

Abstract
Six directed hydrogen bonding (H-bonding) interactions allow for the reversible capture and reduction of dioxygen to a trans-1,2-peroxo within a tripodal zinc(II) framework. Spectroscopic studies of the dizinc peroxides, as well as on model zinc diazides, suggest H-bonding contributions serve a dominant role for the binding/activation of these small molecules.
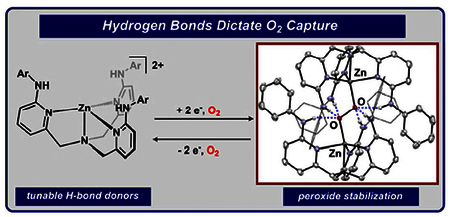
The reversible capture of dioxygen as peroxide (O22−) is required for myriad biological and abiological reactions spanning from oxidases to metal-air batteries.1 Biological systems leverage well-positioned secondary coordination sphere interactions, such as hydrogen bonding (H-bonding), within their active metal site(s) to achieve selective O2 binding, activation, and transfer.2 To emulate this principle, recent synthetic systems have demonstrated that H-bonds can facilitate O2 capture in the superoxo or peroxo state, although binding is typically coupled with a metal-based redox event.3 In contrast, capture of the O2 unit using H-bonds as the primary binding interaction is very rare,4 and was recently enabled by six directed H-bonds within a cryptand-type macrocycle.5 In this case, O2 capture was facilitated by preorganization of a molecular capsule with a binding pocket size-matched for a diatom. Given that host/guest inclusion is highly sensitive to size complementarity,6 the use of a preassembled binding pocket complicates the role that the H-bonds serve in capture and/or stabilization. An open tripodal ligand provides one test of whether preassembly is required to capture O2 using H-bonds without a redox-active metal.
Our group is working to evaluate how the precise structural, electronic, and cooperative modes in the secondary coordination sphere can be used to regulate reactivity.7 Recently, we reported a family of p-substituted tris(6-(p-R-phenylamino-2-pyridylmethyl)amine ligands (LR, R = CF3, H, OMe) that provide electronically tunable −NHAr H-bond donors in the secondary coordination sphere.7f This ligand framework provided the first structural characterization of an H-bonded (trans-1,2-peroxo)dicopper complex in which a combination of six H-bonds and two Cu(II) centers encapsulate the O22− unit. We hypothesized that if the six H-bonds to peroxide were key factors that allowed isolation, then the templating metal and the reductant could be separated. Zinc(II), the ubiquitous redox-inactive d-block metal, was selected to test this hypothesis (Fig. 1). Tripodal ligand scaffolds containing H-bond donors within the secondary coordination sphere have been recently popularized8 and have previously been templated on zinc.9 Zn2O2 fragments remain exceedingly rare and are limited to polymetallic aggregates with greater than two metals.10 Herein, we report dioxygen capture and reduction enabled by H-bonds via (trans-1,2-peroxo)dizinc complexes as well as the reverse—dioxygen release.
Figure 1.

Reversible O2 capture and reduction enabled by H-bonding interactions.
An H-bonded dizinc peroxide was obtained from dioxygen and reductant in the presence of [ZnLH]2+. Saturation of an equimolar MeCN solution of LH and Zn(OTf)2 with O2 followed by rapid addition of bis(cyclopentadienyl)cobalt(II) at room-temperature afforded the (trans-1,2-peroxo)dizinc complex, [(LH)2 Zn2 O2][OTf]2 (1H), in 89% yield (Fig. 2).11 An alternative synthesis of 1H was also developed: sequential addition of H2O2 and NiPr2Et to a MeCN solution of LH and Zn(OTf)2 afforded 1H in 58% yield.12–13 1H is thermally stable in CD3CN for >18 h at 50 °C in an inert atmosphere and is also moisture tolerant.14 However, the addition of a competitive H-bond acceptor (i.e. chloride) induces degradation.15
Figure 2.

Synthesis of 1R and molecular structure of 1H (50% probability ellipsoids) with bond distances of 1R.
The structural metrics of 1H were elucidated by single-crystal X-ray diffraction (XRD) and revealed a trans-1,2-peroxo binding mode with six directed H-bonds to the O22− fragment, establishing the first example of a discrete (trans-1,2-peroxo)dizinc species. For each ‘(LR)Zn’ fragment, one −NHPh group engages in H-bonding interactions with the proximal oxygen (N-Oproximal = 2.738 Å; N-H-O = 158.97°) while two engage the distal oxygen (N-Odistal = 2.837 Å (for both); N-H-O = 176.16 and 169.98°). The N-O distances and N-H-O angles are consistent with moderate-strength H-bonding interactions.16 The N-Cpyr distances range 1.3587(14)-1.3675(14) Å and are consistent with single-bonds where the N-H has not been deprotonated.17 The O2 motif (O1-O1’ = 1.4954(13) Å) is more reduced than dioxygen18 or superoxide,19 and comparable to main-group trans-1,2-peroxides including [O2 (B(C6 F5)3)2]2− (1.488 Å)20 and L2B2O2 21 (1.484 Å; L = subporphyrin).22 Notably, the O-O distance is the same as isostructural [(LH)2 Cu2O2]2+ (1.477(5) Å), which suggests that the ligand scaffold itself may serve a key role in regulating the structure, rather than the metal.
To interrogate the requirement of H-bonding for peroxide capture, we evaluated an isosteric ligand variant, tris(6-phenoxy-2-pyridylmethyl)amine (TPAOPh), that does not contain H-bond donors. In contrast to LH, when TPAOPh and Zn(OTf)2 were combined and treated with H2O2 and iPr2NEt, we observed demetalation, rather than the formation of a (trans-1,2-peroxo)dizinc (see SI). Attempts to synthesize the zinc analogue of Karlin’s [(TPA)2 Cu2 O2]2+ (TPA = tris(2-methylpyridylamine)23 resulted in products derived from [(TPA)Zn(OH)]+.24–25 These results highlight the synergistic effect of H-bonds with the Zn center in 1R to both capture and stabilize O22−.
Given the role of H-bonds for O22− capture, we assessed the extent to which activation of the O–O unit could be tuned by H-bond donor strength. We selected ligand variants that feature identical steric properties surrounding the Zn2O2 core, yet vary in acidity of the NH, and thus H-bond donor strength. Given the highly coupled ligand structure, substituent modification will necessarily alter both H-bond donor strength and ligand electronics.26 For example, an electron-withdrawing aniline (e.g. p-CF3) will afford a better H-bond donor at the expense of a weaker ligand donor strength. Four para-substituted anilines with Hammett substituent (σp) constants ranging −0.83 (R = NMe2) to 0.54 (R = CF3)27 were used to prepare the series of (trans-1,2-peroxo)dizinc complexes (1R; R = NMe2, OMe, CF3; Fig. 2).
The electronic environment provided by each ligand variant in 1R tracked with the methylene resonances (coupled doublets) of the C3-symmetric 1H NMR spectra. For example, electron-deficient 1CF3 exhibits the most downfield resonances (4.22 and 4.08 ppm), while the more electron-rich 1NMe2 features the most upfield resonances (4.00 and 3.86 ppm). These resonances show a good correlation when plotted against Hammett constants (see SI) and provide a descriptive measure of electronic environment provided by each ligand scaffold. In contrast to the direct relationship between ligand electronic environment and the methylene resonances, the −NH resonance involved in H-bonding interactions, which is a composite of −NH—Oproximal and −NH—Odistal, does not exhibit the same trend. 1H displays the most downfield shift (10.21 ppm) while 1OMe and 1NMe2 exhibit the same shift (10.14 ppm). The absence of a clear trend contrasts with the previously reported series of (LR)CuCl,7f where the −NH resonances exhibit a linear correlation with Hammett constants, and suggests the −NH—O interactions in 1 are not adequately described by simple H-bond donor/acceptor contributions, but may also be influenced by the electronics at zinc.
To probe the origin of the 1H NMR discrepancies, the structural metrics of 1R were examined by single-crystal XRD. Complexes 1CF3, 1OMe, and 1NMe2 are isostructural to 1H with six directed H-bonds to the peroxide. The electronic substitutions have negligible consequence on the ability of zinc to dimerize about the O22− unit—the Zn-Zn distances range from 4.719 (1CF3) to 4.784 (1OMe) Å. The Zn-O distance reports on the electronic environment of the TPA-ligand: the most electron-deficient variant, 1CF3, displays the shortest Zn-O distance while the most electron-rich variant, 1NMe2, contains the longest (1.9507(16) and 1.991(3) Å, respectively). The H-bonding interactions within the four species are comparable with N-Oproximal and N-Odistal distances ranging from 2.619–2.741 and 2.811–2.945 Å, respectively.28 Within the series of complexes, the O–O bond length exhibits a variable extent of activation and ranges from 1.483(6) (1NMe2) to 1.524(3) Å (1CF3). Notably, the O–O bond in 1R does not correlate with either the electronic character of the TPA-ligand—as assessed by Hammett constants of para-aniline substitution—or the H-bond donor strength. This directly contrasts the [(LR)2Cu2O2]2+ series whose LMCT-energy correlated to TPA-ligand electronics,7f further suggesting multiple competing factors contribute to the overall description of the Zn2O2 unit.
Oxidation studies were pursued to assess the reversibility of H-bond mediated O2 capture.29 Addition of PhICl2 to 1H cleanly produces [(LH)ZnCl][(OTf)] (76% yield) concomitant with gas evolution. A Clark electrode was used to confirm O2 release during oxidation. Because this analytical technique requires aqueous conditions, the reaction was repeated with a water-soluble oxidant. Injection of 1H into an aqueous solution containing [NH4]2[Ce(NO3)6] and [Bu4N][Cl] caused a rapid increase in dissolved O2, which reached a plateau after 3 minutes, corresponding to approximately 47% yield (Fig. 3, see SI). [(LH)ZnCl][(OTf)] was formed as the (LH)Zn-containing compound in a similar isolated yield as O2 (40%).30 These studies confirm that capture and release is dictated by the direction of electron flow in solutions containing [(LH)Zn]2+.
Figure 3.

A) Oxidative release of dioxygen from 1H. B) Dioxygen evolution trace detected by Clark electrode. C) Molecular structure of [(LH)ZnCl]+ (50% probability ellipsoids).
Azide is a spectroscopic analogue for peroxide due to its similar frontier orbital manifold.31 In contrast to peroxo-units whose vibrational modes can be challenging to identify,32 metal-azides feature intense bands that are sensitive to electronic perturbations. We thus sought to interrogate the electronic and H-bonding contributions imparted on the O22− or N3− unit within a set of isostructural (LR)Zn complexes.33 Octahedral (LR)Zn(N3)2 complexes (2R) were targeted because they feature both axial and equatorial azide environments, and only the axial azide can engage in H-bonding interactions. Complexes 2R were synthesized by treating an acetone solution of LR and Zn(ClO4)2·6H2O with excess NaN3 (Fig. 4). The 1H NMR spectra exhibit downfield −NH resonances, consistent with H-bonding, which are dependent on TPA-ligand electronics. The furthest downfield −NH and methylene resonances (δ = 9.36 and 4.16, respectively) correspond to the most electron-deficient variant, 2CF3, while the furthest upfield −NH and methylene resonances (δ = 8.90 and 4.05 ppm respectively), correspond to the most electron-rich complex, 2NMe2.34 The trend of the −NH resonance position contrasts with the series of 1R (a composite of −NH—Oproximal and −NH—Odistal interactions) but is analogous to the chloride series, (LR)CuCl.7f
Figure 4.

Synthesis of 2R and molecular structure of 2H (30% probability ellipsoids).
Molecular structures for 2R were determined by single-crystal XRD and establish an octahedral geometry in which the axial azide is engaged in H-bonding to the aniline-NH groups. All three H-bonding interactions are directed to the α-nitrogen of the azide; for 2NMe2, the NH-Nazide distances range 2.919–3.049 Å—consistent with moderate-strength H-bonding interactions.16 The Zn-N3(equatorial) bond lengths reflect the electronic perturbations of the TPA-ligand (2CF3 = 2.063(3); 2NMe2 = 2.1357(15) Å); in contrast, minimal variation is displayed within the Zn-N3(axial) bond (2CF3 = 2.055(3); 2NMe2 = 2.0741(15) Å). Furthermore, the bond distances for the previously reported (TPA)Zn(E3)2 (E3 = -N335 or -NCS36) compounds are consistent with the equatorial, but not axial azide units of 2R. This disparity suggests that the polarization of the axial azide ligand in 2R may be governed by H-bonding interactions rather than the overall electronic environment of the TPA-ligand. Vibrational spectroscopy was employed as a complementary metric to decipher azide polarization (similar to gauging CO activation of M-CO species),37 given the low precision in experimentally determined N—N distances.
The independent impact of H-bonding interactions and electronic character in 2R was evident by solid-state IR spectroscopy.38 Each complex displays two distinct ν(N3)asymm modes that were identified by DFT analysis as the ν(N3)-equatorial and ν(N3) -axial.39 In each complex, the H-bonded axial-azide is shifted to higher energy than that of the equatorial azide. Plots of the ν(N3)-equatorial and ν(N3)-axial shift for each complex verses their Hammett substituent constant (Figure 5) exhibit different slopes. The energies of ν(N3)-axial (with H-bonding) are minimally perturbed across the series of complexes (2CF3 = 2076; 2NMe2 = 2069;Δ = 7 cm−1) as compared to the energies of ν(N3)-equatorial (without H-bonding) (2CF3 = 2038; 2NMe2 = 2057; Δ = 19 cm−1) and is consistent with the trend in crystallographically determined Zn–N3(equatorial) bond distances. We propose that the H-bonds in 2R serve as the primary activating interaction for the axial azide, similar to previously reported (LOH)CuF (LOH = tris(6-hydroxy-2-methylpyridyl)amine), where halide (F-) binding was dictated by H-bonds, rather than a Cu(I)-F interaction.40 By extension, the intramolecular H-bonding interactions in 2R attenuate the electronic influence of ligand variation by reducing the covalency between Zn(II) and azide. We propose that this same phenomenon can be applied to rationalize the spectroscopy of 1R. The non-linear correlation of the O-O bond distances as well as the 1H NMR −NH resonances, with respect to Hammett constants, both imply the six H-bonding interactions to the O22− unit play a greater role in substrate activation than the electronic influence of the supporting TPA-ligand acting on the zinc center.
Figure 5.

Linear free energy relationship of ν(N3) (neat, ATR) and Hammett constants for 2R.
We have demonstrated that an H-bond appended tripodal zinc complex assembles to capture peroxide derived from dioxygen and electrons. This is the first example of a discrete (trans-1,2-peroxo)dizinc complex and its isolation was facilitated by H-bonding interactions. The captured peroxide can be liberated as dioxygen via two-electron chemical oxidation. The TPA derivatives, tris(6-(p-R-phenylamino-2-pyridylmethyl)amine, provide tunable secondary sphere H-bond donors that augment the stabilization of otherwise unstable Zn2O2 units. Analysis of a series of related zinc-diazide complexes revealed that H-bonding interactions serve as the primary component responsible for substrate capture and activation in the absence of a redox-active metal.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (1R01GM111486–01A1). N.K.S. is a Camille Dreyfus Teacher–Scholar. X-ray diffractometers were funded by the NSF (CHE 1625543). The authors thank Prof. Charles McCrory for assistance with O2 detection.
ASSOCIATED CONTENT
Supporting Information
Experimental details are available in the Supporting Information free of charge on the ACS Publications website.
The authors declare no competing financial interests.
REFERENCES
- (1).(a) Conte V; Bortolini O: Z. In The Chemistry of Peroxides, Rappoport Z, Ed; John Wiley & Sons: New York, 2006; Vol. 2, Part 2, pp 1053–1128; [Google Scholar]; (b) Dioxygen Activation and Homogeneous Catalytic Oxidation, 1st ed.; Simandi LI; Elsevier Science Publishers B.V.: New York, 1991; [Google Scholar]; (c) Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidation. Meunier B; Springer-Verlag; Berlin Heidelberg: 2000; [Google Scholar]; (d) Fu J; Cano ZP; Park MG; Yu A; Fowler M; Chen Z, Adv. Mater 2017, 29, 1604685. [DOI] [PubMed] [Google Scholar]
- (2).Cook SA; Borovik AS, Acc. Chem. Res 2015, 48, 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB, Chem. Rev 2017, 117, 2059–2107; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Suzuki M, Acc. Chem. Res 2007, 40, 609–617; [DOI] [PubMed] [Google Scholar]; (c) Kim SO; Sastri CV; Seo MS; Kim J; Nam W, J. Am. Chem. Soc 2005, 127, 4178–4179. [DOI] [PubMed] [Google Scholar]
- (4).Takayuki K; Takashi F; Ayumi T; Mitsuru T; Yoshihiro M; Tomoya M, Chem. Lett 2010, 39, 136–137. [Google Scholar]
- (5).Lopez N; Graham DJ; McGuire R Jr.; Alliger GE; Shao-Horn Y; Cummins CC; Nocera DG, Science 2012, 335, 450–453. [DOI] [PubMed] [Google Scholar]
- (6).Chmielewski M; Jurczak J, Tet. Lett 2004, 45, 6007–6010. [Google Scholar]
- (7).(a) Tutusaus O; Ni C; Szymczak NK, J. Am. Chem. Soc 2013, 135, 3403–3406; [DOI] [PubMed] [Google Scholar]; (b) Dahl EW; Szymczak NK, Angew. Chem. Int. Ed 2016, 55, 3101–3105; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tseng K-NT; Kampf JW; Szymczak NK, J. Am. Chem. Soc 2016, 138, 10378–10381; [DOI] [PubMed] [Google Scholar]; (d) Geri JB; Shanahan JP; Szymczak NK, J. Am. Chem. Soc 2017, 139, 5952–5956; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kiernicki JJ; Zeller M; Szymczak NK, J. Am. Chem. Soc 2017, 139, 18194–18197; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dahl EW; Dong HT; Szymczak NK, Chem. Commun 2018, 54, 892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) MacBeth CE; Golombek AP; Young VG; Yang C; Kuczera K; Hendrich MP; Borovik AS, Science 2000, 289, 938–941; [DOI] [PubMed] [Google Scholar]; (b) Syuhei Y; Teppei T; Akira W; Yasuhiro F; Tomohiro O; Koichiro J; Hideki M, Chem. Lett 2007, 36, 842–843; [Google Scholar]; (c) Feng G; Mareque-Rivas JC; Williams NH, Chem. Commun 2006, 1845–1847; [DOI] [PubMed] [Google Scholar]; (d) Tubbs KJ; Fuller AL; Bennett B; Arif AM; Berreau LM, Inorg. Chem 2003, 42, 4790–4791; [DOI] [PubMed] [Google Scholar]; (e) Hart JS; White FJ; Love JB, Chem. Commun 2011, 47, 5711–5713; [DOI] [PubMed] [Google Scholar]; (f) Yamaguchi S; Nagatomo S; Kitagawa T; Funahashi Y; Ozawa T; Jitsukawa K; Masuda H, Inorg. Chem 2003, 42, 6968–6970; [DOI] [PubMed] [Google Scholar]; (g) Taguchi T; Gupta R; Lassalle-Kaiser B; Boyce DW; Yachandra VK; Tolman WB; Yano J; Hendrich MP; Borovik AS, J. Am. Chem. Soc 2012, 134, 1996–1999; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Matson EM; Bertke JA; Fout AR, Inorg. Chem 2014, 53, 4450–4458; [DOI] [PubMed] [Google Scholar]; (i) Ford CL; Park YJ; Matson EM; Gordon Z; Fout AR, Science 2016, 354, 741–743; [DOI] [PubMed] [Google Scholar]; (j) Wallen CM; Palantinus L; Basca J; Scarborough CC Angew. Chem. Int. Ed 2016, 55, 11902–11906. [DOI] [PubMed] [Google Scholar]
- (9).(a) Yamaguchi S; Takahashi T; Wada A; Funahashi Y; Ozawa T; Jitsukawa K; Masuda H, Chem. Lett 2007, 36, 842–843; [Google Scholar]; (b) Saad FA; Knight JC; Kariuki BM; Amoroso AJ, Dalton Trans 2013, 42, 14826–14835; [DOI] [PubMed] [Google Scholar]; (c) Rivas JC; Prabaharan R; de Rosales RT; Metteau L; Parsons S, Dalton Trans 2004, 2800–2807; [DOI] [PubMed] [Google Scholar]; (d) Syuhei Y; Isao T; Yoko W; Yasuhiro F; Koichiro J; Hideki M, Chem. Lett 2003, 32, 406–407; [Google Scholar]; (e) Wallen CM; Bacsa J; Scarborough CC, J. Am. Chem. Soc 2015, 137, 14606–14609. [DOI] [PubMed] [Google Scholar]
- (10).(a) Forbes GC; Kennedy AR; Mulvey RE; Rowlings RB; Clegg W; Liddle ST; Wilson CC, Chem. Commun 2000, 1759–1760; [Google Scholar]; (b) Sobota P; Petrus R; Zelga K; Makolski L; Kubicki D; Lewiński J, Chem. Commun 2013, 49, 10477–10479. [DOI] [PubMed] [Google Scholar]
- (11).This reaction always produces (LH)Zn(OH)(OTf) as a side-product, the relative quantity of which can be diminished during the reaction by rapidly adding cobaltocene. See supporting information.
- (12).1H was the only product observed, even when using <1 equiv NiPr2Et, indicating zinc-hydroperoxide species are not accessible.
- (13).Masuda and coworkers were able to spectroscopically observe a Zn(OOH) with a similar ligand framework: Wada A; Yamaguchi S; Jitsukawa K; Masuda H, Angew. Chem. Int. Ed 2005, 44, 5698–5701.
- (14).Other than the cobaltocene reduction and PhICl2 oxidation, reactions were carried out on the benchtop with no effort to exclude air/moisture. 1H is quantitatively converted to (LH)Zn(OH)(OTf) on exposure to 1000 fold exess of methanol or H2O.
- (15).Adding equimolar [Bu4N][Cl] results in gradual degradation at room temperature and full degradation at 50 °C over 1 hour to unidentified species.
- (16).Steiner T, Angew. Chem. Int. Ed 2002, 41, 48–76. [Google Scholar]
- (17).Kiernicki JJ; Newell BS; Matson EM; Anderson NH; Fanwick PE; Shores MP; Bart SC, Inorg. Chem 2014, 53, 3730–3741. [DOI] [PubMed] [Google Scholar]
- (18).Takamizawa S; Nakata E.-i.; Akatsuka T; Kachi-Terajima C; Miyake R, J. Am. Chem. Soc 2008, 130, 17882–17892. [DOI] [PubMed] [Google Scholar]
- (19).Tao X; Daniliuc CG; Janka O; Pöttgen R; Knitsch R; Hansen MR; Eckert H; Lübbesmeyer M; Studer A; Kehr G; Erker G, Angew. Chem. Int. Ed 2017, 56, 16641–16644. [DOI] [PubMed] [Google Scholar]
- (20).Henthorn JT; Agapie T, Angew. Chem. Int. Ed 2014, 53, 12893–12896. [DOI] [PubMed] [Google Scholar]
- (21).Tsurumaki E; Sung J; Kim D; Osuka A, Angew. Chem. Int. Ed 2016, 55, 2596–2599. [DOI] [PubMed] [Google Scholar]
- (22).The degree of O-O activation in 1H is greater than terminal four-coordinate Zn-OOR examples of the tris(oxazolinyl)borate or β-diketiminate ligand frameworks, see: Mukherjee D; Ellern A; Sadow AD, J. Am. Chem. Soc 2012, 134, 13018–13026;Xu S; Everett WC; Ellern A; Windus TL; Sadow AD, Dalton Trans 2014, 43, 14368–14376.Pietrzak T; Korzyński MD; Justyniak I; Zelga K; Kornowicz A; Ochal Z; Lewiński J, Chem. – Eur. J 2017, 23, 7997–8005.
- (23).Jacobson RR; Tyeklar Z; Farooq A; Karlin KD; Liu S; Zubieta J, J. Am. Chem. Soc 1988, 110, 3690–3692. [Google Scholar]
- (24).Murthy NN; Karlin KD, J. Chem. Soc., Chem. Commun 1993, 1236–1238. [Google Scholar]
- (25).(TPA)Zn(OH) reacts with CO2 in air to form [(TPA)3Zn3(CO3)]4+.
- (26).(a) Lau N; Ziller JW; Borovik AS, Polyhedron 2015, 85, 777–782; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gordon Z; Drummond MJ; Matson EM; Bogart JA; Schelter EJ; Lord RL; Fout AR, Inorg. Chem 2017, 56, 4852–4863; [DOI] [PubMed] [Google Scholar]; (c) Jones JR; Ziller JW; Borovik AS, Inorg. Chem 2017, 56, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hansch C; Leo A; Taft RW, Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (28).The O 22− in 1NMe2 is disordered over two positions and the H-bonding interactions of the minor moiety are discussed in SI.
- (29).Voltammetry of 1H revealed an irreversible oxidation event (Epa = +1.14 V vs. Fc/Fc+).
- (30).Under these aqueous reaction conditions, the formation of [(LH)Zn(OH)]+ is a necessary competing reaction pathway, which diverts the 1H into oxidatively inert Zn(OH), and prevents a higher O2 yield.
- (31).(a) Karlin KD; Hayes JC; Hutchinson JP; Zubieta J, J. Chem. Soc., Chem. Commun 1983, 376–378; [Google Scholar]; (b) Pate JE; Ross PK; Thamann TJ; Reed CA; Karlin KD; Sorrell TN; Solomon EI, J. Am. Chem. Soc 1989, 111, 5198–5209. [Google Scholar]
- (32).In our previous studies of [(LR)2Cu2O2]2+, we noted vibrational spectroscopy did not reveal any 18O active modes—this was again the case for 1R. Raman spectroscopy revealed no changes on 18O labeling, see SI.
- (33).(a) Matson EM; Park YJ; Bertke JA; Fout AR, Dalton Trans 2015, 44, 10377–10384; [DOI] [PubMed] [Google Scholar]; (b) Reid SD; Wilson C; Blake AJ; Love JB, Dalton Trans 2010, 39, 418–425; [DOI] [PubMed] [Google Scholar]; (c) Tchertanov L, Acta Crystallogr. B 1999, 55, 807–809; [DOI] [PubMed] [Google Scholar]; (d) Tchertanov L, Supramol. Chem 2000, 12, 67–91; [Google Scholar]; (e) Xie J; Yikilmaz E; Miller A-F; Brunold TC, J. Am. Chem. Soc 2002, 124, 3769–3774. [DOI] [PubMed] [Google Scholar]
- (34).In solution, these complexes exhibit C3-symmetry and are likely rapidly interconverting between trigonal bipyramidal and octahedral via a hemilabile pyridyl arm. Variable temperature NMR (CD2Cl2) of 2H to −80 °C did not show coalescence to Cs symmetric species. For similar examples: See reference 9B and He Z; Craig DC; Colbran SB, J. Chem. Soc., Dalton Trans 2002, 4224–4235.
- (35).Synthesis and characterization of (TPA)Zn(N3)2 described in supporting information.
- (36).Duboc C; Phoeung T; Jouvenot D; Blackman AG; McClintock LF; Pécaut J; Collomb M-N; Deronzier A, Polyhedron 2007, 26, 5243–5249. [Google Scholar]
- (37).de la Cruz C; Sheppard N, J. Mol. Struct 1990, 224, 141–161. [Google Scholar]
- (38).Solution IR spectroscopy provided the same magnitude of shifts between equitorial and axial azido ligands. See supporting information.
- (39).Asymmetric modes were calculated for (TPA)Zn(N3)2 at 2221 and 2196 cm−1. The difference between the modes, 25 cm−1, is in agreement with experimental, 24.9 cm−1. See SI for details.
- (40).Moore CM; Szymczak NK, Chem. Commun 2015, 51, 5490–5492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.