

Abstract
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the second leading cause of cancer related deaths worldwide. Among others, non-alcoholic steatohepatitis (NASH) and alcoholic steatohepatitis (ASH) are the two major risk factors as both of them may develop cirrhosis and hepatocellular carcinoma (HCC) if left untreated. However, patients with NASH progress to HCC at a rate around 0.5% annually, while 3–10% ASH patients may progress to HCC annually. The present study is to demonstrate the molecular differences in oncogenesis pathway between NASH and ASH. By using immunofluorescence study and quantitating the fluorescence intensity morphometrically in liver biopsied specimens from NASH and ASH patients, the protein expression of candidate molecules within hepatocytes cytoplasm are studied, including two HCC-related molecules FAT10 and FOXO1, and one GPCR pathway related molecule ADRA2A. Compared with the control group patients, the expression levels of all the molecules were upregulated in the ASH group of patients(p<0.001 in all molecules), while FAT10 and ADRA2A were upregulated, FOXO1 did not change in the NASH group of patients.The most important finding is that compared with the ASH group of patients, the expression levels of all three molecules were significantly lower than in the NASH group of patients (p<0.001 in all molecules). These results confirmed our previous finding that there are significant differences of molecules change in ASH compared to NASH. Thus, we conclude that there are significantly different molecules and pathways involved during the pathogenesis of HCC development in ASH compared to NASH which could help explain why the tumorigenic rate is different in ASH and NASH.
Introduction
Liver cancer is the fifth most common cancer and the second leading cause of cancer related deaths worldwide [Ferlay et al., 2012]. The major risk factors include hepatitis B (HBV) and hepatitis C (HCV), obesity/metabolic syndrome, and alcoholism which all may progress to hepatocellular carcinoma (HCC). In the last two decades the rising rates of obesity/metabolic syndrome have led to the increased development of the non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Experimental and epidemic studies have shown relationships between NASH and HCC development. Alcoholic liver disease (ALD) and ASH are the major causes of HCC in US [Morgan et al., 2004][Ramadori et al., 2017][Testino et al., 2014]. NASH has similar histological features with alcoholic hepatitis, such as overt lipid deposition and fat storage in the liver parenchymal cells. Both ASH and NASH may progress to fibrosis, cirrhosis, and ultimately hepatocellular carcinoma (HCC), about 7–10% of NASH may progress to cirrhosis [Scaglioni et al., 2011] and 0.5% to HCC [Lindenmeyer et al., 2018], while 10–20% of ASH progress to cirrhosis [Schwartz et al., 2012][Dam-Larsen et al., 2004] and 7–10% to HCC [Schwartz et al., 2012], annually. Our previous published work showed that there are different molecules protein changes in ASH compared with NASH [Nguyen et al., 2018][ Lu et al., 2018]
The mechanisms by which NASH and ASH progress to HCC are not completely understood and the re are many theories including chromosomal loss of tumor suppressor genes, oxidative stress, decreased liver retinoic acid level, altered DNA methylation, and genetic susceptibility [Morgan et al., 2004][Stickel et al., 2002, 2015]. In our previous studies, we showed that in addition to the TLR/NFKB/CXCR4/7 [Liu et al., 2014][French et al., 2012][Nan et al., 2005][French et al., 2010] and PI3K/AKT/mTORC1 signaling pathways [Afifiyan et al., 2017], Tec kinase signaling pathway connects these two systems together in Mallory-Denk Bodies (MDB) formation both in ASH and NASH [Afifiyan et al., 2017]. During these studies we found that HLA-F-adjacent transcript 10 (FAT10), a ubiquitin-like modifier protein which functions as a proteasomal degradation signal [Schmidtke et al., 2009][Rani et al., 2012][Hipp et al., 2005], plays an important role in MDB formation and tumorigenesis [Oliva et al., 2010][Oliva et al., 2008][French et al., 2012] and we proved that MDB forming cells express preneoplastic phenotypic features [Nan et al., 2006]. To confirm the roles of FAT10 and other related molecules in HCC tumorigenesis in NASH and ASH, we studied the changes of FAT10, FOXO1, and ADRA2A in liver biopsy specimens from NASH and ASH patients and control groups.
Methods
Formalin-fixed paraffin-embedded biopsies of ASH liver (n=39 in FAT10, 50 in FOXO1, 40 in ADRA2A), NASH liver (n=30), and normal liver (n=20) were collected from Harbor-UCLA Medical Center and from the Long Beach Veterans Affairs’ clinical trial in treatment of alcoholic hepatitis. The study was conducted following the principals of the Declaration of Helsinki and was designated as exempt by our institutional ethics review board and the data was analyzed anonymously. The primary antibodies were rabbit anti-FAT10, anti-FOXO1, and anti-ADRA2A purchased (Abcam, Cambridge, MA). For each protein studied, the biopsy sections were stained first with protein-specific primary antibody followed by a secondary fluorescence antibody. Either donkey anti-mouse or anti-rabbit Alex Fluor (Jackson Labs, West Grove, PA) was used as the second antibody. The slides were also stained for ubiquitin (to identify MDBs) using Texas Red (Millipore, Temecula, CA) and nuclear stained by DAPI. The staining of all the specimens was conducted under the same situation to provide accurate comparison between groups. For each candidate molecule, we measured the intensity of the fluorescent staining of the liver cells in at least three different areas on each section with 40× magnifications and 800 ms standard exposure time by using a Nikon 400 fluorescent microscope. On each section area, 10 peak fluorescence intensities were collected and the florescent intensity was quantitated by using the Nikon morphometric system. The mean value, standard error, and statistical differences of data achieved from the Nokia were analyzed by Graph pad statistical software. Controls vs AH, controls vs NASH, and AH vs NASH were compared by unpaired t-test with a p-value of<0.05 considered statistically significant.
Results
The protein expression level of several candidate molecules in specimens from patients with ASH, NASH, and normal controls were compared. Representative data are shown in Fig. 1–3.
Figure 1.
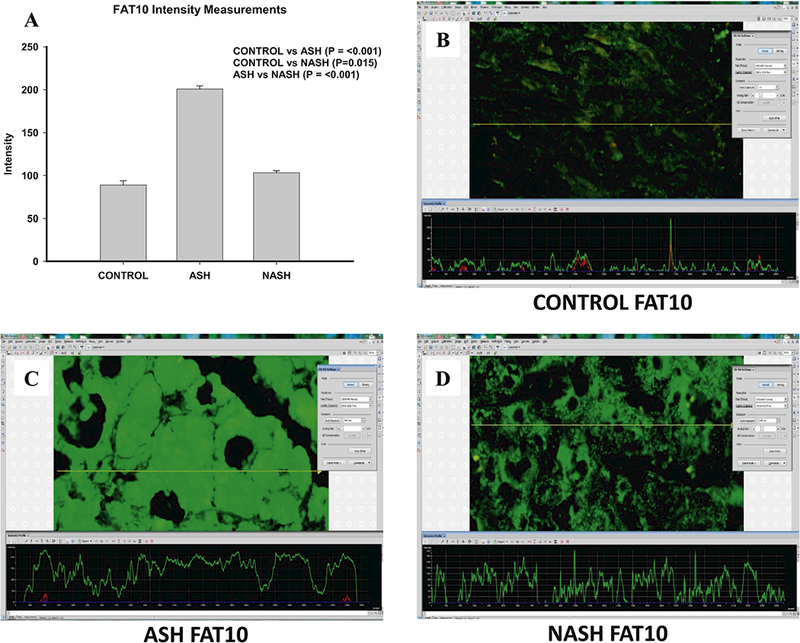
Different Changes of FAT10 in ASH and NASH specimens. (A) Level of expression of FAT10 protein upregulated in ASH, NASH or normal controls. Expression is measured as fluorescence intensity and displayed as mean +/− standard deviation. Representative images of fluorescence intensity to measure FAT10 expression in normal control (B), ASH (C) and NASH (D) liver specimens. A line is drawn through the image to yield a fluorescence intensity graph; the intensity of the ten highest peaks are measured, excluding nuclear regions which are highlighted by DAPI (not shown). Three areas per slide are measured in this manner including Figure 2 and 3.
Figure 3.
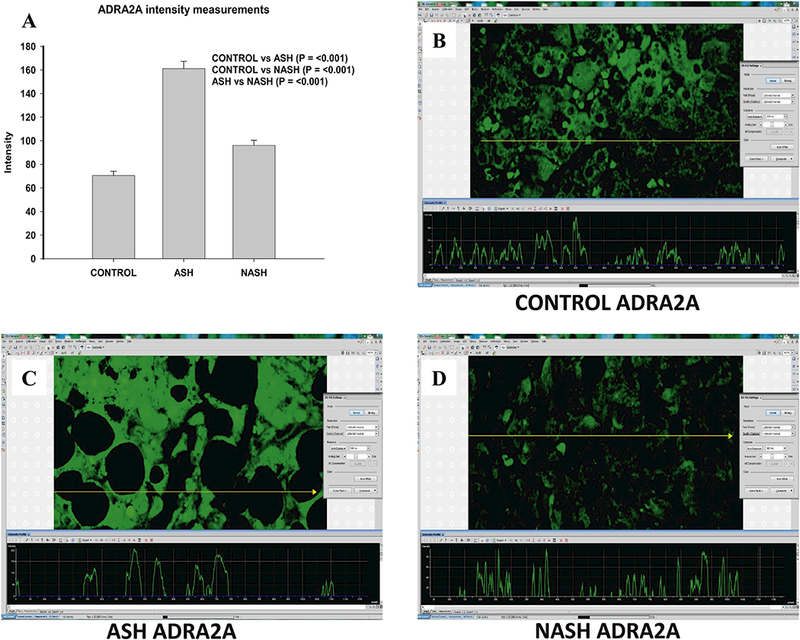
Different Changes of ADRA2A in ASH and NASH specimens. (A) Level of expression of ADRA2A protein upregulated in ASH, NASH or normal controls. Expression is measured as fluorescence intensity and displayed as mean +/− standard deviation. Representative images of fluorescence intensity to measure ADRA2A expression in normal control (B), ASH (C) and NASH (D) liver specimens.
In ASH patients, levels of all tested candidate proteins including FAT10, FOXO1, and ADRA2A (Fig. 1–3) were markedly increased compared with the control group. In NASH patients, FAT10 and ADRA2A were upregulated when compared to control group, while FOXO1 level was not changed (Fig. 2).
Figure 2.
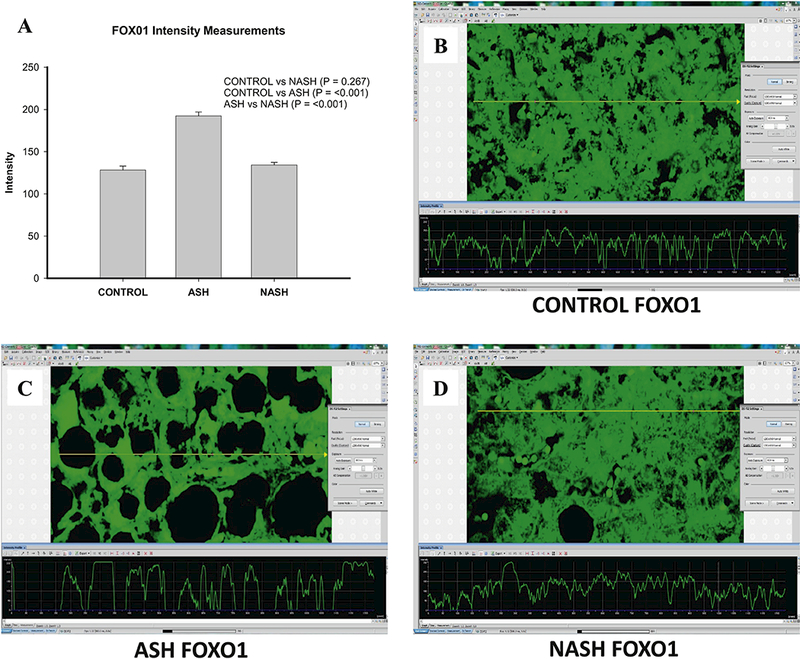
Different Changes of FOXO1 in ASH and NASH specimens. (A) Level of expression of FOXO1 protein upregulated in ASH, NASH or normal controls. Expression is measured as fluorescence intensity and displayed as mean +/− standard deviation. Representative images of fluorescence intensity to measure FOXO1 expression in normal control (B), ASH (C) and NASH (D) liver specimens.
The most interesting finding is that all three molecules, FAT10, FOXO1, and ADRA2A were significant lower in the NASH group specimens compared with the ASH group (Fig. 1–3).
Discussion and Conclusions
The increasing rates of obesity and the metabolic syndrome become a major health problem worldwide which is emphasized by the relationship from NASH to cirrhosis and even HCC. Wong et al. showed that NASH is the most rapid growing risk for liver transplantation in patients with HCC [Wong et al., 2014, 2015]. Chronic (longer than 10 years) alcohol consumption (greater than 80 gram/day) increases the risk for HCC approximately 5 fold [French SW. 2013]. ALD is the most common cause of HCC which accounts for around one-third of all HCC cases in US and Italy [Morgan et al., 2004][Testino et al., 2014]. It is believed that alcohol is a factor in the development of HCC through genotoxic (direct) pathway and cirrhosis (indirect) mechanisms.
Both ASH and NASH could progress to cirrhosis and even hepatocellular carcinoma (HCC), but the mechanisms remain unclear, although there are many theories. It is well accepted that the rates of those progressing to cirrhosis or HCC annually are lower in the NASH if compared with those in ASH. Our published data support that different molecules or pathways may be involved in ASH compared with NASH during the tumorigenesis [Nguyen et al., 2018][Lu et al., 2018] Our work also showed that the TLR/NFKB/CXCR4/7 [Liu et al., 2014][French et al., 2012] [Nan et al., 2010], PI3K/AKT/mTORC1 signaling pathways [Afifiyan et al., 2017], and Tec kinase signaling pathway connected each other during MDB formation both in ASH and NASH [Afifiyan et al., 2017].
FAT10 was found as a proteasomal degradation signal. Recent studies reported that FAT10 is expressed mainly in tissues of the immune system, including the spleen and thymus [Liu et al., 1999][Lee et al., 2003]. In addition, FAT10 is induced by proinflammatory cytokines in various tissues outside the immune system including the liver and colon [Ren et al., 2011][Lukasiak et al., 2008], but the physiological functions of this response remain unknown. However, it is accepted that constitutive induction of FAT10 has deleterious consequences in cellular malignancy development [Gao et al., 2013]. And FAT10 was reported being up-regulated in several tumor types including tumors of the liver and colon [Lee et al., 2003][Lukasiak et al., 2008][Qing et al., 2011][Yan et al., 2010]. Although the mechanism of FAT10 promalignant function remains unclear, many researchers have re-ported that the expression of FAT10 increased with Akt, NF-κB/STAT3 [Gao et al., 2013][ Liu et al., 2014], CXCR4/7[Gao et al., 2013], MAD2 [Theng et al., 2014], p53 [Choi et al., 2014], and β-catenin/TCF4 [Aichem et al., 2016], in addition to its signal for proteasomal degradation [Schmidtke et al., 2014]. Previously, we reported that FAT10 plays an essential role in MDB formation and tumorigenesis [Oliva et al., 2008, 2010][French et al., 2012]. In the present study, we found that FAT10 increased in the ASH and the NASH specimen compared with control group, but significantly decreased in the NASH specimens compared with the ASH group.
Growing evidence strongly supports that FAT10 itself promotes carcinogenesis by directly increasing survival, proliferation, invasion, and metastasis formation of tumor cells. The upregulation of FAT10 expression in a number of different tumor types might be useful as a prognostic marker for the survival rate of the affected patients and for estimating the probability of recurrence. Potential therapeutic avenues aiming at FAT10-inducing pathways such as NF-κB/STAT3 signaling as well as targeting the FAT10 conjugation pathway seem to be promising as the genetic FAT10 knockout mice show less severe phenotypes [Canaan A et al 2006].
FOXO proteins belong to a sub-group of a superfamily of forkhead box (FOX) -containing transcription factors (TFs). Based on the phylogenetic analysis of the sequences of the known chordate FOX proteins, these TFs are classified into 19 subclasses, ranging from FOXA to FOXS [Kaestner et al., 2000][Jackson et al., 2010]. In mammals, four subfamily members of FOXOs have been identified: FOXO1, FOXO3, FOXO4 and FOXO6. FOXO1, FOXO3 and FOXO4 mRNAs are expressed ubiquitously in varying levels in mammals and negatively regulated by the PI3K/Akt pathway [Anderson et al., 1998][Furuyama et al., 2000]. It was thought that FOXO1 was a classic tumor suppressor due to their tumor inhibitory effects, such as repressing the cell cycle, inducing cell apoptosis, suppressing tumorigenesis, and being related with certain cancers via FOXO mutation or translocation [Yadav et al., 2018]. Growing evidence shows that FOXOs also support tumor development and progression [Hornsveld et al., 2018][Shi et al., 2018], including being related to bad prognosis, facilitating/stimulating metastasis, maintaining cellular redox homeostasis and enhancing oxidative stress resistance, and subsequently involving the chemotherapeutic resistance. In summary, the roles of FOXO1 in tumorigenesis is complex, and further studies are needed. In our data, the dramatic increased changes of FOXO1 in ASH specimens compared with control group indicated the FOXO1 may be important in tumorigenesis of HCC in ASH patient. While the no change of FOXO1 in NASH specimen compared with control supported that a different mechanism is involved in HCC development in NASH patients. The similar pattern of protein changes of FAT10 and FOXO1 suggested that FAT10 and FOXO1 may work together in HCC tumorigenesis in ASH patients (Fig. 4). One of the possible putative thoughts is that FAT10 may regulate FOXO1 via ubiquitination and proteasome degradation [Huang et al., 2011]
Figure 4.
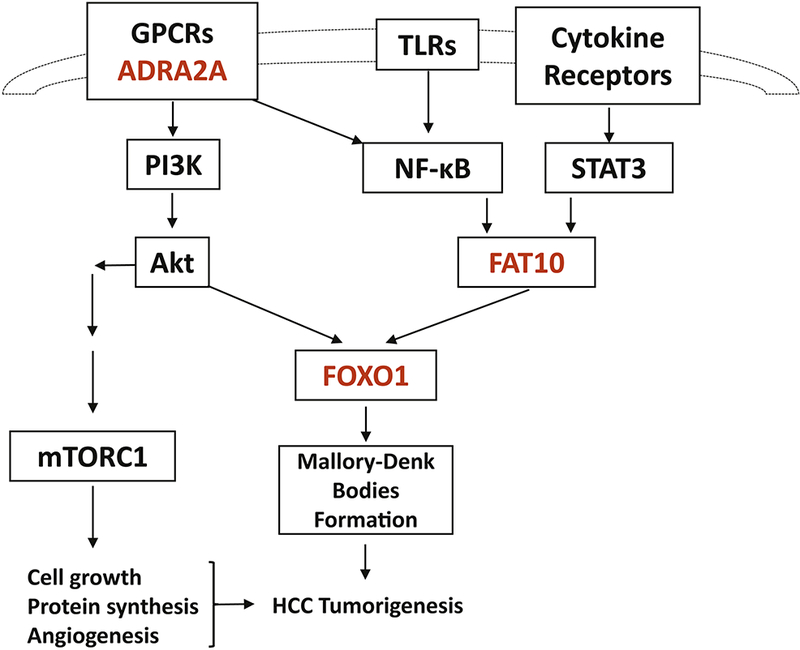
Putative pathway for FAT10, FOXO1, and ADRA2A in the HCC tumorigenesis. The different expression of FAT10, FOXO1, and ADRA2A proteins in ASH and NASH suggest different pathways, including GPCR/PI3K/Akt and/or TLR4/NF-κB/STAT3 pathways in HCC tumorigenesis due to different causes.
ADRA2A (Adrenoceptor A2A) belongs to Adrenoceptors family α-subtypes which are class A G protein-coupled receptors (GPCRs). All three α2-subtypes adrenoceptors (ADRA2) including ADRA2A couple to inhibitory Gi proteins and modulate the downstream PI3K/Akt pathway to regulate multiple physiologic processes, such as neurotransmitter release, platelet aggregation, blood pressure, insulin secretion, and lipolysis [Ahles et al., 2014][Fagerholm et al., 2011]. ADRA2A also plays an important role in modulating the inflammatory process via TLR4/NF-κB pathways [Leong et al., 2010][Wang et al., 2016]. In our published work, we proved the roles of TLR/NF-κB-CXCR4/7 pathway and PI3K/Akt pathway in human ASH and NASH specimen with MDB formation. It is possible that the ADRA2A plays a role in ASH or NASH via GPCR/PI3K/Akt and/or TLR4/NF-κB pathways (Fig.4). Our present data showed that the ADRA2A significantly increased in ASH specimens compared with control but not in NASH specimen. The difference of ADRA2A expression between ASH and NASH specimens may also explain the different rate in HCC tumorigenesis between ASH and NASH patients.
In summary, our present data and previous published studies demonstrates the different expression of proteins which represent different pathways, such as GPCR/PI3K/Akt and/or TLR4/NF-κB/STAT3 pathways, and explains the different rate of progression to fibrosis, cirrhosis, and eventually HCC in ASH and NASH patients. These findings may be very helpful to understand the pathogenesis of HCC and to discover possible therapeutic target points. Obviously, more detailed research is needed.
Acknowledgements
This study was funded by NIH/AAA grant # UO-21898-05.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afifiyan N,Tillman B, French BA, Masouminia M, Samadzadeh S, French SW. 2017. Over expression of proteins that alter the intracellular signaling pathways in the cytoplasm of the liver cells forming Mallory-Denk bodies. Exp Mol Pathol. 102, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afifiyan N, Tillman B, French BA, Sweeny O, Masouminia M, et al. , 2017. The role of Tec kinase signaling pathways in the development of Mallory Denk Bodies in balloon cells in alcoholic hepatitis. Exp Mol Pathol. 103, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahles A, Engelhardt S. 2014. Polymorphic variants of adrenoceptors: pharmacology, physiology, and role in disease. Pharmacol Rev. 66, 598–637. [DOI] [PubMed] [Google Scholar]
- Aichem A, Groettrup M, 2016. The ubiquitin-like modifier FAT10 in cancer development. Int J Biochem Cell Biol. 79, 451–461. [DOI] [PubMed] [Google Scholar]
- Anderson AJ, Viars CS, Czekay S, Cavenee WK, Arden KC, 1998. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily, Genomics 47, 187–199. [DOI] [PubMed] [Google Scholar]
- Canaan A, Yu X, Booth CJ, Lian J, Lazar I, et al. 2006. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol Cell Biol. 26, 5180–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Kim JK, Yoo JY, 2014. NFκB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Mol Oncol. 8, 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, et al. , 2004. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut 53, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerholm V, Haaparanta M, Scheinin M. 2011. α2-adrenoceptor regulation of blood glucose homeostasis. Basic Clin Pharmacol Toxicol. 108, 365–370. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, et al. , 2012. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon: International Agency for Research on Cancer. [Google Scholar]
- French SW. 2013. Epigenetic events in liver cancer resulting from alcoholic liver disease. Alcohol Res. 35, 57–67. [PMC free article] [PubMed] [Google Scholar]
- French SW, Bardag-Gorce F, Li J, French BA, Oliva J. 2010. Mallory-Denk body pathogenesis revisited. World J Hepatol. 2, 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SW, French BA, Oliva J, Li J, Bardag-Gorce F, et al. , FAT10 knock out mice livers fail to develop Mallory-Denk bodies in the DDC mouse model. Exp Mol Pathol. 93, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SW, Lee J, Zhong J, Morgan TR, Buslon V, et al. , 2012. Alcoholic liver disease - Hepatocellular carcinoma transformation. J Gastrointest Oncol. 3, 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama T, Nakazawa T, Nakano I, Mori N, 2000. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-homologues, Biochem. J 349, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Theng SS, Zhuo J, Teo WB, Ren J, Lee CG, et al. , 2013. FAT10, an Ubiquitin-like Protein, Confers Malignant Properties in Non-tumorigenic and Tumorigenic Cells. Carcinogenesis. 35, 923–934. [DOI] [PubMed] [Google Scholar]
- Hipp MS, Kalveram B, Raasi S, Groettrup M, Schmidtke G. 2005. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol Cell Biol. 25, 3483–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsveld M, Dansen TB, Derksen PW, Burgering BMT. 2018. Re-evaluating the role of FOXOs in cancer. Semin Cancer Biol. 50, 90–100. [DOI] [PubMed] [Google Scholar]
- Huang H, Tindall DJ. 2011. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim Biophys Acta. 1813, 1961–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson BC, Carpenter C, Nebert DW, Vasiliou V, 2010 Update of human and mouse forkhead box (FOX) gene families, Hum. Genom 4, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Knochel W, Martinez DE, 2000. Unified nomenclature for the winged helix/forkhead transcription factors, Genes Dev. 14, 142–146. [PubMed] [Google Scholar]
- Lee CG, Ren J, Cheong IS, Ban KH, Ooi LL, et al. , 2003. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene. 22, 2592–2603. [DOI] [PubMed] [Google Scholar]
- Leong J, Zhou M, Jacob A, Wang P., 2010. Aging-related hyperinflammation in endotoxemia is mediated by the alpha2A-adrenoceptor and CD14/TLR4 pathways. Life Sci. 86, 740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenmeyer C, McCullough A, 2018. The natural history of nonalcoholic fatty liver disease-an evolving view. Clin. Liver Dis. 22, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li J, Tillman B, Morgan TR, French BA, French SW. 2014. TLR3/4 signaling is mediated via the NFκB- CXCR4/7 pathway in human alcoholic hepatitis and non-alcoholic steatohepatitis which formed Mallory-Denk bodies. Exp Mol Pathol. 97, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Pan J, Zhang C, Fan W, Collinge M, et al. , 1999. A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc Natl Acad Sci USA. 96, 4313–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JG, Nguyen L, Samadzadeh S, Masouminia M, Mendoza A, et al. 2018. Expression of proteins upregulated in hepatocellular carcinoma in patients with alcoholic hepatitis (AH) compared to non-alcoholic steatohepatitis (NASH): An immunohistochemical analysis of candidate proteins. Exp Mol Pathol. 104, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiak S, Schiller C,Oehlschlaeger P, Schmidtke G, Krause P, et al. 2008. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene. 27, 6068–6074. [DOI] [PubMed] [Google Scholar]
- Morgan TR, Mandayam S, Jamal MM. 2004. Alcohol and hepatocellular carcinoma. Gastroenterology. 127, S87–96. [DOI] [PubMed] [Google Scholar]
- Nan L, Bardag-Gorce F, Wu Y, Li J, French BA, French SW. 2006. Mallory body forming cells express the preneoplastic hepatocyte phenotype. Exp Mol Pathol. 80, 109–118. [DOI] [PubMed] [Google Scholar]
- Nan L, Wu Y, Bardag-Gorce F, Li J, French BA, et al. , 2005. The p105/50 NF-kappaB pathway is essential for Mallory body formation. Exp Mol Pathol. 78, 198–206. [DOI] [PubMed] [Google Scholar]
- Nguyen L, Masouminia M, Mendoza A, Samadzadeh S, Tillman B, et al. , 2018. Alcoholic hepatitis versus non-alcoholic steatohepatitis: Levels of expression of some proteins involved in tumorigenesis. Exp Mol Pathol. 104, 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva J, Bardag-Gorce F, Lin A, French BA, French SW. 2010. The role of cytokines in UbD promoter regulation and Mallory-Denk body-like aggresomes. Exp Mol Pathol. 89, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva J, Bardag-Gorce F, French BA, Li J, McPhaul L, et al. ,2008. Fat10 is an epigenetic marker for liver preneoplasia in a drug-primed mouse model of tumorigenesis. Exp Mol Pathol. 84, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing X, French BA, Oliva J, French SW. 2011. Increased expression of FAT10 in colon benign, premalignant and malignant epithelial neoplasms. Exp Mol Pathol. 90, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadori P, Cubero FJ, Liedtke C,Trautwein C, Nevzorova YA. 2017. Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers (Basel). 9, pii: E130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani N, Aichem A,Schmidtke G, Kreft SG,Groettrup M. 2012. FAT10 and NUB1L bind to the VWA domain of Rpn10 and Rpn1 to enable proteasome-mediated proteolysis. Nat Commun. 3, 749. [DOI] [PubMed] [Google Scholar]
- Ren J, Wang Y, Gao Y, Mehta SB, Lee CG. 2011. FAT10 mediates the effect of TNF-α in inducing chromosomal instability. J Cell Sci. 124, 3665–3675. [DOI] [PubMed] [Google Scholar]
- Scaglioni F, Ciccia S, Marino M, Bedogni G, Bellantani S, 2011. ASH and NASH. Dig. Dis 29, 202–210. [DOI] [PubMed] [Google Scholar]
- Schmidtke G, Aichem A, Groettrup M. 2014. FAT10ylation as a signal for proteasomal degradation. Biochim Biophys Acta. 1843, 97–102. [DOI] [PubMed] [Google Scholar]
- Schmidtke G, Kalveram B, Groettrup M. 2009. Degradation of FAT10 by the 26S proteasome is independent of ubiquitylation but relies on NUB1L. FEBS Lett. 583, 591–594. [DOI] [PubMed] [Google Scholar]
- Schwartz JM, Reinus JF, 2012. Prevalence and natural history of alcoholic liver disease. Clin. Liver Dis 16, 659–666. [DOI] [PubMed] [Google Scholar]
- Shi F, Li T, Liu Z, Qu K, Shi C, et al. , 2018. FOXO1: Another avenue for treating digestive malignancy? Semin Cancer Biol. 50, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickel F 2015. Alcoholic cirrhosis and hepatocellular carcinoma. Adv Exp Med Biol. 815, 113–130. [DOI] [PubMed] [Google Scholar]
- Stickel F, Schuppan D, Hahn EG, Seitz HK. 2002. Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut. 51, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testino G, Leone S, Borro P. 2014. Alcohol and hepatocellular carcinoma: a review and a point of view. World J Gastroenterol. 20, 15943–15954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theng SS, Wang W, Mah WC, Chan C, Zhuo J, et al. , 2014. Disruption of FAT10-MAD2 binding inhibits tumor progression. Proc Natl Acad Sci U S A. 111, E5282–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu S, Yu X, Zhou S, Ge M, et al. , 2016. Dexmedetomidine Protects Rat Liver against Ischemia-Reperfusion Injury Partly by the α2A-Adrenoceptor Subtype and the Mechanism Is Associated with the TLR4/NF-κB Pathway. Int J Mol Sci. 17, pii: E995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RJ, Cheung R, Ahmed A. 2014. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology. 59, 2188–2195. [DOI] [PubMed] [Google Scholar]
- Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, et al. , 2015. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148, 547–555. [DOI] [PubMed] [Google Scholar]
- Yadav RK, Chauhan AS, Zhuang L, Gan B. 2018. FoxO transcription factors in cancer metabolism. Semin Cancer Biol. 50, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan DW, Li DW, Yang YX, Xia J, Wang XL, et al. , 2010. Ubiquitin D is correlated with colon cancer progression and predicts recurrence for stage II-III disease after curative surgery. Br J Cancer. 103, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]