

Abstract
Background
Mitochondrial genomes of flowering plants (angiosperms) are highly dynamic in genome structure. The mitogenome of the earliest angiosperm Amborella is remarkable in carrying rampant foreign DNAs, in contrast to Liriodendron, the other only known early angiosperm mitogenome that is described as ‘fossilized’. The distinctive features observed in the two early flowering plant mitogenomes add to the current confusions of what early flowering plants look like. Expanded sampling would provide more details in understanding the mitogenomic evolution of early angiosperms. Here we report the complete mitochondrial genome of water lily Nymphaea colorata from Nymphaeales, one of the three orders of the earliest angiosperms.
Results
Assembly of data from Pac-Bio long-read sequencing yielded a circular mitochondria chromosome of 617,195 bp with an average depth of 601×. The genome encoded 41 protein coding genes, 20 tRNA and three rRNA genes with 25 group II introns disrupting 10 protein coding genes. Nearly half of the genome is composed of repeated sequences, which contributed substantially to the intron size expansion, making the gross intron length of the Nymphaea mitochondrial genome one of the longest among angiosperms, including an 11.4-Kb intron in cox2, which is the longest organellar intron reported to date in plants. Nevertheless, repeat mediated homologous recombination is unexpectedly low in Nymphaea evidenced by 74 recombined reads detected from ten recombinationally active repeat pairs among 886,982 repeat pairs examined. Extensive gene order changes were detected in the three early angiosperm mitogenomes, i.e. 38 or 44 events of inversions and translocations are needed to reconcile the mitogenome of Nymphaea with Amborella or Liriodendron, respectively. In contrast to Amborella with six genome equivalents of foreign mitochondrial DNA, not a single horizontal gene transfer event was observed in the Nymphaea mitogenome.
Conclusions
The Nymphaea mitogenome resembles the other available early angiosperm mitogenomes by a similarly rich 64-coding gene set, and many conserved gene clusters, whereas stands out by its highly repetitive nature and resultant remarkable intron expansions. The low recombination level in Nymphaea provides evidence for the predominant master conformation in vivo with a highly substoichiometric set of rearranged molecules.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4991-4) contains supplementary material, which is available to authorized users.
Keywords: Mitochondrial genome, Nymphaea, Repeats, Recombination, PacBio sequencing
Background
The advent of high-throughput sequencing technologies has greatly promoted the research for plant mitochondrial (mt) genomes. Most (~ 80%, 176 out of 214) of the plant mitogenomes deposited in the GenBank database (https://www.ncbi.nlm.nih.gov/genome/organelle/) were generated in the past several years (since 2011). Research of plant mitogenomes have also been expanded to cover phylogenetically more diverse organisms from focusing on crops [1]. To date (as of July 2018), 53 bryophyte and 108 vascular plant complete mitogenomes have been reported (https://www.ncbi.nlm.nih.gov/genome/organelle/). Among them, bryophyte mitogenomes show astoundingly conserved structures and stable genome content in its major lineages [2–4], whereas vascular plant mitogenomes vary significantly in both genome structure and content [1, 5], nucleotide substitution rates [6–8], and repeat recombination level [1, 9]. In particular, angiosperm mitogenomes exhibit highly dynamic characters: ranging from 66 Kb [10] to 11.3 Mb [7] with 19 to 64 [10] known genes (not including duplicate genes and ORFs), 5 [10] to 25 [1] introns, and highly variable intergenic regions [11]. The ca. 200-fold range of mitogenome size divergence is primarily due to the variation in non-coding regions, including repeated sequences [12], introns [13], intracellular transferred sequences from plastid [14] and nucleus [13], and horizontal gene transfers from foreign donors [15, 16].
Vascular plant mitogenomes sequenced to date generally contain a large fraction of repeated sequences of unknown origin [1], with some genomes including numerous short dispersed repeats < 100 bp (e.g., Cucurbita pepo [14], Cycas taitungensis, [17]), some containing considerable amount of large repeats > 1000 bp (e.g., Oryza sativa, [18], Zea mays subsp. parviglumis [19]), and some both (e.g., Silene conica, [7], Psilotum nudum [9]). The prevalence and activity of the repeated sequences play a pivotal role in shaping the plant mitogenome structure [20, 21] through participation in pervasive genome rearrangements [22], recombination dependent replication initiations [23], genome sequence duplications, inversions, insertions and deletions [24]. Up to now, mitochondrial homologous recombinations involving repeated sequences have been investigated in about fourteen vascular plant species with high-depth sequencing data [1, 7, 9, 13, 25–32]. Particularly, studies employing quantitative measuring methods unequivocally uncovered positive correlations between repeat length and recombination rate [10]. Although most of these studies detected minor to moderate recombination activities among small (< 100 bp) and medium sized repeats (100~ 1000 bp) [7], large repeat (> 1000 bp) mediated recombinational equilibrium was also frequently observed in a number of species, including the angiosperms Mimulus guttatus [33], Silene latifolia [32], Silene vulgaris [7], Cucumis sativus [13], and the gymnosperm Ginkgo biloba [31]. Recently, the third-generation long-read DNA sequencing technologies have yielded high quality assemblies for plant mitogenomes, which enabled more accurate and sensitive detection for homologous recombinations [30], apparently devoid of false positives introduced by PCR artifacts [20] or insufficient read length in the Next Generation Sequencing (NGS) approaches [10].
Study of early angiosperm mitogenomes would improve the entire view on the evolutionary pattern of plant mitogenomes. Two available mitogenomes of the earliest angiosperms Amborella [16] and Liriodendron [6] show a series of distinctive features. The 3.9-Mb mitochondrial genome of Amborella with a 63 coding gene set, houses massive horizontal gene transfers (HGTs) from a variety of organisms [16], which is unparalleled and extremely unusual, considering the sporadic occurrences of HGTs detected in some vascular plant mitogenomes, such as Gnetum [34], Malpighiales [35], Plantago [36], Viscum [37] and Lophophytum [38]. The 553-Kb mitogenome of Liriodendron with a similar 64 coding gene set, is otherwise described as “fossilized” due to its extremely low synonymous substitution rate, retention of genes that are missing in the other lineages and many ancestral gene clusters [6]. An expanded sampling of the early angiosperm mitogenomes is needed to elucidate the distribution pattern of these features in early angiosperm mitogenomes. Nymphaea (Nymphaeaceae, Nymphaeales), commonly known as water lilies, hold a critical evolutionary status for understanding the origin and early evolution of flowering plants [39]. This pantropical genus belongs to the most species-rich, early diverging flowering plant order Nymphaeales [40], which are deemed as “the first globally diverse clade” [41] within extant angiosperms, compared with the other two early angiosperm lineages, Amborellales and Austrobaileyales, both with limited distribution ranges [42]. In phylogenetic studies, Nymphaeales were resolved as a member of the “ANITA” (Amborella, Nymphaeales, and Illiciales-Trimeniales-Aristolochiales) clades [43], either forming a cluster with Amborella at the base of angiosperms [44–46], or diverging after Amborella as the second paraphyletic lineage of angiosperms [47].
In this study, we presented the complete mitogenome of Nymphaea colorata Peter, a tropical water lily from East Africa [39] to investigate the mitogenomic evolution of early flowering plants. The 617,195-bp mitogenome of Nymphaea encoded a similar 64 coding gene set with 25 group II introns disrupting 10 protein-coding genes, comparable to the other two early angiosperms such as Amborella and Liriodendron. Our study pinpointed the highly repetitive nature of Nymphaea, the resultant remarkable intron expansions in Nymphaea mitogenome, but unexpectedly low homologous recombination.
Results and Discussions
General features of Nymphaea mitogenome
The Nymphaea mitogenome is assembled into a single circular molecule of 617,195 bp (Fig. 1), a size larger than ca. 80% of the currently sequenced vascular plant mitogenomes (as of July 2018). The relatively large size of Nymphaea mtDNA is primarily due to its abundant repetitive sequences, which add up to 301,676 bp and account for nearly half (49%) of the mitogenome, in contrast to most of other vascular plant mitogenomes with repeat ratio generally below 30% (Additional file 1: Table S1). The Nymphaea mitogenome encodes 41 protein genes, three rRNA genes (rrn5, rrn18 and rrn26), and 20 tRNA genes (13 mitochondrial native and seven plastid derived) (Table 1). Intergenic spacers constitute the largest part (519,361 bp, 84%) of the Nymphaea mtDNA, and protein coding sequences comprise only 6% (35,961 bp) of the total length. In general, the gene content of Nymphaea is very similar to the other published angiosperm mitogenomes, especially to Amborella [48] and Liriodendron [6]. Nymphaea mt gene set differs from Amborella only by its presence of the functional protein-coding gene rps10 that is pseudogenized in Amborella, whereas differs from Liriodendron by its presence of plastid derived tRNA gene trnL(CAA)-pt and absence of trnV(TAC). Repeat-induced duplicated genes are widespread in vascular plants [49], such as Nelumbo nucifera possesses six duplicated protein genes [50] and maize (CMS-C) contains 10 duplicated protein genes [51]. In Nymphaea mitogenome, rps19 and atp6 each presents as two copies. The duplicated rps19 are identical, while the two copies of atp6 are different in length, with one copy 114 bp longer at the 3′ terminal. The shorter version of Nymphaea atp6 is still longer than that of Amborella and Liriodendron by 36 bp and 75 bp at the 5′ terminal. Blastn and Blastp searches of the 114-bp nucleotide sequence and the translated amino acid sequence against the NCBI database do not return any hits, suggesting a probably chimeric origin of atp6_D2 (the longer copy), via gene fusion of atp6 with Nymphaea specific intergenic spacer sequence at some evolutionary stage. Considering the majority of the two atp6 copies located in a pair of identical inverted repeats of 3293 bp at a distance of 196 Kb, the identical 882 bp of the two atp6 copies may be indicative of the result of repeat recombination in homogenization of the gene copies carried [7]. We further checked all intergenic spacers for possible pseudogene pieces using 68 annotated Nymphaea coding regions as queries. Altogether, we identified 52 pseudogene fragments ranging from 28 bp to 182 bp, which matched nine protein coding genes (nad5.× 2.× 5, rpl2.× 1, co× 1, atp6, ccmC, nad6, atp8, rrn18, rrn26) with identities ranging from 85 to 100%. Two largest pseudogene pieces of atp6 (182 bp) and rpl2.× 1 (142 bp) formed Nymphaea specific chimeric ORFs with parts of the adjacent intergenic spacer sequences, which, in some cases, may cause cytoplasmic male sterility (CMS) [52]. Blastn search of all these pseudogene fragments against the NCBI nucleotide database yielded much lower similarities with any other species than Nymphaea, indicating the origin of these gene vestiges from intragenomic recombination events [25] rather than horizontal gene transfers from other plants.
Fig. 1.

Mitochondrial genome map of Nymphaea colorata. The total length of the Nymphaea mitogenome is 617,195 bp. Genes (exons are shown as closed boxes) shown on the outside of the circle are transcribed clockwise, whereas those on the inside are transcribed counter-clockwise. Genes from the same protein complex are colored the same, introns are indicated in white boxes, and tRNAs of plastid origin are noted with a ‘-pt’ suffix. The inner circle shows the locations of direct (blue) and inverted (red) repeats (R1 to R10) with evidence for recombination activity (see Methods and Table 2). Numbers on the inner circle represent genome coordinates (Kb)
Table 1.
General features of mitochondrial genomes of Amborella, Liriodendron, and Nymphaea
| Genome feature | Amborella trichopoda | Nymphaea colorata | Liriodendron tulipifera |
|---|---|---|---|
| Accession | KF754799–KF754803 | KY889142 | KC821969 |
| Size (bp) | 3,866,039 | 617,195 | 553,721 |
| GC% | 45.9% | 45.1% | 47.7% |
| Genes | 63 | 64 | 64 |
| tRNAs | 20 | 20 | 20 |
| rRNAs | 3 | 3 | 3 |
| Protein coding | 40 | 41 | 41 |
| Cis-spliced introns | 19 | 19 | 20 |
| Trans-spliced introns | 6 | 6 | 5 |
| Gross length of repeats (> 19 bp) | 914,403 (23.65%) | 301,777 (48.89%) | 85,993 (15.53%) |
| Plastid-derived (bp) | 138,313 (3.58%) | 13,170 (2.13%) | 24,807 (4.48%) |
| Total gene length (bp) | 85,152 (2.20%) | 97,834 (15.85%) | 82,284 (14.86%) |
| Protein exons (bp) | 33,866 (0.88%) | 35,961 (5.83%) | 35,324 (6.38%) |
| Cis-spliced intron length (bp) | 38,896 (1.01%) | 54,840 (8.89%) | 33,631 (6.07%) |
Nymphaea shares 27% (168,686 bp) of its mtDNA with other sequenced plant mitogenomes with nearly half occurred in the genic region, and the other half (95,941 bp) in the intergenic region, accounting for 15% of the mitogenome. Nymphaea shares its intergenic spacer sequences the most with Amborella (58,049 bp), and Liriodendron (28,715 bp), then Phoenix (26,790 bp). As multiple lines of evidence suggested a divergence time of Nymphaea from the rest of angiosperms at 180 Mya (www.timetree.org), the seemingly low level of sequence sharing between Nymphaea and other angiosperms fits well to the regression line generated by analyzing 14 phylogenetically independent seed plant taxa [31], suggesting the generally high divergence nature of angiosperm mitogenomes. For example, Citrullus lanatus [14] shares with Vitis vinifera [49] 72,313 bp of its intergenic spacers despite a divergence time of 105–115 Mya; Carica papaya [53] shares with Nicotiana tabacum [54] 66,327 bp with a divergence time of 110–124 Mya.
The Nymphaea mitogenome contains 25 group II introns, including 19 cis-spliced and six trans-spliced introns (nad1i394g2, nad1i669g2, nad1i728g2, nad2i542g2, nad5i1455g2, nad5i1477g2), which is similar to the intron set of Amborella [48] and Phoenix [55], but differs from Liriodendron by its presence of the trans-splicing nad1i728g2, which is a cis-spliced intron in Liriodendron. It is noteworthy that cox2i373g2 of Nymphaea reaches a length of 11.4 Kb, making it the longest organellar intron reported in plants to date. We checked the coverage of the genome assembly on this intron region. A continual and even coverage of cox2i373 and its cox2 exon regions indicated that the presence of this intron is unlikely yielded from an artifactual assembly result (Additional file 2: Figure S3). We mapped the transcriptomic reads to the mitogenome, but due to the low coverage of the transcriptome data we cannot figure out whether this intron is continually transcribed (Additional file 2: Figure S3). We aligned the Nymphaea cox2i373g2 with that of Triticum timopheevii [56], five out of the six conserved domains of this group II intron were well aligned, except for the domain IV, indicating this domain may expanded in Nymphaea (Additional file 3: Figure S4). Although we recognized Nymphaea cox2i373 as a cis-spliced intron here, we still cannot rule out the possibility that this intron is trans-spliced, but the two parts of the trans-spliced intron happens to locate proximately in the genome and in an orientation consistent with cis-splicing. Besides, intron rpl2i846g2 and nad4i976g2 exceed 6 Kb; intron nad2i1282g2, nad2i156g2, and nad7i917g2 exceed 3 Kb in length. Overall, the total length of the 19 cis-spliced introns add up to 55 Kb, comprising 9% of the whole mitochondrial genome, which is substantially higher than any other angiosperm mitogenomes sequenced to date in both absolute and percentage terms [13]. The highly repetitive nature of the Nymphaea mtDNA accounts for a large portion of its intron size expansion (Fig. 2). About 40% to 80% of the six large introns of the Nymphaea mitogenome (> 3 Kb) are made of repetitive elements, a phenomenon similar to what observed in ferns [9] (Additional file 4: Table S2).
Fig. 2.
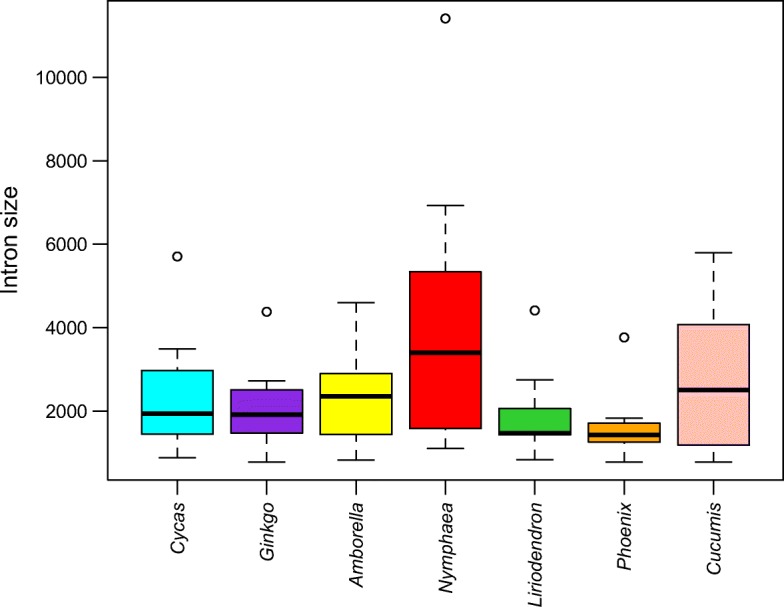
Comparison of the length of 11 introns (see Additional file 4: Table S2) of the Nymphaea mitogenome with repeated sequence inserted with that of some selected seed plant mitogenomes
Repeats and homologous recombinations
Blastn searches identified 1,188,860 repeated sequences that are longer than 30 bp and with unique begin-end coordinates in an overlapping fashion, accounting for nearly half (49%, 301,676 bp) of the Nymphaea mitogenome (Additional file 1: Table S1). These numerous imperfect and partially overlapping repeated sequences in Nymphaea constitute 886,983 repeat pairs, with the length distribution mainly in the range of 100–200 bp and identity distribution mainly between 80 and 95% (Additional file 5: Figure S1). Cd-hit-est as implemented in the cdhit suite [57] recovered 290 families with an identity threshold of 0.8 and a word size of five out of the total repeated sequences using a greedy incremental clustering algorithm method. The representatives of these repeat families were subsequently checked for occurrences using blastn searches against NCBI nucleotide database. Most (252, 87%) of these repeat families are restricted to Nymphaea and are unique in plants, only 38 are shared with other plant mitogenomes, such as 22 with Amborella, 18 with Liriodendron, 11 with Arabidopsis, 11 with Gymnosperms, four with ferns, eight with bryophytes, and nine with charophycean green algae. The observed low repeated sequence sharing of Nymphaea with other plant mitogenomes reflected a commonplace phenomenon of wild divergence of intergenic spacers as has been exemplified by the remarkable intraspecific variation in four mitogenomes of Silene vulgaris [25].
Benefited from the deep sequencing of PacBio long reads (601×, average 7294 bp, Additional file 6: Figure S2), we were able to detect minor recombinations at a frequency as low as 1/1200. A total of 886,983 repeat pairs with length ranging from 30 to 3293 bp and blast identity above 80% were examined for recombination activity (Additional file 5: Figure S1). Unexpectedly, only ten repeat pairs show evidence of recombinations with one to 48 recombined reads detected for each repeat pair (Table 2). Three direct repeats and seven inverted repeats recombined at frequencies ranging from 0.07 to 8.18%, which could possibly give rise to a set of alternative mtDNA configurations and subgenomes via inversions and subdivisions of the master conformations (Fig. 3). According to our observations, a majority of the repeats (R3–R10) recombined at a frequency below 1%; only two repeats yielded more than 10 recombined reads, including the longest inverted repeats of 3293 bp with 48 recombined reads and a self-inverted repeats of 128 bp with 13 recombined reads detected, suggesting alternative conformations (ACs) with a full set of genes rearranged are more abundant than subgenomes with reduced gene set in Nymphaea mitochondria, which resembles that observed in fern Ophioglossum with predominant ACs harboring inversions induced by the longest 4-Kb inverted repeats recombining at a frequency of 24.5% and a small number of subgenomes generated by recombinationally less active medium-sized repeats recombining at a frequency less than 2.5% [9].
Table 2.
Recombination frequency of the mitochondrial genome of Nymphaea colorata related to ten repeat pairs
| Repeat | Length (bp) | Identity (%) | Direction | Position | Reads support master circle conformation | Reads support alternative conformation |
|---|---|---|---|---|---|---|
| R1 | 3293 | 100.00 | + | 153,171–156,463 | 539 (91.82%) | 48 (8.18%) |
| – | 355,403–352,111 | |||||
| R2 | 128 | 82.81 | + | 414,017–414,144 | 1000 (98.72%) | 13 (1.28%) |
| – | 414,144–414,017 | |||||
| R3 | 462 | 100.00 | + | 187,030–187,491 | 874 (99.66%) | 3 (0.34%) |
| + | 307,094–307,555 | |||||
| R4 | 1538 | 97.33 | + | 386,370–387,906 | 822 (99.76%) | 2 (0.24%) |
| – | 389,671–388,149 | |||||
| R5 | 508 | 81.50 | + | 15,484–15,960 | 960 (99.79%) | 2 (0.21%) |
| – | 388,795–388,149 | |||||
| R6 | 399 | 99.75 | + | 39,755–40,153 | 880 (99.77%) | 2 (0.23%) |
| + | 466,916–467,314 | |||||
| R7 | 945 | 98.41 | + | 301,710–302,649 | 915 (99.89%) | 1 (0.11%) |
| + | 357,890–358,834 | |||||
| R8 | 729 | 99.86 | + | 313,110–313,838 | 932 (99.89%) | 1 (0.11%) |
| – | 315,533–314,805 | |||||
| R9 | 692 | 99.86 | + | 80,337–81,028 | 783 (99.87%) | 1 (0.13%) |
| – | 309,412–308,722 | |||||
| R10 | 72 | 97.22 | + | 302,169–302,240 | 1362 (99.93%) | 1 (0.07%) |
| + | 313,560–313,631 |
Fig. 3.

Mitochondrial genome rearrangements and alternative genomic conformations observed from Nymphaea colorata based on repeat-mediated intra-molecular recombination products of three repeat pairs (R1, R2 and R3) that induced recombination with the highest frequencies as listed in Table 2
Recombination involving large repeats generally result in equimolar or nearly equimolar recombined molecules in the genome [58], as exemplified in Silene latifolia, Silene vulgaris [7], Mimulus guttatus [33], and Ginkgo [31]. In our study, two large repeats (out of 2224 repeats) with a length of 3293 bp and 1538 bp show evidence of recombination but with low recombination frequency at only 0.24% and 8.18%. Such low recombination frequency has also been seen in other plant mitogenomes, for example, in Silene conica and Silene noctiflora [7], tens of large repeats induced recombinations at a frequency around 5%; in Ginkgo biloba, a large repeat of 1.5 Kb recombined at a frequency of 9%; in fern Ophioglossum californicum, two large repeats with 4-Kb and 1-Kb induced recombinations at frequency of 24.5% and 0.1%, respectively [9]. In addition to these large repeats, all seven medium sized repeats of the Nymphaea mitogenome recombined even more rarely with recombination frequencies ranging from 0.11 to 0.34%, which is similar to the observation in the gymnosperms Ginkgo biloba and Welwitschia mirabilis [31], ferns [9], the flowering plants Cucumis sativus [13] and Vigna angularis [26], whereas significantly lower than those observed from Viscum scurruloideum [10], Silene latifolia and Silene vulgaris. For example, in Viscum, three medium-sized repeats result in recombination equilibrium, and another two recombined actively at frequencies of 11.2% and 38.7%.
The low recombination level of the Nymphaea mitogenome is further evidenced by a small number of recombined reads detected, i.e., only 74 reads were found to support alternative configurations resulted from repeat-mediated recombinations (Additional file 7: FASTA.fa), accounting for only 0.13% of the total reads (74 out of 56,849), which is apparently lower than those found in other plants, such as 10% in Mimulus guttatus [33], 6.6% in Ophioglossum, and 2.2% in Psilotum. The low level of recombination rate found in Nymphaea suggests the predominant existence of master configuration in vivo in this plant, with a low level of substoichiometric recombinant forms. The latter has been proved to exert profound effect on plant growth, such as cytoplasmic male sterility (CMS) [59–61] and abnormal growth phenotypes [62, 63].
Understanding the paradoxical coexistence of the low recombination and abundant repeats in mitogenomes, such as Nymphaea and Ophioglossum, must take into account the nucleus’ control over the accuracy of the repair of mitochondrial chromosomes by a series of nuclear-encoded and mitochondrial targeted factors [64, 65]. Disruption of these genes could initiate and promote mitochondrial intragenomic recombination [58], as have been documented in Physcomitrella [66] and Arabidopsis [24]. Such nuclear genes may be under different levels of selection pressure, resulting in distinctive stability of mitogenomes in specific plant groups. For example, in each of the major bryophyte lineage, mitochondrial genomes kept a high degree of structural conservation over long period of evolution [2], which is in contrast to the observations in Silene vulgaris [25] and Beta vulgaris [67] with remarkable intraspecific mitogenome rearrangements.
Plastid DNA insertions
The Nymphaea mitogenome possesses 23 fragments of plastid derived sequences ranging from 38 bp to 1878 bp (Table 3) with a total length adding up to 13 Kb. The plastid derived sequences comprise 2% of the mitogenome, which is a typical percentage in angiosperms with the absolute amount of plastid inserts ranging from 4.4 Kb in Arabidopsis [68] to 138 Kb in Amborella [48]. Most (19 out of 23) of these plastid inserts, including those carrying tRNAs, having homologs in other plant mitogenomes, provides a good opportunity to revisit the origin of functional intracellular gene transfers, which remained ambiguous from seed plants [69] or vascular plants [1, 70]. Here we show evidence of the emergence of functional plastid insertions in ferns as exemplified by the presence of plastid derived functional tRNA gene trnN(GTT)-pt in fern Ophioglossum. Specifically, the 97-bp Nymphaea plastid insert carrying trnN(GUU)-pt have a 73-bp homolog in fern Ophioglossum (coverage 90%, identity 90%), in addition to a number of seed plants, suggesting the putative emergence of trnN(GUU)-pt in the ancestor of vascular plants, which is also evidenced by its extremely short flanking sequences measuring only a few bases due to long periods of purifying selections, given its relatively high sequence identity (92%) with their plastid counterparts. The plastid inserts in the Nymphaea mitogenome generally yielded similarities ranging from 74 to 97% (median 84%) while using Nymphaea colorata plastid genome sequence as a reference, indicating that most of the inserted sequences have been streamlined by the mitogenome and have accumulated considerable mutations. Particularly, in the Nymphaea mitogenome, the largest plastid insert of 1878 bp harboring trnL(CAA)-pt show comparatively conserved features with an identity of 96% in its tRNA region, which, however, rapidly declined to 88% and 83% in its up-stream and down-stream flanking sequences. Another two plastid inserts carrying trnF(GAA)-pt and trnW(CAA)-pt–trnP(TGG)-pt, respectively, also show similar degradation patterns in the flanking regions of the functional tRNA genes, as has been observed in Liriodendron [6]. The presence of the two plastid derived tRNAs including trnF(GAA)-pt and trnL(CAA)-pt in Nymphaea, Amborella, Liriodendron, several monocots, and some eudicots could possibly suggest their origin from the ancestor of angiosperms, followed by independent losses and/or gains during the evolution of angiosperms.
Table 3.
Plastid insertions in the mitochondrial genome of Nymphaea colorata
| Length (bp) | Position | Plastid genes contained | Identity (%) |
|---|---|---|---|
| 1878 | 282,896–284,773 | trnL(CAA)–ndhB (partial) | 80.41 |
| 1438 | 328,952–330,389 | psaB (partial) | 84.87 |
| 1428 | 275,870–277,297 | ycf2 (partial) | 87.60 |
| 1426 | 327,426–328,851 | psaB (partial)–psaA (partial) | 89.12 |
| 1194 | 592,277–593,470 | ndhJ–ndhK (partial) | 80.83 |
| 1059 | 299,952–301,010 | petG(partial)–trnW(CCA)–trnP(UGG)–psaJ | 85.62 |
| 857 | 412,364–413,220 | 16S rRNA (partial) | 74.04 |
| 510 | 538,489–538,998 | ycf4 (partial) | 81.88 |
| 447 | 11,109–11,555 | ndhF (partial) | 81.68 |
| 404 | 10,606–11,009 | ndhA (partial) | 90.59 |
| 380 | 330,507–330,886 | psaB (partial) | 78.74 |
| 370 | 278,097–278,466 | none | 80.67 |
| 346 | 79,612–79,957 | 16S rRNA (partial) | 97.11 |
| 331 | 593,561–593,891 | trnF(GAA) | 84.10 |
| 268 | 277,340–277,607 | ycf2 (partial) | 81.48 |
| 258 | 111,292–111,549 | none | 83.02 |
| 104 | 449,246–449,349 | trnQ(UUG) | 95.19 |
| 103 | 436,536–436,638 | trnS(UGA) (partial) | 83.50 |
| 97 | 135,004–135,100 | trnN(GUU) | 91.75 |
| 97 | 395,827–395,923 | 23S rRNA (partial) | 81.63 |
| 78 | 132,465–132,542 | trnM(CAU) | 92.31 |
| 55 | 277,989–278,043 | ycf2 (partial) | 94.55 |
| 38 | 328,916–328,953 | psaB (partial) | 94.87 |
Conserved gene clusters
Plant mitogenomes are highly fluid in genome structure due to the repeat mediated homologous recombinations, sequence duplications, genome expansion and shrinkage, and incorporation of foreign DNAs [71], whereas some gene clusters are conserved across large phylogenetic scale [6, 50, 72]. The relatively low recombination level observed in Nymphaea does not necessarily predict strictly conserved genome arrangement compared with the ‘fossilized’ angiosperm mitogenome of Liriodendron or the other early angiosperm Amborella, as we found 38 and 44 rearrangements between mitogenomes of Nymphaea-Amborella and Nymphaea-Liriodendron, respectively. Nevertheless, in comparison of gene order of Nymphaea with that of the 214 plant mitogenomes, we identified 11 conserved gene clusters in Nymphaea, of which, three (rpl2–rps19–rps3–rpl16, rps13–rps11, and rrn18–rrn5) could be dated back deeply to the origin of mitochondrion from its endosymbiont bacterial ancestor [72]. The cluster trnfM(CAU)–rrn26 is widely distributed in streptophytes. Four clusters (cox3–sdh4, nad3–rps12, rpl5–rps14–cob, and rps10–cox1) emerged since gymnosperms. The cluster trnP(UGG)–sdh3 shows a sporadic distribution pattern in bryophytes, Ginkgo, Cycas and many angiosperms, indicative of the secondary loss of the gene cluster in lycophytes and ferns. The angiosperm conserved cluster trnP(UGG)-pt–trnW(CAA)-pt does not show up in Amborella, suggesting its emergence in Nymphaea or even earlier in the ancestor of angiosperms then secondary loss of the cluster in Amborella. The gene cluster <nad5.× 4.× 5 > <trnE (TTC)–nad7 > is only shared by three angiosperm species, namely, Liriodendron, Nelumbo and Nymphaea, suggesting its emergence in the ancestor of angiosperms followed by fast degeneration as a consequence of extensive genome rearrangements. However, the sporadic distribution of the cluster could more likely indicate a coincidence of independent structural evolutions in the three lineages (Additional file 8: Table S3).
Conclusions
We assembled the complete mitogenome of Nymphaea using the PacBio RSII sequencing technology. Nymphaea mitogenome is similar to that of the Amborella and Liriodendron in the gene and intron contents, but significantly different in its abundant repetitive sequences. Whereas the recombination activity in the Nymphaea mitogenome is relatively quiescent, which evidenced by only a small portion of the examined reads. The length of plastid insertions of Nymphaea falls into the range of that of the other angiosperms, and some plastid derived tRNAs, with their existence in Nymphaea mitogenome, arguing for their earlier emergences in angiosperms than previously postulated. Finally, despite extensive genome rearrangements, 11 conserved gene clusters are identified in Nymphaea, which can be traced back to various stage of mitogenome evolution. This study shed new light on the evolution of mitochondrial genomes in early flowering plants, allowing deeper insights into the repeat-mediated recombination patterns in plant mitogenomes.
Methods
Mitochondrial genome assembly and annotation
The mitochondrial genome of Nymphaea colorata was obtained from the genome project of Nymphaea colorata led by Liangsheng Zhang (unpublished data). The genome sequencing was performed on a PacBio RSII platform (Pacific Biosciences, Menlo Park, CA). The Raw PacBio reads were corrected to accuracy above 99% using the RS_PreAssembler, and then assembled into contigs using the program Canu (github.com/marbl/canu). Two mitochondrial contigs of 527,532 bp and 157,672 bp were identified using the NCBI Blast program with the Liriodendron mitochondrial genome as a reference. The two contigs overlapped with each other at both ends by 34,745 bp and 16,132 bp, and finally formed a circular molecular of 617,195 bp, with an average depth of 601×. RNA-seq data of Nymphaea colorata were also obtained from the genome project of Nymphaea colorata (unpublished data).
The annotation for the Nymphaea mitogenome was performed as previously described [3, 70]. Protein coding genes and rRNA genes were annotated by blastn searches of the non-redundant database at National Center for Biotechnology Information (NCBI). The exact gene and exon/intron boundaries were further confirmed in Geneious software (v.10.0.2, Biomatters, www.geneious.com) by aligning each gene to its orthologs from available annotated plant mitochondrial genomes at the NCBI website (www.ncbi.nlm.nih.gov/genome/organelle). The tRNA genes were detected using tRNAscan-SE 2.0 [73].
Repeats and repeat-mediated homologous recombinations
Repeats identification of Nymphaea and other vascular plant mitogenomes were carried out using NCBI blastn searches by searching the Nymphaea mitogenome sequence against itself with an e-value cut-off of 1e− 6, and a word size of 7 following Guo et al. [9]. All the repeat sequences were subsequently extracted and clustered into difference families using the program cd-hit-est as implemented in cdhit suite v4.6.7 [57], with a word size of 5 and sequence similarity threshold of 0.8. We estimated the number of repeats from the number of unique begin-end coordinates of hits from blastn search according to Alverson et al. [13]. To detect the active repeat-mediated intragenomic recombinations within the PacBio reads, we built up an mt read database using corrected genome sequencing PacBio reads. We used the Nymphaea mitogenome sequence as the reference to blast the total Nymphaea PacBio reads database with an e-value cut-off of 1e− 100 for extraction of mt reads, the resultant mt reads was further searched against Nymphaea plastid sequence with the same parameters to remove putative plastid reads with overall alignment coverage > 85% of the read length. Finally, we got a mitochondrial read database of 75,863 reads with an average length of 7294 bp, and total length 553,358,387 bp.
Repeat-mediated homologous recombinations were evaluated for those repeat pairs ranging from 30 to 3293 bp with blast identity > 80% following Alverson et al. [13]. Specifically, for each repeat pair, we built four or six reference sequences, each with 200 bp up- and down-stream of the two template sequences (original sequences), and the two (for repeat pair with identity =100) or four (for repeat pair with identity < 100) recombined sequences (alternative configurations) constructed from the putative recombination products. Then, we searched the reference sequences against the Nymphaea mt reads database, and count the number of matching reads with a blast identity above 99.5%, and a hit coverage over 200 bp in both flanking regions of each repeat sequence. After that, the templates with evidence of recombination were extracted and elongated in both sides to 2000 bp and searched again to the Nymphaea mt reads database to remove the recombinants with undersized flanking regions. Finally, the best matched reads for all the recombinants were extracted and aligned with the Nymphaea mitogenome in Geneious v10.0.2 (https://www.geneious.com/) to authenticate the accuracy of the recombined reads.
Identification of plastid derived sequences
To identify plastid derived mitochondrial sequences, the Nymphaea mitogenome was searched against the plastid genome of Nymphaea colorata (data unpublished), and all plant mitogenome database with an e-value cut-off of 1e− 6 and a word size of 7, simultaneously. The blastn output was then visualized in Geneious v10.0.2 (https://www.geneious.com/) and each of the identified plastid sequence insert was compared with its co-occurring mt homologs from all other plant mitogenomes to infer the putative origin of the intracellular transfer.
Identification of conserved gene clusters
The gene orders of Nymphaea, Amborella, and Liriodendron were compared with each other using UniMoG [74] to identify rearrangements among three mitogenomes. The conserved gene clusters were identified if they appeared in any two of the three early angiosperms and simultaneously presented in at least one major plant group, e.g., lycophytes, ferns, gymnosperms, or angiosperms.
Additional files
Table S1. Repeat proportions of the mitochondrial genomes of 82 angiosperm species. (PDF 96 kb)
Figure S3. The DNA and RNA coverage plots of the cox2 gene of the mitochondrial genome of Nymphaea colorata. (PDF 139 kb)
Figure S4. The conserved domain alignment of the group II intron cox2i373 of Triticum timopheevii (AP013106) and Nymphaea colorata (KY889142). (PDF 217 kb)
Table S2. Eleven cis-spliced introns of the Nymphaea mitogenome with repeated sequences inserted. (PDF 66 kb)
Figure S1. All the repeat pairs (886,982) evaluated for recombination in our study. The large number of repeats is due to numerous repeats that are partially overlapping with each other in Nymphaea mitochondrial genome. (a) The curve graph shows repeat distribution pattern on sequence identity. (b) The curve graph shows repeat distribution pattern on sequence length. (PDF 171 kb)
Figure S2. The PacBio read depth plot of the mitochondrial genome of Nymphaea colorata. (PDF 96 kb)
FASTA.fa. Seventy-four recombined reads detected for homologous recombination involving ten repeat pairs in our study. (FA 807 kb)
Table S3. Eleven conserved gene clusters in the Nymphaea mitochondrial genome. (PDF 140 kb)
Acknowledgments
We thank Dr. Xingtan Zhang from the Genomic Centre of Fujian Agricultural University for the correction of the PacBio raw reads of Nymphaea colorata.
Funding
This project is funded by the National Natural Science Foundation of China (NSFC31470314, NSFC31600171), Fairy Lake Science Foundation (FLSF2017–03), Shenzhen Urban Management Bureau Fund (201520), Shenzhen Municipal Government of China (JCYJ20150529150409546), and the National Science Foundation grant (DEB-1240045). The funders had no role in the designing the research, data collection, analysis, or manuscript preparation.
Availability of data and materials
The mitochondrial genome of Nymphaea colorata has been submitted to GenBank under the accession number of KY889142. The raw sequence data have been deposited in the Short Read Achieve (SRA) database of NCBI (SAMN08218778). Other supporting results are included within the article and its additional files.
Abbreviations
- CMS
Cytoplasmic male sterility
- HGT
Horizontal gene transfer
- Mitogenome
Mitochondrial genome
- mtDNA
Mitochondrial DNA
- ORFs
Open reading frames
- rRNAs
Ribosomal RNAs
- tRNAs
Transfer RNAs
Authors’ contributions
YL and LZ designed the study. SD, FC, YL carried out most of the experiments. SD & CZ carried out bioinformatics analysis. SD drafted the manuscript. YL & LZ modified the final manuscript and all authors reviewed it. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shanshan Dong, Email: shangrilass@163.com.
Chaoxian Zhao, Email: 1348419384@qq.com.
Fei Chen, Email: frank@fafu.edu.cn.
Yanhui Liu, Email: 591276899@qq.com.
Shouzhou Zhang, Email: shouzhouz@126.com.
Hong Wu, Email: wh@scau.edu.cn.
Liangsheng Zhang, Email: fafuzhang@163.com.
Yang Liu, Email: yang.liu0508@gmail.com.
References
- 1.Mower JP, Sloan DB, Alverson AJ. Plant mitochondrial genome diversity: the genomics revolution. Heidelberg: Springer Vienna; 2012. [Google Scholar]
- 2.Liu Y, Medina R, Goffinet B. 350 my of mitochondrial genome stasis in mosses, an early land plant lineage. Mol Biol Evol. 2014;31(10):2586–2591. doi: 10.1093/molbev/msu199. [DOI] [PubMed] [Google Scholar]
- 3.Xue JY, Liu Y, Li L, Wang B, Qiu YL. The complete mitochondrial genome sequence of the hornwort Phaeoceros laevis: retention of many ancient pseudogenes and conservative evolution of mitochondrial genomes in hornworts. Curr Genet. 2009;56(1):53–61. doi: 10.1007/s00294-009-0279-1. [DOI] [PubMed] [Google Scholar]
- 4.Wang B, Xue J, Li L, Yang L, Qiu YL. The complete mitochondrial genome sequence of the liverwort Pleurozia purpurea, reveals extremely conservative mitochondrial genome evolution in liverworts. Curr Genet. 2009;55(6):601–609. doi: 10.1007/s00294-009-0273-7. [DOI] [PubMed] [Google Scholar]
- 5.Sloan DB. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013;200(4):978–985. doi: 10.1111/nph.12395. [DOI] [PubMed] [Google Scholar]
- 6.Richardson AO, Rice DW, Young GJ, Alverson AJ, Palmer JD. The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013;11(1):29. doi: 10.1186/1741-7007-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, Palmer JD, Taylor DR. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012;10(1):e1001241. doi: 10.1371/journal.pbio.1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu A, Guo W, Jain K, Mower JP. Unprecedented heterogeneity in the synonymous substitution rate within a plant genome. Mol Biol Evol. 2014;31(5):1228–1236. doi: 10.1093/molbev/msu079. [DOI] [PubMed] [Google Scholar]
- 9.Guo W, Zhu A, Fan W, Mower JP. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 2017;213(1):391–403. doi: 10.1111/nph.14135. [DOI] [PubMed] [Google Scholar]
- 10.Skippington E, Barkman TJ, Rice DW, Palmer JD. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 2015;112(27):E3515–E3524. doi: 10.1073/pnas.1504491112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burger G, Gray MW, Franz Lang B. Mitochondrial genomes: anything goes. Trends in Genet. 2003;19(12):709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Lilly JW, Havey MJ. Small, repetitive dnas contribute significantly to the expanded mitochondrial genome of cucumber. Genetics. 2001;159(1):317–328. doi: 10.1093/genetics/159.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alverson AJ, Rice DW, Dickinson S, Barry K, Palmer JD. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell. 2011;23(7):2499–2513. doi: 10.1105/tpc.111.087189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alverson AJ, Wei X, Rice DW, Stern DB, Barry K, Palmer JD. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae) Mol Biol Evol. 2010;27(6):1436–1448. doi: 10.1093/molbev/msq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bock R. Witnessing genome evolution: experimental reconstruction of endosymbiotic and horizontal gene transfer. Annu Rev Genet. 2017;51:1–22. doi: 10.1146/annurev-genet-120215-035329. [DOI] [PubMed] [Google Scholar]
- 16.Rice DW, Alverson AJ, Richardson AO, Young GJ, Sanchezpuerta MV, Munzinger J, Barry K, Boore JL, Zhang L, DePamphilis CW, et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science. 2013;342(6165):1468–1473. doi: 10.1126/science.1246275. [DOI] [PubMed] [Google Scholar]
- 17.Chaw SM, Shih AC, Wang D, Wu YW, Liu SM, Chou TY. The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol Biol Evol. 2008;25(3):603–615. doi: 10.1093/molbev/msn009. [DOI] [PubMed] [Google Scholar]
- 18.Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Gen Genomics. 2002;268(4):434–445. doi: 10.1007/s00438-002-0767-1. [DOI] [PubMed] [Google Scholar]
- 19.Darracq A, Varre JS, Touzet P. A scenario of mitochondrial genome evolution in maize based on rearrangement events. BMC Genomics. 2010;11:233. doi: 10.1186/1471-2164-11-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One. 2011;6(1):e16404. doi: 10.1371/journal.pone.0016404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gualberto JM, Mileshina D, Wallet C, Niazi AK, Weber-Lotfi F, Dietrich A. The plant mitochondrial genome: dynamics and maintenance. Biochimie. 2014;100:107–120. doi: 10.1016/j.biochi.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 22.André C, Levy A, Walbot V. Small repeated sequences and the structure of plant mitochondrial genomes. Perspectives. 1992;8(4):128–32. [DOI] [PubMed]
- 23.Cheng N, Lo YS, Ansari MI, Ho KC, Jeng ST, Lin NS, Dai H. Correlation between mtDNA complexity and mtDNA replication mode in developing cotyledon mitochondria during mung bean seed germination. New Phytol. 2017;213(2):751–763. doi: 10.1111/nph.14158. [DOI] [PubMed] [Google Scholar]
- 24.Davila JI, Arrietamontiel MP, Wamboldt Y, Cao J, Hagmann J, Shedge V, Xu Y, Weigel D, Mackenzie SA. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 2011;9(1):64. doi: 10.1186/1741-7007-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sloan DB, Muller K, McCauley DE, Taylor DR, Storchova H. Intraspecific variation in mitochondrial genome sequence, structure, and gene content in Silene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 2012;196(4):1228–1239. doi: 10.1111/j.1469-8137.2012.04340.x. [DOI] [PubMed] [Google Scholar]
- 26.Naito K, Kaga A, Tomooka N, Kawase M. De novo assembly of the complete organelle genome sequences of azuki bean (Vigna angularis) using next-generation sequencers. Breed Sci. 2013;63(2):176–182. doi: 10.1270/jsbbs.63.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogihara Y, Yamazaki Y, Murai K, Kanno A, Terachi T, Shiina T, Miyashita N, Nasuda S, Nakamura C, Mori N, et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005;33(19):6235–6250. doi: 10.1093/nar/gki925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecht J, Grewe F, Knoop V. Extreme RNA editing in coding islands and abundant microsatellites in repeat sequences of Selaginella moellendorffii mitochondria: the root of frequent plant mtDNA recombination in early tracheophytes. Genome Biol Evol. 2011;3:344–358. doi: 10.1093/gbe/evr027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Puerta MV, Zubko MK, Palmer JD. Homologous recombination and retention of a single form of most genes shape the highly chimeric mitochondrial genome of a cybrid plant. New Phytol. 2015;206(1):381–396. doi: 10.1111/nph.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shearman JR, Sonthirod C, Naktang C, Pootakham W, Yoocha T, Sangsrakru D, Jomchai N, Tragoonrung S, Tangphatsornruang S. The two chromosomes of the mitochondrial genome of a sugarcane cultivar: assembly and recombination analysis using long PacBio reads. Sci Rep. 2016;6:31533. doi: 10.1038/srep31533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo W, Grewe F, Fan W, Young GJ, Knoop V, Palmer JD, Mower JP. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol Biol Evol. 2016;33(6):1448–1460. doi: 10.1093/molbev/msw024. [DOI] [PubMed] [Google Scholar]
- 32.Sloan DB, Alverson AJ, Storchova H, Palmer JD, Taylor DR. Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evol Biol. 2010;10(1):274. doi: 10.1186/1471-2148-10-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mower JP, Case AL, Floro ER, Willis JH. Evidence against equimolarity of large repeat arrangements and a predominant master circle structure of the mitochondrial genome from a monkeyflower (Mimulus guttatus) lineage with cryptic CMS. Genome Biol Evol. 2012;4(5):670–686. doi: 10.1093/gbe/evs042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Won H, Renner SS. Horizontal gene transfer from flowering plants to Gnetum. Proc Natl Acad Sci U S A. 2003;100(19):10824–10829. doi: 10.1073/pnas.1833775100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis CC, Wurdack KJ. Host-to-parasite gene transfer in flowering plants: phylogenetic evidence from malpighiales. Science. 2004;305(5684):676–678. doi: 10.1126/science.1100671. [DOI] [PubMed] [Google Scholar]
- 36.Mower JP, Stefanovic S, Young GJ, Palmer JD. Gene transfer from parasitic to host plants. Nature. 2004;432(7014):165–166. doi: 10.1038/432165b. [DOI] [PubMed] [Google Scholar]
- 37.Skippington E, Barkman TJ, Rice DW, Palmer JD. Comparative mitogenomics indicates respiratory competence in parasitic Viscum despite loss of complex I and extreme sequence divergence, and reveals horizontal gene transfer and remarkable variation in genome size. BMC Plant Biol. 2017;17(1):49. doi: 10.1186/s12870-017-0992-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez-Puerta MV, Garcia LE, Wohlfeiler J, Ceriotti LF. Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant. New Phytol. 2017;214(1):376–387. doi: 10.1111/nph.14361. [DOI] [PubMed] [Google Scholar]
- 39.Chen F, Liu X, Yu C, Chen Y, Tang H, Zhang L. Water lilies as emerging models for Darwin's abominable mystery. Hort Res. 2017;4:17051. doi: 10.1038/hortres.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soltis DE, Bell CD, Kim S, Soltis PS. Origin and early evolution of angiosperms. Ann N Y Acad Sci. 2008;1133:3–25. doi: 10.1196/annals.1438.005. [DOI] [PubMed] [Google Scholar]
- 41.Borsch T, Soltis PS. Nymphaeales – the first globally diverse clade? Taxon. 2008;57(4):1051. [Google Scholar]
- 42.Löhne C, Yoo MJ, Borsch T, Wiersema J, Wilde V, Bell CD, Barthlott W, Soltis DE, Soltis PS. Biogeography of nymphaeales: extant patterns and historical events. Taxon. 2008;57(4):1123–1146. [Google Scholar]
- 43.Qiu YL, Lee J, Bernasconi-Quadroni F, Soltis DE, Soltis PS, Zanis M, Zimmer EA, Chen Z, Savolainen V, Chase MW. The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature. 1999;402(6760):404–407. doi: 10.1038/46536. [DOI] [PubMed] [Google Scholar]
- 44.Barkman TJ, Chenery G, McNeal JR, Lyons-Weiler J, Ellisens WJ, Moore G, Wolfe AD, dePamphilis CW. Independent and combined analyses of sequences from all three genomic compartments converge on the root of flowering plant phylogeny. Proc Natl Acad Sci U S A. 2000;97(24):13166–13171. doi: 10.1073/pnas.220427497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xi Z, Liu L, Rest JS, Davis CC. Coalescent versus concatenation methods and the placement of Amborella as sister to water lilies. Syst Biol. 2014;63(6):919–931. doi: 10.1093/sysbio/syu055. [DOI] [PubMed] [Google Scholar]
- 46.Edwards SV, Xi Z, Janke A, Faircloth BC, McCormack JE, Glenn TC, Zhong B, Wu S, Lemmon EM, Lemmon AR, et al. Implementing and testing the multispecies coalescent model: a valuable paradigm for phylogenomics. Mol Phylogenet Evol. 2016;94:447–462. doi: 10.1016/j.ympev.2015.10.027. [DOI] [PubMed] [Google Scholar]
- 47.Simmons MP. Mutually exclusive phylogenomic inferences at the root of the angiosperms: Amborella is supported as sister and observed variability is biased. Cladistics. 2016;0(2016):1–25. doi: 10.1111/cla.12177. [DOI] [PubMed] [Google Scholar]
- 48.Bergthorsson U, Richardson AO, Young GJ, Goertzen LR, Palmer JD. Massive horizontal transfer of mitochondrial genes from diverse land plant donors to the basal angiosperm Amborella. Proc Natl Acad Sci U S A. 2004;101(51):17747–17752. doi: 10.1073/pnas.0408336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goremykin VV, Salamini F, Velasco R, Viola R. Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol Biol Evol. 2009;26(1):99–110. doi: 10.1093/molbev/msn226. [DOI] [PubMed] [Google Scholar]
- 50.Gui S, Wu Z, Zhang H, Zheng Y, Zhu Z, Liang D, Ding Y. The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci Rep. 2016;6:30158. doi: 10.1038/srep30158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen JO, Fauron CM, Minx P, Roark L, Oddiraju S, Lin GN, Meyer L, Sun H, Kim K, Wang C, et al. Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics. 2007;177(2):1173–1192. doi: 10.1534/genetics.107.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanson MR, Bentolila S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell. 2004;16(Suppl 1):S154–S169. doi: 10.1105/tpc.015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magee AM, Aspinall S, Rice DW, Cusack BP, Semon M, Perry AS, Stefanovic S, Milbourne D, Barth S, Palmer JD, et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010;20(12):1700–1710. doi: 10.1101/gr.111955.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li F, Yang A, Lv J, Gong D, Sun Y. The complete mitochondrial genome sequence of Sua-type cytoplasmic male sterility of tobacco (Nicotiana tabacum) Mitochondrial DNA. 2016;27(4):2929–2930. doi: 10.3109/19401736.2015.1060445. [DOI] [PubMed] [Google Scholar]
- 55.Fang Y, Wu H, Zhang T, Yang M, Yin Y, Pan L, Yu X, Zhang X, Hu S, Al-Mssallem IS, et al. A complete sequence and transcriptomic analyses of date palm (Phoenix dactylifera L.) mitochondrial genome. PLoS ONE. 2012;7(5):e37164. doi: 10.1371/journal.pone.0037164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farré J-C, Araya A. RNA splicing in higher plant mitochondria: determination of functional elements in group II intron from a chimeric cox II gene in electroporated wheat mitochondria. Plant J. 2002;29(2):203–213. doi: 10.1046/j.1365-313x.2002.01207.x. [DOI] [PubMed] [Google Scholar]
- 57.Huang Y, Niu B, Gao Y, Fu L, Li W. Cd-hit suite. Bioinformatics. 2010;26(5):680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marechal A, Brisson N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010;186(2):299–317. doi: 10.1111/j.1469-8137.2010.03195.x. [DOI] [PubMed] [Google Scholar]
- 59.Woloszynska M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes – though this be madness, yet there's method in't. J Exp Bot. 2010;61(3):657–671. doi: 10.1093/jxb/erp361. [DOI] [PubMed] [Google Scholar]
- 60.Tang M, Chen Z, Grover CE, Wang Y, Li S, Liu G, Ma Z, Wendel JF, Hua J. Rapid evolutionary divergence of Gossypium barbadense and G. hirsutum mitochondrial genomes. BMC Genomics. 2015;16(1):770. doi: 10.1186/s12864-015-1988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J, Guan R, Chang S, Du T, Zhang H, Xing H. Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS One. 2011;6(3):e17662. doi: 10.1371/journal.pone.0017662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakamoto W, Kondo H, Murata M, Motoyoshi F. Altered mitochondrial gene expression in a maternal distorted leaf mutant of Arabidopsis induced by chloroplast mutator. Plant Cell. 1996;8(8):1377–1390. doi: 10.1105/tpc.8.8.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abdelnoor RV, Yule R, Elo A, Christensen AC, Meyer-Gauen G, Mackenzie SA. Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc Natl Acad Sci U S A. 2003;100(10):5968–5973. doi: 10.1073/pnas.1037651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dietrich A, Wallet C, Janica S, Gualberto JM. Mitochondrial DNA recombination, repair and segregation: recent scientific data and perspectives. J WMS. 2016;2(2):2023. [Google Scholar]
- 65.Gualberto JM, Newton KJ. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu Rev Plant Biol. 2017;68:225–252. doi: 10.1146/annurev-arplant-043015-112232. [DOI] [PubMed] [Google Scholar]
- 66.Odahara M, Kuroiwa H, Kuroiwa T, Sekine Y. Suppression of repeat-mediated gross mitochondrial genome rearrangements by RecA in the moss Physcomitrella patens. Plant Cell. 2009;21(4):1182–1194. doi: 10.1105/tpc.108.064709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kubo T, Nishizawa S, Mikami T. Alterations in organization and transcription of the mitochondrial genome of cytoplasmic male sterile sugar beet (Beta vulgaris L.) Mol Gen Genet. 1999;262(2):283–290. doi: 10.1007/s004380051085. [DOI] [PubMed] [Google Scholar]
- 68.Giegé P, Brennicke A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci U S A. 1999;96(26):15324–15329. doi: 10.1073/pnas.96.26.15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang D, Wu YW, Shih AC, Wu CS, Wang YN, Chaw SM. Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol Biol Evol. 2007;24(9):2040–2048. doi: 10.1093/molbev/msm133. [DOI] [PubMed] [Google Scholar]
- 70.Li L, Wang B, Liu Y, Qiu YL. The complete mitochondrial genome sequence of the hornwort Megaceros aenigmaticus shows a mixed mode of conservative yet dynamic evolution in early land plant mitochondrial genomes. J Mol Evol. 2009;68(6):665–678. doi: 10.1007/s00239-009-9240-7. [DOI] [PubMed] [Google Scholar]
- 71.Palmer JD, Adams KL, Cho Y, Parkinson CL, Qiu YL, Song K. Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc Natl Acad Sci. 2000;97:6960–6966. doi: 10.1073/pnas.97.13.6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takemura M, Oda K, Yamato K, Ohta E, Nakamura Y, Nozato N, Akashi K, Ohyama K. Gene clusters for ribosomal proteins in the mitochondrial genome of a liverwort, Marchantia polymorpha. Nucleic Acids Res. 1992;20(12):3199–3205. doi: 10.1093/nar/20.12.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lowe TM, Chan PP. tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44(W1):W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hilker R, Sickinger C, Pedersen CN, Stoye J. UniMoG – a unifying framework for genomic distance calculation and sorting based on DCJ. Bioinformatics. 2012;28(19):2509–2511. doi: 10.1093/bioinformatics/bts440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Repeat proportions of the mitochondrial genomes of 82 angiosperm species. (PDF 96 kb)
Figure S3. The DNA and RNA coverage plots of the cox2 gene of the mitochondrial genome of Nymphaea colorata. (PDF 139 kb)
Figure S4. The conserved domain alignment of the group II intron cox2i373 of Triticum timopheevii (AP013106) and Nymphaea colorata (KY889142). (PDF 217 kb)
Table S2. Eleven cis-spliced introns of the Nymphaea mitogenome with repeated sequences inserted. (PDF 66 kb)
Figure S1. All the repeat pairs (886,982) evaluated for recombination in our study. The large number of repeats is due to numerous repeats that are partially overlapping with each other in Nymphaea mitochondrial genome. (a) The curve graph shows repeat distribution pattern on sequence identity. (b) The curve graph shows repeat distribution pattern on sequence length. (PDF 171 kb)
Figure S2. The PacBio read depth plot of the mitochondrial genome of Nymphaea colorata. (PDF 96 kb)
FASTA.fa. Seventy-four recombined reads detected for homologous recombination involving ten repeat pairs in our study. (FA 807 kb)
Table S3. Eleven conserved gene clusters in the Nymphaea mitochondrial genome. (PDF 140 kb)
Data Availability Statement
The mitochondrial genome of Nymphaea colorata has been submitted to GenBank under the accession number of KY889142. The raw sequence data have been deposited in the Short Read Achieve (SRA) database of NCBI (SAMN08218778). Other supporting results are included within the article and its additional files.