

Abstract
HPV variants from the same type can be classified into lineages and sublineages based on the complete genome differences and the phylogenetic topologies. We examined nucleotide variations of twelve HPV types within the species Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61) by analyzing 1,432 partial sequences and 181 complete genomes from multiple geographic populations. The inter-lineage and inter-sublineage mean differences of HPV variants ranged between 0.9%−7.3% and 0.3%−0.9%, respectively. The heterogeneity and phylogenies of HPV isolates indicate an independent evolutionary history for each type. The noncoding regions were the most variable regions whereas the capsid proteins were relatively conserved. Certain variant lineages and/or sublineages were geographically-associated. These data provide the basis to further classify HPV variants and should foster future studies on the evolution of HPV genomes and the associations of HPV variants with cancer risk.
Keywords: Human papillomavirus, Cervical cancer, Variant, Classification, Evolution
Introduction
Human papillomavirus (HPV) infections are very common and viral DNA can be detected from skin, oral and anogenital samples from all human populations. Currently, over 200 types of HPV have been fully characterized and predominantly assigned into three genera: Alphapapillomavirus, Betapapillomavirus and Gammapapillomavirus (Bernard et al., 2010; de Villiers et al., 2004). Among the 65 HPV types belonging to Alphapapillomavirus (Alpha-HPV types), a limited set of viruses (e.g., HPV types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59) are considered oncogenic and associated with the development of cervical cancer and its precursor lesions (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2012; Schiffman et al., 2016). Cervical cancer is the fourth most common cancer among women worldwide and is of particular importance in developing countries based on the lack of proper cervical cancer screening programs (Catarino et al., 2015; Forman et al., 2012; Torre et al., 2017). The oncogenic HPVs causing essentially all cervical cancers are phylogenetically clustered in one clade composed of species Alpha-5, Alpha-6, Alpha-7, Alpha-9 and Alpha-11 (see Figure 1) (Burk et al., 2009; Schiffman et al., 2005).
Figure 1.
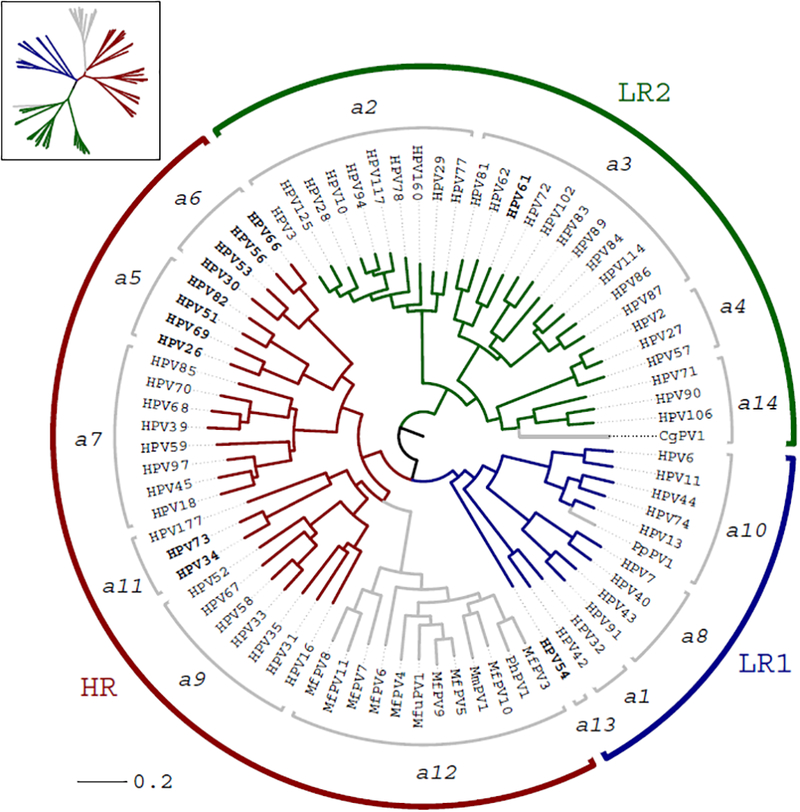
Phylogenetic tree of Alphapapillomavirus. The maximum likelihood tree was constructed using RAxML based on the nucleotide sequence alignment of concatenated 6 ORFs (E6, E7, E1, E2, L2 and L1) and the URR region. Saimiri sciureus papillomavirus types 1/2/3 (SscPV1/2/3) were used as the outgroup taxa. The numbers just within the outer brackets represent the species groups (e.g. ‘α6’ contains HPV30, 53, 56 and 66). At least 3 ancestral papillomaviruses are responsible for the current heterogeneous groups of genital HPV genomes including LR1 (α1, 8, 10 and 13), LR2 (α 2, 3, 4 and 14) and HR (α 5, 6, 7, 9 and 11). The HPV type variants sequenced in this study are indicated in bold. The types joined by grey lines represent non-human primate PVs within Alphapapillomavirus. HR = High-risk; LR = low-risk.
It is still unclear why only a small proportion of oncogenic HPV infections progresses to precancer and cancer (Burk et al., 2009; Schiffman et al., 2016). Besides the pathogenic heterogeneity of distinct HPV types, previous studies indicate that HPV variants are also associated with different risks of cancer progression. For example, HPV type 16 (HPV16) variants can be divided into four main lineages and have been shown to correlate with different degrees of cancer risk (Burk et al., 2013; Mirabello et al., 2016; Xi et al., 2006). Particularly, D2 sublineage had the strongest increased risk of cervical intraepithelial neoplasia grade 3 (CIN3) (OR=6.2) and cancer (OR=28.5). HPV31 lineages A/B have elevated risks for cervical precancers, i.e., cervical intraepithelial neoplasia grades 2–3 (CIN2/3) when compared with lineage C (Schiffman et al., 2010; Xi et al., 2014; Xi et al., 2012). Similarly, HPV58 E7 T20I/G63S variants (sublineage A3) are more frequently detected in East Asia and have been associated with a 7–9 fold higher risk for cervical cancer when compared with other HPV58 variants (Chan et al., 2002; Chan et al., 2011; Chan et al., 2013). These data indicate that HPV variants have different phenotypic characteristics including carcinogenicity.
Distinct HPV types are defined based on the L1 open reading frame (ORF) genetic sequence; those differing from all other characterized types by at least 10% are considered a “novel” HPV type and if the genome is isolated it can be curated and assigned a new number (http://www.nordicehealth.se/hpvcenter/) (Bernard et al., 2010; de Villiers et al., 2004). Isolates of the same HPV type are referred to variant lineages and sublineages when the pairwise nucleotide sequences of their complete genomes differ by approximately 1.0%−10.0% and 0.5%−1.0%, respectively (Burk et al., 2013; Chen et al., 2011, 2013). We have employed this nomenclature system to classify HPV variants within the species Alpha-7 (represented by HPV18) and Alpha-9 (represented by HPV16). A comprehensive classification system of HPV variants facilitates investigation of genotype-phenotype associations of clinical and biological characteristics by comparing data across different epidemiological and molecular studies (see Burk et al, 2013 for review). In addition, studies of HPV variants at the population level provide unique perspectives on host evolution (Chen et al., 2017). For example, the deep phylogenetic separation between HPV16 lineage A and BCD variants implies ancient codivergence between viruses and archaic hominins; a subset of HPV variants (e.g., HPV16 lineage A) could be the descendants of sexual transmission of viruses from Neanderthals/Denisovans to modern human populations through interbreeding (Pimenoff et al., 2017).
In this study, we focused on HPV variants from types within the species Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), and Alpha-11 (HPV34, 73) that cluster into an oncogenic clade (Burk et al., 2009; Schiffman et al., 2005), and from HPV54 and HPV61, two nononcogenic types highly prevalent in our study cohorts. The data presented in this report provide basic information and reference variant sequences for future investigation of viral-host evolution and viral pathogenesis.
Results
Alpha Papillomaviruses
The genus Alphapapillomavirus is a group of PVs that have been predominantly, although not exclusively isolated from the cervicovaginal anatomic region of humans and other non-human primates. We utilized a set of representative Alphapapillomavirus genomes, including 65 papillomavirus types from humans, 12 from macaques, 1 from colobus monkey and 1 from chimpanzee, to construct a phylogenetic tree shown in Figure 1. This group of viruses constitutes 14 species groups (Bernard et al., 2010). Of particular medical interest is the group of papillomaviruses constituting the high-risk (HR) clade that encompasses all the human papillomaviruses associated with cervical cancer and the topology of the tree suggests they have a common origin. In addition, there is a large set of PVs (within the species Alpha-12) that were isolated from the cervicovaginal region of macaques and have also been associated with cervical cancers (Chen et al., 2009b; Wood et al., 2007). Our interest has been to characterize the extent of HPV variability within this important HR clade and, in this report, we focus on types within the species Alpha-5, 6, 11 in addition to a few common or interesting non-HR types, HPV54 (Alpha-13) and HPV61 (Alpha-3) from the low-risk (LR) clades depicted in Figure 1.
HPV variant distribution, lineage classification and nomenclature
HPV isolates in this study were selected from a large set of samples previously typed for HPV and further analyzed for viral variants by sequencing partial segments of the upstream regulatory region (URR) and/or E6 open reading frame (ORF) (Table 1). The complete genome sequences of HPV variants obtained in this study (n=141) and those identified in NCBI/GenBank (n=40) were compared to reveal the extent of viral genomic heterogeneity (isolates listed in Table S1). In addition, phylogenetic trees were created for each type (Figure 2) and the topologies were examined to define major lineages and sublineages, with inter-lineage and inter-sublineage mean differences ranging between 0.9% - 7.3% and 0.3% - 0.9%, respectively (Table S2 and Figure S1). The prototype isolate was always assigned into the “A” lineage or “A1” sublineage. Previously defined “subtype” designation (AE2 was a subtype of HPV82, AE9 was a subtype of HPV54, and HPV64 was a subtype of HPV34) was replaced by assigning a variant lineage name to each taxon.
Table 1.
Summary of HPV isolates, genomic variability and variant lineages.
| Genus | HPV type | No. of URR/E 6 screene da | No. of CG sequence db | No. of CG from NCB Ic | Genome size (bp)d | GC conten t(%) | Cp G site | Variant lineage / sublineage |
|---|---|---|---|---|---|---|---|---|
| Alpha-5 | HPV26 | 19 | 3 | 1 | 7855–7855 | 38.6–38.6 | 145 – 146 | A |
| HPV51 | 233 | 22 | 7 | 7808–7816 | 38.9–39.2 | 140 – 145 | A1-A4, B1-B2 | |
| HPV69 | 21 | 6 | 1 | 7700–7705 | 38.7–38.9 | 130 – 136 | A1-A4 | |
| HPV82 | 58 | 17 | 3 | 7870–7912 | 39.9–40.2 | 135 – 153 | A1-A3, B1-B2, C1-C5 | |
| Alpha-6 | HPV30 | 23 | 14 | 2 | 7843–7881 | 40.2–40.5 | 149 – 157 | A1-A5, B |
| HPV53 | 362 | 22 | 5 | 7856–7892 | 40.0–40.2 | 142 – 148 | A, B, C, D1-D4 | |
| HPV56 | 260 | 6 | 6 | 7790–7866 | 37.9–38.0 | 129 – 134 | A1-A2, B | |
| HPV66 | 146 | 10 | 4 | 7816–7824 | 38.3–38.5 | 128 – 136 | A, B1-B2 | |
| Alpha-11 | HPV34 | 25 | 14 | 1 | 7723–7790 | 37.8–38.2 | 118 – 125 | A1-A2, B, C1-C2 |
| HPV73 | 57 | 11 | 5 | 7697–7730 | 36.2–36.3 | 106 – 109 | A1-A2, B | |
| Alpha-13 | HPV54 | 121 | 8 | 3 | 7701–7760 | 41.8–42.0 | 142 – 154 | A1-A2, B, C1-C2 |
| Alpha-3 | HPV61 | 107 | 8 | 2 | 7989–8030 | 45.9–46.4 | 198 – 207 | A1-A2, B,C |
Number of isolates characterized by sequencing the partial regions of URR and/or E6;
Number of complete genome (CG) variants the authors’ group sequenced;
Number of complete genome (or near complete genome) variants previously published or available on NCBI/GenBank including the prototype. See Table S1 for the list of isolates with accession numbers.
Near complete genomes were not included.
Figure 2.
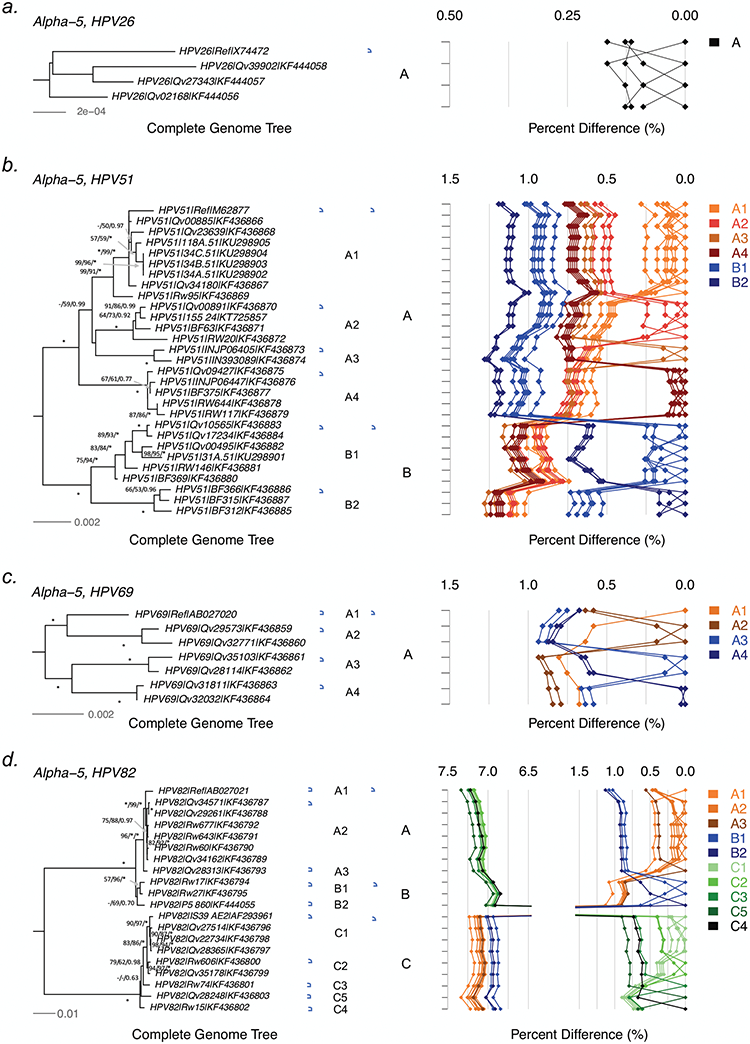
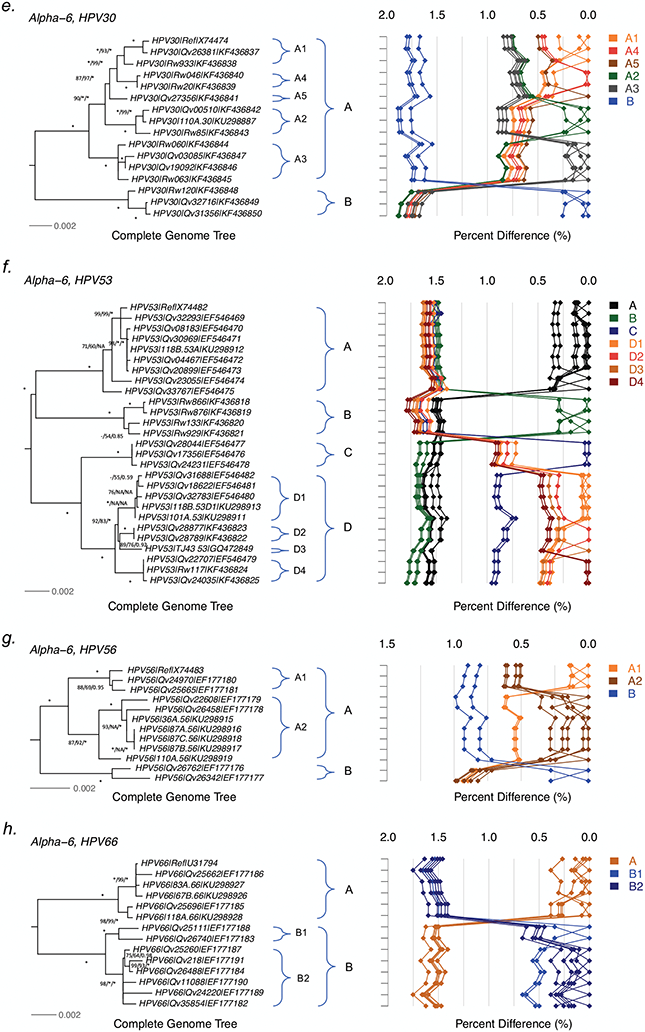
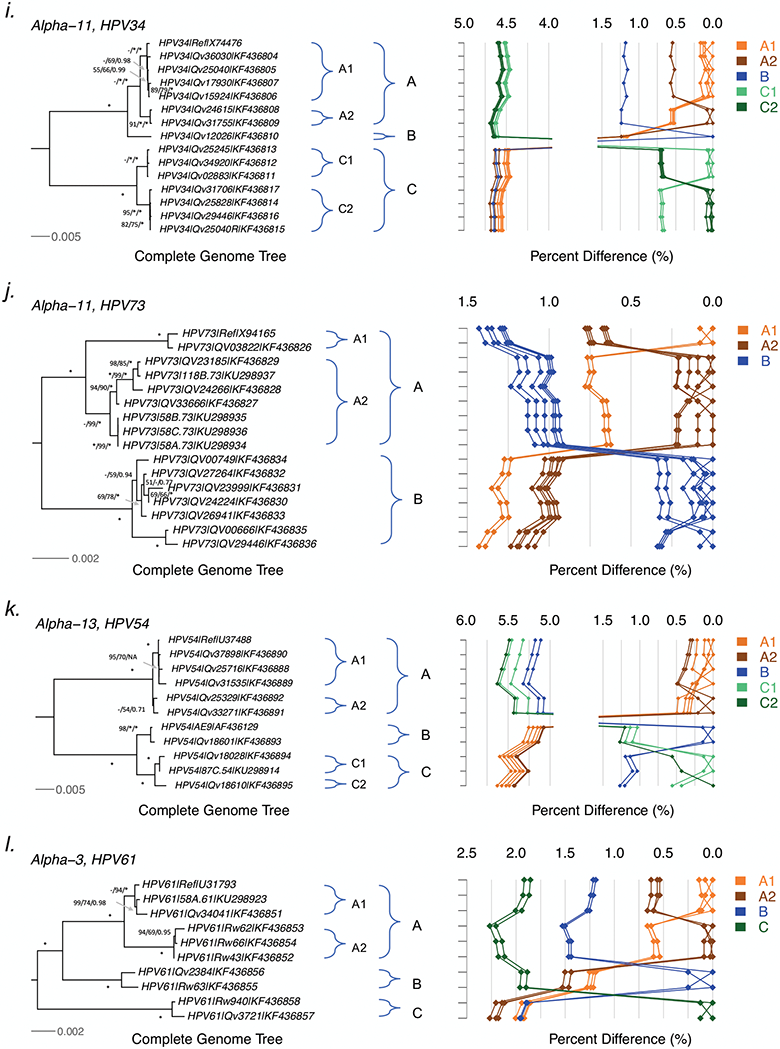
Tree topologies and pairwise comparisons of complete genome variants from individual types. Phylogenetic trees were inferred from a global alignment of complete genome nucleotide sequences of the following variants from each of these types: (a) HPV26, (b) HPV69, (c) HPV51, (d) HPV82, (e) HPV30, (f) HPV53, (g) HPV56, (h) HPV66, (i) HPV34, (j) HPV73, (k) HPV54 and (l) HPV61. Numbers on or near branches indicate support indices in the following order: maximum likelihood (ML) bootstrap percentages using RAxML and PhyML, and Bayesian credibility value percentage using MrBayes. An asterisk (*) indicates 100% agreement between methods. “NA” indicates disagreement between a method and the reference RAxML tree at a given node. Thus, one tree is shown, but three different methods of tree construction were used to estimate the support of the provided tree, as explained above. Distinct variant lineages (i.e., termed A, B, and C) are classified according to the topology and nucleotide sequence differences from > 1% to < 10%; distinct sublineages (e.g., termed A1 and A2) were also inferred from the tree topology and nucleotide sequence differences in the > 0.5% to < 1% range. The bar indicates the nucleotide substitution of unit changes (i.e., 0.002) per site. The percent nucleotide differences for each isolate compared to all other isolates (i.e., 1 × 1 comparisons) are shown in the panel to the right of the phylogeny. Values for each comparison of a given isolate are connected by lines and the comparison to self is indicated by the 0.0% difference point. Different colored lines are used to distinguish each lineage and sublineage.
During review of the prototype genome sequences of HPV51, 53, 54, 56, 69 and 82, we noted a number of probable “errors” in the original clones and have modified the reference genome sequences for these types as shown in Table S3. Most errors interrupted or shifted well-characterized ORFs, while some may be present in the genomes that were directly isolated from the original materials. Since these “errors” are unlikely representative and propagated as circulating viruses, a “repaired” genome is recommended to serve as a revised prototype reference, though the original clones were not re-sequenced in this study.
Genomic diversity of Alpha-5 variants
HPV26 variants.
HPV26 isolates were highly conserved in the sampled cohorts. Three isolates were selected for complete genome sequencing based on the SNP patterns within the partial region of URR. A total of 18 nucleotide changes across the complete genomes was observed; 4 were nonsynonymous within the E1, E2 and L1 ORFs (Tables 2 and S4a). No lineage or sublineage was assigned (Figures 2a).
Table 2.
Genomic diversity of HPV complete genome variants.
| Number of variable nt positionsb | Variable nt codon positionsc | Max aa pairwise difference | Number of aa sequences | Number of variable aa positionsd | ||||
|---|---|---|---|---|---|---|---|---|
| no. | % | 1st | 2nd | 3rd | no. | % | ||
| 1 | 0.2% | 0 | 0 | 1 | 0.0% | 150 | 0 | 0.0% |
| 1 | 0.3% | 1 | 0 | 0 | 0.0% | 104 | 0 | 0.0% |
| 1 | 0.1% | 0 | 0 | 1 | 0.2% | 638 | 1 | 0.2% |
| 3 | 0.3% | 1 | 0 | 2 | 0.3% | 375 | 1 | 0.3% |
| 0 | 0.0% | 0 | 0 | 0 | 0.0% | 106 | 0 | 0.0% |
| 0 | 0.0% | - | - | - | - | - | - | - |
| 0 | 0.0% | 0 | 0 | 0 | 0.0% | 85 | 0 | 0.0% |
| 0 | 0.0% | - | - | - | - | - | - | - |
| 0 | 0.0% | 0 | 0 | 0 | 0.0% | 472 | 0 | 0.0% |
| 5 | 0.3% | 2 | 0 | 3 | 0.4% | 503 | 2 | 0.4% |
| 7 | 0.8% | - | - | - | - | - | - | - |
| 18 | 0.2% | - | - | - | - | 2433 | 4 | 0.2% |
| 6 | 1.3% | 0 | 3 | 3 | 2.0% | 151 | 3 | 2.0% |
| 15 | 4.9% | 5 | 4 | 6 | 6.9% | 101 | 10 | 9.9% |
| 33 | 1.7% | 7 | 4 | 22 | 0.8% | 634 | 10 | 1.6% |
| 33 | 3.1% | 7 | 11 | 15 | 3.1% | 358 | 20 | 5.6% |
| 7 | 2.7% | 1 | 4 | 2 | 4.6% | 87 | 4 | 4.6% |
| 4 | 6.7% | - | - | - | - | - | - | - |
| 9 | 3.5% | 5 | 2 | 2 | 7.1% | 84 | 8 | 9.5% |
| 4 | 16.0% | - | - | - | - | - | - | - |
| 46 | 3.3% | 14 | 3 | 29 | 1.7% | 469 | 19 | 4.1% |
| 52 | 3.4% | 7 | 9 | 36 | 1.4% | 504 | 15 | 3.0% |
| 34 | 3.9% | - | - | - | - | - | - | - |
| 235 | 3.0% | - | - | - | - | 2388 | 85 | 3.6% |
| 7 | 1.5% | 2 | 3 | 2 | 2.0% | 151 | 5 | 3.3% |
| 2 | 0.6% | 0 | 1 | 1 | 1.9% | 104 | 2 | 1.9% |
| 18 | 0.9% | 4 | 2 | 12 | 1.0% | 634 | 8 | 1.3% |
| 23 | 2.1% | 6 | 2 | 15 | 1.9% | 368 | 9 | 2.4% |
| 8 | 2.6% | 2 | 3 | 3 | 5.0% | 101 | 7 | 6.9% |
| 2 | 28.6% | - | - | - | - | - | - | - |
| 9 | 3.0% | 4 | 4 | 1 | 8.2% | 98 | 8 | 8.2% |
| 0 | 0.0% | - | - | - | - | - | - | - |
| 22 | 1.6% | 4 | 5 | 13 | 2.0% | 467 | 9 | 1.9% |
| 20 | 1.3% | 5 | 4 | 11 | 1.2% | 508 | 8 | 1.6% |
| 17 | 2.3% | - | - | - | - | - | - | - |
| 119 | 1.5% | - | - | - | - | 2431 | 56 | 2.3% |
| 16 | 3.5% | 5 | 4 | 7 | 5.3% | 151 | 8 | 5.3% |
| 24 | 7.9% | 9 | 4 | 11 | 11.7% | 100 | 13 | 13.0% |
| 172 | 8.9% | 32 | 24 | 116 | 6.6% | 642 | 55 | 8.6% |
| 87 | 8.1% | 20 | 17 | 50 | 10.0% | 359 | 39 | 10.9% |
| 25 | 9.4% | 8 | 8 | 9 | 15.9% | 88 | 15 | 17.0% |
| 10 | 14.7% | - | - | - | - | - | - | - |
| 21 | 8.2% | 7 | 3 | 11 | 9.8% | 84 | 9 | 10.7% |
| 3 | 10.7% | - | - | - | - | - | - | - |
| 124 | 8.7% | 24 | 13 | 87 | 5.7% | 473 | 30 | 6.3% |
| 149 | 9.9% | 25 | 11 | 113 | 5.0% | 503 | 29 | 5.8% |
| 120 | 12.7% | - | - | - | - | - | - | - |
| 719 | 9.1% | - | - | - | - | 2384 | 201 | 8.4% |
| 6 | 1.3% | 2 | 1 | 3 | 1.3% | 153 | 4 | 2.6% |
| 5 | 1.6% | 2 | 0 | 3 | 1.9% | 105 | 2 | 1.9% |
| 67 | 3.5% | 15 | 7 | 45 | 2.6% | 631 | 20 | 3.2% |
| 31 | 2.7% | 7 | 4 | 20 | 2.7% | 380 | 15 | 3.9% |
| 13 | 3.5% | 2 | 6 | 5 | 5.2% | 154 | 10 | 6.5% |
| 6 | 7.4% | - | - | - | - | - | - | - |
| 9 | 3.1% | 5 | 1 | 3 | 3.1% | 96 | 4 | 4.2% |
| 1 | 2.1% | - | - | - | - | - | - | - |
| 46 | 3.3% | 3 | 6 | 37 | 2.2% | 463 | 12 | 2.6% |
| 50 | 3.3% | 7 | 9 | 34 | 2.0% | 501 | 15 | 3.0% |
| 32 | 3.9% | - | - | - | - | - | - | - |
| 254 | 3.2% | - | - | - | - | 2471 | 81 | 3.3% |
| 27 | 5.8% | 8 | 7 | 12 | 5.3% | 154 | 11 | 7.1% |
| 3 | 0.9% | 1 | 0 | 1 | 1.9% | 105 | 2 | 1.9% |
| 55 | 2.9% | 16 | 11 | 28 | 2.6% | 636 | 28 | 4.4% |
| 44 | 3.8% | 13 | 10 | 21 | 3.6% | 384 | 27 | 7.0% |
| 17 | 3.8% | 1 | 9 | 7 | 4.0% | 150 | 12 | 8.0% |
| 4 | 3.8% | - | - | - | - | - | - | - |
| 13 | 5.0% | 3 | 5 | 5 | 5.9% | 85 | 8 | 9.4% |
| 3 | 7.0% | - | - | - | - | - | - | - |
| 54 | 3.9% | 2 | 8 | 44 | 1.5% | 463 | 12 | 2.6% |
| 58 | 3.9% | 12 | 3 | 43 | 1.6% | 499 | 15 | 3.0% |
| 39 | 4.8% | - | - | - | - | - | - | - |
| 295 | 3.7% | - | - | - | - | 2476 | 116 | 4.7% |
| 6 | 1.3% | 2 | 1 | 3 | 2.6% | 155 | 4 | 2.6% |
| 7 | 2.2% | 2 | 0 | 5 | 2.9% | 105 | 4 | 3.8% |
| 23 | 1.2% | 11 | 3 | 9 | 1.6% | 636 | 13 | 2.0% |
| 21 | 1.9% | 7 | 5 | 8 | 2.7% | 371 | 12 | 3.2% |
| 6 | 1.5% | 1 | 1 | 4 | 3.0% | 135 | 4 | 3.0% |
| 4 | 1.1% | 0 | 2 | 2 | 2.7% | 120 | 2 | 1.7% |
| 24 | 1.7% | 3 | 2 | 19 | 0.9% | 464 | 6 | 1.3% |
| 26 | 1.6% | 3 | 3 | 20 | 0.9% | 534 | 7 | 1.3% |
| 24 | 2.6% | - | - | - | - | - | - | - |
| 134 | 1.7% | - | - | - | - | 2520 | 51 | 2.0% |
| 6 | 1.3% | 4 | 0 | 2 | 1.3% | 155 | 2 | 1.3% |
| 9 | 2.8% | 4 | 2 | 3 | 4.8% | 105 | 5 | 4.8% |
| 48 | 2.5% | 10 | 11 | 26 | 1.7% | 630 | 21 | 3.3% |
| 25 | 2.3% | 6 | 5 | 14 | 2.2% | 369 | 11 | 3.0% |
| 12 | 2.3% | 3 | 5 | 4 | 3.4% | 170 | 7 | 4.1% |
| 8 | 5.2% | - | - | - | - | - | - | - |
| 11 | 4.3% | 2 | 4 | 5 | 5.9% | 85 | 6 | 7.1% |
| 3 | 14.3% | - | - | - | - | - | - | - |
| 40 | 2.9% | 5 | 10 | 25 | 1.5% | 464 | 14 | 3.0% |
| 49 | 3.2% | 7 | 15 | 27 | 1.9% | 503 | 18 | 3.6% |
| 21 | 2.7% | - | - | - | - | - | - | - |
| 218 | 2.8% | - | - | - | - | 2519 | 82 | 3.3% |
| 23 | 5.1% | 7 | 7 | 9 | 5.4% | 148 | 11 | 7.4% |
| 9 | 3.1% | 5 | 1 | 3 | 3.1% | 96 | 4 | 4.2% |
| 1 | 2.1% | - | - | - | - | - | - | - |
| 46 | 3.3% | 3 | 6 | 37 | 2.2% | 463 | 12 | 2.6% |
| 50 | 3.3% | 7 | 9 | 34 | 2.0% | 501 | 15 | 3.0% |
| 32 | 3.9% | - | - | - | - | - | - | - |
| 254 | 3.2% | - | - | - | - | 2471 | 81 | 3.3% |
| 27 | 5.8% | 8 | 7 | 12 | 5.3% | 154 | 11 | 7.1% |
| 3 | 0.9% | 1 | 0 | 1 | 1.9% | 105 | 2 | 1.9% |
| 55 | 2.9% | 16 | 11 | 28 | 2.6% | 636 | 28 | 4.4% |
| 44 | 3.8% | 13 | 10 | 21 | 3.6% | 384 | 27 | 7.0% |
| 17 | 3.8% | 1 | 9 | 7 | 4.0% | 150 | 12 | 8.0% |
| 4 | 3.8% | - | - | - | - | - | - | - |
| 13 | 5.0% | 3 | 5 | 5 | 5.9% | 85 | 8 | 9.4% |
| 3 | 7.0% | - | - | - | - | - | - | - |
| 54 | 3.9% | 2 | 8 | 44 | 1.5% | 463 | 12 | 2.6% |
| 58 | 3.9% | 12 | 3 | 43 | 1.6% | 499 | 15 | 3.0% |
| 39 | 4.8% | - | - | - | - | - | - | - |
| 295 | 3.7% | - | - | - | - | 2476 | 116 | 4.7% |
| 6 | 1.3% | 2 | 1 | 3 | 2.6% | 155 | 4 | 2.6% |
| 7 | 2.2% | 2 | 0 | 5 | 2.9% | 105 | 4 | 3.8% |
| 23 | 1.2% | 11 | 3 | 9 | 1.6% | 636 | 13 | 2.0% |
| 21 | 1.9% | 7 | 5 | 8 | 2.7% | 371 | 12 | 3.2% |
| 6 | 1.5% | 1 | 1 | 4 | 3.0% | 135 | 4 | 3.0% |
| 4 | 1.1% | 0 | 2 | 2 | 2.7% | 120 | 2 | 1.7% |
| 24 | 1.7% | 3 | 2 | 19 | 0.9% | 464 | 6 | 1.3% |
| 26 | 1.6% | 3 | 3 | 20 | 0.9% | 534 | 7 | 1.3% |
| 24 | 2.6% | - | - | - | - | - | - | - |
| 134 | 1.7% | - | - | - | - | 2520 | 51 | 2.0% |
| 6 | 1.3% | 4 | 0 | 2 | 1.3% | 155 | 2 | 1.3% |
| 9 | 2.8% | 4 | 2 | 3 | 4.8% | 105 | 5 | 4.8% |
| 48 | 2.5% | 10 | 11 | 26 | 1.7% | 630 | 21 | 3.3% |
| 25 | 2.3% | 6 | 5 | 14 | 2.2% | 369 | 11 | 3.0% |
| 12 | 2.3% | 3 | 5 | 4 | 3.4% | 170 | 7 | 4.1% |
| 8 | 5.2% | - | - | - | - | - | - | - |
| 11 | 4.3% | 2 | 4 | 5 | 5.9% | 85 | 6 | 7.1% |
| 3 | 14.3% | - | - | - | - | - | - | - |
| 40 | 2.9% | 5 | 10 | 25 | 1.5% | 464 | 14 | 3.0% |
| 49 | 3.2% | 7 | 15 | 27 | 1.9% | 503 | 18 | 3.6% |
| 21 | 2.7% | - | - | - | - | - | - | - |
| 218 | 2.8% | - | - | - | - | 2519 | 82 | 3.3% |
| 23 | 5.1% | 7 | 7 | 9 | 5.4% | 148 | 11 | 7.4% |
| 8 | 2.7% | 2 | 4 | 2 | 5.2% | 97 | 6 | 6.2% |
| 88 | 4.5% | 21 | 11 | 56 | 3.9% | 647 | 27 | 4.2% |
| 44 | 4.2% | 13 | 6 | 25 | 4.9% | 345 | 18 | 5.2% |
| 9 | 3.8% | 3 | 2 | 4 | 5.1% | 78 | 4 | 5.1% |
| 15 | 12.5% | - | - | - | - | - | - | - |
| 21 | 9.3% | 7 | 1 | 13 | 6.8% | 74 | 5 | 6.8% |
| 5 | 8.6% | - | - | - | - | - | - | - |
| 99 | 7.0% | 27 | 13 | 59 | 6.8% | 473 | 37 | 7.8% |
| 96 | 6.4% | 13 | 8 | 75 | 3.4% | 502 | 18 | 3.6% |
| 66 | 8.1% | - | - | - | - | - | - | - |
| 466 | 6.0% | - | - | - | - | 2364 | 126 | 5.3% |
| 8 | 1.8% | 3 | 2 | 3 | 3.4% | 148 | 6 | 4.1% |
| 5 | 1.7% | 2 | 1 | 2 | 3.1% | 97 | 3 | 3.1% |
| 36 | 1.8% | 11 | 7 | 18 | 1.5% | 650 | 18 | 2.8% |
| 24 | 2.3% | 8 | 3 | 13 | 2.9% | 350 | 14 | 4.0% |
| 7 | 3.0% | 0 | 5 | 2 | 3.9% | 78 | 5 | 6.4% |
| 2 | 0.9% | 1 | 0 | 1 | 1.4% | 74 | 1 | 1.4% |
| 2 | 4.4% | - | - | - | - | - | - | - |
| 25 | 1.8% | 5 | 1 | 19 | 1.7% | 475 | 9 | 1.9% |
| 25 | 1.7% | 2 | 1 | 22 | 0.6% | 503 | 4 | 0.8% |
| 33 | 4.0% | - | - | - | - | - | - | - |
| 159 | 2.1% | - | - | - | - | 2375 | 59 | 2.5% |
| 27 | 6.2% | 9 | 6 | 12 | 9.7% | 144 | 15 | 10.4% |
| 18 | 6.3% | 5 | 7 | 6 | 7.7% | 95 | 10 | 10.5% |
| 111 | 5.8% | 25 | 12 | 73 | 4.9% | 635 | 36 | 5.7% |
| 74 | 6.7% | 27 | 16 | 31 | 8.9% | 367 | 44 | 12.0% |
| 30 | 7.4% | 4 | 14 | 12 | 14.7% | 134 | 18 | 13.4% |
| 24 | 16.7% | - | - | - | - | - | - | - |
| 10 | 4.9% | 3 | 1 | 6 | 7.6% | 67 | 6 | 9.0% |
| 7 | 8.0% | - | - | - | - | - | - | - |
| 113 | 8.0% | 22 | 12 | 79 | 6.8% | 470 | 34 | 7.2% |
| 85 | 5.4% | 21 | 12 | 52 | 4.6% | 525 | 26 | 5.0% |
| 57 | 7.3% | - | - | - | - | - | - | - |
| 520 | 6.7% | - | - | - | - | 2372 | 186 | 7.8% |
| 16 | 3.6% | 4 | 2 | 10 | 3.5% | 146 | 6 | 4.1% |
| 7 | 2.4% | 2 | 0 | 5 | 4.3% | 95 | 4 | 4.2% |
| 59 | 3.0% | 16 | 7 | 36 | 2.2% | 654 | 21 | 3.2% |
| 30 | 2.6% | 14 | 7 | 9 | 4.0% | 382 | 19 | 5.0% |
| 11 | 3.5% | 2 | 1 | 8 | 2.9% | 104 | 3 | 2.9% |
| 5 | 3.9% | - | - | - | - | - | - | - |
| 7 | 3.1% | 1 | 4 | 2 | 3.6% | 75 | 4 | 5.3% |
| 10 | 5.1% | - | - | - | - | - | - | - |
| 48 | 3.5% | 10 | 5 | 33 | 3.1% | 459 | 15 | 3.3% |
| 42 | 2.8% | 15 | 2 | 25 | 2.4% | 505 | 13 | 2.6% |
| 33 | 3.9% | - | - | - | - | - | - | - |
| 259 | 3.2% | - | - | - | - | 2345 | 81 | 3.5% |
| Type/Genea | Max nt pairwise difference | Number of nt sequences | Number of variable nt positionsb | Variable nt codon positionsc | Max aa pairwise difference | Number of aa sequences | Number of variable aa positionsd | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| no. | % | 1st | 2nd | 3rd | no. | % | |||||
| Alpha-6, HPV30 (N=16) | |||||||||||
| E6 | 0.7% | 462 | 6 | 1.3% | 2 | 1 | 3 | 1.3% | 153 | 4 | 2.6% |
| E7 | 1.3% | 318 | 5 | 1.6% | 2 | 0 | 3 | 1.9% | 105 | 2 | 1.9% |
| El | 2.2% | 1896 | 67 | 3.5% | 15 | 7 | 45 | 2.6% | 631 | 20 | 3.2% |
| E2 | 1.6% | 1143 | 31 | 2.7% | 7 | 4 | 20 | 2.7% | 380 | 15 | 3.9% |
| E4 | 2.2% | 375 | 13 | 3.5% | 2 | 6 | 5 | 5.2% | 154 | 10 | 6.5% |
| NCR1 | 5.1% | 81 | 6 | 7.4% | - | - | - | - | - | - | - |
| E5 | 2.4% | 291 | 9 | 3.1% | 5 | 1 | 3 | 3.1% | 96 | 4 | 4.2% |
| NCR2 | 2.1% | 47 | 1 | 2.1% | - | - | - | - | - | - | - |
| L2 | 2.4% | 1392 | 46 | 3.3% | 3 | 6 | 37 | 2.2% | 463 | 12 | 2.6% |
| L1 | 2.3% | 1506 | 50 | 3.3% | 7 | 9 | 34 | 2.0% | 501 | 15 | 3.0% |
| URR | 2.4% | 828 | 32 | 3.9% | - | - | - | - | - | - | - |
| CG | 1.9% | 7890 | 254 | 3.2% | - | - | - | - | 2471 | 81 | 3.3% |
| Alpha-6, HPV53 (N=27) | |||||||||||
| E6 | 3.2% | 465 | 27 | 5.8% | 8 | 7 | 12 | 5.3% | 154 | 11 | 7.1% |
| E7 | 0.9% | 318 | 3 | 0.9% | 1 | 0 | 1 | 1.9% | 105 | 2 | 1.9% |
| E1 | 1.7% | 1911 | 55 | 2.9% | 16 | 11 | 28 | 2.6% | 636 | 28 | 4.4% |
| E2 | 1.7% | 1155 | 44 | 3.8% | 13 | 10 | 21 | 3.6% | 384 | 27 | 7.0% |
| E4 | 2.2% | 453 | 17 | 3.8% | 1 | 9 | 7 | 4.0% | 150 | 12 | 8.0% |
| NCR1 | 3.5% | 104 | 4 | 3.8% | - | - | - | - | - | - | - |
| E5 | 3.1% | 258 | 13 | 5.0% | 3 | 5 | 5 | 5.9% | 85 | 8 | 9.4% |
| NCR2 | 7.0% | 43 | 3 | 7.0% | - | - | - | - | - | - | - |
| L2 | 2.3% | 1392 | 54 | 3.9% | 2 | 8 | 44 | 1.5% | 463 | 12 | 2.6% |
| L1 | 2.1% | 1500 | 58 | 3.9% | 12 | 3 | 43 | 1.6% | 499 | 15 | 3.0% |
| URR | 2.9% | 818 | 39 | 4.8% | - | - | - | - | - | - | - |
| CG | 1.8% | 7892 | 295 | 3.7% | - | - | - | - | 2476 | 116 | 4.7% |
| Alpha-6, HPV56 (N=12) | |||||||||||
| E6 | 1.1% | 468 | 6 | 1.3% | 2 | 1 | 3 | 2.6% | 155 | 4 | 2.6% |
| E7 | 1.9% | 318 | 7 | 2.2% | 2 | 0 | 5 | 2.9% | 105 | 4 | 3.8% |
| E1 | 0.8% | 1911 | 23 | 1.2% | 11 | 3 | 9 | 1.6% | 636 | 13 | 2.0% |
| E2 | 1.4% | 1116 | 21 | 1.9% | 7 | 5 | 8 | 2.7% | 371 | 12 | 3.2% |
| E4 | 1.5% | 408 | 6 | 1.5% | 1 | 1 | 4 | 3.0% | 135 | 4 | 3.0% |
| E5 | 0.8% | 363 | 4 | 1.1% | 0 | 2 | 2 | 2.7% | 120 | 2 | 1.7% |
| L2 | 0.9% | 1395 | 24 | 1.7% | 3 | 2 | 19 | 0.9% | 464 | 6 | 1.3% |
| L1 | 1.1% | 1605 | 26 | 1.6% | 3 | 3 | 20 | 0.9% | 534 | 7 | 1.3% |
| URR | 2.0% | 926 | 24 | 2.6% | - | - | - | - | - | - | - |
| CG | 1.0% | 7922 | 134 | 1.7% | - | - | - | - | 2520 | 51 | 2.0% |
| Alpha-6, HPV66 (N=14) | |||||||||||
| E6 | 1.1% | 468 | 6 | 1.3% | 4 | 0 | 2 | 1.3% | 155 | 2 | 1.3% |
| E7 | 2.5% | 318 | 9 | 2.8% | 4 | 2 | 3 | 4.8% | 105 | 5 | 4.8% |
| E1 | 1.7% | 1893 | 48 | 2.5% | 10 | 11 | 26 | 1.7% | 630 | 21 | 3.3% |
| E2 | 1.6% | 1110 | 25 | 2.3% | 6 | 5 | 14 | 2.2% | 369 | 11 | 3.0% |
| E4 | 1.9% | 513 | 12 | 2.3% | 3 | 5 | 4 | 3.4% | 170 | 7 | 4.1% |
| NCR1 | 5.2% | 155 | 8 | 5.2% | - | - | - | - | - | - | - |
| E5 | 3.9% | 258 | 11 | 4.3% | 2 | 4 | 5 | 5.9% | 85 | 6 | 7.1% |
| NCR2 | 10.0% | 21 | 3 | 14.3% | - | - | - | - | - | - | - |
| L2 | 1.9% | 1395 | 40 | 2.9% | 5 | 10 | 25 | 1.5% | 464 | 14 | 3.0% |
| L1 | 2.0% | 1512 | 49 | 3.2% | 7 | 15 | 27 | 1.9% | 503 | 18 | 3.6% |
| URR | 1.5% | 769 | 21 | 2.7% | - | - | - | - | - | - | - |
| CG | 1.8% | 7827 | 218 | 2.8% | - | - | - | - | 2519 | 82 | 3.3% |
| Alpha-11, HPV34 (N=15) | |||||||||||
| E6 | 4.0% | 447 | 23 | 5.1% | 7 | 7 | 9 | 5.4% | 148 | 11 | 7.4% |
| E7 | 2.0% | 294 | 8 | 2.7% | 2 | 4 | 2 | 5.2% | 97 | 6 | 6.2% |
| El | 4.0% | 1944 | 88 | 4.5% | 21 | 11 | 56 | 3.9% | 647 | 27 | 4.2% |
| E2 | 3.5% | 1038 | 44 | 4.2% | 13 | 6 | 25 | 4.9% | 345 | 18 | 5.2% |
| E4 | 4.1% | 237 | 9 | 3.8% | 3 | 2 | 4 | 5.1% | 78 | 4 | 5.1% |
| NCR1 | 17.8% | 120 | 15 | 12.5% | - | - | - | - | - | - | - |
| E5 | 8.9% | 225 | 21 | 9.3% | 7 | 1 | 13 | 6.8% | 74 | 5 | 6.8% |
| NCR2 | 14.3% | 58 | 5 | 8.6% | - | - | - | - | - | - | - |
| L2 | 5.6% | 1422 | 99 | 7.0% | 27 | 13 | 59 | 6.8% | 473 | 37 | 7.8% |
| L1 | 5.4% | 1509 | 96 | 6.4% | 13 | 8 | 75 | 3.4% | 502 | 18 | 3.6% |
| URR | 6.4% | 819 | 66 | 8.1% | - | - | - | - | - | - | - |
| CG | 4.7% | 7828 | 466 | 6.0% | - | - | - | - | 2364 | 126 | 5.3% |
| Alpha-11, HPV73 (N=16) | |||||||||||
| E6 | 1.6% | 447 | 8 | 1.8% | 3 | 2 | 3 | 3.4% | 148 | 6 | 4.1% |
| E7 | 1.4% | 294 | 5 | 1.7% | 2 | 1 | 2 | 3.1% | 97 | 3 | 3.1% |
| E1 | 1.2% | 1953 | 36 | 1.8% | 11 | 7 | 18 | 1.5% | 650 | 18 | 2.8% |
| E2 | 1.7% | 1053 | 24 | 2.3% | 8 | 3 | 13 | 2.9% | 350 | 14 | 4.0% |
| E4 | 2.1% | 237 | 7 | 3.0% | 0 | 5 | 2 | 3.9% | 78 | 5 | 6.4% |
| E5 | 0.9% | 225 | 2 | 0.9% | 1 | 0 | 1 | 1.4% | 74 | 1 | 1.4% |
| NCR2 | 4.4% | 45 | 2 | 4.4% | - | - | - | - | - | - | - |
| L2 | 1.3% | 1428 | 25 | 1.8% | 5 | 1 | 19 | 1.7% | 475 | 9 | 1.9% |
| L1 | 1.2% | 1512 | 25 | 1.7% | 2 | 1 | 22 | 0.6% | 503 | 4 | 0.8% |
| URR | 2.8% | 829 | 33 | 4.0% | - | - | - | - | - | - | - |
| CG | 1.4% | 7733 | 159 | 2.1% | - | - | - | - | 2375 | 59 | 2.5% |
| Alpha-13, HPV54 (N=11) | |||||||||||
| E6 | 5.1% | 435 | 27 | 6.2% | 9 | 6 | 12 | 9.7% | 144 | 15 | 10.4% |
| E7 | 4.5% | 288 | 18 | 6.3% | 5 | 7 | 6 | 7.7% | 95 | 10 | 10.5% |
| E1 | 4.8% | 1908 | 111 | 5.8% | 25 | 12 | 73 | 4.9% | 635 | 36 | 5.7% |
| E2 | 5.0% | 1104 | 74 | 6.7% | 27 | 16 | 31 | 8.9% | 367 | 44 | 12.0% |
| E4 | 6.2% | 405 | 30 | 7.4% | 4 | 14 | 12 | 14.7% | 134 | 18 | 13.4% |
| NCR1 | 16.7% | 144 | 24 | 16.7% | - | - | - | - | - | - | - |
| E5 | 3.5% | 204 | 10 | 4.9% | 3 | 1 | 6 | 7.6% | 67 | 6 | 9.0% |
| NCR2 | 8.0% | 87 | 7 | 8.0% | - | - | - | - | - | - | - |
| L2 | 7.4% | 1413 | 113 | 8.0% | 22 | 12 | 79 | 6.8% | 470 | 34 | 7.2% |
| L1 | 5.0% | 1578 | 85 | 5.4% | 21 | 12 | 52 | 4.6% | 525 | 26 | 5.0% |
| URR | 7.0% | 111 | 57 | 7.3% | - | - | - | - | - | - | - |
| CG | 5.6% | 7799 | 520 | 6.7% | - | - | - | - | 2372 | 186 | 7.8% |
| Alpha-3, HPV61 (N=10) | |||||||||||
| E6 | 2.3% | 441 | 16 | 3.6% | 4 | 2 | 10 | 3.5% | 146 | 6 | 4.1% |
| E7 | 2.4% | 288 | 7 | 2.4% | 2 | 0 | 5 | 4.3% | 95 | 4 | 4.2% |
| E1 | 2.3% | 1965 | 59 | 3.0% | 16 | 7 | 36 | 2.2% | 654 | 21 | 3.2% |
| E2 | 1.8% | 1149 | 30 | 2.6% | 14 | 7 | 9 | 4.0% | 382 | 19 | 5.0% |
| E4 | 2.5% | 315 | 11 | 3.5% | 2 | 1 | 8 | 2.9% | 104 | 3 | 2.9% |
| NCR1 | 1.6% | 129 | 5 | 3.9% | - | - | - | - | - | - | - |
| E5 | 2.9% | 228 | 7 | 3.1% | 1 | 4 | 2 | 3.6% | 75 | 4 | 5.3% |
| NCR2 | 4.6% | 196 | 10 | 5.1% | - | - | - | - | - | - | - |
| L2 | 2.8% | 1380 | 48 | 3.5% | 10 | 5 | 33 | 3.1% | 459 | 15 | 3.3% |
| L1 | 2.0% | 1518 | 42 | 2.8% | 15 | 2 | 25 | 2.4% | 505 | 13 | 2.6% |
| URR | 2.4% | 851 | 33 | 3.9% | - | - | - | - | - | - | - |
| CG | 2.3% | 8037 | 259 | 3.2% | - | - | - | - | 2345 | 81 | 3.5% |
| Alpha-11, HPV34 (N=15) | |||||||||||
| E6 | 4.0% | 447 | 23 | 5.1% | 7 | 7 | 9 | 5.4% | 148 | 11 | 7.4% |
| E7 | 2.0% | 294 | 8 | 2.7% | 2 | 4 | 2 | 5.2% | 97 | 6 | 6.2% |
| E1 | 4.0% | 1944 | 88 | 4.5% | 21 | 11 | 56 | 3.9% | 647 | 27 | 4.2% |
| E2 | 3.5% | 1038 | 44 | 4.2% | 13 | 6 | 25 | 4.9% | 345 | 18 | 5.2% |
| E4 | 4.1% | 237 | 9 | 3.8% | 3 | 2 | 4 | 5.1% | 78 | 4 | 5.1% |
| NCR1 | 17.8% | 120 | 15 | 12.5% | - | - | - | - | - | - | - |
| E5 | 8.9% | 225 | 21 | 9.3% | 7 | 1 | 13 | 6.8% | 74 | 5 | 6.8% |
| NCR2 | 14.3% | 58 | 5 | 8.6% | - | - | - | - | - | - | - |
| L2 | 5.6% | 1422 | 99 | 7.0% | 27 | 13 | 59 | 6.8% | 473 | 37 | 7.8% |
| L1 | 5.4% | 1509 | 96 | 6.4% | 13 | 8 | 75 | 3.4% | 502 | 18 | 3.6% |
| URR | 6.4% | 819 | 66 | 8.1% | - | - | - | - | - | - | - |
| CG | 4.7% | 7828 | 466 | 6.0% | - | - | - | - | 2364 | 126 | 5.3% |
| Alpha-11, HPV73 (N=16) | |||||||||||
| E6 | 1.6% | 447 | 8 | 1.8% | 3 | 2 | 3 | 3.4% | 148 | 6 | 4.1% |
| E7 | 1.4% | 294 | 5 | 1.7% | 2 | 1 | 2 | 3.1% | 97 | 3 | 3.1% |
| E1 | 1.2% | 1953 | 36 | 1.8% | 11 | 7 | 18 | 1.5% | 650 | 18 | 2.8% |
| E2 | 1.7% | 1053 | 24 | 2.3% | 8 | 3 | 13 | 2.9% | 350 | 14 | 4.0% |
| E4 | 2.1% | 237 | 7 | 3.0% | 0 | 5 | 2 | 3.9% | 78 | 5 | 6.4% |
| E5 | 0.9% | 225 | 2 | 0.9% | 1 | 0 | 1 | 1.4% | 74 | 1 | 1.4% |
| NCR2 | 4.4% | 45 | 2 | 4.4% | - | - | - | - | - | - | - |
| L2 | 1.3% | 1428 | 25 | 1.8% | 5 | 1 | 19 | 1.7% | 475 | 9 | 1.9% |
| L1 | 1.2% | 1512 | 25 | 1.7% | 2 | 1 | 22 | 0.6% | 503 | 4 | 0.8% |
| URR | 2.8% | 829 | 33 | 4.0% | - | - | - | - | - | - | - |
| CG | 1.4% | 7733 | 159 | 2.1% | - | - | - | - | 2375 | 59 | 2.5% |
| Alpha-13, HPV54 (N=11) | |||||||||||
| E6 | 5.1% | 435 | 27 | 6.2% | 9 | 6 | 12 | 9.7% | 144 | 15 | 10.4% |
| E7 | 4.5% | 288 | 18 | 6.3% | 5 | 7 | 6 | 7.7% | 95 | 10 | 10.5% |
| E1 | 4.8% | 1908 | 111 | 5.8% | 25 | 12 | 73 | 4.9% | 635 | 36 | 5.7% |
| E2 | 5.0% | 1104 | 74 | 6.7% | 27 | 16 | 31 | 8.9% | 367 | 44 | 12.0% |
| E4 | 6.2% | 405 | 30 | 7.4% | 4 | 14 | 12 | 14.7% | 134 | 18 | 13.4% |
| NCR1 | 16.7% | 144 | 24 | 16.7% | - | - | - | - | - | - | - |
| E5 | 3.5% | 204 | 10 | 4.9% | 3 | 1 | 6 | 7.6% | 67 | 6 | 9.0% |
| NCR2 | 8.0% | 87 | 7 | 8.0% | - | - | - | - | - | - | - |
| L2 | 7.4% | 1413 | 113 | 8.0% | 22 | 12 | 79 | 6.8% | 470 | 34 | 7.2% |
| L1 | 5.0% | 1578 | 85 | 5.4% | 21 | 12 | 52 | 4.6% | 525 | 26 | 5.0% |
| URR | 7.0% | 111 | 57 | 7.3% | - | - | - | - | - | - | - |
| CG | 5.6% | 7799 | 520 | 6.7% | - | - | - | - | 2372 | 186 | 7.8% |
| Alpha-3, HPV61 (N=10) | |||||||||||
| E6 | 2.3% | 441 | 16 | 3.6% | 4 | 2 | 10 | 3.5% | 146 | 6 | 4.1% |
| E7 | 2.4% | 288 | 7 | 2.4% | 2 | 0 | 5 | 4.3% | 95 | 4 | 4.2% |
| E1 | 2.3% | 1965 | 59 | 3.0% | 16 | 7 | 36 | 2.2% | 654 | 21 | 3.2% |
| E2 | 1.8% | 1149 | 30 | 2.6% | 14 | 7 | 9 | 4.0% | 382 | 19 | 5.0% |
| E4 | 2.5% | 315 | 11 | 3.5% | 2 | 1 | 8 | 2.9% | 104 | 3 | 2.9% |
| NCR1 | 1.6% | 129 | 5 | 3.9% | - | - | - | - | - | - | - |
| E5 | 2.9% | 228 | 7 | 3.1% | 1 | 4 | 2 | 3.6% | 75 | 4 | 5.3% |
| NCR2 | 4.6% | 196 | 10 | 5.1% | - | - | - | - | - | - | - |
| L2 | 2.8% | 1380 | 48 | 3.5% | 10 | 5 | 33 | 3.1% | 459 | 15 | 3.3% |
| LI | 2.0% | 1518 | 42 | 2.8% | 15 | 2 | 25 | 2.4% | 505 | 13 | 2.6% |
| URR | 2.4% | 851 | 33 | 3.9% | - | - | - | - | - | - | - |
| CG | 2.3% | 8037 | 259 | 3.2% | - | - | - | - | 2345 | 81 | 3.5% |
CG, complete genome; NCR1, noncoding region 1 between the E2 and E5 ORFs; NCR2, noncoding region 2 between the E5 and L2 ORFs; URR, upstream regulatory region.
Each insert or deletion event was counted as one variation.
The first, second, and third nucleotide positions in a codon.
Amino acid changes (nonsynonymous changes). Each insert or deletion event was counted as one variation.
HPV51 variants.
We screened 233 HPV51 isolates for viral variation by sequencing the partial regions of URR and/or E6, among which 22 variants were characterized for complete genome sequences. In addition, the prototype sequence and 6 complete genomes from a Brazilian study were also included in this report (Table S1). The overall nucleotide variability was 3.0% (235/7822); 85 of 2388 amino acid sites (3.6%) of 8 ORFs were variable (Tables 2 and S4b). The noncoding regions (NCR1, NCR2 and URR) were more variable than the coding ORFs, while E7 and E5 were the two most diversified proteins with overall amino acid differences of 9.9% and 9.5%, respectively. No insertion or deletion (indel) changes were observed within the ORFs. Phylogenetic topology based on the complete genome alignment clustered HPV51 variants into 2 lineages designated A and B (Figure 2b). The A lineage was further divided into 4 sublineages (A1-A4), and B into 2 sublineages (B1-B2). The inter-lineage and inter-sublineage mean differences ranged between 0.52%−0.74% and 0.92%−1.24%, respectively, and the intrasublineage mean differences were less than 0.20±0.03% (Table S2). All HPV51 isolates from Asia were assigned into the A lineage, whereas B variants were more commonly detected in African women (74%−100%) (Table 3). Both A and B lineage isolates were found in women from Costa Rica while the B2 sublineage was only detected in Africa.
Table 3.
Prevalence of HPV variants of each type in samples from Costa Rica, Taiwan, China, Thailand, Rwanda and Burkina Faso.
| Costa Rica | Taiwan, China | Thailand | Rwanda | Burkina Faso | Total | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type/Lineage | Count | % within Cohort | % within Lineage* | Count | % within Cohort | % within Lineage | Count | % within Cohort | % within Lineage | Count | % within Cohort | % within Lineage | Count | % within Cohort | % within Lineage | Count | % within Cohorts |
| Alpha-5, HPV26 | |||||||||||||||||
| A | 19 | 100.0% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 19 | 100.0% |
| Total | 19 | 100.0% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 19 | 100.0% |
| Alpha-5, HPV51 | |||||||||||||||||
| A1 | 80 | 58.0% | 41.5% | 1 | 12.5% | 8.9% | 18 | 36.0% | 25.8% | 9 | 33.3% | 23.8% | - | - | - | 108 | 46.4% |
| A2 | 12 | 8.7% | 8.7% | 3 | 37.5% | 37.5% | 20 | 40.0% | 40.0% | 1 | 3.7% | 3.7% | 1 | 10.0% | 10.0% | 37 | 15.9% |
| A3 | - | - | - | - | - | - | 2 | 4.0% | 100.0% | - | - | - | - | - | - | 2 | 0.9% |
| A4 | 3 | 2.2% | 2.2% | 4 | 50.0% | 51.6% | 10 | 20.0% | 20.6% | 4 | 14.8% | 15.3% | 1 | 10.0% | 10.3% | 22 | 9.4% |
| B1 | 43 | 31.2% | 26.1% | - | - | - | - | - | - | 13 | 48.1% | 40.4% | 4 | 40.0% | 33.5% | 60 | 25.8% |
| B2 | - | - | - | - | - | - | - | - | - | - | - | - | 4 | 40.0% | 100.0% | 4 | 1.7% |
| Total | 138 | 100.0% | 20.0% | 8 | 100.0% | 20.0% | 50 | 100.0% | 20.0% | 27 | 100.0% | 20.0% | 10 | 100.0% | 20.0% | 233 | 100.0% |
| Alpha-5, HPV69 A1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A2 | 2 | 9.5% | 9.5% | - | - | - | - | - | - | - | - | - | - | - | - | 2 | 9.5% |
| A3 | 7 | 33.3% | 33.3% | - | - | - | - | - | - | - | - | - | - | - | - | 7 | 33.3% |
| A4 | 12 | 57.1% | 57.1% | - | - | - | - | - | - | - | - | - | - | - | - | 12 | 57.1% |
| Total | 21 | 100.0% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 21 | 100.0% |
| Alpha-5, HPV82 A1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A2 | 5 | 10.9% | 30.3% | - | - | - | - | - | - | 3 | 25.0% | 69.7% | - | - | - | 8 | 13.8% |
| A3 | 1 | 2.2% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 1.7% |
| B1 | - | - | - | - | - | - | - | - | - | 3 | 25.0% | 100.0% | - | - | - | 3 | 5.2% |
| B2 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| C1 | 38 | 82.6% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 38 | 65.5% |
| C2 | 1 | 2.2% | 11.5% | - | - | - | - | - | - | 2 | 16.7% | 88.5% | - | - | - | 3 | 5.2% |
| C3 | - | - | - | - | - | - | - | - | - | 1 | 8.3% | 100.0% | - | - | - | 1 | 1.7% |
| C4 | - | - | - | - | - | - | - | - | - | 3 | 25.0% | 100.0% | - | - | - | 3 | 5.2% |
| C5 | 1 | 2.2% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 1.7% |
| Total | 46 | 100.0% | 50.0% | - | - | - | - | - | - | 12 | 100.0% | 50.0% | - | - | - | 58 | 100.0% |
| Alpha-6, HPV30 | |||||||||||||||||
| A1 | 1 | 10.0% | 56.5% | - | - | - | - | - | - | 1 | 7.7% | 43.5% | - | - | - | 2 | 8.7% |
| A4 | - | - | - | - | - | - | - | - | - | 5 | 38.5% | 100.0% | - | - | - | 5 | 21.7% |
| A5 | 1 | 10.0% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 4.3% |
| A2 | 1 | 10.0% | 39.4% | - | - | - | - | - | - | 2 | 15.4% | 60.6% | - | - | - | 3 | 13.0% |
| A3 | 3 | 30.0% | 56.5% | - | - | - | - | - | - | 3 | 23.1% | 43.5% | - | - | - | 6 | 26.1% |
| B | 4 | 40.0% | 72.2% | - | - | - | - | - | - | 2 | 15.4% | 27.8% | - | - | - | 6 | 26.1% |
| Total | 10 | 100.0% | 50.0% | - | - | - | - | - | - | 13 | 100.0% | 50.0% | - | - | - | 23 | 100.0% |
| Alpha-6, HPV53 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A | 65 | 20.2% | 22.0% | 2 | 40.0% | 43.7% | - | - | - | 11 | 31.4% | 34.3% | - | - | - | 78 | 21.5% |
| B | - | - | - | - | - | - | - | - | - | 8 | 22.9% | 100.0% | - | - | - | 8 | 2.2% |
| C | 32 | 9.9% | 17.0% | 2 | 40.0% | 68.4% | - | - | - | 3 | 8.6% | 14.6% | - | - | - | 37 | 10.2% |
| D1 | 221 | 68.6% | 55.8% | 1 | 20.0% | 16.3% | - | - | - | 12 | 34.3% | 27.9% | - | - | - | 234 | 64.6% |
| D2 | 2 | 0.6% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 2 | 0.6% |
| D3 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| D4 | 2 | 0.6% | 17.9% | - | 10 | - | - | - | - | 1 | 2.9% | 82.1% | - | - | - | 3 | 0.8% |
| Total | 322 | 100.0% | 33.3% | 5 | 100.0% | 33.3% | - | - | - | 35 | 0.0% | 33.3% | - | - | - | 362 | 100.0% |
| Alpha-6, HPV56 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A1 | 81 | 38.4% | 75.4% | - | - | - | 2 | 12.5% | 24.6% | - | - | - | - | - | - | 83 | 31.9% |
| A2 | 77 | 36.5% | 12.5% | 2 | 66.7% | 22.9% | 9 | 56.3% | 19.3% | 18 | 81.8% | 28.1% | 4 | 50.0% | 17.2% | 110 | 42.3% |
| B | 53 | 25.1% | 15.9% | 1 | 33.3% | 21.1% | 5 | 31.3% | 19.8% | 4 | 18.2% | 11.5% | 4 | 50.0% | 31.7% | 67 | 25.8% |
| Total | 211 | 100.0% | 20.0% | 3 | 100.0% | 20.0% | 16 | 100.0% | 20.0% | 22 | 100.0% | 20.0% | 8 | 100.0% | 20.0% | 260 | 100.0% |
| Alpha-6, HPV66 | |||||||||||||||||
| A | 68 | 52.3% | 45.1% | - | - | - | - | - | - | 7 | 63.6% | 54.9% | - | - | - | 75 | 51.4% |
| B1 | 11 | 8.5% | 3.5% | 1 | 100.0% | 40.8% | - | - | - | 4 | 36.4% | 14.9% | 4 | 100.0% | 40.8% | 20 | 13.7% |
| B2 | 51 | 2% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 51 | 9% |
| Total | 130 | 100.0% | 25.0% | 1 | 100.0% | 25.0% | - | - | - | 11 | 100.0% | 25.0% | 4 | 100.0% | 25.0% | 146 | 100.0% |
| Alpha-11, HPV34 | |||||||||||||||||
| A1 | 11 | 45.8% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 11 | 44.0% |
| A2 | 2 | 8.3% | 7.7% | - | - | - | - | - | - | - | - | - | 1 | 100.0% | 92.3% | 3 | 12.0% |
| B | 1 | 4.2% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 4.0% |
| C1 | 4 | 16.7% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 4 | 16.0% |
| C2 | 6 | 25.0% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 6 | 24.0% |
| Total | 24 | 100.0% | 50.0% | - | - | - | - | - | - | - | - | - | 1 | 100.0% | 50.0% | 25 | 100.0% |
| Alpha-11, HPV73 | |||||||||||||||||
| A1 | 2 | 3.70% | 100.00% | - | - | - | - | - | - | - | - | - | - | - | - | 2 | 3.5% |
| A2 | 24 | 44.44% | 100.00% | - | - | - | - | - | - | - | - | - | - | - | - | 24 | 42.1% |
| B | 28 | 51.85% | 34.15% | - | - | - | - | - | - | 3 | 100.0% | 65.9% | - | - | - | 31 | 54.4% |
| Total | 54 | 100.00% | 50.00% | - | - | - | - | - | - | 3 | 100.0% | 50.0% | - | - | - | 57 | 100.0% |
| Alpha-13, HPV54 | |||||||||||||||||
| A1 | 60 | 50.4% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 60 | 49.6% |
| A2 | 43 | 36.1% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 43 | 35.5% |
| B | 14 | 11.8% | 100.0% | - | - | - | - | - | - | - | - | - | - | - | - | 14 | 11.6% |
| C1 | 1 | 0.8% | 1.7% | - | - | - | - | - | - | - | - | - | 1 | 50.0% | 98.3% | 2 | 1.7% |
| C2 | 1 | 0.8% | 1.7% | - | - | - | - | - | - | - | - | - | 1 | 50.0% | 98.3% | 2 | 1.7% |
| - | 11 | 100 | 50. | - | - | - | - | - | - | - | - | - | - | 100.0 | 50. | 12 | 100 |
| Total | 9 | .0% | 0% | - | - | - | - | - | - | - | - | - | 2 | % | 0% | 1 | .0% |
| Alpha-3, HPV61 | |||||||||||||||||
| A1 | 85 | 91.40% | 100.00% | - | - | - | - | - | - | - | - | - | - | - | - | 85 | 79.4% |
| A2 | 1 | 1.08% | 2.45% | - | - | - | - | - | - | 6 | 42.9% | 97.6% | - | - | - | 7 | 6.5% |
| B | 1 | 1.08% | 2.45% | - | - | - | - | - | - | 6 | 42.9% | 97.6% | - | - | - | 7 | 6.5% |
| - | - | 6.4 | 31.11 | - | - | - | - | - | - | - | 14. | 68. | - | - | - | - | 7.5 |
| C | 6 | 5% 100 | % 50. | - | - | - | - | - | - | 2 | 3% 10 | 9% | - | - | - | 8 | % |
| Total | 93 | .00% | 00% | - | - | - | - | - | - | 14 | 0.0% | 50.0% | - | - | - | 107 | 100.0% |
The percentage within lineage of HPV variant is taken as the proportion within cohort divided by the total proportion within cohort of variants.
HPV69 variants.
Six variants from 21 HPV69-containing Costa Rican women were selected for complete genome sequencing. We identified a total of 119 nucleotide sites amongst the 7705 bp HPV69 genome that were variable (1.5%). Of the 2431 encoded amino acids in the 8 ORFs, 56 (2.3%) showed variations (Tables 2 and S4c). The complete genome pairwise differences and tree topology support the assignment of HPV69 isolates into 4 sublineages (A1-A4) (Figure 2c). The inter-sublineage mean differences ranged between 0.61±0.08% and 0.90±0.09% (Table S2); the maximum pairwise difference was 0.9% as observed between isolates of Qv32771 and Qv35103. Hence, all sampled HPV69 variants were clustered into one lineage.
HPV82 variants.
The isolate AE2 (NCBI accession AF293961) was initially assigned as a “subtype” of the prototype HPV82. Among 58 screened HPV82 isolates, 46 (79.3%) shared a pattern with the AE2 genome. We selected 9 samples from the Costa Rica study and 8 from the Rwanda study representing unique SNP patterns for complete genome sequencing. Additionally three genomes were downloaded from GenBank (Tables 1 and S1). The maximum pairwise difference of any two HPV82 isolates was 7.3% across the complete genome and 7.8% within the L1 ORF. Overall, 9.1% (719/7931) of nucleotide sites and 8.4% (201/2384) of amino acids were variable (Tables 2 and S4d). The tree topology clustered the HPV82 variants into two deeply separated clades, termed A/B and C, with mean differences of 6.89%−7.33% across the complete genomes (Figure 2d). The lineages A and B were relatively close, sharing mean differences of 0.83%−1.13% to each other. The “subtype” AE2 was designated the C lineage. In addition, each lineage was further divided into multiple sublineages (A1-A3, B1-B2 and C1-C5), with inter-sublineage mean differences ranging between 0.35±0.06% and 0.85±0.11%. Most isolates from Costa Rica (85%) sorted to the C lineage, whereas equal portions of Rwanda variants mapped to the A/B and C clades, respectively (Table 3).
Genomic diversity of Alpha-6 variants
HPV30 variants.
Twenty-three HPV30 isolates had the URR/E6 region sequenced. These sequences clustered into two main clades, from which we selected 14 samples for complete genome analysis (Tables 1 and S1). A total of 254/7890 (3.2%) nucleotide positions showed variations (Tables 2 and S4f). Similarly, there were 81/2471 (3.3%) variable amino acids across the 8 ORFs. Two distinct groups of HPV30 complete genomes were clustered, with inter-lineage mean differences of 1.66%−1.83% (Figure 2e). The A lineage was more diversified than the B lineage (intra-lineage mean distance of 0.59±0.07% and 0.18±0.04%, respectively, p<0.001) and further divided into 5 sublineages (A1-A5). The inter-sublineage mean differences ranged between 0.40% and 0.84%. Insertions were detected within the E2/E4 ORF (6-bp) and the URR region (21-bp and 11-bp) of A3 and B variants (Table S4f).
HPV53 variants.
PCR amplification and sequencing of the URR and/or E6 partial regions of 362 HPV53-containing samples from Costa Rica, China and Rwanda clustered isolates into four clades, from which we characterized 22 complete genomes, in addition to 6 sequences available in GenBank (Table S1). In total, 295 (3.7%) nucleotide variable positions and 116 (4.7%) encoded amino acid changes were detected across the complete genomes and the 8 ORFs, respectively (Tables 2 and S4g). Indels were observed within the noncoding regions only (NCR1 and URR). Phylogenetic trees inferred from HPV53 complete genomes assigned isolates into four groups of variant lineages, A, B, C and D (Figure 2f). The maximum pairwise nucleotide difference was 1.8% between two variants from B and D. The C and D variants were more related, with mean differences of 0.81%−0.91% when compared with the differences to other lineages (1.51%−1.73%). The D variants were further divided into 4 sublineages (D1-D4), sharing mean differences of 0.29%−0.45% to each other. Isolates differed between geographic regions: B variants were only observed in Rwanda; whereas, Costa Rica had more D variants (70%) than did Taiwan (20%) or Rwanda (37%) (Table 3).
HPV56 variants.
The SNP patterns within the partial regions of the URR and/or E6 from 260 sampled HPV56 isolates revealed limited variation. We selected 6 variants for complete genome sequencing, and incorporated 6 previously published genomes for analysis (Tables 1 and S1). The overall nucleotide and amino acid variable positions of the complete genomes were 1.7% (134 sites among 7922 nt) and 2.0% (51 sites among 2520 aa), respectively (Tables 2 and S4h). The maximum likelihood trees inferred from the 12 HPV56 complete genomes clustered variants into two lineages (A and B); the A lineage was further divided into two sublineages (A1-A2) (Figure 2g). The inter-sublineage and inter-lineage mean differences were 0.55±0.05% and 0.87±0.10%, respectively, with a 1.0% maximum pairwise nucleotide difference between isolates from A2 and B. The B variants were 66-bp longer than A due to an insertion within the URR region. HPV56 isolates from Costa Rica mapped to A1 (38%, 81/211), A2 (37%, 77/211) or B (25%, 53/211) lineages; whereas, more than half of samples from Asia and Africa sorted to A2. No A1 isolates were detected in samples from either Rwanda or Burkina Faso (Table 3).
HPV66 variants.
The URR/E6 sequences of 146 HPV66 isolates were clustered into two distant clades; 10 isolates capturing the maximum viral genomic heterogeneity were sequenced. In total, 218 variable nucleotide positions were identified within the 7827 bp HPV66 genome (2.8%). There were 82/2519 (3.3%) variable amino acids within the 8 ORFs (Tables 2 and S4i). Phylogenetic topologies and pairwise differences classified HPV66 complete genome variants into two lineages (A and B) and two sublineages (B1-B2) (Figure 2h). The inter-lineage and inter-sublineage mean differences were 1.55±0.13% and 0.53±0.06%, respectively. The majority of HPV66 isolates (130/146) were from Costa Rica samples, and equally sorted to A (52%) and B (48%) lineages. We did not detect B2 isolates in African populations (Rwanda or Burkina Faso) (Table 3).
Genomic diversity of Alpha-11 variants
HPV34 variants.
Screening the partial URR and/or E6 regions of 25 HPV34-positive samples from patients in Costa Rica and Burkina Faso identified 60% of isolates (15/25) related to the HPV34 prototype, and 40% (10/25) related to previously named HPV64 (now recognized as a “subtype” of HPV34). A total of 15 complete genomes (7 from each clade) including the reference sequence were analyzed in this report (Tables 1, and S1). The maximum pairwise difference of any two HPV34 variants was 4.7% across the complete genome and 5.4% within the L1 ORF (Tables 2, S4j and S4k). We identified 6.0% (466/7828) of nucleotide sites and 5.3% (126/2364) of amino acids variable. The tree topology assigned HPV34 prototype-related variants into A and B lineages (inter-lineage mean differences of 1.18%−1.24%), and HPV64-related isolates into the C lineage (Figure 2i). The A and C lineages were composed of 2 sublineages (A1-A2, C1-C2), with inter-sublineage mean differences of 0.55±0.08% and 0.69±0.09%, respectively. Only one HPV34 A2 isolate was observed in samples from Burkina Faso, whereas the remainder were from Costa Rica and mapped to A1 (11/24), A2 (2/24), B (1/24), C1 (4/24) and C2 (6/24) (Table 3).
HPV73 variants.
Of the 57 HPV73 isolates sequenced for the partial regions of URR and/or E6, 11 were selected for complete genome characterization (Tables 1 and S1). The overall nucleotide and amino acid sequence variations were 2.1% (159/7733) and 2.5% (59/2375), respectively (Tables 2 and S4l). The maximum pairwise nucleotide difference was 1.4%, supporting the assignment of HPV73 variants into two distinct lineages (A and B), which is in consistent with the deep separation between A and B lineages in the maximum likelihood tree (Figure 2j). The A lineage was divided into two sublineages (A1-A2), with inter-sublineage mean differences of 0.69±0.09%. All B variants formed one single lineage, sharing genomic differences less than 0.20±0.03%. Interestingly, when compared with the A lineage, the B variants had an amino acid deletion within the E2 ORF (Cysteine, aa 251) and the E4 ORF (Valine, aa 57), while most of them had a 19-bp or 23-bp insertion within the URR region (Table S4l). Inspection of the geographic distribution of 57 HPV73 variants indicated that all 3 isolates from Rwanda were of the B lineage, whereas 48% (26/54) and 52% of Costa Rica samples were A and B variants, respectively (Table 3).
Genomic diversity of HPV54 variants
The majority of HPV54 variants screened for the partial regions of URR and/or E6 were related to the prototype (85%, 103/121). Two isolates from Burkina Faso and sixteen isolates from Costa Rica (15%, 18/121) were more similar to the previously characterized AE9 subtype genome (NCBI accession AF436129). Including two reference sequences and one variant (87C.54) from a Brazilian study, we sequenced 8 complete genomes from the Costa Rican cohort to characterize the genomic diversity (Tables 1 and S1). In total, 520 amongst 7799 nucleotide sites were variable (6.7%) across the HPV54 complete genome. Of the 2372 encoded amino acids, 186 (7.8%) were variable (Tables 2, S4m and S4n). The maximum pairwise nucleotide diversity of the complete genomes was 5.6%, as observed between isolates from the prototype and AE9 clades. The maximum L1 diversity was 5.0%. The complete genome phylogeny separated HPV54 variants into two distantly divided groups. The prototype group was assigned to lineage A, which was further divided into two sublineages (A1-A2). The AE9 related isolates formed two lineages (B and C), and the C lineage was comprised of two sublineages (C1-C2) (Figure 2k). The mean differences between “prototypes” and “subtypes” (HPV54 A vs B/C) was 5.1%−5.6%, similar to the maximum diversity of HPV34 (4.5%−4.7%, A/B vs C) and HPV82 isolates (6.9%−7.3%, A/B vs C) (Figure S1). The variants from Costa Rica mapped to lineages A (87%), B (12%) and C (2%), whereas isolates from Burkina Faso sorted to C only, implying the C lineage was more common in Africa. However, variant screening in other countries/regions to validate the geographic dispersion of HPV54 and other types is warranted (Table 3).
Genomic diversity of HPV61 variants
We sequenced the partial regions of URR and/or E6 of 107 HPV61 variants from Costa Rica (n=93) and Rwanda (n=14). The nucleotide alignments identified three potential clusters; from each cluster, isolates representing unique variation patterns (n=8) were selected for complete genome sequencing (Tables 1 and S1). A total of 259/8037 (3.2%) nucleotide positions were changed and there were 81/2345 (3.5%) variable amino acid residues (Tables 2 and S4o). The maximum nucleotide pairwise difference between the most dissimilar isolates was 2.3%; the noncoding regions (NCR1, NCR2 and URR) were the most variable regions. Phylogenetic trees generated from the complete genomes clustered HPV61 variants into three distinct lineages designated A, B and C (Figure 2l). The A and B lineages were relatively closer (mean differences of 1.23%−1.47%), and more distant to the C variants (1.92%−2.19%). Two A sublineages (A1-A2) were assigned, with differences of 0.59±0.08%. Isolates from A lineage were highly abundant in Costa Rica (93%, 86/93) while B variants were more common in Rwanda (43%, 6/14). The C variants were relatively rare in the targeted populations (6.5% in Costa Rica, 14.3% in Rwanda) (Table 3).
HPV variant genomic diversity and evolution
The pairwise mean differences calculated from the complete genomes revealed different strata between distinct types: HPV34, 54 and 82 containing previously termed “subtypes” were most diversified, followed by HPV61, 30, 53, 66, 73, 51 and 56 that were designated with at least two main lineages. HPV69 was composed of one single lineage while HPV26 was the least heterogeneous (Tables 2 and S2, Figures 2 and S1). To investigate the relationship between isolates of different types and species, we took representative variant genomes from each lineage and sublineage from species Alpha-3, 5, 6, 7, 9, 10, 11 and 13 as reported in this study to construct a phylogeny and plotted the percent differences based on the global alignment (Figure 3a) (Chen et al., 2011, 2013; Jelen et al., 2014; Jelen et al., 2016; Smith et al., 2011). Using a Bayesian Markov Chain Monte Carlo (MCMC) method, we estimated the divergence times of Alphapapillomavirus types and species (Figure 3b). In addition, the correlation between sequence differences and divergence times was estimated when different HPV types/species (between type) (Figure 3c) or variants of the same type (within type) (Figure 3d) were compared. Multiple strata of percent differences were apparent, for example, isolates from one species are approximately 35–45% different from viruses within other species (inter-species difference). This difference corresponds to divergence times spanning 22 – 38 million years ago (mya). Most distinct HPV types emerged from their most recent common ancestors (MRCAs) around 6 – 15 mya and share more than 70% sequence identity with members from the same species group (intra-species difference). In contrast, variants diverged on estimated 2.5 mya and many split from their MRCAs approximately 400 – 600 thousand years ago (kya). This later divergence time is approximately three times longer than modern Homo sapiens’ divergence times but well within the era of separation between ancestral modern humans and archaic Neanderthal/Denisovans (Pimenoff et al., 2017; Prufer et al., 2014; Stringer and Barnes, 2015). Nevertheless, the time estimates using unknown variables (e.g., generation times) could be subject to error. However, there is a strong correlation between divergence times for both types and variants with a slight falloff for the most divergent types (Figs 3C and 3D).
Figure 3.
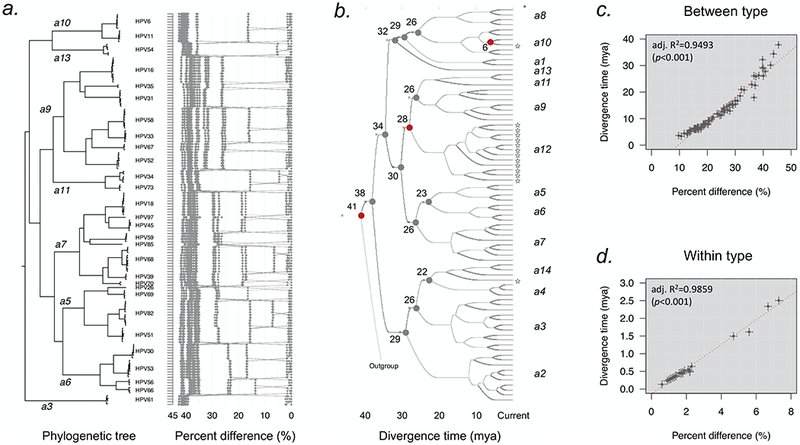
Divergence time estimation of Alphapapillomavirus HPV types and variants. (a) Phylogenetic tree and pairwise comparisons of representative Alphapapillomavirus lineages and sublineages show multiple strata of genomic differences. A maximum likelihood (ML) tree was constructed using RAxML inferred from the global alignment of complete genome nucleotide sequences. The percent nucleotide sequence differences are shown in the panel to the right of the phylogenetic tree as described in Fig. 2. The Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61) variants from this report, and Alpha-7 (HPV18, 39, 45, 59, 68, 70, 85, 97), Alpha-9 (HPV16, 31, 33, 35, 52, 58, 67) and Alpha-10 (HPV6, 11) variants from previous publications were included. (b) Divergence time estimation of HPV types using a Bayesian MCMC method. The nodes in the tree show split times of distinct species groups, with gray bars indicating the 95% highest posterior density for the corresponding divergence age. The nodes in red represent three main co-divergence events between viruses and host speciation (human and chimpanzee, human-chimpanzee and macaque, and New World and New World monkeys). The stars indicate non-human primate PVs within Alphapapillomavirus. (c) Correlation between genomic diversity (X-axis) and divergence time (Y-axis) of HPV types and species (between type). The divergence time was cited from each node in Figure 3b, and the percent difference was the maximum pairwise diversity of PV types within each node calculated based on the global alignment. (d) Correlation between genomic diversity (X-axis) and divergence time (Y-axis) of HPV variants (within type). The divergence time was the initial split time of variants of each type estimated using a Bayesian MCMC method, and the percent difference inferred from the maximum pairwise diversity of variant of each type.
Diversity of ORFs and viral regions
We evaluated all Alphapapillomavirus HPV variants (as listed in Figure 3a) to examine the genomic diversity across the predicted ORFs. The E5 and E4 ORFs showed the largest variability when compared to the other genes (Figure S2). The capsid proteins (L1 and L2) and the multipurpose DNA helicase E1 were the most conserved ORFs in terms of amino acid diversity. Nevertheless, lineage fixation of genetic changes is observed throughout all genes/regions. Lineage fixation is the property of genomes that do not undergo recombination whereby variable sites are highly correlated with and inseparable from other changes within genomes from the same sublineage and lineage, and may represent adapted changes in natural selection when variants split from their MRCAs (Chen et al., 2005). For each surveyed HPV type, we identify short regions that contain lineage- and sublineage-specific single nucleotide polymorphisms (SNPs) characteristic of lineage fixation (Figure S3). For example, a 136-bp fragment within the E1 gene of HPV51 (nt. 1329 – 1464) is able to discriminate different variant sublineages based on unique SNP patterns that are correlated with changes within a partial URR region (nt. 7466 – 7547). Although variation patterns differ by genes/regions and types, they may facilitate characterization of HPV variants for simple assays. For computational access, we packaged the complete genome alignment in a fasta format and called the SNPs and indels of variants across the complete genomes of each type in a variant call format (VCF) as supplemental data in this work.
Discussion
In this report, we describe the complete genomes of HPV variants from the species Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61). This work expands the study of viral heterogeneity and provides a taxonomy of variants based on comparison of the complete genome pairwise similarity and the phylogenetic topologies within a specified HPV type. These genomes were sampled from over 12,000 women residing in Central America (Costa Rica), Africa (Rwanda, Burkina Faso) and Asia (Thailand, Taiwan) whose specimens had been tested and typed for HPV infections. We screened this set of HPV types by sequencing a small region of the genome to identify isolates for further classification that represent circulating variants in the population. Based on the complete genome sequences, we created phylogenies and genome comparisons to define and name variant lineages and sublineages as previously described (Burk et al., 2013; Chen et al., 2011, 2013). In a monograph from International Agency for Research on Cancer (IARC), 12 HPV types (HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59) were classified as “carcinogenic to humans” (Group 1), and another 8 types (HPV68, 26, 53, 66, 67, 70, 73, 82) as “probably carcinogenic to humans” (Group 2) (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2012). Thus, this report combines with our previous work (Chen et al., 2011, 2013) and provides a complete picture of the variation of essentially all HPV genomes associated with cervical cancer.
Differential divergence times, genomic diversity and geographic dispersals of HPV variants indicate independent evolutionary histories for each HPV type. We detected a wide range of variation between HPV types: the maximum pairwise nucleotide sequence difference of HPV82 variants was 7.3% whereas HPV69 variants differed by less than 0.9%. The divergence time estimation in this work and other studies suggest ancient hominin-virus codivergence of HPV variants when ancestral modern humans split from the most recent common ancestors of archaic Neanderthal/Denisovans or other hominins (Chen et al., 2017; Pimenoff et al., 2017). Recent host switch, for example, interbreeding between modern humans and archaic Neanderthal/Denisovans, may be responsible largely for the viral transmission of HPV variants and the differential distribution in prevalence and geography. Some HPV variants, such as HPV26 and HPV35, showed less genomic diverse possibly due to the bottleneck of viral transmission or extinction of host migration shaping the radiation we observe in the phylogenetic tree of extant HPV variants. Alternatively, there could be isolated populations with other variants not seen in our samples. HPV genomic diversity has no direct correlation with viral carcinogenesis, for example, HPV61 variants with no evidence of oncogenicity have nearly twice the sequence diversity than does the carcinogenic HPV51 (maximum pairwise difference of 2.3% vs 1.3%, respectively), it is possible that HPV variants and/or types with higher rates of persistence that maintain production of infectious viral particles may accumulate random mutations perhaps under natural selection over millions of years of evolution (Chen et al., 2009a; Chen et al., 2005). In contrast, HPV variants could also be targeted and induced partly by host immune pressure, such as the innate antiviral activity of APOBEC3 cytosine deaminases (Mirabello et al., 2017; Vartanian et al., 2008; Warren et al., 2015). These acquired mutations, if not lethal for the virus, may be accumulated through selection and convergent evolution that affect the success of fitness and niche adaptation potentially contributing to HPV-associated cancer. Moreover, it is remarkable that the HPV types studied in this report seem to diverge over million years ago. The exact events resulting in the pattern seen in the phylogeny and the time frame need to be considered in light of the divergence of non-human primate PVs (e.g., types within the species Alpha-12), as these are the time periods encompassing host speciation (Shah et al., 2010).
Viral genomic variation was not evenly distributed through the whole genome. The capsid proteins, particularly L1 ORF, were the least heterogeneous, probably reflecting the constraints of maintaining an icosahedral structure similar for all papillomaviruses. Such structurally confined phenotypes would be expected to be under negative Darwinian selection, consistent with the observations of the papillomavirus predicted ORF encoding regions. In line with this reasoning, the E1 and L2 ORFs must also serve a highly conserved function whereby its structure is necessary for the viral life cycle (Burk et al., 2009; Doorbar et al., 2012). The more adaptive encoding regions of the genome would include the E2, E4, E5, E6 and E7 ORFs, based on the predicted amino acid sequence diversity of Alphapapillomavirus HPV variants.
The strengths of this study are based on the careful selection and complete genome sequencing of isolates from various populations. We used overlapping PCR and Sanger sequencing in all cases and confirmed any ambiguous sequences, although variations from potential PCR or sequencing errors remain unavoidable. Nevertheless, this study provides sufficient information to gain a picture of the viral variants across the HPV spectrum. The availability of samples was based on studies performed in the Burk lab over the years and the sharing of samples from collaborators. Although we believe that essentially all variant lineages from the surveyed populations have been described, it is possible that future studies will reveal unexpected variants lineages. Certainly, not all viral variant isolates for a given type have been described. The sample size for many countries in this study was small and the number of isolates selected for sequencing was not exhaustive and the geographic patterns should not be interpreted as definitive. Lastly, this was not meant to be an exhaustive study of viral evolution since a comprehensive genome dataset based on random and large sampling is warranted in future studies to better interpret the complex evolution of HPV variants and types. Nevertheless, the findings in this work provide sufficient information to gain a picture of the viral variants described in the context of a larger story.
Implementation of new technologies such as Next-Generation Sequencing (NGS) facilitates the sequencing of large numbers of HPV complete genomes (Cullen et al., 2015; Mirabello et al., 2016; Siqueira et al., 2016). A high-throughput, ultra-deep coverage method permits more detailed examination of genotype-phenotype relationships between viral isolates and clinical outcomes, including carcinogenesis (Mirabello et al., 2017). Given the large differences in pathogenic consequence associated with HPV variants, a coherent and well-defined classification of genotypic variants will make it possible to incorporate multicenter studies and/or meta-analyses and determine specific SNPs, sublineages or lineages responsible for unique carcinogenicity, geographic dispersion, virus-host interaction and human evolution.
Conclusion
Persistent infection with oncogenic human papillomaviruses (HPVs) has been associated with cervical cancer and precancer. Among the 65 genital HPV types, we still do not know clearly why there are only a limited set of HPV viruses that progress to cancer. HPV variants have different phenotypic characteristics including carcinogenicity. In this study, we focus on the heterogeneity, classification, evolution and dispersion of variants for 12 HPV types. We provide a comprehensive classification that will facilitate our understanding of the clinical and biological roles the sequence variations play. It will also allow HPV researchers to discuss and compare the properties of HPV variants across studies without having to describe sets of nucleotide changes to define a group of HPV variants. The findings in this work provide the basis to study pathogenesis, viral evolution, epidemiology, pathogenesis and preventative/therapeutic interventions of HPV infection and the associated diseases.
Materials and Methods
Ethics Statement.
The studies providing cervical samples for this work have been IRB approved by each ethics committee. Only subjects older than 18 years were included in this work. Written informed consent was given to each participator. All samples tested in this study were anonymized without individual identifying information. In details, Rwanda: The Rwanda National Ethics Committee and the Institutional Review Board of Montefiore Medical Center, Bronx NY approved the study protocol and the consent process. Burkina Faso: The Yerelon Cohort Research Programme was approved by the Ethical Committee of the Centre Muraz, Bobo Dioulasso, the National Ethical Committee of the Ministry of Health, Burkina Faso, and the Ethics Committee of the London School of Hygiene and Tropical Medicine. Thailand: The study protocols were reviewed and approved by the committees on human subject research at Johns Hopkins Bloomberg School of Public Health, Baltimore, MD; Merck & Co., Inc., West Point, PA, each participating recruitment site, and the Institutional Review Board of the Thailand Ministry of Health (MOH), Thailand. Taiwan, China: The study was approved by the Institution Review Board of the National Taiwan University University College of Public Health. Costa Rica: The study and informed consent forms were approved by Institutional Review Boards of Costa Rica and the U.S. National Cancer Institute. Other sources: Previously published HPV complete genome sequences by other research groups (NCBI accession numbers listed in Table S1).
Clinical specimens, identification of novel HPV variants and whole genome sequencing.
All samples containing HPV isolates had been tested previously as reported in the following studies- Costa Rica (Herrero et al., 2005), Taiwan, China (Liaw et al., 1995), Thailand (Marks et al., 2011; Wongworapat et al., 2008), Rwanda (Singh et al., 2009) and Burkina Faso (Didelot-Rousseau et al., 2006). The methods for sample collection, DNA extraction and HPV genotyping are provided in the references for each study. The number of samples analyzed for variant screening is shown in Table 1. The HPV variants were initially classified by sequencing a small fragment of the URR region (approximately 300–600 bp) from PCR products as described (Chen et al., 2011, 2013). Isolates were further subject to the E6 ORF if the URR region did not yield data. The sequences of isolates for each type were aligned separately and the preliminary phylogenetic trees were constructed using a maximum likelihood algorithm to identify samples that likely contained divergent viral genomes. We selected type-specific viral isolates for complete genome sequencing that (1) represented novel variant clades or (2) had 2 or more isolates that contained at least 2 unique sequence variations in common (e.g., single nucleotide polymorphisms (SNPs)) not present in other isolates within the URR/E6 regions.
The complete viral genomes were amplified by PCR using type-specific primer sets in 2 to 3 overlapping fragments, as described in previous publications (Chen et al., 2011, 2013). The Sanger sequence reads from the PCR products were assembled using the prototype sequences as a reference employing Geneious R9.1.7 (Kearse et al., 2012). Comparison of repeat sequencing of PCR products from the same isolates resulted in a difference of less than one change per 8,000 bp; whereas, comparison of the cloned genomes gave a difference of approximately one difference per 5,000 bp. For discrepancies between sequences, we used the sequence of the PCR product as the valid sequence. To supplement the number of genomes in the current study, a GenBank search at the time of initial analysis (April 2017) identified an addition of 39 complete genomes or near complete genomes (including the prototype references) previously published (Burk et al., 2013; Delius and Hofmann, 1994; Kino et al., 2000; Lungu et al., 1991; Narechania et al., 2005; Siqueira et al., 2016; Volter et al., 1996; Wu et al., 2010). The accession numbers of all sequences analyzed in this study are listed in Table S1.
Evolutionary analyses and phylogenetic tree construction.
The complete viral genome sequences were linearized at the first ATG of the E1 ORF and globally aligned using the program MAFFT v7.221 (Katoh and Toh, 2010). The codon sequences from each ORF were aligned based on the aligned amino acids using MUSCLE v3.8.31 (Edgar, 2004). Based on the concept of a single ancestor for each type, a unique genome size was assigned to each HPV type based on the global alignment; the variation in genome sizes of isolated variants is the result of insertions and deletions (indels). Each indel was counted as one event. The assignment of position number for each nucleotide change is based on the nucleotide numbering of the prototype reference sequence under investigation.
Maximum likelihood (ML) trees were constructed using RAxML MPI v8.2.9 (Stamatakis, 2006) and PhyML MPI v3.0 (Guindon and Gascuel, 2003) with optimized parameters based on the aligned complete genome nucleotide sequences. Data were bootstrap resampled 1,000 times in RAxML and PhyML. MrBayes v3.2.6 (Ronquist and Huelsenbeck, 2003) with 10,000,000 cycles for the Markov chain Monte Carlo (MCMC) algorithm was used to generate Bayesian trees. A 10% discarded burn-in was set to eliminate iterations at the beginning of the MCMC run. The average standard deviation of split frequencies was check to confirm the independent analyses approach stationarity when the convergence diagnostic approached <0.001 as runs converge. For Bayesian tree construction, the computer program ModelTest v3.7 (Posada and Crandall, 1998) was used to identify the best evolutionary model; the identified GTR model was set for among-site rate variation and allowed substitution rates of aligned sequences to be different. The CIPRES Science Gateway (Miller et al., 2010) was accessed to facilitate RAxML and MrBayes high-performance computation.
Nucleotide sequence and amino acid variations were determined based on the global complete genome alignment and the codon alignment of each ORF using scripts developed in R v3.3.2 (Team, 2014). Inter- and intra-lineage and sublineage nucleotide differences (± standard errors) of each type were calculated from the global sequence alignment using the p-distance algorithm in MEGA7 with 1,000 bootstraps (Tamura et al., 2011). Wilcoxon-Mann-Whitney U test was used to determine the significance of pairwise sequence identity between the defined lineage or sublineage groups.
Divergence time estimation.
We used a Bayesian Markov Chain Monte Carlo (MCMC) method implemented by BEAST2 v2.4.5 (Drummond and Rambaut, 2007) and the previously published PV evolutionary rates (Rector et al., 2007) to estimate the divergence times of Alphapapillomavirus. A phylogenetic tree was generated using a coalescent Bayesian skyline model, with the assumption of an uncorrelated lognormal distribution (UCLD) molecular clock of variation rate among branches. This model was tested to be the “best” choose by a posterior simulation-based analogue of Akaike’s Information Criterion for MCMC samples (AICM), as implemented in Tracer v.1.6 (data not shown) (Baele et al., 2012). Meantime, we chose the General Time Reversible (GTR) sequence revolution model with the gamma-distributed rate heterogeneity among sites and a proportion of invariant sites (GTR+G+I) determined by the best-fit model approach of Modeltest v3.7 (Posada and Crandall, 1998). The concatenated nucleotide sequence partitions of six ORFs (E6, E7, E1, E2, L2 and L1) of Alphapapillomavirus types with variable rates of substitution over time were used: 2.39 × 10−8 (95% confidence interval 1.70 – 3.26 × 10−8) substitutions per site per year for the E6 gene, 1.44 × 10−8 (0.97 – 2.00 × 10−8) for the E7 gene, 1.76 × 10−8 (95% CI: 1.20 – 2.31 × 10−8) for the E1 gene, 2.11 × 10−8 (95% CI: 1.52 – 2.81 × 10−8) for the E2 gene, 2.13 × 10−8 (95% CI: 1.46 – 2.76 × 10−8) for the L2 gene, and 1.84 × 10−8 (95% CI: 1.27 – 2.35 × 10−8) for the L1 gene (Rector et al., 2007). In order to calibrate the divergence times, we introduced three time points inside and at the root of the Alpha-PV tree, with assumption of coevolved histories between primate papillomaviruses (humans and non-humans) and their hosts: (1) the node between HPV13 and PpPV1 (Pan paniscus papillomavirus 1) at 7 million years ago (mya) (95% CI, 6–8 mya) matching the split between hominin and chimpanzee ancestors; (2) the node between the species Alpha-12 (represented by Macaca mulatta papillomavirus 1) and Alpha-9/11 (represented by HPV16) at 28 mya (25–31 mya) matching the speciation between hominin and macaque ancestors; and (3) the node between Alphapapillomavirus and Dyoomikronpapillomavirus (represented by Saimiri sciureus papillomavirus 1) at 49 mya (41–58 mya) matching the divergence between Old World and New World monkey ancestors (Perez et al., 2013). The Saimiri sciureus papillomaviruses (SscPV1–3, accession numbers of JF304765-JF304767) were used as outgroup taxa for phylogenetic tree analysis and divergence time estimation. In order to estimate the initial divergence of HPV variants of each type from their most recent common ancestor, we used the complete genome alignments and the HPV16 variant evolutionary rate of 1.84 × 10−8 (95% CI: 1.43 – 2.21 × 10−8) substitutions per site per year, following the Hominin-host-switch (HHS) topology without time point calibrations as previously published (Pimenoff et al., 2017). The MCMC analyses were run for 100,000,000 steps, with a subsampling every 10,000 generations and a discarded burn-in of the first 10% steps. Effective sample sizes (ESS) of all parameters are >300 (Alphapapillomavirus tree) and >2000 (HPV variant trees of each type), indicating that all Bayesian chains were well sampled and have converged. The tree files and the log files were analyzed in Tracer v.1.6. A consensus phylogeny tree with divergence times was inferred using TreeAnnotater v2.4.5 and visualized using FigTree v1.4.2. The linear model (lm) function in R was used to estimate the correlation between sequence diversity and divergence time of HPV types and variants.
Supplementary Material
Figure S1. Heatmap display of HPV variant pairwise sequence differences. Percent differences calculated based on the complete genome nucleotide sequence alignment of each type were measured and scaled such that complete identity (0.0% difference) is displayed as yellow and a maximum difference (2.3%) as blue. Phylogenetic trees indicating the position and name of each lineage and sublineage from the same species are shown besides of the heatmap.
Figure S2. Accumulated amino acid diversity of Alphapapillomavirus variants. The percentages were calculated based on the overall amino acid variable positions across each open reading frame (ORF) (E6, E7, E1, E2, E4, E5, L2, L1) or the combined 8 ORFs of variants from the same HPV type. Box plots are shown indicating the median, 25%–75% and 5%–95%. Asterisks represent the actual diversity for variants of each HPV type.
Figure S3. Proposed short regions containing lineage- and sublineage-specific single nucleotide polymorphisms (SNPs) of each HPV type. Only sites conserved in each lineage/sublineage are displayed, with dots indicating sites matched with reference prototype. Dashes indicate insertion/deletion events.
Research Highlights:
Define variant lineages and sublineages for 12 distinct Alphapapillomavirus HPV types.
The heterogeneity and phylogenies of HPV isolates indicate an independent evolutionary history for each type.
A comprehensive classification will facilitate our understanding of the clinical and biological roles the sequence variations play.
Acknowledgments
We thank members of the Burk lab for performing HPV DNA genotyping analyses. This work was supported in part by the National Cancer Institute (CA78527) (RDB), the Einstein-Montefiore Center for AIDS funded by the NIH (AI-51519) (RDB) and the Einstein Cancer Research Center (P30CA013330) from the National Cancer Institute (RDB). ZC was supported in part by the Research Grants Council of Hong Kong SAR (ECS 2191114) and the Health and Medical Research Fund of Food and Health Bureau of the Hong Kong SAR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV, 2012. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol 29, 2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM, 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk RD, Chen Z, Van Doorslaer K, 2009. Human papillomaviruses: genetic basis of carcinogenicity. Public Health Genomics 12, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk RD, Harari A, Chen Z, 2013. Human papillomavirus genome variants. Virology 445, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino R, Petignat P, Dongui G, Vassilakos P, 2015. Cervical cancer screening in developing countries at a crossroad: Emerging technologies and policy choices. World J Clin Oncol 6, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PK, Lam CW, Cheung TH, Li WW, Lo KW, Chan MY, Cheung JL, Cheng AF, 2002. Association of human papillomavirus type 58 variant with the risk of cervical cancer. J Natl Cancer Inst 94, 1249–1253. [DOI] [PubMed] [Google Scholar]
- Chan PK, Luk AC, Park JS, Smith-McCune KK, Palefsky JM, Konno R, Giovannelli L, Coutlee F, Hibbitts S, Chu TY, Settheetham-Ishida W, Picconi MA, Ferrera A, De Marco F, Woo YL, Raiol T, Pina-Sanchez P, Cheung JL, Bae JH, Chirenje MZ, Magure T, Moscicki AB, Fiander AN, Di Stefano R, Cheung TH, Yu MM, Tsui SK, Pim D, Banks L, 2011. Identification of human papillomavirus type 58 lineages and the distribution worldwide. J Infect Dis 203, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PK, Zhang C, Park JS, Smith-McCune KK, Palefsky JM, Giovannelli L, Coutlee F, Hibbitts S, Konno R, Settheetham-Ishida W, Chu TY, Ferrera A, Alejandra Picconi M, De Marco F, Woo YL, Raiol T, Pina-Sanchez P, Bae JH, Wong MC, Chirenje MZ, Magure T, Moscicki AB, Fiander AN, Capra G, Young Ki E, Tan Y, Chen Z, Burk RD, Chan MC, Cheung TH, Pim D, Banks L, 2013. Geographical distribution and oncogenic risk association of human papillomavirus type 58 E6 and E7 sequence variations. Int J Cancer 132, 2528–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, DeSalle R, Schiffman M, Herrero R, Burk RD, 2009a. Evolutionary dynamics of variant genomes of human papillomavirus types 18, 45, and 97. J Virol 83, 1443–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ho WCS, Boon SS, Law PTY, Chan MCW, DeSalle R, Burk RD, Chan PKS, 2017. Ancient Evolution and Dispersion of Human Papillomavirus 58 Variants. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Schiffman M, Herrero R, Desalle R, Anastos K, Segondy M, Sahasrabuddhe VV, Gravitt PE, Hsing AW, Burk RD, 2011. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 6, e20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Schiffman M, Herrero R, DeSalle R, Anastos K, Segondy M, Sahasrabuddhe VV, Gravitt PE, Hsing AW, Burk RD, 2013. Evolution and taxonomic classification of alphapapillomavirus 7 complete genomes: HPV18, HPV39, HPV45, HPV59, HPV68 and HPV70. PLoS ONE 8, e72565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Terai M, Fu L, Herrero R, DeSalle R, Burk RD, 2005. Diversifying selection in human papillomavirus type 16 lineages based on complete genome analyses. J Virol 79, 7014–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, van Doorslaer K, DeSalle R, Wood CE, Kaplan JR, Wagner JD, Burk RD, 2009b. Genomic diversity and interspecies host infection of alpha12 Macaca fascicularis papillomaviruses (MfPVs). Virology 393, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen M, Boland JF, Schiffman M, Zhang X, Wentzensen N, Yang Q, Chen Z, Yu K, Mitchell J, Roberson D, Bass S, Burdette L, Machado M, Ravichandran S, Luke B, Machiela MJ, Andersen M, Osentoski M, Laptewicz M, Wacholder S, Feldman A, Raine-Bennett T, Lorey T, Castle PE, Yeager M, Burk RD, Mirabello L, 2015. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res 1, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H, 2004. Classification of papillomaviruses. Virology 324, 17–27. [DOI] [PubMed] [Google Scholar]
- Delius H, Hofmann B, 1994. Primer-directed sequencing of human papillomavirus types. Curr Top Microbiol Immunol 186, 13–31. [DOI] [PubMed] [Google Scholar]
- Didelot-Rousseau MN, Nagot N, Costes-Martineau V, Valles X, Ouedraogo A, Konate I, Weiss HA, Van de Perre P, Mayaud P, Segondy M, 2006. Human papillomavirus genotype distribution and cervical squamous intraepithelial lesions among high-risk women with and without HIV-1 infection in Burkina Faso. Br J Cancer 95, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA, 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30 Suppl 5, F55–70. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A, 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S, 2012. Global burden of human papillomavirus and related diseases. Vaccine 30 Suppl 5, F12–23. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O, 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Herrero R, Castle PE, Schiffman M, Bratti MC, Hildesheim A, Morales J, Alfaro M, Sherman ME, Wacholder S, Chen S, Rodriguez AC, Burk RD, 2005. Epidemiologic profile of type-specific human papillomavirus infection and cervical neoplasia in Guanacaste, Costa Rica. J Infect Dis 191, 1796–1807. [DOI] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2012. Biological agents. Volume 100 B. A review of human carcinogens. IARC monographs on the evaluation of carcinogenic risks to humans / World Health Organization, International Agency for Research on Cancer 100, 1–441. [PMC free article] [PubMed] [Google Scholar]
- Jelen MM, Chen Z, Kocjan BJ, Burt FJ, Chan PK, Chouhy D, Combrinck CE, Coutlee F, Estrade C, Ferenczy A, Fiander A, Franco EL, Garland SM, Giri AA, Gonzalez JV, Groning A, Heidrich K, Hibbitts S, Hosnjak L, Luk TN, Marinic K, Matsukura T, Neumann A, Ostrbenk A, Picconi MA, Richardson H, Sagadin M, Sahli R, Seedat RY, Seme K, Severini A, Sinchi JL, Smahelova J, Tabrizi SN, Tachezy R, Tohme S, Uloza V, Vitkauskiene A, Wong YW, Zidovec Lepej S, Burk RD, Poljak M, 2014. Global genomic diversity of human papillomavirus 6 based on 724 isolates and 190 complete genome sequences. J Virol 88, 7307–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelen MM, Chen Z, Kocjan BJ, Hosnjak L, Burt FJ, Chan PK, Chouhy D, Combrinck CE, Estrade C, Fiander A, Garland SM, Giri AA, Gonzalez JV, Groning A, Hibbitts S, Luk TN, Marinic K, Matsukura T, Neumann A, Ostrbenk A, Picconi MA, Sagadin M, Sahli R, Seedat RY, Seme K, Severini A, Sinchi JL, Smahelova J, Tabrizi SN, Tachezy R, Tohme Faybush S, Uloza V, Uloziene I, Wong YW, Zidovec Lepej S, Burk RD, Poljak M, 2016. Global Genomic Diversity of Human Papillomavirus 11 Based on 433 Isolates and 78 Complete Genome Sequences. J Virol 90, 5503–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H, 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A, 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino N, Sata T, Sato Y, Sugase M, Matsukura T, 2000. Molecular cloning and nucleotide sequence analysis of a novel human papillomavirus (Type 82) associated with vaginal intraepithelial neoplasia. Clin Diagn Lab Immunol 7, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw K-L, Hsing A, Chen C-J, Schiffman M, Zhang T, Hsieh C-Y, Greer C, You S-L, Huang T, Wu T, O’Leary T, Seidman J, Blot W, Meinert C, Manos M, 1995. Human papillomavirus and cervical neoplasia: a case-control study in Taiwan. Int. J. Cancer 62, 565–571. [DOI] [PubMed] [Google Scholar]
- Lungu O, Crum CP, Silverstein S, 1991. Biologic properties and nucleotide sequence analysis of human papillomavirus type 51. J Virol 65, 4216–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks M, Gravitt PE, Gupta SB, Liaw KL, Kim E, Tadesse A, Phongnarisorn C, Wootipoom V, Yuenyao P, Vipupinyo C, Rugpao S, Sriplienchan S, Celentano DD, 2011. The association of hormonal contraceptive use and HPV prevalence. Int J Cancer. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T, 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees, Gateway Computing Environments Workshop (GCE), 2010. IEEE, pp. 1–8. [Google Scholar]
- Mirabello L, Yeager M, Cullen M, Boland JF, Chen Z, Wentzensen N, Zhang X, Yu K, Yang Q, Mitchell J, Roberson D, Bass S, Xiao Y, Burdett L, Raine-Bennett T, Lorey T, Castle PE, Burk RD, Schiffman M, 2016. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J Natl Cancer Inst 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Yeager M, Yu K, Clifford GM, Xiao Y, Zhu B, Cullen M, Boland JF, Wentzensen N, Nelson CW, Raine-Bennett T, Chen Z, Bass S, Song L, Yang Q, Steinberg M, Burdett L, Dean M, Roberson D, Mitchell J, Lorey T, Franceschi S, Castle PE, Walker J, Zuna R, Kreimer AR, Beachler DC, Hildesheim A, Gonzalez P, Porras C, Burk RD, Schiffman M, 2017. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 170, 1164–1174 e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narechania A, Chen Z, DeSalle R, Burk RD, 2005. Phylogenetic incongruence among oncogenic genital alpha human papillomaviruses. J Virol 79, 15503–15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SI, Tejedor MF, Novo NM, Aristide L, 2013. Divergence Times and the Evolutionary Radiation of New World Monkeys (Platyrrhini, Primates): An Analysis of Fossil and Molecular Data. PLoS ONE 8, e68029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimenoff VN, de Oliveira CM, Bravo IG, 2017. Transmission between Archaic and Modern Human Ancestors during the Evolution of the Oncogenic Human Papillomavirus 16. Mol Biol Evol 34, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA, 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. [DOI] [PubMed] [Google Scholar]
- Prufer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, Li H, Mallick S, Dannemann M, Fu Q, Kircher M, Kuhlwilm M, Lachmann M, Meyer M, Ongyerth M, Siebauer M, Theunert C, Tandon A, Moorjani P, Pickrell J, Mullikin JC, Vohr SH, Green RE, Hellmann I, Johnson PL, Blanche H, Cann H, Kitzman JO, Shendure J, Eichler EE, Lein ES, Bakken TE, Golovanova LV, Doronichev VB, Shunkov MV, Derevianko AP, Viola B, Slatkin M, Reich D, Kelso J, Paabo S, 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector A, Lemey P, Tachezy R, Mostmans S, Ghim SJ, Van Doorslaer K, Roelke M, Bush M, Montali RJ, Joslin J, Burk RD, Jenson AB, Sundberg JP, Shapiro B, Van Ranst M, 2007. Ancient papillomavirus-host co-speciation in Felidae. Genome Biol 8, R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, Stanley MA, Franceschi S, 2016. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2, 16086. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Herrero R, Desalle R, Hildesheim A, Wacholder S, Rodriguez AC, Bratti MC, Sherman ME, Morales J, Guillen D, Alfaro M, Hutchinson M, Wright TC, Solomon D, Chen Z, Schussler J, Castle PE, Burk RD, 2005. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 337, 76–84. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Rodriguez AC, Chen Z, Wacholder S, Herrero R, Hildesheim A, Desalle R, Befano B, Yu K, Safaeian M, Sherman ME, Morales J, Guillen D, Alfaro M, Hutchinson M, Solomon D, Castle PE, Burk RD, 2010. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res 70, 3159–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SD, Doorbar J, Goldstein RA, 2010. Analysis of host-parasite incongruence in papillomavirus evolution using importance sampling. Mol Biol Evol 27, 1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Anastos K, Hoover DR, Burk RD, Shi Q, Ngendahayo L, Mutimura E, Cajigas A, Bigirimani V, Cai X, Rwamwejo J, Vuolo M, Cohen M, Castle PE, 2009. Human Papillomavirus Infection and Cervical Cytology in HIV-Infected and HIV-Uninfected Rwandan Women. J Infect Dis 199, 1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siqueira JD, Alves BM, Prellwitz IM, Furtado C, Meyrelles AR, Machado ES, Seuanez HN, Soares MA, Soares EA, 2016. Identification of novel human papillomavirus lineages and sublineages in HIV/HPV-coinfected pregnant women by next-generation sequencing. Virology 493, 202–208. [DOI] [PubMed] [Google Scholar]
- Smith B, Chen Z, Reimers L, van Doorslaer K, Schiffman M, Desalle R, Herrero R, Yu K, Wacholder S, Wang T, Burk RD, 2011. Sequence imputation of HPV16 genomes for genetic association studies. PLoS ONE 6, e21375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stringer CB, Barnes I, 2015. Deciphering the Denisovans. Proc Natl Acad Sci U S A 112, 15542–15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S, 2011. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C., 2014. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: 2013 ISBN 3-900051-07-0. [Google Scholar]
- Torre LA, Islami F, Siegel RL, Ward EM, Jemal A, 2017. Global Cancer in Women: Burden and Trends. Cancer Epidemiol Biomarkers Prev 26, 444–457. [DOI] [PubMed] [Google Scholar]
- Vartanian JP, Guetard D, Henry M, Wain-Hobson S, 2008. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320, 230–233. [DOI] [PubMed] [Google Scholar]
- Volter C, He Y, Delius H, Roy-Burman A, Greenspan JS, Greenspan D, de Villiers EM, 1996. Novel HPV types present in oral papillomatous lesions from patients with HIV infection. Int J Cancer 66, 453–456. [DOI] [PubMed] [Google Scholar]
- Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, Lambert PF, Santiago ML, Pyeon D, 2015. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol 89, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongworapat K, Keawvichit R, Sirirojn B, Dokuta S, Ruangyuttikarn C, Sriplienchan S, Sontirat A, Kla KT, Gravitt PE, Celentano DD, 2008. Detection of human papillomavirus from self-collected vaginal samples of women in Chiang Mai, Thailand. Sex Transm Dis 35, 172–173. [DOI] [PubMed] [Google Scholar]
- Wood CE, Chen Z, Cline JM, Miller BE, Burk RD, 2007. Characterization and experimental transmission of an oncogenic papillomavirus in female macaques. J Virol 81, 6339–6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XL, Zhang CT, Zhu XK, Wang YC, 2010. Detection of HPV types and neutralizing antibodies in women with genital warts in Tianjin City, China. Virologica Sinica 25, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi LF, Kiviat NB, Hildesheim A, Galloway DA, Wheeler CM, Ho J, Koutsky LA, 2006. Human papillomavirus type 16 and 18 variants: race-related distribution and persistence. J Natl Cancer Inst 98, 1045–1052. [DOI] [PubMed] [Google Scholar]
- Xi LF, Schiffman M, Koutsky LA, Hughes JP, Winer RL, Mao C, Hulbert A, Lee SK, Shen Z, Kiviat NB, 2014. Lineages of oncogenic human papillomavirus types other than type 16 and 18 and risk for cervical intraepithelial neoplasia. J Natl Cancer Inst 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi LF, Schiffman M, Koutsky LA, Hulbert A, Lee SK, Defilippis V, Shen Z, Kiviat NB, 2012. Association of human papillomavirus type 31 variants with risk of cervical intraepithelial neoplasia grades 2–3. Int J Cancer. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Heatmap display of HPV variant pairwise sequence differences. Percent differences calculated based on the complete genome nucleotide sequence alignment of each type were measured and scaled such that complete identity (0.0% difference) is displayed as yellow and a maximum difference (2.3%) as blue. Phylogenetic trees indicating the position and name of each lineage and sublineage from the same species are shown besides of the heatmap.
Figure S2. Accumulated amino acid diversity of Alphapapillomavirus variants. The percentages were calculated based on the overall amino acid variable positions across each open reading frame (ORF) (E6, E7, E1, E2, E4, E5, L2, L1) or the combined 8 ORFs of variants from the same HPV type. Box plots are shown indicating the median, 25%–75% and 5%–95%. Asterisks represent the actual diversity for variants of each HPV type.
Figure S3. Proposed short regions containing lineage- and sublineage-specific single nucleotide polymorphisms (SNPs) of each HPV type. Only sites conserved in each lineage/sublineage are displayed, with dots indicating sites matched with reference prototype. Dashes indicate insertion/deletion events.