

Abstract
Trauma-induced hemorrhagic shock (HS) plays a decisive role in the development of immune, coagulation, and organ dysfunction often resulting in a poor clinical outcome. Imbalanced complement activation is intricately associated with the molecular danger response and organ damage after HS. Thus, inhibition of the central complement component C3 as turnstile of both inflammation and coagulation is hypothesized as a rational strategy to improve the clinical course after HS.
Applying intensive care conditions, anaesthetized, monitored, and protectively ventilated non-human primates (NHP; cynomolgus monkeys) received a pressure-controlled severe HS (60 min at MAP 30 mmHg) with subsequent volume resuscitation. Thirty min after HS, animals were randomly treated with either an analog of the C3 inhibitor compstatin (i.e., Cp40) in saline (n=4) or with saline alone (n=4). The observation period lasted 300 min after induction of HS.
We observed improved kidney function in compstatin Cp40-treated animals after HS as determined by improved urine output, reduced damage markers and a tendency of less histopathological signs of acute kidney injury. Sham-treated animals revealed classical signs of mucosal edema, especially in the ileum and colon reflected by worsened microscopic intestinal injury scores. In contrast, Cp40-treated HS animals exhibited only minor signs of organ edema and significantly less intestinal damage. Furthermore, early systemic inflammation and coagulation dysfunction were both ameliorated by Cp40.
The data suggest that therapeutic inhibition of C3 is capable to significantly improve immune, coagulation and organ function and to preserve organ-barrier integrity early after traumatic HS. C3-targeted complement inhibition may therefore reflect a promising therapeutic strategy in fighting fatal consequences of HS.
INTRODUCTION
Hemorrhagic shock (HS) is a major pathophysiological driver of systemic inflammation, organ barrier break down, organ dysfunction and lethal outcome after severe injury (1, 2). The role of innate immunity during HS, and the complement system in particular has been recognized for at least half a century (3). There is increasing experimental and clinical evidence of excessive complement activation and depletion, with resulting development of complementopathy, during hemorrhagic shock (4). After severe tissue trauma, early induction of the alternative pathway of complement activation seems to be the predominant culprit, being associated with injury severity, tissue hypoperfusion and poor clinical outcome (5). Furthermore, an increased C3a/C3 ratio was found in plasma of trauma patients and proposed as trigger for septic complications (6). Moreover, in critically ill patients enhanced plasma levels of C3a during the early phase of intensive care were predictive for the onset of adult respiratory distress syndrome (ARDS) (7). From a therapeutic aspect, early recognition of HS and innovative resuscitation strategies are crucial to improve organ function and the outcome of shock patients (8, 9). However, resuscitation protocols employing distinct blood products at various ratios often disregard blood-derived complement activation products as additional, and highly potent, inflammatory drivers - thus possibly replacing one harmful product with another. Consequently, specific complement inhibitory strategies are needed.
In experimental murine HS, absence of C3 reduced signs of HS-induced hepatic injury and systemic inflammation (10). In rat HS, C3 depletion by cobra venom factor (CVF) resulted in improved hemodynamic parameters post resuscitation (11) and vascular reactivity to norepinephrine (12). In line with these observations, blockade of C3 convertases by decay accelerating factor (DAF) in a porcine HS model appeared to significantly reduce fluid requirements, intestinal-, lung- and kidney damage, and of note, also early mortality (13).
Despite broad experimental evidence corroborating beneficial effects of targeted complement intervention after traumatic hemorrhagic shock, no valid preclinical study to investigate the early effects of specific C3 blockade during HS has so far been conducted. For an effective and translational complement inhibition at the level of C3, which marks the point of convergence of all complement activation pathways and major mediator of effector and cross-talk functions, the C3 inhibitor compstatin and its analogs have emerged as promising candidate. The compstatin analog Cp40, a cyclic, non-immunogenic peptide of 14 amino acids with strong and exclusive affinity for human and NHP C3 (14), has shown strong efficacy both in NHP models of disease, e.g. in the setting of sepsis, hemodialysis or periodontal disease (15, 16). Moreover, several compstatin derivatives are currently in clinical development and evaluation for the treatment of various diseases.
Therefore, we hypothesized that effective C3 blockade by compstatin Cp40 in a resuscitated NHP model of hemorrhagic shock may lead to early organ protection and improved clinical outcome.
MATERIALS AND METHODS
Compstatin Cp40
The Cp40 analog of the peptidic C3 inhibitor compstatin with the sequence dTyr-Ile-[Cys-Val-(1Me)Trp-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-mIle (with brackets marking the disulfide-bridged cycle; Sar, sarcosine; mIle, N-methyl isoleucine) was produced by solid-phase peptide synthesis as described before (14). The compound was attested a purity of more than 98.9% and was tested negative for endotoxins. For all experiments, lyophilized Cp40 was dissolved in saline shortly before administration.
Experimental design
Eight male cynomolgus monkeys (Macaca fascicularis) weighing 4.8 ± 0.4 kg, aged 4-5 years were divided into 2 groups (treatment and vehicle control). The study was approved by the IACUC of the Simian Conservation Breeding and Research Center in Makati City, Philippines (Study SIC-120 approval 2013-02). All experiments were performed under the conditions described in the Guide for the Care and Use of Laboratory Animals as defined by the National Institutes of Health and following the ARRIVE guidelines. The animals were quarantined for 3 months prior to the study. They were fasted overnight before start of the experiment, but had access to water ad libitum. Housing was performed at 26 ± 4°C with a relative humidity of 75 ± 25% (natural humidity) and a 12-hour light-dark cycle.
Anesthesia was induced by intramuscular injection of 10 mg/kg BW ketamine and 1.0 mg/kg BW Xylazine. The animals were intubated and ventilated in a volume-controlled mode using a tidal volume of 6 ml/kg, I:E=1:2, PEEP=5 cmH2O and a respiratory rate of 18-20 breaths/min (Servo 900B, Maquet) in order to maintain an arterial partial pressure of CO2 at 35–45 mmHg. The fraction of inspired oxygen was set at 0.30 and adjusted, if necessary, according to blood gas analyses. Body temperature was kept at 37 °C.
Catheters were placed in both right and left femoral arteries; one for blood pressure and heart rate measurements and one for blood withdrawal. The left femoral vein was cannulated for blood withdrawal. A central arterial catheter (Pulsion Medical Systems, Feldkirchen, Germany) was placed in the right femoral artery. Finally, a urinary catheter was inserted through the urethra.
After 15 minutes stabilization time (Figure 1A), hemorrhagic shock was induced by constantly withdrawing blood from one femoral artery. This was done until a mean arterial pressure (MAP) of 30 mmHg or until 45% of the calculated total blood volume was withdrawn. The MAP of 30 mmHg was maintained for 1 hour (Figure 1A). If necessary, more blood was withdrawn within this 1-hour timeframe, unless the threshold of 45% total blood volume had been reached. After 1 hour of HS, the animals were resuscitated with four times the withdrawn blood volume as ringer’s lactated solution within 30 minutes (Figure 1A). Thereafter, intravenous infusion of ringer’s lactated solution was maintained at 10mL/kg/h. If the MAP fell below 60 mmHg, norepinephrine was infused. If blood sugar levels were below 80 mg/dL, ringer’s solution was replaced by D5-Ringer’s lactated solution until a glucose level between 80 and 120 mg/dL was reached.
Figure 1.
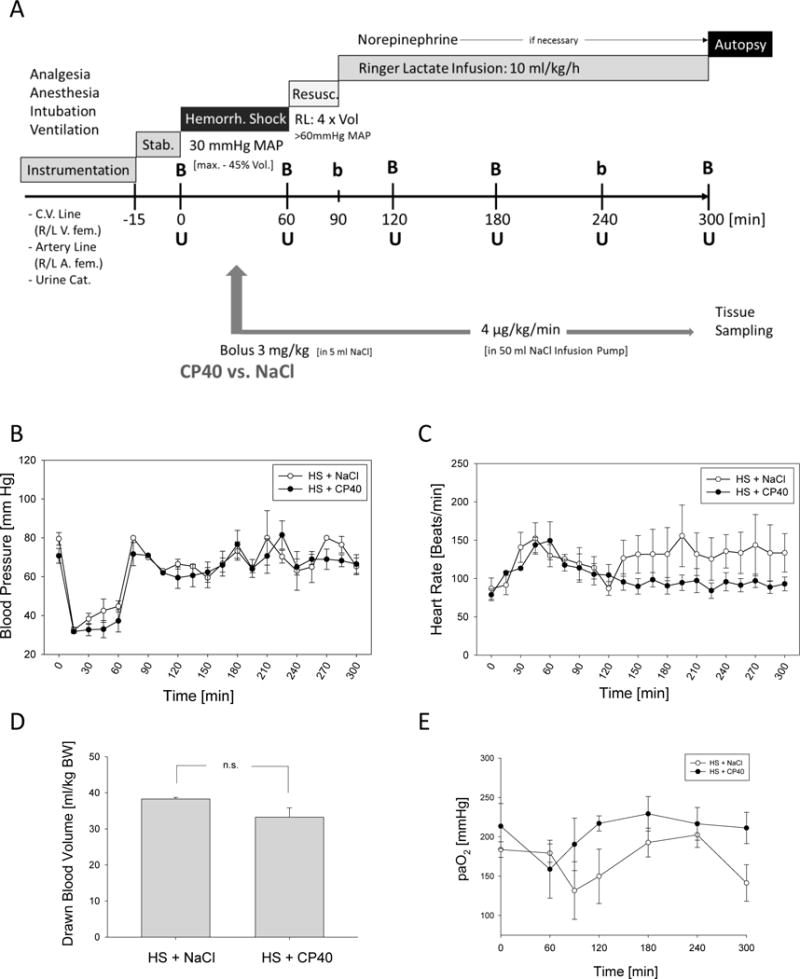
A. Experimental design. B=Blood drawing including blood gas analysis, b=blood gas analysis only, U=urine sampling, Resusc.=resuscitation, Stab=stabilization phase, RL=Ringer´s lactate, R/L=right/left, fem=femoralis. After induction of hemorrhagic shock, animals received either the compstatin analogue Cp40 or vehicle. Hemodynamic monitoring during the experimental period, B. intra-arterial blood pressure measurements, C. heart rate. D. Volume of the total blood drawn for induction of the hemorrhagic shock. E. Blood gas analysis: paO2. Mean ± SEM; n=4/group.
To mimic a first therapeutic window of opportunity, e.g. in the emergency room, 30 minutes after induction of HS, treatment was started with either Cp40 or vehicle (Figure 1A). An initial bolus of 5 ml 0.9% NaCl, with addition of 3 mg/kg Cp40 in the treatment group, was given. Subsequently, a continuous infusion of 4 μg/kg/min Cp40 in 0.9% NaCl was administered via an infusion pump. A total volume of 50 mL was administered until the end of the experiment, i.e. 5 hours after induction of HS. In the vehicle group, the same volume of 0.9% NaCl without Cp40 was infused in the same time.
Blood pressure, heart rate, international normalized ratio (INR), and activated partial thromboplastin time (aPTT) were measured every 30 minutes. Rotational thromboelastometry (ROTEM; TEM GmbH, Munich, Germany) was performed only at baseline and after resuscitation due to limited instrument capacity. Blood gases, lactate, glucose, electrolytes, interleukin (IL)-6, macrophage migration inhibitory factor (MIF), urine output, serum creatinine, urine neutrophil gelatinase-associated lipocalin (NGAL), serum sodium, and intestinal fatty acid-binding protein (iFABP) were measured every 60 minutes (Figure 1A). At the end of observation period, anaesthetized animals were euthanized by intracardial injection of 150 mg/kg BW KCl. Post mortem, the small intestine and kidneys were collected for further microscopic analysis.
Inflammation
Blood was collected and centrifuged at 2000 × g for 15 minutes at 4°C. Serum samples were stored at −80°C until batch sample analysis for IL-6, MIF, macrophage inflammatory protein (MIP)‐1α, and iFABP concentrations using the corresponding Quantikine ELISA kits (R&D systems, USA) according to manufacturer`s instructions. Monkey multiplex cytokine serum analyses were performed in accordance to the manufacturer’s protocol (Life Technology, Frederick, MD, USA).
Coagulation
Citrated blood was used for the coagulation tests. Prothrombin time (PT) and aPTT were measured using standard clinical laboratory equipment. The INR (international normalized ratio) was calculated as the PT ratio from the test sample to a control sample.
ROTEM analysis was performed immediately after sample collection. For each animal of the treatment or vehicle control groups, two cups (EXTEM and FIBTEM) were used. EXTEM is an extrinsically activated assay using recombinant tissue factor and FIBTEM is an extrinsically activated test using recombinant tissue factor with cytochalasin D, which blocks the platelet skeleton and thereby inhibits platelet function. Thus, this test provides information on the fibrin component of the clot. Maximum clot firmness (MCF [mm], the peak strength of the clot, resulting from the interaction of fibrin, activated platelets and factor XIII [FXIII]) were measured. The platelet component of clot strength was calculated by subtracting FIBTEM MCF from EXTEM MCF (MCFplatelet=MCFEXTEM-MCFFIBTEM).
Kidney function
Urine output was determined every 60 min (Figure 1A) and NGAL was determined by ELISA (R&D systems). Serum creatinine was measured using the Jaffe reaction. Serum sodium and potassium was obtained from the blood gas measurements.
Histology
Kidney and small intestinal tissue samples were fixed in 3.7% formaldehyde (Fishar) and embedded in paraffin. Four-μm paraffin sections were cut and subsequently stained with Gill`s hematoxylin and eosin (H&E) (Morphisto). Slides were visualized using a Zeiss Axio Imager A1 microscope. The lens was a 10× objective and the fields evaluated were 3,150,000 μm2.
Sections of small intestine were scored as described by Cho et al. (17) in a blinded fashion. At least 80 villi per section were analyzed and means were calculated for each animal. Goblet Cells (GCs) along the epithelial lining of villi were counted and length of analyzed section of epithelium was measured with Axio Vision Software (Edition 4.9; Zeiss, Germany). The number of GCs was divided by the length of epithelium in μm and is depicted as fraction ratio. In addition, an immuno-histochemical analysis was performed on small intestine tissue sections staining for cleaved caspase-3 (Cell Signaling Technology, Germany).
In kidney sections, thirty glomeruli per specimen were analyzed for dilation of bowman’s capsule, precipitations in the urinary space and neutrophilic infiltration. Results are depicted as fraction of positive findings among all glomeruli evaluated.
Per sample, 5 fields of view (200× magnification) were scored for tubular injury and neutrophilic infiltration. Necrosis of epithelial cells and dilation of tubules were graded as follows: 1) findings in less than 5%, 2) findings in 5-25%, 3) findings in 25-50%, 4) findings in 50-75%, 5) findings in more than 75% of respective tubules. Neutrophilic Infiltration was graded as follows: 1) no infiltration present, 2) single focus of marked infiltration, 3) multiple foci and/or widespread infiltration.
Statistical Analysis
For statistical analysis SigmaStat version 3.5 (Systat Software, Inc.) was used. All values are expressed as means ± SEM. Data were assumed to be normally distributed and were therefore analyzed by one-way ANOVA followed by Student-Newman Keuls post-hoc testing. To analyze differences between Cp40 treatment and vehicle in a kinetic way, two-way ANOVA with Sidak correction for multiple comparisons was performed. Differences were always considered significant when p ≤ 0.05.
RESULTS
Physiology
Hemorrhagic shock could be successfully induced in all animals. In none of the animals, the maximum amount of 45% total blood volume was needed to be withdrawn. There was no difference between the two groups concerning withdrawn blood volume (Figure 1D). All animals reached an MAP of 30 mmHg within 10 min, which was stable until the beginning of the resuscitation (Figure 1B). Concomitantly, the heart rate increased to 150 bpm (Figure 1C) indicating HS status. This was confirmed by low base excess, low hemoglobin concentration and low hematocrit (data not shown). Thus, at the beginning of administration of Cp40 or vehicle, the animals were similar in their physiological status.
Treatment with Cp40 did not result in differences in blood pressure over the entire observation time when compared to the vehicle control group. Upon resuscitation, MAP returned to normal levels, and no differences between the two groups could be observed (Figure 1B). Significantly different kinetics (p<0.0001) were, however, measured for heart frequency. Animals receiving Cp40 treatment returned to normal frequency within 60 min (Figure 1C). In contrast, heart rates remained high until the end of the experiment in the vehicle-treated animals (Figure 1C). Interestingly, both the hemoglobin and hematocrit values were significantly higher during the last two hours of the experiment when animals were treated with Cp40. Of note, their values were almost in the lower normal range (data not shown). paO2 values stayed normal with Cp40 treatment, whereas they decreased at the end of the observation period in the vehicle-treated animals (Figure 1E). Finally, the kinetics of paO2 values was significantly different between the Cp40- and vehicle-treated animals (p=0.0086).
Inflammation
The inflammatory markers IL-6 and MIF showed a continuous and significant increase until the end of the experiment in vehicle-treated animals. Whereas Cp40-treated animals also had an increase in these marker, the change was much more moderate. During the last 2 hours of the observation period, inflammatory marker concentrations were significantly higher in vehicle control animals when compared to Cp40-treated HS animals (Figures 2A and 2B). A similar pattern was found by trend for other key inflammatory mediators such as IL-1 receptor antagonist (IL-1RA), regulated on activation normal T cell expressed and secreted (RANTES), macrophage inflammatory protein-1α (MIP-1α), monocyte chemoattractant protein-1 (MCP-1), and interferon γ (IFNγ) (Supplementary Figure 1).
Figure 2.
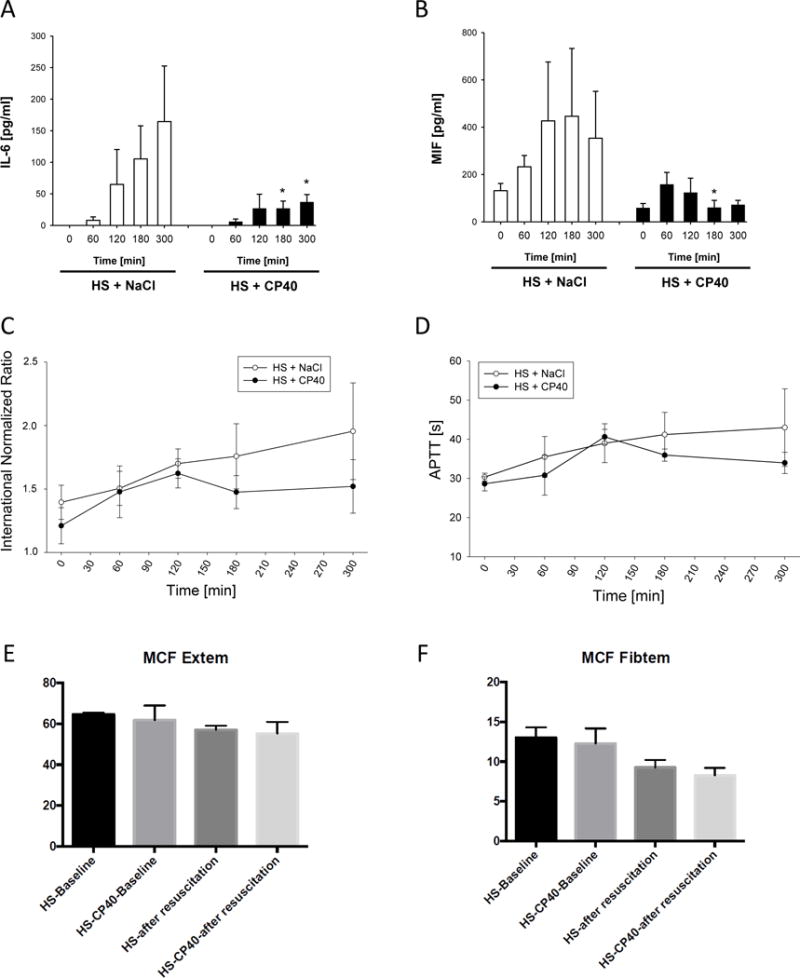
Inflammatory and coagulatory response during HS in absence or presence of C3 blockade by Cp40. Serum concentrations of A. interleukin IL-6 and B. macrophage inhibitory factor (MIF). Coagulation was monitored by C. international normalized ration (INR) and D. activated prothrombin time (APTT) as well as ROTEM-analyses: mean clot firmness in E. Extem and F. Fibtem. Mean ± SEM; n=4/group; *p<0.05 Cp40 vs vehicle-treatment.
Coagulation
INR showed a slight increase of 0.3 in both groups until 60 min after beginning of resuscitation (Figure 2C), which can be attributed to HS. Interestingly, Cp40-treated animals maintained an INR of around 1.4 whereas vehicle treatment led to a further increase of the INR by 0.43 ± 0.03 until the end of the experiment (Figure 2C). A similar, though less pronounced, difference of kinetics between both groups was seen for the aPTT measurements (Figure 2D). ROTEM analysis for MCF in both EXTEM and FIBTEM indicated some reduction after resuscitation (Figure 2E and 2F), with no differences observed between the two experimental groups.
Kidney
Cp40 treatment protected the kidneys from damage as seen in the vehicle-treated animals (Figure 3). Urine output kinetics were significantly different when comparing Cp40 with vehicle treatment (p=0.0009). The urine output of animals receiving Cp40 remained significantly improved (p<0.0001) over vehicle-treated animals, even after 240 min (p<0.01; Figure 3A). In fact, vehicle-treated animals hardly produced any urine from this time point on. In contrast, no significant differences could be detected for creatinine concentrations in serum (Figure 3B). They remained within the normal range in both groups.
Figure 3.
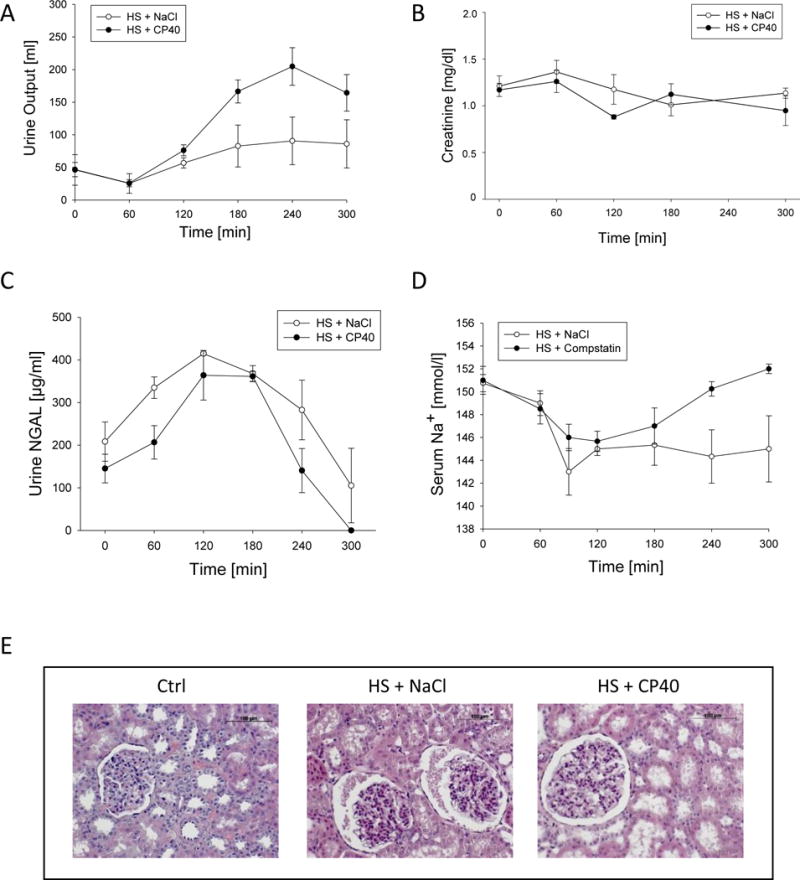
Renal function and morphology in animals suffering from HS treated with either Cp40 or vehicle. A. Urine Output, B. serum creatinine concentrations, C. urine neutrophil gelatinase-associated lipocalin (NGAL), D. serum sodium concentrations, and E. hematoxylin eosin staining of representative kidney sections (magnification 200×). Mean ± SEM; n=4/group; *p<0.05 Cp40 vs vehicle-treatment.
NGAL concentrations in urine displayed similar kinetics in both groups with a trend of lower concentrations in presence of Cp40 (Figure 3C). Concentrations increased until 2 hours after induction of HS and remained stable until 180 min. Thereafter, concentrations decreased below baseline values until the end of the experiment (Figure 3C).
Serum sodium concentrations initially dropped in both groups during HS until the beginning of resuscitation (Figure 3D). From that time on, concentrations in Cp40-treated animals returned to baseline levels, whereas serum sodium stayed at minimum levels in vehicle control animals until the end of HS (Figure 3D). The kinetics between the two groups were significantly different (p<0.01). Moreover, from 240 min onwards, the single sodium concentrations were significantly different between Cp40- and vehicle-treated animals (p<0.05). Therefore, it is likely that Cp40 treatment reversed the impaired sodium retention in the kidneys as observed in the vehicle-treated animals. Potassium levels didn’t differ between groups and time and was in mean 4.2 ± 0.1 mmol/L.
Histopathologic features observed in the kidney are presented by Figure 3E. Overall, kidney histology of Cp40-treated animals revealed a weak amelioration of the significant morphological alterations induced by HS in vehicle-treated animals, such as dilation of the glomerular capsule and signs of beginning protein casts (Figure 3), which were confirmed to contain albumin using immunohistochemistry (data not shown). Histological assessment of HE- and PAS-stained sections revealed a loss of tubular brush borders after HS, which was ameliorated although statistically not significant upon Cp40 treatment (Table 1).
Table 1.
Histological findings of kidney tissue obtained after HS, treated with Cp40 or vehicle.
| Parameter | Ctrl | HS + NaCl | HS + CP40 | Significance NaCl vs CP40 |
|---|---|---|---|---|
| mean ± SEM | mean ± SEM | mean ± SEM | ||
| Glomeruli | ||||
| Dilation | 0.11 ± 0.06 | 0.73 ± 0.03* | 0.65 ± 0.07* | n.s. |
| Precipitation | 0.11 ± 0.08 | 0.16 ± 0.10 | 0.05 ± 0.03 | n.s. |
| Infiltration | 0 ± 0 | 0 ± 0 | 0 ± 0 | n.s. |
| Proximal Tubulus | ||||
| Apoptosis | 1.07 ± 0.07 | 1.40 ± 0.20 | 1.15 ± 0.05 | n.s. |
| Dilation | 1.53 ± 0.24 | 3.73 ± 0.37* | 3.75 ± 0.38* | n.s. |
| Distal Tubulus | ||||
| Apoptosis | 1.07 ± 0.07 | 1.13 ± 0.07 | 1.20 ± 0.12 | n.s. |
| Dilation | 1.13 ± 0.07 | 2.07 ± 0.18* | 2.40 ± 0.29* | n.s. |
| Neutrophil Infiltration | ||||
| 1.00 ± 0.00 | 1.07 ± 0.07 | 1.10 ± 0.10 | n.s. | |
| Brush Border Loss | ||||
| 1.20 ± 0.2 | 4.00 ± 0.33* | 3.40 ± 1.35* | n.s. | |
p<0.05 versus Ctrl (ANOVA, post-hoc SNK test)
Small intestine
Cp40 could also reduce pathological changes in the small intestine in comparison to vehicle treatment (Figure 4). This was already macroscopically observed during the autopsy. Almost no intestinal edema nor petechial bleedings were detected when the animals had been treated with Cp40 (Figure 4A). Animals without Cp40 presented massive swelling of the small intestine with loss of macroscopic discernibility of villi (Figure 4A), and their tissue did show petechial bleedings.
Figure 4.
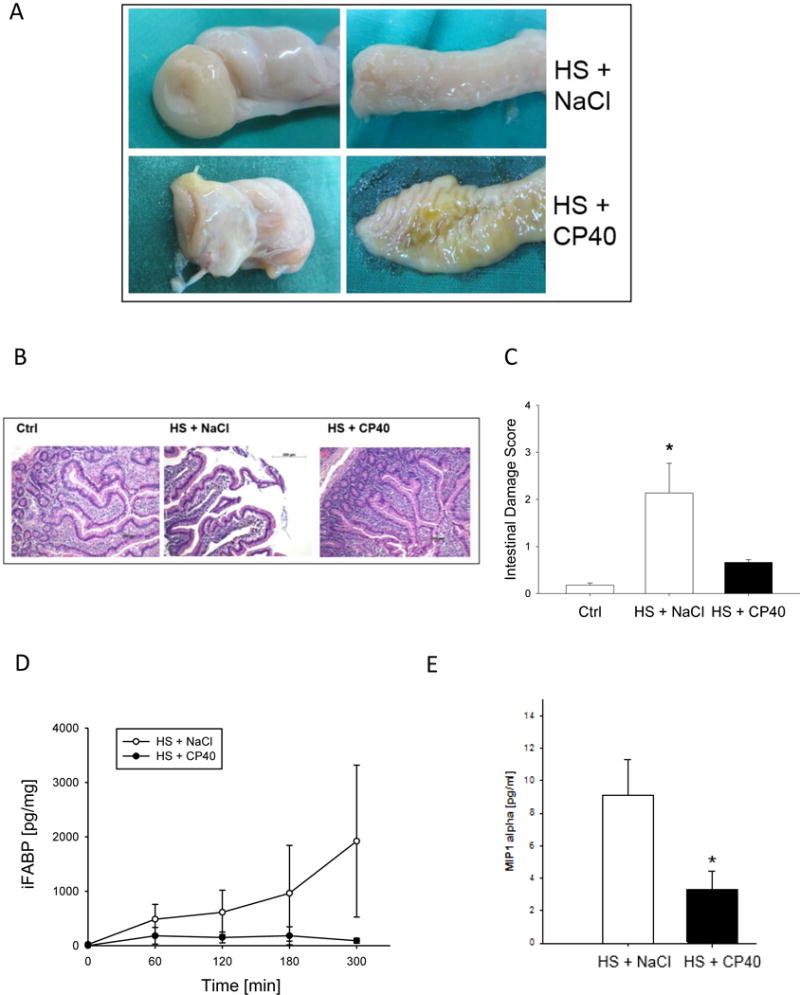
Intestinal macro- and microscopical findings after HS in non-human primates treated with either Cp40 or vehicle. A. Autopsy findings of small bowel, B. hematoxylin eosin staining of ileum sections (magnification 100×), C. intestinal damage score according to Chiu. Corresponding biochemical marker D. in serum: intestinal fatty acid binding protein (iFABP) and E. in peritoneal fluid: macrophage inflammatory protein (MIP-1α). Mean ± SEM; n=4/group; *p<0.05 Cp40 vs vehicle-treatment.
The GC ratio (number of GCs divided by length of intestinal epithelium) was 0.6 ± 0.004 in sham animals and decreased to 0.3 ± 0.01 after HS. Upon Cp40 treatment, the GC ratio slightly but insignificantly rose to 0.4 ± 0.01 (data not displayed). Villi damage was confirmed in histological samples of vehicle-treated animals, whereas Cp40 treatment resulted in preservation of the villi structures including the crypts (Figure 4B). This was confirmed by a worsened intestinal damage score (Chiu) in vehicle control animals after HS, indicating significant small intestinal damage (Figure 4C). In contrast, the Chiu score was not significantly different in Cp40-treated HS animals when compared to sham animals (Figure 4C). Furthermore, the Cp40 treatment reduced by trend immunohistochemical signs of apoptotic events within the small intestine (Supplementary Figure 2).
These observations were biochemically confirmed by measuring iFABP levels (Figure 4D), which maintained normal (close to the detection limit) in presence of Cp40 treatment. A continuous and significant increase was determined in vehicle-treated animals until the end of the observation period (Figure 4D). Furthermore, MIP1α concentrations in peritoneal fluid after HS was significantly decreased in animals treated with Cp40 (Figure 4E).
DISCUSSION
Hemorrhagic shock (HS), and the resulting early onset of complications, remain a scientific and clinical challenge. Despite the emergence of various resuscitation protocols, a mechanistically rational therapy remains challenging. To address HS-induced early immune, coagulation and organ dysfunction, a specific therapeutic strategy targeting central complement components could be promising. In this study, we therefore applied the compstatin analog Cp40 during HS to effectively and specifically block C3 as a hub for complement and coagulation activation, danger molecule and pathogen clearance, and microbial immune evasion (15, 18). Of note, both animal and human phase I trial data have demonstrated that Cp40 is safe for inhibiting complement (reviewed in (19)). HS was induced in NHP in a pressure-controlled manner, and Cp40 was applied after the acute shock phase with a therapeutic delay of 30 min that mimics the clinical situation (Fig. 1A). It is noteworthy, that a 30 min delay of intervention does by far not cover all trauma patients, but certainly could be acutely applied in many cases at the scene or in the hospital in cases of acute bleeding such as upper gastrointestinal bleeding. Furthermore, it could be argued that the positive effects of Cp40 may be due to a slightly (although not significant) smaller blood volume withdrawn. However, despite this by tendency smaller withdrawal of blood, the MAP in the Cp40 treated group was lower during the hemorrhagic shock period compared to control, suggesting that the Cp40 effects observed post resuscitation on the organ functions are highly relevant. In this therapeutic setting of a resuscitated hemorrhagic shock, we observed a marked effect of complement-targeted therapy on several outcome parameters. Concerning hemodynamics, the mean MAP course was rather superimposable in the control and Cp40 group, whereas the heart rate returned to normal values after the shock phase in the presence of Cp40, but remained elevated in the control group (Fig. 1B and C). In baboons with E.coli-induced severe sepsis, a similar approach with a previously developed compstatin analog was capable to improve the hemodynamic alterations even when applied in a delayed fashion (16). Similarly, in a rodent model of HS, comprehensive C3 depletion by CVF resulted in improved post resuscitation hemodynamics (11). Furthermore, a rapid normalization of the HS-caused drop in paO2 levels could be detected in presence of Cp40 post resuscitation, but not in absence of C3 inhibition (Fig. 1E). Calculation of the Horovitz coefficient revealed an early development of a mild acute respiratory distress syndrome (ARDS) with paO2/FiO2 <300 mmHg for the first hour after HS in the control group but not in the Cp40-treated group. This is in accordance with reported improved early signs of ARDS in septic baboons by compstatin treatment resulting in reduced capillary leak and leucocyte infiltration (16), and also in reduced pulmonary fibrogenic events within the lungs (20). Regarding systemic effects, HS expectably caused an early enhanced cytokine and chemokine release, which was significantly ameliorated by Cp40 infusion (Fig. 2A and B). The used Cp40 concentrations were measured and verified to be sufficient to completely block systemic C3 levels (data not shown). Based on the intense complement-coagulation crosstalk (19), we also expected some systemic effects on the HS-induced hemostasis. Although not altering mean clot firmness, Cp40 was capable to beneficially improve disturbances of the coagulatory response, especially of the tissue factor pathway as indicated by improved prothrombin time (INR values) (Fig. 2C-F). Again, these data are supported by improved coagulation tests (e.g. normalized aPTT) in septic baboons upon C3 inhibition (16). Although not shown here, it is likely that C3 inhibition results in reduced thrombus formation. Indeed, C3 has previously been proposed as a focal point in the formation of thrombi, and C3 deficiency seems to be associated with a lower incidence of thrombosis, smaller thrombus size and reduced fibrin deposition (21, 22).
Owing to the kidneys’ relevance as an important shock organ, we could detect early protective effects after delayed C3 blockade using Cp40, as indicated by an enhanced urine output, a trend in reduction of NGAL serum levels, normalized sodium concentrations and a tendency to less dilatation of the glomeruli, proximal and distal tubules as well as less brush border loss (Fig. 3, Table 1). The creatinine serum levels did not significantly rise early after HS and did not differ between the treatment groups up to 4 hours after HS and thus might not be a reliable early renal damage marker. In contrast, NGAL serum levels peaked 60 min after resuscitation and were less pronounced, although not significantly, at each time point in the Cp40 group. In accordance, earlier studies of renal ischemia/reperfusion injury in pigs showed that creatinine levels only rose beyond day 1 and were to some extent less pronounced when the classical pathway was inhibited by a C1q inhibitor (23). Similar findings were published for creatinine levels in non-human primates with E. coli-induced sepsis (16). In experimental HS and synchronic LPS challenge, a deposition of C3 cleavage products was reported in the kidney, known as a frequent early failing organ (24). Concomitantly, after murine renal ischemia/reperfusion injury, C3b accumulated in the brush borders of the proximal tubules (25). Even though histological changes such as acute tubular necrosis or glomerular changes are hard to detect as early as 4 hours after shock, some morphological alterations were evident. A tendency of improved early signs of morphological kidney alterations were noted in the Cp40 group, although an assessment of the development of full acute kidney injury after HS would require a longer observation period. The exact mechanism how Cp40 improves kidney function is not yet known and needs to be addressed in future analyses.
Importantly, the gastrointestinal tract has been considered a critical engine of HS-related multiple-organ-failure. Not only protease release (26) but also epithelial damage and gut-barrier-failure with subsequent bacterial translocation has been proposed as pathophysiological drivers of HS towards multiple organ dysfunction syndrome (MODS). In the past, complement inhibition on the level of C5 activation revealed some protective effects on HS-induced intestinal damage (27). Recently, TLR2-dependent intestinal expression and deposition of C3 has been proposed as central pathomechanism for intestinal damage and systemic inflammatory response after HS (28). In the present study, blinded assessment by an independent pathologist found significant signs of mucosal edema in the control group, predominantly in the small intestine, which were completely abolished in the case of Cp40 treatment. The macroscopic findings were reflected by the histological (intestinal damage score) and biochemical data (iFABP in serum and inflammatory mediators in peritoneal fluids), indicating protection of HS-induced intestinal tissue damage by Cp40 (Fig. 4). Accordingly, in a porcine pressure-controlled model of HS, indirect C3 targeting by C1 inhibition (29) or by DAF (13) revealed a dose-dependent protection against HS-associated intestinal and pulmonary damage. These protective effects on the intestine and various other organs by C1 inhibition were still present when the pigs were objected to further multiple injuries in addition to HS (30). In rodent HS models, the soluble complement receptor 1 was capable to reduce small bowel injury and intestinal neutrophil influx (31), to enhance compromised mucosal blood flow, to prevent post resuscitation vasoconstriction and thereby gut ischemia, and to improve intestinal endothelial function (32). In this context, it had been proposed that HS may engage a vicious cycle that leads to activated complement-dependent bacterial translocation from the gut and results in subsequent endotoxemia (33). Notably, C3 targeting has been suggested to reduce endotoxin levels after HS by improving the gut-blood barrier through a hitherto undescribed mechanism (34). As recently shown, intestinal ischemia/reperfusion injury in mice led to mucosal injury as early as 30 min after ischemia, which was associated with an increased appearance of C3, or C3 fragments, within intestinal epithelial cells (35). The reperfusion injury was significantly reduced in C3-deficient mice or by C3 depletion (35).
The study presented here has some intrinsic limitations that need to be considered. First, the number of animals per group was kept rather small. As a consequence, we have assumed normal distribution, and some of the observed effects might have been underestimated. Second, to allow for a rational use of local resources, the observation period was limited to 5 hours after induction of HS and thus might have been too short for manifestation of organ failure, thereby not enabling an assessment of potential long-term effects of Cp40 therapy. Third, no dose-escalation protocol could be applied without significantly expanding the number of experimental animals (see above). However, we have used an established dose, based on previous dose findings studies, which could completely inhibit circulating C3 as confirmed by serum complement analyses (see above).
Overall, present data suggest that C3 inhibition by Cp40 may improve early organ dysfunction in a NHP model of HS and holds a promising translational potential for an improved clinical outcome in HS patients.
Supplementary Material
Acknowledgments
We thank Sonja Braumüller, Bettina Klohs and Anne Rittlinger for excellent technical support.
SOURCES OF FUNDING
This study was supported by institutional funds to MHL and MvG, and by grants from the National Institutes of Health (AI030040, AI068730) and funding provided by the seventh framework programme of the European Union (FP7 DIREKT) to JDL. The authors are responsible for the contents of this publication.
Footnotes
DISCLOSURES
The authors declare there are no commercial or financial conflicts of interests related to the studies. J.D. Lambris is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs such as Cp40). J.D. Lambris and D. Ricklin are inventors of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. J.D. Lambris is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (i.e., 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives).
References
- 1.Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36:691–709. doi: 10.1016/j.injury.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 2.Schochl H, Schlimp CJ, Maegele M. Tranexamic acid, fibrinogen concentrate, and prothrombin complex concentrate: data to support prehospital use? Shock. 2014;41(Suppl 1):44–46. doi: 10.1097/SHK.0000000000000093. [DOI] [PubMed] [Google Scholar]
- 3.Levy MN, Blattberg B, Barlow JL. Effects of hemorrhagic shock upon the properdin system of the dog. J Lab Clin Med. 1960;56:105–109. [PubMed] [Google Scholar]
- 4.Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, Bruckner U, Stahl GL, Blom AM, Perl M, et al. Early complementopathy after multiple injuries in humans. Shock. 2012;37(4):348–354. doi: 10.1097/SHK.0b013e3182471795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, Pittet JF. Role of the alternative pathway in the early complement activation following major trauma. Shock. 2007;28(1):29–34. doi: 10.1097/shk.0b013e3180342439. [DOI] [PubMed] [Google Scholar]
- 6.Hecke F, Schmidt U, Kola A, Bautsch W, Klos A, Kohl J. Circulating complement proteins in multiple trauma patients–correlation with injury severity, development of sepsis, and outcome. Crit Care Med. 1997;25(12):2015–2024. doi: 10.1097/00003246-199712000-00019. [DOI] [PubMed] [Google Scholar]
- 7.Gama de Abreu M, Kirschfink M, Quintel M, Albrecht DM. White blood cell counts and plasma C3a have synergistic predictive value in patients at risk for acute respiratory distress syndrome. Crit Care Med. 1998;26(6):1040–1048. doi: 10.1097/00003246-199806000-00025. [DOI] [PubMed] [Google Scholar]
- 8.Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, del Junco DJ, Brasel KJ, Bulger EM, Callcut RA, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471–482. doi: 10.1001/jama.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbee RW, Reynolds PS, Ward KR. Assessing shock resuscitation strategies by oxygen debt repayment. Shock. 2010;33(2):113–122. doi: 10.1097/SHK.0b013e3181b8569d. [DOI] [PubMed] [Google Scholar]
- 10.Cai C, Gill R, Eum HA, Cao Z, Loughran PA, Darwiche S, Edmonds RD, Menzel CL, Billiar TR. Complement factor 3 deficiency attenuates hemorrhagic shock-related hepatic injury and systemic inflammatory response syndrome. Am J Physiol Regul Integr Comp Physiol. 2010;299(5):R1175–1182. doi: 10.1152/ajpregu.00282.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Younger JG, Sasaki N, Waite MD, Murray HN, Saleh EF, Ravage ZB, Hirschl RB, Ward PA, Till GO. Detrimental effects of complement activation in hemorrhagic shock. J Appl Physiol (1985) 2001;90(2):441–446. doi: 10.1152/jappl.2001.90.2.441. [DOI] [PubMed] [Google Scholar]
- 12.Chen D, Song MQ, Liu YJ, Xue YK, Cheng P, Zheng H, Chen LB. Inhibition of complement C3 might rescue vascular hyporeactivity in a conscious hemorrhagic shock rat model. Microvasc Res. 2016;105:23–29. doi: 10.1016/j.mvr.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Dalle Lucca JJ, Li Y, Simovic MO, Slack JL, Cap A, Falabella MJ, Dubick M, Lebeda F, Tsokos GC. Decay-accelerating factor limits hemorrhage-instigated tissue injury and improves resuscitation clinical parameters. J Surg Res. 2013;179(1):153–167. doi: 10.1016/j.jss.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, Tzekou A, DeAngelis RA, Resuello RR, Lupu F, et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218(4):496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, Lupu F, Nilsson B, Risitano AM, Ricklin D, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45(4):423–440. doi: 10.1111/eci.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, Ivanciu L, Lupu C, Mollnes TE, Taylor FB, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116(6):1002–1010. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho SS, Rudloff I, Berger PJ, Irwin MG, Nold MF, Cheng W, Nold-Petry CA. Remifentanil ameliorates intestinal ischemia-reperfusion injury. BMC Gastroenterol. 2013;13:69. doi: 10.1186/1471-230X-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. 2016;274(1):33–58. doi: 10.1111/imr.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiegner R, Chakraborty S, Huber-Lang M. Complement-coagulation crosstalk on cellular and artificial surfaces. Immunobiology. 2016;221(10):1073–1079. doi: 10.1016/j.imbio.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Silasi-Mansat R, Zhu H, Georgescu C, Popescu N, Keshari RS, Peer G, Lupu C, Taylor FB, Pereira HA, Kinasewitz G, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med. 2015;19(11):2549–2563. doi: 10.1111/jcmm.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norgaard I, Nielsen SF, Nordestgaard BG. Complement C3 and High Risk of Venous Thromboembolism: 80517 Individuals from the Copenhagen General Population Study. Clin Chem. 2016;62(3):525–534. doi: 10.1373/clinchem.2015.251314. [DOI] [PubMed] [Google Scholar]
- 22.Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, Wenzel P, Langer F, Reinhardt C, Ruf W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129(16):2291–2302. doi: 10.1182/blood-2016-11-749879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delpech PO, Thuillier R, SaintYves T, Danion J, Le Pape S, van Amersfoort ES, Oortwijn B, Blancho G, Hauet T. Inhibition of complement improves graft outcome in a pig model of kidney autotransplantation. J Transl Med. 2016;14(1):277. doi: 10.1186/s12967-016-1013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmermann T, Laszik Z, Nagy S, Kaszaki J, Joo F. The role of the complement system in the pathogenesis of multiple organ failure in shock. Prog Clin Biol Res. 1989;308:291–297. [PubMed] [Google Scholar]
- 25.Hirano M, Ma BY, Kawasaki N, Oka S, Kawasaki T. Role of interaction of mannan-binding protein with meprins at the initial step of complement activation in ischemia/reperfusion injury to mouse kidney. Glycobiology. 2012;22(1):84–95. doi: 10.1093/glycob/cwr107. [DOI] [PubMed] [Google Scholar]
- 26.Bieth J. Problems in intensive care posed by imbalance in the protease–protease inhibitor system. Ann Anesthesiol Fr. 1980;21(6):647–652. [PubMed] [Google Scholar]
- 27.Fleming SD, Phillips LM, Lambris JD, Tsokos GC. Complement component C5a mediates hemorrhage-induced intestinal damage. J Surg Res. 2008;150(2):196–203. doi: 10.1016/j.jss.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goering J, Pope MR, Fleming SD. TLR2 Regulates Complement-Mediated Inflammation Induced by Blood Loss During Hemorrhage. Shock. 2016;45(1):33–39. doi: 10.1097/SHK.0000000000000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalle Lucca JJ, Li Y, Simovic M, Pusateri AE, Falabella M, Dubick MA, Tsokos GC. Effects of C1 inhibitor on tissue damage in a porcine model of controlled hemorrhage. Shock. 2012;38(1):82–91. doi: 10.1097/SHK.0b013e31825a3522. [DOI] [PubMed] [Google Scholar]
- 30.Campbell JC, Li Y, van Amersfoort E, Relan A, Dubick M, Sheppard F, Pusateri A, Niemeyer D, Tsokos GC, Dalle Lucca JJ. C1 Inhibitor Limits Organ Injury and Prolongs Survival in Swine Subjected to Battlefield Simulated Injury. Shock. 2016;46(3 Suppl 1):177–188. doi: 10.1097/SHK.0000000000000677. [DOI] [PubMed] [Google Scholar]
- 31.Spain DA, Fruchterman TM, Matheson PJ, Wilson MA, Martin AW, Garrison RN. Complement activation mediates intestinal injury after resuscitation from hemorrhagic shock. J Trauma. 1999;46(2):224–233. doi: 10.1097/00005373-199902000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Fruchterman TM, Spain DA, Wilson MA, Harris PD, Garrison RN. Complement inhibition prevents gut ischemia and endothelial cell dysfunction after hemorrhage/resuscitation. Surgery. 1998;124(4):782–791. doi: 10.1067/msy.1998.91489. discussion 791–782. [DOI] [PubMed] [Google Scholar]
- 33.Szebeni J, Baranyi L, Savay S, Gotze O, Alving CR, Bunger R, Mongan PD. Complement activation during hemorrhagic shock and resuscitation in swine. Shock. 2003;20(4):347–355. doi: 10.1097/01.shk.0000082444.66379.17. [DOI] [PubMed] [Google Scholar]
- 34.Wei YJ, Wang RL, Li GP. Influence on the concentration of plasma endotoxin by inhibition of complement activation in traumatic hemorrhagic shock rats. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2006;18(3):180–183. [PubMed] [Google Scholar]
- 35.Satyam A, Kannan L, Matsumoto N, Geha M, Lapchak PH, Bosse R, Shi GP, Dalle Lucca JJ, Tsokos MG, Tsokos GC. Intracellular Activation of Complement 3 Is Responsible for Intestinal Tissue Damage during Mesenteric Ischemia. J Immunol. 2017;198(2):788–797. doi: 10.4049/jimmunol.1502287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.