAM evolution
The distribution of CAM plants in diverse phylogenetic lineages indicates CAM has evolved from C3 via convergent evolution [23]. This wide-spread convergent evolution could lead to a hypothesis that C3-to-CAM evolution is relatively easy, not requiring whole-genome-scale changes. In support of this hypothesis, the results in this study indicate that CAM evolution required genes that are shared across C3, C4, and CAM lineages to act as regulatory agents, whereas the core metabolic CAM machinery predates the CAM-C4 divergence and is shared by NVP, C3, CAM, and C4 lineages. The data presented here have shown that genes encoding enzymes required for C3 and C4 carboxylation, decarboxylation, and carbohydrate processing, as well as membrane transporters required for intercellular trafficking of metabolites required for CAM, were present across all the plant lineages examined. Thus, comparative analysis of protein sequences revealed that the core metabolic CAM machinery predates the CAM-C4 divergence and is shared by non-vascular plants (NVP), C3, CAM, and C4 lineages. Such data is also consistent with recent reports that PEPC had shared origins in C4 and CAM lineages before the divergence of these two pathways from a C3 progenitor [24].
As a means of focusing in on the evolution of the regulatory components required for the diel re-programming of metabolism that defines CAM, gene co-expression network analysis was used to reveal modules with distinctive diel patterns of abundance. One gene module in particular, designated M11, was identified as providing a molecular signature for the temporal re-programming of metabolism underpinning CAM. Gene module M11, showed abundant expression in mature leaves at night and contained PPCK1, which regulates the temporal activation of nocturnal CO2 uptake by PEPC [25, 26]. Module M11 was also over-represented by biological processes relevant to stomatal movement and signal transduction. In particular, gene ontology terms related to the ABA signaling pathway were significantly enriched in M11 (Table 1). Such genes are commonly known for their key roles in stomatal regulation and responses to stress [27]. It is also noteworthy that sucrose and jasmonic acid signaling pathways were also enriched in M11, suggesting the involvement of hormones and/or metabolites in the regulation of nocturnal CO2 uptake in CAM. The genes in M11, along with multiple transcription factors, were over-represented in the ortholog clades shared across C3, C4 and the Agave/CAM lineages. Therefore, it can be hypothesized that a limited number of key regulators can drive the C3-to-CAM transition. To test this hypothesis, future studies should focus on detailed functional characterization of the transcription factors in module M11, which belong to the ortholog clade shared across C3, C4, and CAM lineages. Such approaches will be critical for accelerating efforts designed to engineer CAM into C3 crops [13, 16].
Regulation of stomatal movement is critical for CAM evolution
The altered night/day opening/closing of stomata is a core feature of CAM which requires coordination between mesophyll and guard cells [28]. Genes related to stomatal movement were found to be enriched in module M11, alongside genes relevant for nocturnal carboxylation (Table 1). Guard cell inward-rectifying K+ channel AKT2 plays an important role in light-induced stomatal opening in Arabidopsis [29]. Our analysis revealed that the peak expression of AKT2 was shifted to the night in Agave (CAM), as compared with day-time peak expression in Arabidopsis (C3) (Fig. 4a; Fig. 6b), suggesting that AKT2 is involved in nocturnal stomatal opening in Agave. In terms of determining the signals that might be responsible for shifting the timing of AKT2 expression in CAM, it has been suggested that photosynthetic metabolism in the mesophyll cells could contribute to the regulation of guard cell function [30]. Interactions between mesophyll photosynthesis and guard cell regulation have been revealed in many non-CAM species [31]. In particular, signals driven by sugar and malate content in the mesophyll appear to have central roles in controlling stomatal aperture [31]. In C3 plants, malate metabolism in the mesophyll and malate transport from mesophyll to guard cells has been shown to play a central role in regulating stomatal responses over the day/night cycle [16]. By analogy, it can be hypothesized that in CAM plants, the diel turnover and transport of malate across the vacuolar tonoplast membrane will play a critical role in stomatal regulation [16]. In Arabidopsis, tonoplast dicarboxylate transporter (TDT) imports malate into the vacuoles [32]. In Agave, the peak expression of TDT was shifted to the night, as compared to peak expression during the day in Arabidopsis (Fig. 4a; Fig. 6c), implying that the Agave TDT is responsible for the transport of malate into the vacuole during the dark period. Future molecular genetic experiments are needed to confirm the function of AKT2 and TDT, and consequently shed new light on identifying signals which integrate carboxylation processes with stomatal movement in CAM.
Diversification of circadian clock genes in CAM
Our results suggest that circadian shifts in gene transcription underpin the CAM-defining diel patterns of stomatal conductance, malate transport, carbohydrate processing and supply, and demand for ATP and reducing power in Agave. The circadian clock has been proposed to control the extensive re-synchronization of metabolism that distinguishes CAM from C3 and C4 photosynthesis [4]. Circadian rhythms are ubiquitous in eukaryotes and many features of the circadian clock are conserved across plant lineages [33]. Genes in the circadian system have been well-studied in Arabidopsis and can be divided into three functional groups: signal input (e.g., PHOT2), clock oscillation (e.g., CCA1, TOC1), and regulatory output (e.g., RVE1) [34]. We identified homologs in Agave (CAM plant) for all the known clock genes in Arabidopsis (C3) and found that the diel expression patterns of all the clock genes are conserved between CAM and C3, except for RVE1 (a Myb-like transcription factor) in the output subset (Fig. 5), suggesting that C3 and CAM plants share the same core circadian oscillator, with diversification occurring in the regulatory output from the core clock, such as RVE1. It was previously reported that RVE1 integrates the circadian clock and auxin pathways to coordinate plant growth with changes in environmental time cues in Arabidopsis [35]. Here we hypothesize that RVE1 is a key a node from which the C3 and CAM clock diverge and rewiring the diel expression of RVE1 is one of the necessary steps to switch from C3 to CAM.
Besides expression analysis, changes in the protein sequences of circadian clock components may also contribute to the diversification of clock functions. We performed 3D structural modeling to understand the function of protein domains containing sites of positive selection (reflected by Ka/Ks ratio > 1). Our structure-modeling revealed characteristics of positive-selection in the intrinsically disordered protein regions in some circadian clock proteins (Additional file 19: Figure S6). It has become clear that a certain protein may not have a well-defined and compactly folded three-dimensional (3D) structure under physiological conditions and such proteins are often termed natively unfolded protein [36] or an intrinsically disordered protein (IDP) [37]. Many intrinsically disordered protein regions (IDPRs) have been known to carry out important biological functions [38]. The clock proteins Late Elongated Hypocotyl (LHY), Early Flowering 3 (ELF3), and Lux Arrhythmo (LUX) play key roles in the plant circadian oscillation [39]. The Ka/Ks profile of each of these circadian rhythm proteins showed multiple positive selection regions with significant fluctuations in Agave (data not shown). Interestingly, these proteins were found to exhibit a high ratio of IDPRs with high PONDR scores (Additional file 20: Table S14). The IDPRs of the clock proteins may lead to high flexibility in both structure and function, which in turn, could favor novel interactions with nucleic acids and/or other proteins. Further molecular genetics studies are needed to gain deeper understanding of the functions encoded in the apparent disordered state of these clock proteins and their role in the evolution of CAM.
CAM-specific genes and evolution
Multiple lineages of CAM photosynthesis plants have evolved independently from C3 photosynthesis ancestors [13, 40]. Recently, a comparative study using four genera in subfamily Agavoideae with CAM, weak CAM and C3, has suggested that gene family analysis together with expression profiling is informative in understanding the divergence of CAM [41]. However, a broader sampling of diverse CAM lineages is still necessary. It can be hypothesized that there are two types of CAM-specific genes: 1) lineage-specific CAM genes shared by multiple closely related-species (e.g., Agave spp.) and 2) conserved CAM-specific genes shared by multiple independent lineages of CAM plants. To test this hypothesis, it would be useful in the future to expand this work by including several independently evolved CAM lineages in order to separate evolutionary phenomena unique to the CAM Agave from ones shared by different CAM lineages.
Conclusions
Evidence is presented that the genetic components of core CAM machinery in Agave have an ancient origin traceable to non-vascular plant lineages, and that regulatory proteins, which are shared between C3, CAM, and C4 species, were essential to the C3-to-CAM transition. The evolution of CAM in Agave from C3 photosynthesis also required positive selection in protein sequences of enzymes and transporters implicated in metabolism and signaling associated with CAM, as well as diel re-programming of gene expression related to key biological processes, such as circadian rhythms, redox homeostasis, and carbohydrate metabolism (Fig. 6). These results provide a set of new candidate genes for engineering increased water-use efficiency in crop plants experiencing water-limiting conditions via synthetic biology approaches.
Methods
Representative protein model per locus in Agave species
The transcript sequences of A. americana [14] (Additional file 21) were first filtered by CD-HIT-EST [42, 43] with a sequence identity threshold of 0.98 and the alignment coverage for the shorter sequence set as 0.5. This was an optimal setting based on the test with various combinations of sequence identity (0.90, 0.91, …, 1.0), and the alignment coverage for the shorter sequence (0.4, 0.5, …, 0.8), using the Arabidopsis genome annotation (TAIR10). To assign the representative protein model per locus with high-confidence, the protein sequences corresponding to the representative transcript sequences in A. americana, obtained from CD-HIT-EST clustering, as well as the non-redundant representative protein models of A. deserti and A. tequilana [44], were mapped onto the aforementioned draft genome assembly of A. tequilana using BLAT [45] with a minimum coverage (i.e., minimum fraction of query that must be aligned) of 60% and a minimum identity of 90%. Only the “best match” position was selected as the genomic location for each query protein sequence. If multiple proteins mapped to the overlapping genome locations and they shared significant sequence similarity, as determined by BLASTp [46, 47] with E-value cutoff of 1e-5, the longest protein sequence was selected as the representative protein model for the gene locus, resulting in 55,451, 31,761, and 31,799 representative protein sequences in A. americana, A. deserti and A. tequilana, respectively (Additional files 22, 23 and 24).
Comparative analysis of protein sequences
The protein sequences of 15 plant species, including the aforementioned representative protein sequences in three CAM species of Agave (Agave americana, A. deserti and A. tequilana), and 12 non-CAM plant species downloaded from public databases, which included three non-vascular plant species Chlamydomonas reinhardtii (www.Phytozome.net; Phytozome v9.0), Physcomitrella patens (Phytozome v9.0), Selaginella moellendorffii (Phytozome v9.0); three C4 plant species Sorghum bicolor (Phytozome v9.0), Setaria italica (Phytozome v9.0) and Zea mays (Phytozome v9.0); three C3 monocot plant species Brachypodium distachyon (Phytozome v9.0e), Oryza sativa (Phytozome v9.0), and Musa acuminata (version 1; http://banana-genome.cirad.fr); three C3 dicot species Arabidopsis thaliana (v10; www.Arabidopsis.org), Populus trichocarpa (Phytozome v9.0), and Solanum tuberosum (DM_v3.4; potatogenomics.plantbiology.msu.edu). The longest protein sequence was selected in case of multiple transcripts annotated for one gene locus. The ortholog groups (OGs) were constructed using OrthoMCL [48] with default parameters (a BLASTp E-value cutoff of 1e-5 and percent match cutoff of 50%). Also, the protein sequences used for ortholog analysis were clustered into tribes using TRIBE-MCL [19], with a BLASTp E-value cutoff of 1e-5 and an inflation value of 1.5. To identify conserved CAM-specific genes, the A. americana genes in both the CAM-only ortholog groups and CAM-only tribes were then compared with an extended list of three independent CAM lineages and 21 non-CAM species using BLASTp [46, 47] with an E-value cutoff of 1e-5. The three independent CAM lineages are Kalanchoë fedtschenkoi [49], Ananas comosus [50] and Phalaenopsis equestris [51]. The 21 non-CAM species are Amborella trichopoda (PLAZA 3.0 [52]; available at http://bioinformatics.psb.ugent.be/plaza/), Arabidopsis thaliana (PLAZA 3.0), Beta vulgaris (PLAZA 3.0), Brachypodium distachyon (PLAZA 3.0), Carica papaya (PLAZA 3.0), Citrus sinensis (PLAZA 3.0), Eucalyptus grandis (PLAZA 3.0), Fragaria vesca (PLAZA 3.0), Medicago truncatula (PLAZA 3.0), Mimulus guttatus (PLAZA 3.0), Musa acuminata (PLAZA 3.0), Oryza sativa (PLAZA 3.0), Populus trichocarpa (PLAZA 3.0), Prunus persica (PLAZA 3.0), Setaria italica (PLAZA 3.0), Solanum lycopersicum (PLAZA 3.0), Solanum tuberosum (PLAZA 3.0), Sorghum bicolor (PLAZA 3.0), Theobroma cacao (PLAZA 3.0), Vitis vinifera (PLAZA 3.0), Zea may (PLAZA 3.0).
Co-expression network
A total of 47,677 transcripts that were detected in at least 4 of the 15 samples, with an average expression level of 5 RPKM or higher (Additional file 25), were utilized to construct a weighted gene co-expression network using the R package WGCNA [53]. The gene expression data were log2 transformed. The dynamic tree-cut algorithm was used to identify co-expression modules with a minimum module size of 30 and a height cut of 0.25.
Comparative analysis of gene expression patterns between CAM and C3 plants
The Arabidopsis–Agave orthologous gene pairs were identified through the combination of both OrthoMCL strategies and the reciprocal best hits (RBH) based on BLASTp with an E-value cutoff of 1e-5. The diurnal expression data for Arabidopsis thaliana were obtained from Mockler et al. (2007) [9]. Both Arabidopsis and Agave plants were grown under a photoperiod of 12 h light:12 h dark cycle. The Arabidopsis expression data were collected at 0, 4, 8, 12, 16, 20, and 24 h, whereas the Agave data were collected at 0, 3, 6, 9, 12, 15, 18, and 21 h after the start of the light period [14]. The cubic interpolation algorithm implemented in Matlab (Mathworks, Inc.) was used to simulate the gene expression levels at additional time points, so that both time-course data sets consisted of the same time points: 0, 3, 4, 6, 8, 9, 12, 15, 16, 18, 20, and 21 h after the start of the light period. The gene expression data were normalized by Z score transformation. The hierarchical clustering of gene expression was performed using the Bioinformatics Toolbox in Matlab (Mathworks, Inc.).
Gene ontology analysis
Whole-genome GO term annotation was performed using Blast2GO with a BLASTp E-value hit filter of 1 × 10–6, an annotation cutoff value of 55, and GO weight of 5. GO enrichment analysis for the ortholog clades was performed using BiNGO [54]. In addition, GO enrichment analysis was performed on each of the 16 co-expression modules using ClueGO [55] to interpret functionally grouped gene ontology annotation networks. The right-sided hypergeometric enrichment test was performed at a medium network specificity selection, and p-value correction was performed using the Benjamini-Hochberg method. The selected GO tree levels were a minimum of 3 and a maximum of 8, while each cluster was set to a minimum of between 3 and 4% genes. The GO term grouping setting was selected to minimize GO term redundancy, and the highest significance term enriched was used as the representative term for each functional cluster. The GO terms with p-values less than or equal to 0.05 were considered significantly enriched.
Annotation of pathway and transcription factors
Pathway annotation for the protein sequences was performed on the KEGG Automatic Annotation Server KAAS [56], using the BBH (bi-directional best hit) method to assign orthologs. Transcription factors were identified from the protein sequences using the online tool PlantTFcat [57].
Nonsynonymous (Ka) to synonymous (Ks) substitution ratio and positively selected sites
The orthologous gene pairs between two species were identified through the combination of both Best Reciprocal Hits (BRH) and OrthoMCL strategies. The coding sequences were aligned using PAL2NAL [58], guided by protein sequence alignment generated by MAFFT (linsi; version 7.045b) [59], and gaps in the alignment were removed. The gapless coding sequence alignments were used for Ka/Ks ratio calculation using the Bioinformatics Toolbox in Matlab (Mathworks, Inc.) with a 50-codon sliding window. For identifying positively selected sites, coding sequences from Arabidopsis, maize, rice and Agave were aligned by Translatorx [60] using the standalone script. The HyPhy package were used to identify positively selected sites as described [61], and the tests of FUBAR and REL models as implemented in Datamonkey webserver were used with default settings [62]. Since we used a sliding window to study the regions of protein with positive selection, we calculated the probabilities of Ka/Ks positive regions to a null hypothesis that Ka/Ks equals to one by one-sided t-test, as described by Schmid and Yang (2008) [63].
Protein structure modeling
Protein structure models were built using the iterative threading assembly refinement (I-TASSER, version 3.0) methods [64]. The structure-based annotation tool COFACTOR [65] was adopted to predict the potential function and the cofactor binding site of the models. Disordered region(s) were analyzed using the PONDR VL-XT program [66–68].
Phylogenetic tree construction
The multiple sequence alignment of protein sequences was created using MAFFT [59]. The phylogenetic tree was constructed from the protein sequence alignment using the Neighbor-Joining method [69] implemented in MEGA7 [70], with the percentage of replicate trees calculated by the bootstrap test (100 replicates). For trees with three sequences, no bootstrap value was given due to the lack of phylogeny tests of branch. All ambiguous positions were removed for each sequence pair.
Additional files
Figure S1. Overview charts of the gene ontology (GO) biological processes over-represented in individual co-expression modules. (PDF 464 kb)
Table S1. Percentage of the gene set in each individual species distributed into different ortholog clades. (PDF 39 kb)
Table S2. Biological processes over-represented (p < 1E-20) in ortholog clade NVP:C3:CAM:C4 in Agave americana. NVP:C3:CAM:C4 represents orthologs shared by C3, CAM, C4 and NVP (i.e. non-vascular plants). (PDF 39 kb)
Table S3. Biological processes over-represented in ortholog clade C3:CAM:C4 in Agave americana. C3:CAM:C4 represents orthologs shared only by C3, CAM and C4 species. (PDF 17 kb)
Table S4. Ortholog clades of C4 cycle genes & related transporters. (PDF 118 kb)
Table S5. Distribution of co-expression modules in each individual ortholog clade in Agave americana. (PDF 90 kb)
Table S6. List of Agave americana genes that have homologs in other three independent CAM lineages but not in 21 non-CAM plant species. (PDF 31 kb)
Figure S2. The diel expression pattern of conserved CAM-specific genes in Agave. (PDF 111 kb)
Table S7. List of Agave americana genes with Ka/Ks ratio greater than one in Agave-Arabidopsis, Agave-Oryza and Agave-Zea pairs. (PDF 353 kb)
Table S8. Genes implicated in the CAM pathway undergoing positive selection as revealed from Ka/Ks ratio calculated from Agave-Arabidopsis, Agave-Oryza and Agave-Zea orthologous gene pairs. (PDF 91 kb)
Table S9. List of Agave americana genes with Ka/Ks ratio greater than one in Agave-Arabidopsis and Agave-Oryza but not Agave-Zea pairs. (PDF 47 kb)
Table S10. The positively selected sites of candidates with Ka/Ks > 1 from Table S9. (PDF 304 kb)
Figure S3. Positive selection region in chloroplast β-amylase (CT-BMY). (PDF 171 kb)
Table S11. List of Agave americana genes with morning-to-night shift in expression pattern as compared with the orthologous genes in Arabidopsis. (PDF 70 kb)
Table S12. List of Agave americana genes with afternoon-to-night shift in expression pattern as compared with the orthologous genes in Arabidopsis. (PDF 69 kb)
Figure S4. Phylogenetic trees and diel gene expression patterns of multi-gene ortholog groups (i.e., with a total of more than two Agave and Arabidopsis genes) listed Fig. 4a. (PDF 164 kb)
Figure S5. Phylogenetic trees and diel gene expression patterns of multi-gene ortholog groups (i.e., with a total of more than two Agave and Arabidopsis genes) listed Fig. 4b. (PDF 156 kb)
Table S13. List of circadian clock genes in Agave americana and Arabidopsis thaliana. (PDF 18 kb)
Figure S6. Intrinsically disordered protein regions (IDPRs) and structural models of the circadian clock proteins in Agave americana. (PDF 153 kb)
Table S14. Intrinsically disordered protein region (IDPR) in protein sequences under positive selection. (PDF 33 kb)
Agave americana transcript sequences of less than 200 bp. (FASTA 5010 kb)
Representative protein sequences in Agave americana. (FASTA 9640 kb)
Representative protein sequences in Agave deserti. (FASTA 11600 kb)
Representative protein sequences in Agave tequilana. (FASTA 11400 kb)
Expression data of Agave americana transcripts detected in at least 4 of the 15 samples, with an average expression level of 5 RPKM or higher. (XLSX 14100 kb)
Acknowledgments
The authors would like to thank J.L. Labbe for critical review and clarifying comments on the manuscript. We are grateful to anonymous reviewers for comments to improve the manuscript.
Funding
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Genomic Science Program (under award number DE-SC0008834), the DOE Center for Bioenergy Innovation (CBI), and the Laboratory Directed Research and Development (LDRD) Program (Project ID: 5801) of Oak Ridge National Laboratory. Oak Ridge National Laboratory is managed by UT-Battelle, LLC for the US DOE under Contract Number DE–AC05–00OR22725. Work conducted at the US Department of Energy Joint Genome Institute (JGI) was supported by the Office of Science of the US Department of Energy under contract no. DE-AC02-05CH11231.
Availability of data and materials
All data of this work are available from links of the manuscript. The supporting files are available from the Dryad Digital Repository: 10.5061/dryad.37g7v3v.
Abbreviations
- CAM
Crassulacean acid metabolism
- CCA1
Circadian clock associated 1
- CT-BMY
Chloroplast beta-amylase
- ELF3
Early flowering 3
- FUBAR
Fast unconstrained bayesian approximation
- I-TASSER
Iterative threading assembly refinement
- LHY
Late elongated hypocotyl
- LUX
Lux arrhythmo
- NVP
Non-vascular plant
- PEPC
Phosphoenolpyruvate carboxylase.
- PHOT2
Phototropin-2
- PPCK1
phosphoenolpyruvate carboxylase kinase 1
- RBH
Reciprocal best hits
- REL
Random effects likelihood
- RuBisCO
Ribulose-1,5-bisphosphate carboxylase/oxygenase
- RVE1
Reveille 1
- TOC1
Timing of cab expression1
Authors’ contributions
HY and SJ prepared samples and RNA sequencing. HG, DW, PR, and XY carried out data processing, assembly and gene family analysis. PA, JW, HG, RH analyzed gene models and protein sequences. SG, ZW and AX performed comparative analyses in other agave species. SW, AB, GT, TT, DW and XY interpreted the data and supervised the project. HY and XY drafted the manuscript and all authors participated in revision. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing interests.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hengfu Yin, Email: hfyin@sibs.ac.cn.
Hao-Bo Guo, Email: guohaobo@gmail.com.
David J. Weston, Email: westondj@ornl.gov
Anne M. Borland, Email: anne.borland@newcastle.ac.uk
Priya Ranjan, Email: ranjanp@ornl.gov.
Paul E. Abraham, Email: abrahampe@ornl.gov
Sara S. Jawdy, Email: jawdys@ornl.gov
James Wachira, Email: james.wachira@morgan.edu.
Gerald A. Tuskan, Email: tuskanga@ornl.gov
Timothy J. Tschaplinski, Email: tschaplinstj@ornl.gov
Stan D. Wullschleger, Email: wullschlegsd@ornl.gov
Hong Guo, Email: hguo1@utk.edu.
Robert L. Hettich, Email: hettichrl@ornl.gov
Stephen M. Gross, Email: smgross@mac.com
Zhong Wang, Email: zhongwang@lbl.gov.
Axel Visel, Email: avisel@lbl.gov.
Xiaohan Yang, Email: yangx@ornl.gov.
References
- 1.Ehleringer J, Monson R. Evolutionary and ecological aspects of photosynthetic pathway variation. Annu Rev Ecol Syst. 1993;24:411–439. doi: 10.1146/annurev.es.24.110193.002211. [DOI] [Google Scholar]
- 2.Yamori W, Hikosaka K, Way DA. Temperature response of photosynthesis in C3, C4, and CAM plants: temperature acclimation and temperature adaptation. Photosynth Res. 2014;119:101–117. doi: 10.1007/s11120-013-9874-6. [DOI] [PubMed] [Google Scholar]
- 3.Borland AM, Griffiths H, Hartwell J, Smith JA. Exploiting the potential of plants with crassulacean acid metabolism for bioenergy production on marginal lands. J Exp Bot. 2009;60:2879–2896. doi: 10.1093/jxb/erp118. [DOI] [PubMed] [Google Scholar]
- 4.Hartwell J. The co-ordination of central plant metabolism by the circadian clock. Biochem Soc Trans. 2005;33:945–948. doi: 10.1042/BST0330945. [DOI] [PubMed] [Google Scholar]
- 5.Nimmo HG. How to tell the time: the regulation of phosphoenolpyruvate carboxylase in crassulacean acid metabolism (CAM) plants. Biochem Soc Trans. 2003;31:728–730. doi: 10.1042/bst0310728. [DOI] [PubMed] [Google Scholar]
- 6.Nimmo HG. The regulation of phosphoenolpyruvate carboxylase in CAM plants. Trends Plant Sci. 2000;5:75–80. doi: 10.1016/S1360-1385(99)01543-5. [DOI] [PubMed] [Google Scholar]
- 7.Boxall SF, Foster JM, Bohnert HJ, Cushman JC, Nimmo HG, Hartwell J. Conservation and divergence of circadian clock operation in a stress-inducible crassulacean acid metabolism species reveals clock compensation against stress. Plant Physiol. 2005;137:969–982. doi: 10.1104/pp.104.054577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallona I, Egea-Cortines M, Weiss J. Conserved and divergent rhythms of crassulacean acid metabolism-related and core clock gene expression in the cactus Opuntia ficus-indica. Plant Physiol. 2011;156:1978–1989. doi: 10.1104/pp.111.179275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mockler TC, Michael TP, Priest HD, Shen R, Sullivan CM, Givan SA, et al. The DIURNAL project: DIURNAL and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb Symp Quant Biol. 2007;72:353–363. doi: 10.1101/sqb.2007.72.006. [DOI] [PubMed] [Google Scholar]
- 10.Kapralov MV, Filatov DA. Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol Biol. 2007;7:73. doi: 10.1186/1471-2148-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapralov MV, Kubien DS, Andersson I, Filatov DA. Changes in Rubisco kinetics during the evolution of C4 photosynthesis in Flaveria (Asteraceae) are associated with positive selection on genes encoding the enzyme. Mol Biol Evol. 2011;28:1491–1503. doi: 10.1093/molbev/msq335. [DOI] [PubMed] [Google Scholar]
- 12.Escamilla-Trevino L. Potential of plants from the genus Agave as bioenergy crops. Bioenergy Research. 2012;5:1–9. doi: 10.1007/s12155-011-9159-x. [DOI] [Google Scholar]
- 13.Yang X, Cushman JC, Borland AM, Edwards EJ, Wullschleger SD, Tuskan GA, et al. A roadmap for research on crassulacean acid metabolism (CAM) to enhance sustainable food and bioenergy production in a hotter, drier world. New Phytol. 2015;207:491–504. doi: 10.1111/nph.13393. [DOI] [PubMed] [Google Scholar]
- 14.Abraham PE, Yin H, Borland AM, Weighill D, Lim SD, De Paoli HC, et al. Transcript, protein and metabolite temporal dynamics in the CAM plant Agave. Nature Plants. 2016;2:16178. doi: 10.1038/nplants.2016.178. [DOI] [PubMed] [Google Scholar]
- 15.Davis S, Dohleman F, Long S. The global potential for Agave as a biofuel feedstock. Global Change Biology Bioenergy. 2011;3:68–78. doi: 10.1111/j.1757-1707.2010.01077.x. [DOI] [Google Scholar]
- 16.Borland AM, Hartwell J, Weston DJ, Schlauch KA, Tschaplinski TJ, Tuskan GA, et al. Engineering crassulacean acid metabolism to improve water-use efficiency. Trends Plant Sci. 2014;19:327–338. doi: 10.1016/j.tplants.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.West-Eberhard MJ, Smith JA, Winter K. Plant science. Photosynthesis, reorganized. Science. 2011;332:311–312. doi: 10.1126/science.1205336. [DOI] [PubMed] [Google Scholar]
- 18.Hartwell J, Dever LV, Boxall SF. Emerging model systems for functional genomics analysis of crassulacean acid metabolism. Curr Opin Plant Biol. 2016;31:100–108. doi: 10.1016/j.pbi.2016.03.019. [DOI] [PubMed] [Google Scholar]
- 19.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Q, Han Z, Jiang H, Tian D, Yang S. Strong positive selection drives rapid diversification of R-genes in Arabidopsis relatives. J Mol Evol. 2010;70:137–148. doi: 10.1007/s00239-009-9316-4. [DOI] [PubMed] [Google Scholar]
- 21.Looman C, Abrink M, Mark C, Hellman L. KRAB zinc finger proteins: an analysis of the molecular mechanisms governing their increase in numbers and complexity during evolution. Mol Biol Evol. 2002;19:2118–2130. doi: 10.1093/oxfordjournals.molbev.a004037. [DOI] [PubMed] [Google Scholar]
- 22.Ihnatowicz A, Pesaresi P, Varotto C, Richly E, Schneider A, Jahns P, et al. Mutants for photosystem I subunit D of Arabidopsis thaliana: effects on photosynthesis, photosystem I stability and expression of nuclear genes for chloroplast functions. Plant J. 2004;37:839–852. doi: 10.1111/j.1365-313X.2004.02011.x. [DOI] [PubMed] [Google Scholar]
- 23.Edwards E, Ogburn R. Angiosperm responses to a low-CO2 world: CAM and C4 photosynthesis as parallel evolutionary trajectories. Int J Plant Sci. 2012;173:724–733. doi: 10.1086/666098. [DOI] [Google Scholar]
- 24.Christin PA, Arakaki M, Osborne CP, Brautigam A, Sage RF, Hibberd JM, et al. Shared origins of a key enzyme during the evolution of C4 and CAM metabolism. J Exp Bot. 2014;65:3609–3621. doi: 10.1093/jxb/eru087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartwell J, Gill A, Nimmo GA, Wilkins MB, Jenkins GI, Nimmo HG. Phosphoenolpyruvate carboxylase kinase is a novel protein kinase regulated at the level of expression. Plant J. 1999;20:333–342. doi: 10.1046/j.1365-313X.1999.t01-1-00609.x. [DOI] [PubMed] [Google Scholar]
- 26.Owen NA, Griffiths H. A system dynamics model integrating physiology and biochemical regulation predicts extent of crassulacean acid metabolism (CAM) phases. New Phytol. 2013;200:1116–1131. doi: 10.1111/nph.12461. [DOI] [PubMed] [Google Scholar]
- 27.Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR. Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol. 2010;61:651–679. doi: 10.1146/annurev-arplant-042809-112122. [DOI] [PubMed] [Google Scholar]
- 28.Lee JS. Stomatal opening mechanism of CAM plants. Journal of Plant Biology. 2010;53:19–23. doi: 10.1007/s12374-010-9097-8. [DOI] [Google Scholar]
- 29.Ivashikina N, Deeken R, Fischer S, Ache P, Hedrich R. AKT2/3 subunits render guard cell K+ channels Ca2+ sensitive. J Gen Physiol. 2005;125:483–492. doi: 10.1085/jgp.200409211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luttge U. Ecophysiology of crassulacean acid metabolism (CAM) Ann Bot. 2004;93:629–652. doi: 10.1093/aob/mch087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawson T, Blatt MR. Stomatal size, speed, and responsiveness impact on photosynthesis and water use efficiency. Plant Physiol. 2014;164:1556–1570. doi: 10.1104/pp.114.237107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emmerlich V, Linka N, Reinhold T, Hurth MA, Traub M, Martinoia E, et al. The plant homolog to the human sodium/dicarboxylic cotransporter is the vacuolar malate carrier. Proc Natl Acad Sci U S A. 2003;100:11122–11126. doi: 10.1073/pnas.1832002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Neill JS, van Ooijen G, Dixon LE, Troein C, Corellou F, Bouget FY, et al. Circadian rhythms persist without transcription in a eukaryote. Nature. 2011;469:554–558. doi: 10.1038/nature09654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McWatters HG, Devlin PF. Timing in plants--a rhythmic arrangement. FEBS Lett. 2011;585:1474–1484. doi: 10.1016/j.febslet.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 35.Rawat R, Schwartz J, Jones MA, Sairanen I, Cheng Y, Andersson CR, et al. REVEILLE1, a Myb-like transcription factor, integrates the circadian clock and auxin pathways. Proc Natl Acad Sci U S A. 2009;106:16883–16888. doi: 10.1073/pnas.0813035106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uversky VN, Gillespie JR, Fink AL. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 38.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 1804;2010:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Staiger D, Shin J, Johansson M, Davis SJ. The circadian clock goes genomic. Genome Biol. 2013;14:208. doi: 10.1186/gb-2013-14-6-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silvera K, Neubig KM, Whitten WM, Williams NH, Winter K, Cushman JC. Evolution along the crassulacean acid metabolism continuum. Funct Plant Biol. 2010;37:995–1010. doi: 10.1071/FP10084. [DOI] [Google Scholar]
- 41.Heyduk K, Ray JN, Ayyampalayam S, Leebens-Mack J. Shifts in gene expression profiles are associated with weak and strong crassulacean acid metabolism. Am J Bot. 2018;105:587–601. doi: 10.1002/ajb2.1017. [DOI] [PubMed] [Google Scholar]
- 42.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 44.Gross SM, Martin JA, Simpson J, Abraham-Juarez MJ, Wang Z, Visel A. De novo transcriptome assembly of drought tolerant CAM plants, Agave deserti and Agave tequilana. BMC Genomics. 2013;14:563. doi: 10.1186/1471-2164-14-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang X, Hu R, Yin H, Jenkins J, Shu S, Tang H, et al. The Kalanchoë genome provides insights into convergent evolution and building blocks of crassulacean acid metabolism. Nat Commun. 2017;8:1899. doi: 10.1038/s41467-017-01491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ming R, VanBuren R, Wai CM, Tang H, Schatz MC, Bowers JE, et al. The pineapple genome and the evolution of CAM photosynthesis. Nat Genet. 2015;47:1435–1442. doi: 10.1038/ng.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai J, Liu X, Vanneste K, Proost S, Tsai W-C, Liu K-W, et al. The genome sequence of the orchid Phalaenopsis equestris. Nat Genet. 2015;47:65–72. doi: 10.1038/ng.3149. [DOI] [PubMed] [Google Scholar]
- 52.Proost S, Van Bel M, Vaneechoutte D, Van de Peer Y, Inze D, Mueller-Roeber B, et al. PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 2015;43:D974–D981. doi: 10.1093/nar/gku986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 55.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dai X, Sinharoy S, Udvardi M, Zhao PX. PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics. 2013;14:321. doi: 10.1186/1471-2105-14-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 62.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmid K, Yang Z. The trouble with sliding windows and the selective pressure in BRCA1. PLoS One. 2008;3:e3746. doi: 10.1371/journal.pone.0003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40:W471–W477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Informatics. 1999;10:30–40. [PubMed] [Google Scholar]
- 67.Romero P, Obradovic Z, Dunker AK. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Informatics. 1997;8:110–124. [PubMed] [Google Scholar]
- 68.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 69.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 70.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overview charts of the gene ontology (GO) biological processes over-represented in individual co-expression modules. (PDF 464 kb)
Table S1. Percentage of the gene set in each individual species distributed into different ortholog clades. (PDF 39 kb)
Table S2. Biological processes over-represented (p < 1E-20) in ortholog clade NVP:C3:CAM:C4 in Agave americana. NVP:C3:CAM:C4 represents orthologs shared by C3, CAM, C4 and NVP (i.e. non-vascular plants). (PDF 39 kb)
Table S3. Biological processes over-represented in ortholog clade C3:CAM:C4 in Agave americana. C3:CAM:C4 represents orthologs shared only by C3, CAM and C4 species. (PDF 17 kb)
Table S4. Ortholog clades of C4 cycle genes & related transporters. (PDF 118 kb)
Table S5. Distribution of co-expression modules in each individual ortholog clade in Agave americana. (PDF 90 kb)
Table S6. List of Agave americana genes that have homologs in other three independent CAM lineages but not in 21 non-CAM plant species. (PDF 31 kb)
Figure S2. The diel expression pattern of conserved CAM-specific genes in Agave. (PDF 111 kb)
Table S7. List of Agave americana genes with Ka/Ks ratio greater than one in Agave-Arabidopsis, Agave-Oryza and Agave-Zea pairs. (PDF 353 kb)
Table S8. Genes implicated in the CAM pathway undergoing positive selection as revealed from Ka/Ks ratio calculated from Agave-Arabidopsis, Agave-Oryza and Agave-Zea orthologous gene pairs. (PDF 91 kb)
Table S9. List of Agave americana genes with Ka/Ks ratio greater than one in Agave-Arabidopsis and Agave-Oryza but not Agave-Zea pairs. (PDF 47 kb)
Table S10. The positively selected sites of candidates with Ka/Ks > 1 from Table S9. (PDF 304 kb)
Figure S3. Positive selection region in chloroplast β-amylase (CT-BMY). (PDF 171 kb)
Table S11. List of Agave americana genes with morning-to-night shift in expression pattern as compared with the orthologous genes in Arabidopsis. (PDF 70 kb)
Table S12. List of Agave americana genes with afternoon-to-night shift in expression pattern as compared with the orthologous genes in Arabidopsis. (PDF 69 kb)
Figure S4. Phylogenetic trees and diel gene expression patterns of multi-gene ortholog groups (i.e., with a total of more than two Agave and Arabidopsis genes) listed Fig. 4a. (PDF 164 kb)
Figure S5. Phylogenetic trees and diel gene expression patterns of multi-gene ortholog groups (i.e., with a total of more than two Agave and Arabidopsis genes) listed Fig. 4b. (PDF 156 kb)
Table S13. List of circadian clock genes in Agave americana and Arabidopsis thaliana. (PDF 18 kb)
Figure S6. Intrinsically disordered protein regions (IDPRs) and structural models of the circadian clock proteins in Agave americana. (PDF 153 kb)
Table S14. Intrinsically disordered protein region (IDPR) in protein sequences under positive selection. (PDF 33 kb)
Agave americana transcript sequences of less than 200 bp. (FASTA 5010 kb)
Representative protein sequences in Agave americana. (FASTA 9640 kb)
Representative protein sequences in Agave deserti. (FASTA 11600 kb)
Representative protein sequences in Agave tequilana. (FASTA 11400 kb)
Expression data of Agave americana transcripts detected in at least 4 of the 15 samples, with an average expression level of 5 RPKM or higher. (XLSX 14100 kb)
Data Availability Statement
All data of this work are available from links of the manuscript. The supporting files are available from the Dryad Digital Repository: 10.5061/dryad.37g7v3v.