

Acinetobacter baumannii has become an important pathogen in hospitals worldwide, where the incidence of these infections has been increasing. A. baumannii infections have become exceedingly difficult to treat due to a rapid increase in the frequency of multidrug- and pan-resistant isolates. This has prompted the World Health Organization to list A. baumannii as the top priority for the research and development of new antibiotics. This study reports for the first time a detailed analysis of aminoglycoside heteroresistance in A. baumannii. We define the mechanistic basis for heteroresistance, where the aadB(ant2″)Ia gene encoding an aminoglycoside adenylyltransferase becomes highly amplified in a RecA-dependent manner. Remarkably, this amplification of 20 to 40 copies occurs stochastically in 1/200 cells in the absence of antibiotic selection. In addition, we provide evidence for a second RecA-independent mechanism for aminoglycoside heteroresistance. This study reveals that aminoglycoside resistance in A. baumannii is far more complex than previously realized and has important implications for the use of aminoglycosides in treating A. baumannii infections.
KEYWORDS: Acinetobacter baumannii, aminoglycoside-modifying enzymes, gene amplification, heteroresistance
ABSTRACT
Heteroresistance is a phenomenon where a subpopulation of cells exhibits higher levels of antibiotic resistance than the general population. Analysis of tobramycin resistance in Acinetobacter baumannii AB5075 using Etest strips demonstrated that colonies with increased resistance arose at high frequency within the zone of growth inhibition. The presence of a resistant subpopulation was confirmed by population analysis profiling (PAP). The tobramycin-resistant subpopulation was cross resistant to gentamicin but not amikacin. The increased tobramycin resistance phenotype was highly unstable, and cells reverted to a less resistant population at frequencies of 60 to 90% after growth on nonselective media. Furthermore, the frequency of the resistant subpopulation was not increased by preincubation with subinhibitory concentrations of tobramycin. The tobramycin-resistant subpopulation was shown to replicate during the course of antibiotic treatment, demonstrating that these were not persister cells. In A. baumannii AB5075, a large plasmid (p1AB5075) carries aadB, a 2″-nucleotidyltransferase that confers resistance to both tobramycin and gentamicin but not amikacin. The aadB gene is part of an integron and is carried adjacent to four additional resistance genes that are all flanked by copies of an integrase gene. In isolates with increased resistance, this region was highly amplified in a RecA-dependent manner. However, in a recA mutant, colonies with unstable tobramycin resistance arose by a mechanism that did not involve amplification of this region. These data indicate that tobramycin heteroresistance occurs by at least two mechanisms in A. baumannii, and future studies to determine its effect on patient outcomes are warranted.
IMPORTANCE Acinetobacter baumannii has become an important pathogen in hospitals worldwide, where the incidence of these infections has been increasing. A. baumannii infections have become exceedingly difficult to treat due to a rapid increase in the frequency of multidrug- and pan-resistant isolates. This has prompted the World Health Organization to list A. baumannii as the top priority for the research and development of new antibiotics. This study reports for the first time a detailed analysis of aminoglycoside heteroresistance in A. baumannii. We define the mechanistic basis for heteroresistance, where the aadB(ant2″)Ia gene encoding an aminoglycoside adenylyltransferase becomes highly amplified in a RecA-dependent manner. Remarkably, this amplification of 20 to 40 copies occurs stochastically in 1/200 cells in the absence of antibiotic selection. In addition, we provide evidence for a second RecA-independent mechanism for aminoglycoside heteroresistance. This study reveals that aminoglycoside resistance in A. baumannii is far more complex than previously realized and has important implications for the use of aminoglycosides in treating A. baumannii infections.
INTRODUCTION
Antibiotic heteroresistance occurs when subpopulations of an isogenic bacterial strain exhibit decreased susceptibility to a particular antibiotic (1). Although the clinical significance of heteroresistance has been the subject of debate, resistant subpopulations of otherwise susceptible strains have been demonstrated to mediate treatment failure in animal models (2) and have been associated with treatment failure in human patients (3–6). Heteroresistance has been reported to a wide variety of antibiotics, including β-lactams, glycopeptides, and antimicrobial peptides (1, 7–9). However, reports of aminoglycoside heteroresistance are uncommon. In 1947, it was reported that populations of type b Haemophilus influenzae contained rare cells with increased streptomycin resistance, although the mechanism responsible for the formation of these cells was not investigated (10). More recently, decreased expression of the porin gene ompC was associated with nonmutational kanamycin-resistant subpopulations in Salmonella enterica (11). However, to our knowledge, heteroresistance to tobramycin or gentamicin has not been previously reported.
Acinetobacter baumannii is a Gram-negative, nosocomial, opportunistic pathogen (12–14). Widespread antibiotic resistance in this species recently led the World Health Organization to name carbapenem-resistant A. baumannii as its most critical priority pathogen for research and development of new interventions (15). Aminoglycoside resistance in A. baumannii has been associated with the acquisition, increased expression, and/or gene amplification of aminoglycoside-modifying enzymes and efflux pumps (13, 16, 17). The multidrug-resistant isolate AB5075 carries a number of antibiotic resistance genes, many of which are carried on the large plasmid p1AB5075. This plasmid includes an integron-like structure encoding four aminoglycoside-modifying enzymes, including the tobramycin resistance gene aadB, and a chloramphenicol resistance transporter (18). The plasmid also carries an additional tobramycin-modifying enzyme gene, aacA4 (18, 19). A recent study of loci required for tobramycin resistance in AB5075 showed that in addition to aadB and aacA4, 34 chromosomal genes also contribute to resistance to this drug (19).
AB5075 is resistant to tobramycin and gentamicin, meaning that its MIC values for these drugs are above the CLSI breakpoints (19, 20). Here, we report that AB5075 also exhibits tobramycin and gentamicin heteroresistance, as it produces subpopulations of cells that grow at concentrations of these drugs that are higher than the MIC for the general population. The subpopulations with increased resistance were shown to be unstable. We demonstrated that the integron-like structure of five adjacent antibiotic resistance genes, including aadB, becomes amplified to 20 to 40 copies in this resistant subpopulation. While this amplification was RecA dependent, colonies with increased tobramycin resistance could also be selected in a recA::Tc mutant. These resistant isolates did not contain amplifications of the region containing aadB, indicating that tobramycin heteroresistance can occur by at least two distinct mechanisms.
RESULTS
An AB5075 subpopulation exhibits increased tobramycin and gentamicin resistance.
When performing tobramycin Etest assays with the A. baumannii strain AB5075, we observed colonies arising at a high frequency within the zone of inhibition, consistent with a phenomenon termed heteroresistance (Fig. 1A). Heteroresistance was not observed during Etest assays with colistin, rifampin, or tetracycline (data not shown). In order to characterize the population with increased tobramycin resistance, colonies representative of this subpopulation were isolated by plating AB5075 on agar plates with various inhibitory concentrations of tobramycin. In general, colonies representing the resistant subpopulation were heterogenous in size on tobramycin plates but exhibited normal size on medium without drug. The colony size differences in the presence of tobramycin likely reflect differences in the levels of resistance. The frequency of resistant colonies decreased with increasing drug concentrations, so we chose a concentration twice the baseline MIC to determine the frequency. In three independent experiments, colonies with increased resistance arose at an average frequency of 0.52% ± 0.24%. Among the resistant subpopulation, both virulent opaque (VIR-O) and avirulent translucent (AV-T) colony variants were observed, as described previously (21, 22). Because VIR-O and AV-T variants were previously reported to exhibit subtle differences in tobramycin resistance (21, 23), both VIR-O and AV-T tobramycin-resistant colonies were selected for further characterization. These resistant isolates were designated hetR-O2, hetR-O3, hetR-T1, and hetR-T4.
FIG 1 .
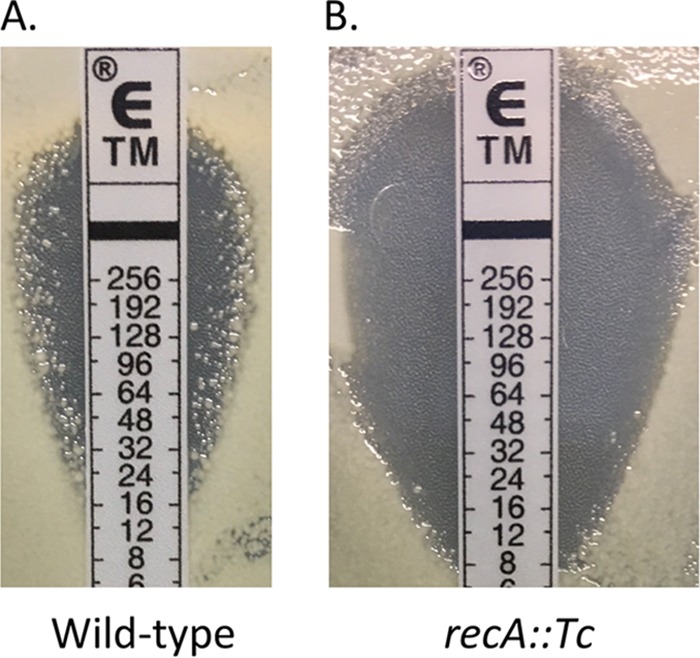
AB5075 produces a subpopulation of cells with increased resistance to tobramycin. Virulent opaque (VIR-O) wild-type (A) or recA::Tc (B) cells of AB5075 were grown to 1 × 107 CFU/ml, and 100 µl was plated on LB agar to obtain a lawn of growth. After plating, a tobramycin Etest strip was added, and the plate was incubated for 16 h at 37°C.
The subpopulation with increased tobramycin resistance was analyzed for cross-resistance to other antimicrobials. Etest assays were used to compare antimicrobial susceptibilities of the tobramycin-resistant isolates to those of wild-type VIR-O and AV-T variants with baseline levels of resistance. Etest assays for a variety of antimicrobials were conducted, revealing that hetR-O2, hetR-O3, hetR-T1, and hetR-T4 all exhibited cross-resistance to gentamicin but not amikacin (Table 1). In addition, cross-resistance to nonaminoglycosides was not observed (Table 1).
TABLE 1 .
The tobramycin-resistant subpopulation exhibits increased cross-resistance to gentamicin but not other antimicrobials
| Direct comparison of strains |
MIC (µg/ml) of druga: |
||||||
|---|---|---|---|---|---|---|---|
| TOB | CST | RIF | TET | AMK | CHL | GEN | |
| Comparison 1 | |||||||
| VIR-O | 48 | 1 | 4 | 3 | 192 | 256 | 96 |
| hetR-O2 | 128 | 1 | 4 | 3 | 192 | >256 | 192 |
| Comparison 2 | |||||||
| VIR-O | 64 | 1 | 4 | 3 | 96 | 96 | 32 |
| hetR-O3 | 192 | 1 | 4 | 3 | 96 | 128 | 128 |
| Comparison 3 | |||||||
| AV-T | 48 | 1 | 4 | 2 | 128 | 256 | 96 |
| hetR-T1 | 256 | 1 | 4 | 2 | 128 | 192 | >256 |
| Comparison 4 | |||||||
| AV-T | 32 | 1 | 4 | 3 | 64 | 96 | 32 |
| hetR-T4 | 384 | 1 | 4 | 3 | 64 | 96 | 128 |
Abbreviations: TOB, tobramycin; CST, colistin; RIF, rifampin; TET, tetracycline; AMK, amikacin; CHL, chloramphenicol; GEN, gentamicin. MICs were measured after 16 (TOB, CST, RIF, and TET) or 5 (AMK, CHL, and GEN) h of growth at 37°C. MICs were determined using Etest strips. Direct comparisons between isolates were performed by inoculating two cultures at an optical density A600 of 0.1 along the same Etest strip. Two independent experiments were performed for each comparison to assess reproducibility; data from a single representative experiment are shown.
The tobramycin-resistant subpopulation is not induced by tobramycin and is not composed of persister cells.
Further phenotypic characterization of the tobramycin-resistant subpopulation was performed. The ability of subinhibitory concentrations of tobramycin to induce the formation of the tobramycin-resistant subpopulation was first investigated. Population analysis profile (PAP) results for treated and untreated cultures of AB5075 were identical (Fig. 2A), suggesting that the formation of the tobramycin-resistant subpopulation was not increased by the presence of tobramycin. To confirm that the tobramycin-resistant subpopulation is capable of growing in the presence of antibiotic, a killing assay with AB5075 was conducted. This assay clearly demonstrated that the tobramycin-resistant subpopulation was capable of growing in high tobramycin concentrations, as the CFU counts increased during treatment (Fig. 2B). These data show that AB5075 forms a subpopulation with increased resistance to tobramycin that is distinct from persister cells, which are antibiotic tolerant at the expense of metabolic activity and active growth (24). Although exposure to a subinhibitory concentration of tobramycin does not increase the frequency of the resistant subpopulation, selection with a high level of drug kills off the majority of susceptible cells, allowing the resistant subpopulation to dominate the culture.
FIG 2 .

Characterization of tobramycin heteroresistance in AB5075. (A) Preincubation of AB5075 with a subinhibitory concentration of tobramycin had no effect on the population analysis profile (PAP) for this strain, indicating that increased resistance is not induced by exposure to this drug. (B) Incubation of AB5075 in liquid culture with 64 µg/ml of tobramycin resulted in killing of the majority of the population, while the CFU of the tobramycin-resistant subpopulation continued to increase. This indicates that the tobramycin-resistant subpopulation is distinct from persisters, which would be unable to replicate in the presence of drug. (C) Increased tobramycin resistance is unstable in liquid cultures. Following exposure of AB5075 to 64 µg/ml tobramycin, the majority of the surviving population exhibited increased tobramycin resistance. However, when tobramycin selection was removed, most of the cells in the population lost their increased resistance. (D) Twenty-four-hour stability of the tobramycin resistance phenotype in colonies was examined using stocks of hetR-O2 and hetR-T1. Colonies were plated from stock onto plates supplemented with tobramycin. Individual colonies were picked and passaged onto plates with 40 µg/ml tobramycin or with no tobramycin. After 24 h of growth, individual colonies were resuspended and the percentages of resistant cells within each colony were determined. Data represent the means and standard errors of means for four (hetR-T1) or three (hetR-O2) independent replicates. (E) Forty-eight-hour stability of the tobramycin resistance phenotype was assessed for stocks of hetR-O2 and hetR-T1. The experiment was performed as described for panel D but with colonies incubated on plates with and without 40 µg/ml of tobramycin for 48 h. Data represent the means and standard errors of means for two independent replicates.
Increased tobramycin resistance is an unstable phenotype.
The stability of the increased tobramycin resistance phenotype was first tested in liquid culture. Cultures containing a mix of VIR-O and AV-T cells were grown, treated with tobramycin, and then subcultured without drug. Serial dilutions of cultures at each step indicated that following subculture without drug, the population reverted from being almost entirely tobramycin resistant to containing only about 10% resistant cells (Fig. 2C). To determine whether VIR-O and AV-T representatives of the tobramycin-resistant subpopulation behave similarly in terms of their resistance stability, the hetR-O2 and hetR-T1 stocks were examined for loss of resistance on agar plates. Colonies of hetR-O2 and hetR-T1 were passaged from plates containing tobramycin onto plates with and without drug. After 24 h and 48 h of growth, individual colonies were resuspended and serially diluted onto plates with and without tobramycin to determine the percent resistance within individual colonies (Fig. 2D and E). Interestingly, hetR-O2 and hetR-T1 exhibited differing levels of resistance stability. At 24 h, the percent resistance values of hetR-T1 were similar for colonies grown with and without tobramycin, with about half of the cells from each condition exhibiting resistance. In contrast, hetR-O2 colonies grown without tobramycin for 24 h lost their increased resistance, whereas the majority of cells taken from colonies grown with tobramycin remained resistant (Fig. 2D). In colonies at 48 h, the frequency of resistant cells for both hetR-O2 and hetR-T1 continued to drop (Fig. 2E). Taken together, these results show that increased tobramycin resistance in AB5075 is an unstable phenotype, although the level of instability varies within the tobramycin-resistant subpopulation.
Tobramycin-resistant subpopulations exhibit increased expression of aadB.
The mechanism of tobramycin heteroresistance in AB5075 was investigated by performing quantitative reverse transcriptase PCR (qRT-PCR) on hetR-O2 and hetR-T1, as well as the parental AB5075 VIR-O and AV-T variants not exposed to tobramycin. Two genes, aadB and aacA4, known to confer tobramycin resistance were selected for gene expression analysis (19). Differences in aadB or aacA4 expression between the wild-type VIR-O and AV-T variants were not observed (Fig. 3A and B). However, hetR-O2 and hetR-T1 exhibited 5- and 15-fold-increased expression of aadB, respectively, compared to VIR-O (Fig. 3A). No differences in expression were observed for aacA4 (Fig. 3B). These results are consistent with the aminoglycoside resistance profiles of hetR-O2 and hetR-T1, as aadB is associated with resistance to both tobramycin and gentamicin but not amikacin (25).
FIG 3 .

Increased expression of aadB in cells with increased tobramycin resistance. Expression of aminoglycoside resistance genes aadB (A) and aacA4 (B) was quantified by qRT-PCR. Data are presented as the averages and standard errors of means of three independent biological replicates (*, P < 0.05, and ****, P < 0.0001, relative to AB5075 VIR-O by one-way analysis of variance with Dunnett’s posttest).
Tobramycin-resistant subpopulations contain a highly amplified region that includes aadB.
To determine whether the preexisting tobramycin-resistant subpopulation in AB5075 could be due to gene amplification, we measured gene copy number of the region surrounding aadB. The aadB gene is found on the large plasmid p1AB5075 and is carried adjacent to four other resistance genes that are all flanked by two copies of an integrase (intI) gene in the same orientation (Fig. 4). We hypothesized that if an amplification event were occurring, it would be within the interval flanked by the intI genes, possibly facilitated by recombination between the intI genes on adjacent plasmids during DNA replication. Gene copy number was measured by quantitative PCR (qPCR) using genomic DNA (gDNA) from the hetR-O2 and hetR-T1 isolates, as well as gDNA generated from VIR-O cells not exposed to tobramycin. The relative copy numbers of the strB and aadB genes (located immediately inside the interval flanked by intI) and the ABUW_4052 and ABUW_RS19335 genes (located immediately outside the intI region) were normalized to levels of aacA4, which is carried outside this region and served as a control for changes in plasmid copy number. In both the hetR-O2 and hetR-T1 isolates, the strB and aadB genes were highly amplified, whereas the ABUW_4052 and ABUW_RS19335 genes were not amplified (Table 2). As presented previously in Fig. 2D and E, cells lost tobramycin resistance when grown in the absence of antibiotic. Consistent with this loss of resistance, the levels of aadB amplification were reduced in colonies at 24 h in the absence of selection (Table 3). These results strongly suggest that tobramycin heteroresistance in AB5075 is due to the amplification of aadB.
FIG 4 .

Amplified region in p1AB5075 that includes aadB. The genes surrounding aadB on p1AB5075 are shown. Gene annotations are based on the most recent sequence annotation of p1AB5075 available on NCBI (accession NZ_CP008707.1); some small annotated open reading frames are not shown. Based on qPCR results, open reading frames annotated in green were amplified in the tobramycin-resistant subpopulation, whereas open reading frames annotated in blue were not changed. The two integrase genes annotated in gray were not examined by qPCR.
TABLE 2 .
hetR-O2 and hetR-T1 exhibit gene amplifications that include aadB
| Strain | Relative copy no. (mean ± SD) of genea: |
|||
|---|---|---|---|---|
| ABUW_4052 | strB | aadB | ABUW_RS19335 | |
| VIR-O | 1.31 ± 0.09 | 2.94 ± 0.07 | 2.45 ± 0.06 | 0.72 ± 0.01 |
| hetR-O2 | 1.80 ± 0.53 | 20.26 ± 6.18* | 17.23 ± 6.80* | 0.83 ± 0.31 |
| hetR-T1 | 1.44 ± 0.04 | 55.62 ± 9.27* | 44.07 ± 8.26* | 0.64 ± 0.03 |
Relative copy number of genes surrounding aadB measured in three biological replicates. Relative copy numbers were measured by qPCR using normalization to aacA4, a presumed single-copy gene located on the same plasmid. Copy numbers of aadB and strB were significantly increased in hetR-O2 and hetR-T1 relative to VIR-O not exposed to tobramycin (*, P < 0.001 relative to VIR-O by two-way analysis of variance with Dunnett’s posttest).
TABLE 3 .
aadB copy number in the presence and absence of antibiotic selection
| Strain | Copy no. (mean ± SD) of genea: |
|
|---|---|---|
| aadB | ABUW_RS19335 | |
| hetR-O2 + Tob | 40.35 ± 0.62 | 0.72 ± 0.16 |
| hetR-O2 − Tob | 16.75 ± 0.16* | 0.71 ± 0.03 |
| hetR-T1 + Tob | 55.12 ± 4.35 | 0.72 ± 0.05 |
| hetR-T1 − Tob | 40.38 ± 10.71 | 0.77 ± 0.08 |
Copy numbers of aadB and ABUW_RS19335 relative to aacA4 in hetR-O2 and hetR-T1 colonies grown with (+) or without (−) tobramycin for 24 h. The means and standard deviations of two biological replicates are shown. The copy number of aadB was significantly decreased in hetR-O2 grown without tobramycin compared to this strain grown in the presence of drug (*, P < 0.05 by paired two-tailed t test).
To confirm the role of aadB in heteroresistance, we obtained three independent aadB::T26 transposon insertion mutants from the University of Washington library. However, PCR analysis of the mutants revealed that all three had two copies of aadB, a wild-type copy and a T26-disrupted copy (data not shown). Similarly, in our wild-type VIR-O AB5075 parent strain, qPCR analysis indicated that there were two copies of strB and aadB relative to the aacA4 gene (Table 2), which is in contrast to the published genome sequence, where a single copy of each gene is present (18). Southern blot analysis confirmed that the region between the intI genes was duplicated in our AB5075 parental strain (data not shown). Due to the duplication of aadB in our parental strain and the presence of a wild-type copy of aadB in the University of Washington library mutants, we have been unable to construct and test a defined aadB mutant.
Tobramycin heteroresistance can occur by RecA-dependent and -independent mechanisms.
To determine if the above amplification event between duplicated copies of the intI gene required homologous recombination, tobramycin heteroresistance was examined in a recA::Tc mutant. The recA::Tc mutant exhibited intrinsic levels of tobramycin resistance that were lower than the wild-type parent (Fig. 1B and Table 4). Although this strain still appeared heteroresistant, the frequency of tobramycin-resistant colonies arising in the zone of clearing was lower than in wild-type cells (Fig. 1B). Introduction of the wild-type recA gene into the recA::Tc mutant partially restored heteroresistance (see Fig. S1 in the supplemental material). Interestingly, when six tobramycin-resistant isolates from the recA::Tc mutant were tested for amplification of aadB, the copy number was similar to the recA::Tc parent strain (Table 4). This indicates that increased resistance arose in these isolates by a mechanism that did not involve aadB amplification. When three of these resistant isolates, 1-2, 1-4, and 1-10, were cultured for approximately 30 generations in the absence of tobramycin, the frequency of cells retaining tobramycin resistance was 11%, 23%, and 35%, respectively, demonstrating that these isolates are not stable mutants. The resistant subpopulation in the recA::Tc mutant consisted of both VIR-O and AV-T cells (data not shown).
TABLE 4 .
Relative copy number of aadB in recA::Tc isolates with increased tobramycin resistance
| Strain | aadB relative copy no.b | MIC (µg/ml) |
|---|---|---|
| recA::Tc | 2.49a | 8 |
| hetR-recA-1-1 | 3.34 | 16 |
| hetR-recA-1-2 | 3.15 | 64 |
| hetR-recA-1-3 | 2.85 | 16 |
| hetR-recA-1-4 | 2.64 | 32 |
| hetR-recA-1-5 | 2.63 | 16 |
| hetR-recA-1-10 | 2.72 | 96 |
The recA::Tc control sample was used as a control in three independent experiments; the value shown represents the average relative copy number across the three experiments.
Relative copy number of aadB normalized to aacA4 was measured using gDNA isolated from a recA transposon mutant and 6 independently isolated derivatives with increased tobramycin resistance. Data shown are from a single sample for each strain. None of the tobramycin-resistant recA::Tc isolates exhibit increased copy number of aadB, suggesting that gene amplification of aadB in wild-type cells is RecA dependent.
Complementation of recA leads to increased heteroresistance in the recA::Tc mutant. The recA::Tc strain was complemented by introducing the recA gene on a multicopy plasmid. Lawns were inoculated by spreading 2 × 106 CFU of each strain on an LB plate. Lawns were overlaid with an Etest strip, incubated at 37°C for 16 h, and photographed. Download FIG S1, PDF file, 0.5 MB (546KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To confirm that the tobramycin-resistant subpopulation in the recA::Tc mutant was capable of growing in the presence of antibiotic, a tobramycin killing assay was conducted (Fig. S2A). This indicated that the tobramycin-resistant subpopulation was capable of growing in the presence of drug and was not composed of persister cells. In addition, PAP analysis demonstrated that pretreatment with tobramycin did not alter the frequency of cells that became resistant to tobramycin in the recA::Tc background (Fig. S2B).
Characterization of tobramycin heteroresistance in a recA mutant. (A) Incubation of the recA::Tc mutant in liquid culture with 16 µg/ml of tobramycin resulted in growth of the resistant subpopulation over time. (B) Population analysis profile (PAP) of the recA::Tc mutant with or without preincubation (3 h) in tobramycin. Download FIG S2, PDF file, 0.1 MB (105.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
This study demonstrates that subpopulations of A. baumannii AB5075 exhibit heterogeneous levels of resistance to aminoglycosides. The subpopulation with increased tobramycin resistance can be visualized using both an Etest MIC assay and PAP analysis. Application of selective concentrations of tobramycin resulted in the outgrowth of the tobramycin-resistant subpopulation, allowing it to dominate the surviving culture. However, once selective pressure was removed, the majority of the subpopulation returned to baseline levels of resistance. Independent isolates from the resistant subpopulation were found to exhibit increased expression and amplification of the 2″-aminoglycoside nucleotidyltransferase gene aadB. This gene is likely involved in heteroresistance as the substrate profile of this enzyme matches the resistance profile observed, i.e., resistance to tobramycin and gentamicin but not amikacin.
The role of phenotypic heterogeneity in the formation of subpopulations that are able to survive antibiotic treatment is well appreciated. This has been best studied in the case of dormant persister cells and slow-growing small-colony variants (SCVs), both of which exhibit increased tolerance to antibiotics, including aminoglycosides (26–29). Both persistence and SCV formation confer antibiotic tolerance at the expense of normal growth. In contrast, heteroresistance involves the formation of a subpopulation of cells with increased antibiotic resistance, which maintain the ability to actively grow during antibiotic exposure (1). Although heteroresistance has been reported in many species, including A. baumannii (30, 31), reports of aminoglycoside heteroresistance are exceedingly rare. This study is the first report of aminoglycoside heteroresistance in A. baumannii and to our knowledge only the second definitive report of this phenomenon in any species. In Salmonella enterica, aminoglycoside heteroresistance has been shown to occur when a subpopulation of cells expresses decreased levels of the porin gene ompC. This limits uptake of kanamycin into the cells, causing increased resistance (11).
This study demonstrates that tobramycin heteroresistance can arise by the extensive amplification (20 to 40 copies) of a region carried on the large p1AB5075 plasmid that includes five resistance genes in tandem, including aadB, flanked by copies of an integrase (intI) gene. The mechanism by which the aadB-containing region gets extensively amplified is unclear, but our work has established that RecA is required. Duplications can occur by nonequal recombination between directly repeated regions on adjacent replicons (32). Gene amplification of the aminoglycoside-modifying enzyme gene aphA1 has previously been reported to cause unstable tobramycin resistance in A. baumannii AB0057 and a clinical isolate (16). However, in that study, amplifications were dependent on prior exposure to tobramycin and were likely selected for by the antibiotic. Heteroresistance resulting from gene amplification of a chromosomal locus has also been reported in Salmonella enterica, where amplification of a region containing pmrD conferred colistin heteroresistance (33). Our data suggest that extensive amplifications preexist in 1/200 cells (i.e., the frequency of the resistant subpopulation) in the absence of any selective tobramycin pressure. As extensive gene amplifications typically require growth in the presence of selective pressures, this suggests that additional mechanisms, such as the rolling-circle-dependent generation of tandem arrays, may contribute to amplification of this region in A. baumannii (32). In a recA::Tc mutant, cells with increased resistance still arose, but none of the isolates examined contained duplications of the aadB gene (Table 4). Therefore, heteroresistance can occur by at least one additional mechanism. A recent study by Gallagher et al. demonstrated that at least 32 chromosomally carried genes in AB5075 function to maintain intrinsic tobramycin resistance (19). In principle, amplification of any of these genes could potentially lead to increased tobramycin resistance and may account for the resistant subpopulation that does not contain amplification of aadB. However, if this amplification is occurring, it does not appear to require RecA.
When the stability of the tobramycin-resistant subpopulation was examined, contrasting results were found for the virulent opaque (VIR-O) and avirulent translucent (AV-T) isolates (Fig. 2D). As long as selection was maintained, the majority of hetR-O2 cells remained tobramycin resistant; however, resistance was lost in approximately 90% of the population in colonies grown without drug for 24 h. In contrast, cells of hetR-T1 lost their increased resistance at roughly the same rate regardless of whether selection was maintained, with approximately 50% of the population in a 24-h colony maintaining the increased resistance state. However, in both isolates the loss of heteroresistance at high rates is consistent with the unstable nature of extensive duplications (32). The increased stability of heteroresistance in hetR-T1 in the absence of selection may be due to the larger number of tandem repeats that includes aadB (Table 2).
The clinical relevance of the aminoglycoside heteroresistance phenomenon described here remains to be determined. The plasmid-borne aadB gene is common in A. baumannii, and strains carrying this gene should exhibit clinically relevant resistance to tobramycin and gentamicin (34–37). Further research is needed to determine whether A. baumannii strains identified as being aminoglycoside sensitive also exhibit heteroresistance, which could pose problems for appropriately treating these infections. It is possible that strains lacking aadB could still exhibit heteroresistance by an aadB-independent mechanism, similarly to the recA::Tc mutant discussed in this work. The data presented here illustrate that the full picture of antibiotic resistance in A. baumannii is more complicated than has been traditionally recognized, with both the acquisition of resistance determinants and phenotypic heterogeneity contributing to resistance.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strains of A. baumannii were maintained at −80°C in 15% glycerol. Pure stocks of opaque and translucent variants were prepared as previously described (23). Liquid cultures were prepared in sterile LB broth, supplemented as needed with tobramycin (Sigma-Aldrich, St. Louis, MO) at the concentrations indicated. Resistance stability experiments and experiments to select isolates with increased resistance were performed using 0.5× LB supplemented with 0.8% agar. All other experiments were performed using regular LB supplemented with 1.5% agar. Plates were supplemented with tobramycin as indicated.
A T26 insertion mutant in recA was obtained from the AB5075 transposon mutant library maintained at the University of Washington (18). A culture of this strain was grown overnight at 37°C with shaking and used to prepared genomic DNA (gDNA) as outlined below. A culture of VIR-O AB5075 was grown at 37°C with shaking to late log phase and used to prepare electrocompetent cells by washing three times with 10% glycerol. The recA::T26 mutant DNA was electroporated into these cells, and transformants were selected on LB supplemented with 10 µg/ml tetracycline. A single colony was isolated and designated the recA::Tc strain. The presence of the recA mutation was confirmed by PCR.
Isolation of tobramycin-resistant subpopulations was conducted by plating serial dilutions of an early-log-phase culture of the wild-type or recA::Tc VIR-O variant on 0.5× LB and 0.5× LB containing tobramycin (Sigma-Aldrich, St. Louis, MO) at 2.5, 5, 10, 15, 20, 25, and 30 µg/ml. Colonies exhibiting increased resistance were apparent at 15 µg/ml tobramycin for wild type and at 5 µg/ml for the recA::Tc mutant. Resistant colonies were restreaked on 0.5× LB with tobramycin and examined under a stereomicroscope with oblique lighting to determine whether they were opaque or translucent variants.
Oligonucleotides.
All oligonucleotides used in this study are listed in Table S1 in the supplemental material.
Oligonucleotides used in this study. Download TABLE S1, PDF file, 0.1 MB (136.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Complementation of the recA mutant.
The wild-type recA gene was amplified by PCR using the primers oSA77 (5′-GCTCATCGTTTCGTTTGAAC-3′) and oSA78 (5′-GAATAAAAACGTCGAGTTGTG-3′) (Table S1). This fragment was then cloned into the SmaI site of pQF1266Blue, a derivative of pQF50 (38) where a hygromycin resistance gene has been cloned into the bla gene encoding β-lactamase. In addition, this plasmid contains an origin of replication from pWH1266 (39) cloned into the NcoI site. The resulting plasmid was designated precA.
MIC assays.
MICs of different antibiotics were measured using Etest strips (bioMérieux, Marcy-l’Etoile, France). For the tobramycin MICs used to visualize heteroresistance, AB5075 wild-type or recA::Tc VIR-O cells were inoculated into LB broth and grown at 37°C with shaking to a concentration of 1.1 × 107 to 1.3 × 107 CFU/ml. Lawns were inoculated by spreading 100 µl of culture onto an LB plate, followed by application of the Etest strip. The plate was photographed following incubation for 16 h at 37°C.
For MIC experiments reported in Table 1, strains were inoculated into LB broth, grown overnight statically at room temperature, grown at 37°C with shaking to an optical density at 600 nm (OD600) of 0.1, and stored at 4°C for use later in the day. Etest strips were placed on LB plates, and strains were inoculated next to each strip by spotting 10 µl of culture next to the bottom of the strip, tilting the plate so that the culture spread up the side of the strip, and removing excess culture at the top of the strip by pipetting. Two strains were inoculated on each side of each strip to facilitate a direct comparison of susceptibility. MIC values were recorded after incubation for 5 h or 16 h at 37°C, as noted. MIC experiments were performed two independent times to confirm the reproducibility of trends.
For the recA::Tc mutant, colonies that were growing in the zone of tobramycin inhibition were placed into a small vial of 20% glycerol and stored at −80°C. MIC experiments were performed by growing cells from the −80°C glycerol stock for several hours in LB and performing Etest assays as outlined above for the data presented in Table 1.
PAP.
Population analysis profile (PAP) analysis was performed by growing bacteria overnight to stationary phase and then plating serial dilutions on LB agar with or without various concentrations of tobramycin (Spectrum, New Brunswick, NJ). Plates were then incubated at 37°C, and CFU were enumerated after 24 h. Percent tobramycin resistance was calculated as the number of bacterial colonies that grew on tobramycin plates divided by the number of bacteria that grew on LB alone without drug.
Tobramycin killing assays.
Briefly, AB5075 was grown overnight to stationary phase in LB medium and serially diluted to 1 × 106 CFU/ml. Tobramycin (Spectrum) was added at a concentration of 64 µg/ml. One-hundred-microliter aliquots were taken at desired time points, serially diluted, and plated on LB medium alone (to quantify total CFU) or LB plates containing 64 µg/ml tobramycin (to quantify resistant CFU).
Stability measurements of the resistant subpopulation.
For experiments conducted with broth cultures, AB5075 was grown overnight to stationary phase in LB medium. The bacteria were then serially diluted and plated on LB agar plates with and without 64 µg/ml tobramycin (Spectrum) to enumerate total and resistant CFU for the pretreatment group (day 1). A subculture (1:1,000) was then grown overnight in LB supplemented with 64 µg/ml tobramycin, serially diluted, and plated on LB agar with or without 64 µg/ml tobramycin to enumerate total and resistant CFU for the treatment group (day 2). This process was repeated in LB broth without antibiotics (day 3), and dilutions were plated on agar plates with and without 64 µg/ml tobramycin to enumerate total and resistant CFU.
For experiments with hetR-O2 and hetR-T1 colonies, cells from the two subpopulations were struck from freezer stocks onto 0.5× LB supplemented with 20 µg/ml tobramycin (Sigma-Aldrich). Plates were incubated overnight at 37°C, and single colonies were resuspended in 1 ml LB and struck onto 0.5× LB agar with and without 40 µg/ml tobramycin. After 24 h or 48 h of incubation at 37°C, individual colonies were resuspended in 1 ml LB and serial dilutions were plated in duplicate on 0.5× LB with and without 40 µg/ml tobramycin. Plates were incubated for up to 48 h at 37°C, and colonies were enumerated to determine the percent tobramycin resistance by comparing the CFU on plates with and without tobramycin.
RNA isolation.
Cultures of different A. baumannii AB5075 subpopulations were grown in LB medium at 37°C with shaking to an OD600 of 0.5. Cells were pelleted by centrifugation, and RNA was isolated using the MasterPure RNA purification kit according to the manufacturer’s protocol (Epicentre, Madison, WI). Contaminating DNA was degraded by two treatments with Turbo DNA-free according to the manufacturer’s protocol (Invitrogen, Waltham, MA). DNA contamination was evaluated by PCR with purified RNA as the template, and RNA concentration was measured with a NanoDrop ND-1000 spectrophotometer.
qRT-PCR.
Total RNA (1 µg) was used to prepare cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) with random primers and either iScript or SuperScript III (Invitrogen, Waltham, MA) reverse transcriptase. Reactions with mixtures lacking reverse transcriptase were also performed as a control for the presence of contaminating DNA. Incubation conditions for cDNA synthesis were 25°C for 5 min, 42°C for 45 min, and 85°C for 5 min. cDNA reaction mixtures and controls were then diluted 1:10 with sterile water and used as a template for reverse transcriptase quantitative PCR (qRT-PCR). Oligonucleotide primer pairs for qRT-PCR were designed to amplify approximately 150-bp fragments from each gene of interest and were generated using the Primer-BLAST program available at https://www.ncbi.nlm.nih.gov/tools/primer-blast. qRT-PCR was performed using iQ Sybr green Supermix (Bio-Rad, Hercules, CA) on a Bio-Rad CFX Connect cycler. Cycle parameters were as follows: 95°C for 3 min, followed by 40 cycles of 95°C for 10 s, 55°C for 10 s, and 72°C for 20 s. Melt curve data were then collected to confirm the specificity of the oligonucleotide primer pairs. Data were generated using cDNA prepared from three independent RNA isolations, and qRT-PCRs were performed in technical triplicate to ensure accuracy. Fold changes in gene expression relative to the control strain (VIR-O) and a control gene (clpX) were determined using the threshold cycle (2−ΔΔCT) method (40).
Genomic DNA isolation.
For experiments presented in Table 2, cultures of different AB5075 subpopulations were grown in LB medium with shaking at 37°C to an OD600 of 0.5. For experiments presented in Table 3, colonies were grown for 24 h on 0.5× LB with or without 40 µg/ml tobramycin; individual colonies were then resuspended in 1 ml of LB. In both cases, cells were pelleted by centrifugation and resuspended in Tris-EDTA (TE). Cells were lysed by incubation with 0.5% SDS and 400 µg/ml proteinase K for 1 h at 37°C. Following lysis, NaCl was added to a final concentration of 0.7 M, and DNA was extracted twice with equal volumes of phenol-chloroform-isoamyl alcohol. DNA was precipitated by mixing with 1.5 volumes of 95% ethanol until a precipitate formed. DNA pellets were collected by centrifugation, washed twice with 75% ethanol, dried, and resuspended in molecular-grade water.
qPCR.
Concentrations of gDNA samples were determined using a NanoDrop ND-100 spectrophotometer. Samples were diluted to a concentration of 15 µg/ml, and qPCR was performed as outlined above for cDNA samples. Standard curves using 10-fold serial dilutions of wild-type gDNA were used to ensure that primers exhibited similar efficiencies. Relative gene copy numbers normalized to aacA4 were determined using the equation 2−ΔCT (40).
Statistical analyses.
Statistical analyses were performed with Prism 7 (GraphPad Software, Inc., La Jolla, CA).
ACKNOWLEDGMENTS
This study was supported by a VA Merit award I01 BX001725 and a Research Career Scientist Award (IK6BX004470) from the Department of Veterans Affairs to P.N.R. D.S.W. is supported by a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease award, VA Merit award I01 BX002788, and National Institutes of Health (NIH) grant AI098800. S.E.A. and E.X.S. were both supported by T32 training grant AI106699 from the National Institutes of Health.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs.
We have no conflicts of interest to disclose.
REFERENCES
- 1.El-Halfawy OM, Valvano MA. 2015. Antimicrobial heteroresistance: an emerging field in need of clarity. Clin Microbiol Rev 28:191–207. doi: 10.1128/CMR.00058-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Band VI, Crispell EK, Napier BA, Herrera CM, Tharp GK, Vavikolanu K, Pohl J, Read TD, Bosinger SE, Trent MS, Burd EM, Weiss DS. 2016. Antibiotic failure mediated by a resistant subpopulation in Enterobacter cloacae. Nat Microbiol 1:16053. doi: 10.1038/nmicrobiol.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fusco DN, Alexander EL, Weisenberg SA, Mediavilla JR, Kreiswirth BN, Schuetz AN, Jenkins SG, Rhee KY. 2009. Clinical failure of vancomycin in a dialysis patient with methicillin-susceptible vancomycin-heteroresistant S. aureus. Diagn Microbiol Infect Dis 65:180–183. doi: 10.1016/j.diagmicrobio.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin S-Y, Chen T-C, Chen F-J, Chen Y-H, Lin Y-I, Siu LK, Lu P-L. 2012. Molecular epidemiology and clinical characteristics of hetero-resistant vancomycin intermediate Staphylococcus aureus bacteremia in a Taiwan medical center. J Microbiol Immunol Infect 45:435–441. doi: 10.1016/j.jmii.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Sola C, Lamberghini RO, Ciarlantini M, Egea AL, Gonzalez P, Diaz EG, Huerta V, Gonzalez J, Corso A, Vilaro M, Petiti JP, Torres A, Vindel A, Bocco JL. 2011. Heterogeneous vancomycin-intermediate susceptibility in a community-associated methicillin-resistant Staphylococcus aureus epidemic clone, in a case of infective endocarditis in Argentina. Ann Clin Microbiol Antimicrob 10:15. doi: 10.1186/1476-0711-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charles PGP, Ward PB, Johnson PDR, Howden BP, Grayson ML. 2004. Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 38:448–451. doi: 10.1086/381093. [DOI] [PubMed] [Google Scholar]
- 7.Napier BA, Band V, Burd EM, Weiss DS. 2014. Colistin heteroresistance in Enterobacter cloacae is associated with cross-resistance to the host antimicrobial lysozyme. Antimicrob Agents Chemother 58:5594–5597. doi: 10.1128/AAC.02432-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiramatsu K, Aritaka N, Hanaki H, Kawasaki S, Hosoda Y, Hori S, Fukuchi Y, Kobayashi I. 1997. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350:1670–1673. doi: 10.1016/S0140-6736(97)07324-8. [DOI] [PubMed] [Google Scholar]
- 9.Hung KH, Wang MC, Huang AH, Yan JJ, Wu JJ. 2012. Heteroresistance to cephalosporins and penicillins in Acinetobacter baumannii. J Clin Microbiol 50:721–726. doi: 10.1128/JCM.05085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander HE, Leidy G. 1947. Mode of action of streptomycin on type b Hemophilus influenzae: II. Nature of resistant variants. J Exp Med 85:607–621. doi: 10.1084/jem.85.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sánchez-Romero MA, Casadesús J. 2014. Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc Natl Acad Sci U S A 111:355–360. doi: 10.1073/pnas.1316084111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CR, Lee JH, Park M, Park KS, Bae IK, Kim YB, Cha CJ, Jeong BC, Lee SH. 2017. Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol 7:55. doi: 10.3389/fcimb.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baumann P. 1968. Isolation of Acinetobacter from soil and water. J Bacteriol 96:39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organization 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. http://who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 16.McGann P, Courvalin P, Snesrud E, Clifford RJ, Yoon EJ, Onmus-Leone F, Ong AC, Kwak YI, Grillot-Courvalin C, Lesho E, Waterman PE. 2014. Amplification of aminoglycoside resistance gene aphA1 in Acinetobacter baumannii results in tobramycin therapy failure. mBio 5:e00915-14. doi: 10.1128/mBio.00915-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon EJ, Balloy V, Fiette L, Chignard M, Courvalin P, Grillot-Courvalin C. 2016. Contribution of the ade resistance-nodulation-cell division-type efflux pumps to fitness and pathogenesis of Acinetobacter baumannii. mBio 7:e00697-16. doi: 10.1128/mBio.00697-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, Huse HK, Zurawski DV, Brittnacher MJ, Manoil C. 2015. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol 197:2027–2035. doi: 10.1128/JB.00131-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallagher LA, Lee SA, Manoil C. 2017. Importance of core genome functions for an extreme antibiotic resistance trait. mBio 8:e01655-17. doi: 10.1128/mBio.01655-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs AC, Thompson MG, Black CC, Kessler JL, Clark LP, McQueary CN, Gancz HY, Corey BW, Moon JK, Si Y, Owen MT, Hallock JD, Kwak YI, Summers A, Li CZ, Rasko DA, Penwell WF, Honnold CL, Wise MC, Waterman PE, Lesho EP, Stewart RL, Actis LA, Palys TJ, Craft DW, Zurawski DV. 2014. AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. mBio 5:e01076-14. doi: 10.1128/mBio.01076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tipton KA, Dimitrova D, Rather PN. 2015. Phase-variable control of multiple phenotypes in Acinetobacter baumannii strain AB5075. J Bacteriol 197:2593–2599. doi: 10.1128/JB.00188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chin CY, Tipton KA, Farokhyfar M, Burd EM, Weiss DS, Rather PN. 2018. A high-frequency phenotypic switch links bacterial virulence and environmental survival in Acinetobacter baumannii. Nat Microbiol 3:563–569. doi: 10.1038/s41564-018-0151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tipton KA, Farokhyfar M, Rather PN. 2017. Multiple roles for a novel RND-type efflux system in Acinetobacter baumannii AB5075. MicrobiologyOpen 6(2). doi: 10.1002/mbo3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol 6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw KJ, Rather PN, Hare RS, Miller GH. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massey RC, Buckling A, Peacock SJ. 2001. Phenotypic switching of antibiotic resistance circumvents permanent costs in Staphylococcus aureus. Curr Biol 11:1810–1814. doi: 10.1016/S0960-9822(01)00507-3. [DOI] [PubMed] [Google Scholar]
- 27.Edwards AM. 2012. Phenotype switching is a natural consequence of Staphylococcus aureus replication. J Bacteriol 194:5404–5412. doi: 10.1128/JB.00948-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shan Y, Lazinski D, Rowe S, Camilli A, Lewis K. 2015. Genetic basis of persister tolerance to aminoglycosides in Escherichia coli. mBio 6:e00078-15. doi: 10.1128/mBio.00078-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li P, Huang Y, Yu L, Liu Y, Niu W, Zou D, Liu H, Zheng J, Yin X, Yuan J, Yuan X, Bai C. 2017. Isolation and whole-genome sequence analysis of the imipenem heteroresistant Acinetobacter baumannii clinical isolate HRAB-85. Int J Infect Dis 62:94–101. doi: 10.1016/j.ijid.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, Liolios L. 2006. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50:2946–2950. doi: 10.1128/AAC.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersson DI, Hughes D. 2009. Gene amplification and adaptive evolution in bacteria. Annu Rev Genet 43:167–195. doi: 10.1146/annurev-genet-102108-134805. [DOI] [PubMed] [Google Scholar]
- 33.Hjort K, Nicoloff H, Andersson DI. 2016. Unstable tandem gene amplification generates heteroresistance (variation in resistance within a population) to colistin in Salmonella enterica. Mol Microbiol 102:274–289. doi: 10.1111/mmi.13459. [DOI] [PubMed] [Google Scholar]
- 34.Karah N, Dwibedi CK, Sjöström K, Edquist P, Johansson A, Wai SN, Uhlin BE. 2016. Novel aminoglycoside resistance transposons and transposon-derived circular forms detected in carbapenem-resistant Acinetobacter baumannii clinical isolates. Antimicrob Agents Chemother 60:1801–1818. doi: 10.1128/AAC.02143-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aliakbarzade K, Farajnia S, Karimi Nik A, Zarei F, Tanomand A. 2014. Prevalence of aminoglycoside resistance genes in Acinetobacter baumannii isolates. Jundishapur J Microbiol 7:e11924. doi: 10.5812/jjm.11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamidian M, Nigro SJ, Hall RM. 2012. Variants of the gentamicin and tobramycin resistance plasmid pRAY are widely distributed in Acinetobacter. J Antimicrob Chemother 67:2833–2836. doi: 10.1093/jac/dks318. [DOI] [PubMed] [Google Scholar]
- 37.Nigro SJ, Hall RM. 2016. Loss and gain of aminoglycoside resistance in global clone 2 Acinetobacter baumannii in Australia via modification of genomic resistance islands and acquisition of plasmids. J Antimicrob Chemother 71:2432–2440. doi: 10.1093/jac/dkw176. [DOI] [PubMed] [Google Scholar]
- 38.Farinha MA, Kropinski AM. 1990. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol 172:3496–3499. doi: 10.1128/jb.172.6.3496-3499.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunger M, Schmucker R, Kishan V, Hillen W. 1990. Analysis and nucleotide sequence of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 87:45–51. doi: 10.1016/0378-1119(90)90494-C. [DOI] [PubMed] [Google Scholar]
- 40.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complementation of recA leads to increased heteroresistance in the recA::Tc mutant. The recA::Tc strain was complemented by introducing the recA gene on a multicopy plasmid. Lawns were inoculated by spreading 2 × 106 CFU of each strain on an LB plate. Lawns were overlaid with an Etest strip, incubated at 37°C for 16 h, and photographed. Download FIG S1, PDF file, 0.5 MB (546KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Characterization of tobramycin heteroresistance in a recA mutant. (A) Incubation of the recA::Tc mutant in liquid culture with 16 µg/ml of tobramycin resulted in growth of the resistant subpopulation over time. (B) Population analysis profile (PAP) of the recA::Tc mutant with or without preincubation (3 h) in tobramycin. Download FIG S2, PDF file, 0.1 MB (105.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Oligonucleotides used in this study. Download TABLE S1, PDF file, 0.1 MB (136.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.