

Abstract
Neuroplasticity in the dorsal horn following peripheral nerve damage contributes critically to the establishment of chronic pain. The neurosecretory protein VGF (non-acronymic) is rapidly and robustly upregulated following nerve injury, and therefore peptides generated from it are positioned to serve as signals for peripheral damage. The goal of this project was to understand the spinal modulatory effects of the C-terminal VGF derived peptide TLQP-62 at the cellular level and gain insight into the function of the peptide in the development of neuropathic pain. In a rodent model of neuropathic pain, we demonstrate that endogenous levels of TLQP-62 increased in spinal cord, and its immunoneutralization led to prolonged attenuation of the development of nerve injury-induced hypersensitivity. Using multiphoton imaging of submaximal glutamate-induced Ca2+ responses in spinal cord slices, we demonstrate the ability of TLQP-62 to potentiate glutamatergic responses in the dorsal horn. We further demonstrate that the peptide selectively potentiates responses of high threshold spinal neurons to mechanical stimuli in singe-unit in vivo recordings. These findings are consistent with a function of TLQP-62 in spinal plasticity that may contribute to central sensitization following nerve damage.
Introduction
Neuropathic pain has proven resistant to current therapeutics, incurs a significant loss to quality of life, and is hallmarked by mechanical hypersensitivity (allodynia) [5,17,18]. The establishment of chronic pain involves changes in the synaptic function of circuitry in the dorsal horn of the spinal cord [22,29,29,35,47,50]. This maladaptive plasticity facilitates the transmission of nociceptive information to the brain. However, details of the signaling mechanisms initiating these neuroplastic events remain largely unknown.
In the context of peripheral neuropathic pain, it is rational to hypothesize that the initiating factors for maladaptive plasticity may derive from injured primary afferent neurons. Previously, we and others demonstrated rapid and persistent upregulation of the neurosecretory protein VGF (non-acronymic) in primary afferent neurons following peripheral nerve injury [30,37,41]. VGF undergoes proteolytic processing to yield multiple bioactive peptides [32] that may act as initiators of maladaptive neuroplasticity in the spinal cord following nerve injury.
Several peptides that contain the VGF C-terminus induce hyperalgesia or allodynia in the absence of nocifensive behaviors when administered intrathecally [14,37], suggesting that they contribute to central sensitization. However, their presence in spinal cord and their function in neuropathic pain have not been examined. The C-terminal peptide TLQP-62 is of particular relevance to maladaptive mechanisms of chronic pain as it is a known mediator of neuroplasticity in the hippocampus affecting learning, memory, depression, and adult neurogenesis [1,3,4,23,33,48,49]. Moreover, bath application of exogenous TLQP-62 has been shown to change the excitability of dorsal horn neurons [37]. In the present study, we examined the involvement of endogenous C-terminal VGF peptides in the development of nerve injury-induced hypersensitivity. We also addressed the hypothesis that TLQP-62 contributes to spinal neuroplasticity by examining its effects on glutamatergic signaling ex vivo and on responses of dorsal horn neurons to peripheral stimulation via in vivo recordings of single-unit activity.
Materials and Methods
Animals
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Minnesota Institutional Animal Care and Use Committee. Adult male ICR mice (20–30 g; Envigo) were used for behavioral, physiological, immunofluorescent, and biochemical studies. Some immunofluorescent studies were conducted in adult male Pirt-GCamp3 mice (20–25 g) (gift from Dr. Xizhong Dong, [27]). Physiological studies (imaging of calcium sensitive fluorescent dye, in vivo electrophysiology) were also conducted in adult male Sprague-Dawley rats (150–450 g). In vivo electrophysiology was performed in rats because of higher survival rates during prolonged in vivo recording, relative to mice.
Reagents
The generation and validation of the anti-AQEE30 antibody has been previously described [33,41]. All VGF peptides were synthesized by AAPPTec (Louisville, KY).
Behavioral analysis
Mechanical sensory assessment
Behavioral responses to mechanical stimulation were assessed using an electronic von Frey aesthesiometer (IITC Life Sciences, Woodland Hills, USA) by an experimenter blinded to the treatment. Mice were placed in glass enclosures on an elevated mesh screen and allowed to acclimate for 15–30 minutes. The probe was gently applied to the plantar surface of each hind paw until a brisk withdrawal response terminated application of pressure, which was recorded by the instrument. Mechanical withdrawal thresholds were measured before and at 1, 2, 3, 7, 12 and 19 days after surgery.
Spared Nerve Injury (SNI)
SNI was induced in mice according to the method described previously [43]. The left sciatic nerve and its three terminal branches were exposed under isoflurane anesthesia. The common peroneal and tibial nerves were ligated with a 5-0 silk suture and sectioned distal to the ligation. The sural nerve was left intact. Sham surgeries consisted of exposure of the sciatic nerve branches without any further manipulation.
Intrathecal injections of IgG
Within 1 h prior to SNI surgery, Protein A purified anti-AQEE30 [33], or control IgG, (0.5 or 1 μg / 5 μL) was delivered intrathecally (i.t.) by direct lumbar puncture in conscious adult male ICR outbred mice as described [13,21]. The mice were gently gripped by the iliac crest, and a 30-gauge, 0.5 inch needle connected to a 50 μL Luer-hub Hamilton syringe was used to deliver 5 μL of injectate into the subarachnoid space over 1–3s at the level of the cauda equina between the L5/L6 vertebrae. The duration of the procedure was approximately 15–30 s per mouse. All injections were confirmed by observation of a tail flick upon entry into the intrathecal space. Intrathecal injections of TLQP-62 were carried out in the same way (see Figure, Supplemental Digital Content 1).
Western blot
Mouse spinal cords were harvested by hydraulic extrusion. The lumbar enlargement was isolated and divided into ipsi- and contralateral halves before being homogenized in solubilization buffer (Tris-buffered saline, pH 7.4 containing 1% Triton X-100, 10 mM EGTA, 10 mM EDTA) containing protease inhibitors (Complete Mini, EDTA-free (Roche)) using a glass bead homogenizer. The homogenate was centrifuged at 14,000 G for 15 min at 4°C and the supernatant was collected. Samples were mixed with sample buffer (NuPage 4X sample buffer (Invitrogen, Grand Island, NY)) and heated to 95°C for 10 min. Samples from naïve mice were loaded at 45 μg protein per lane. For comparison a sample containing 25 μg protein was spiked with 0.125 pmol of TLQP-62. Samples from sham and SNI spinal cords were loaded with 35 μg protein per lane. Samples were loaded onto a 12% gel (NuPage Novex bis-tris, Invitrogen) and run at 150 volts for 1 hr. After removal from the cassette, the gel was incubated in 10% glutaraldehyde for 1 hr and rinsed with PBS before being transferred to PVDF membranes (Immobilin-FL, Millipore) using 120 mA current overnight at 4°C. Membranes were incubated in blocking buffer (LiCor Odyssey Blocking buffer (Invitrogen)) for 1 hr at room temperature, then incubated in primary antisera 48 hours at 4°C (guinea pig anti-AQEE30 serum [41], 1:10,000; mouse anti-actin 1:15,000 (Cell Signaling)). After rinsing with PBS containing 0.1% Tween-20, the membranes were incubated for 1 hr in secondary antisera (IR-Dye donkey anti-guinea pig 800cw or donkey anti-mouse 680rd, LiCor). After rinsing with PBS-Tween-20 the blot was placed in PBS until it was imaged using the LiCor Odyssey Imager. Quantification of band density was performed using the Gel Analysis tool in Fiji [42]. Results normalized to actin were averaged within group across two experiments, n = 3 animals per group per experiment.
Immunofluorescence
Analysis of VGF labeling in spinal primary afferent processes in naïve mice
To quantify the proportion of VGF found in spinal primary afferent processes, we used Pirt-GCamp3 mice that express a GFP variant in >96% of primary afferents [27]. Mice, were deeply anesthetized with isoflurane and perfused via the heart with calcium-free Tyrode’s solution (in mM: 116 NaCl, 5.4 KCl, 1.6 MgCl2·6H20, 0.4 MgSO4·7H2O, 1.4 NaH2PO4, 5.6 glucose, and 26 Na2HCO3) followed by fixative (4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer, pH 6.9). The spinal cord was removed, cryoprotected in 10% sucrose, and cryostat-sectioned into 14 μm sections that were slide-mounted. Sections were preabsorbed in blocking buffer (PBS containing 0.3% Triton-X 100, 1% BSA, 1% normal donkey serum) for 30 min without or with IB4 (5 μg/ml, Sigma-Aldrich, St. Louis, MO), followed by incubation overnight at 4°C with appropriate primary antibodies: chicken anti-GFP (1:1000 Abcam, ab13970, Cambridge, UK), guinea pig anti-AQEE30 primary antibodies (1:300, neat serum [41]), rabbit anti-CGRP (1:1000, ImmunoStar, Hudson, WI), goat anti-IB4 (1:500, Vector Laboratories, Burlingame, CA). After rinsing, the sections were then incubated overnight at 4°C with appropriate secondary antibodies Alexa Fluor 488 conjugated donkey-anti-chicken, Cy5 conjugated donkey-anti-guinea pig, Cy3 conjugated donkey-anti-rabbit, Alexa Fluor 488 conjugated donkey-anti-goat secondary antibodies (1:1000, Jackson ImmunoResearch). High resolution (pixel size: 250 nm x 250 nm) laser scanning confocal images were taken with a Nikon A1R (objective: 25X, NA 1.1) and captured digitally using Nikon Elements software (Nikon, Melville, NY; University Imaging Centers; University of Minnesota, Minneapolis, MN). Confocal images (optical slice = 1.08 μm; 5 slices/animal, 6 animals) were analyzed for the distribution of anti-AQEE30 immunoreactivity with relation to labeling for GFP, CGRP, and IB4 by auto thresholding each channel using Fiji’s default threshold algorithm and analyzing the number of overlapping and non-overlapping positive pixels.
Analysis of VGF labeling in spinal cord and L4 dorsal root ganglia (DRG) of SNI and sham-operated mice
For quantification of anti-AQEE30 immunoreactivity among sham- and SNI-operated mice, tissues from adult male ICR mice were prepared, processed and imaged as above with the exception that DRG tissues were stained with NeuroTrace (1:1000, Invitrogen). All analyses was performed by trained observers blinded to the experimental groups using Fiji. For quantification of labeling in DRG, NeuroTrace-labeled neuronal profiles with nuclei were outlined in 5–6 evenly spaced sections per DRG, and the mean grey value of anti-AQEE30 immunoreactivity in each profile was measured. The threshold intensity for identifying anti-AQEE30 immunoreactive neurons was defined as 3 standard deviations from the average mean grey value of three unlabeled DRG neurons within the image. Analysis of spinal cord dorsal horn anti-AQEE30 immunoreactivity consisted of calculating the mean grey value of anti-AQEE30 immunoreactivity following auto thresholding using Fiji’s default threshold algorithm. These values were used to calculate a ratio of anti-AQEE30 immunoreactivity between ipsilateral and contralateral dorsal horns which was then averaged and compared between groups.
Measurement of intracellular free calcium transients and analysis
Adult male rats and mice were deeply anesthetized with isoflourane (10%) and perfused transcardially with ice-cold high-sucrose artificial cerebral spinal fluid (aCSF) bubbled with (95% O2 / 5% CO2) and consisting of, in mM: NaCl 95; KCl 1.8; KH2PO4 1.2; CaCl2 0.5; MgSO4 7; NaHCO3 26; glucose 15; sucrose 50; kynurenic acid 1, at a rate of 8 ml/min. While maintaining perfusion at 4 ml/min the spinal cord was removed following laminectomy and transferred to a dissection chamber containing ice-cold, bubbled, high-sucrose aCSF. Meninges were removed and the roots trimmed. The lumbar enlargement was mounted on a 4% agarose block and 400 μm transverse sections were made on a vibratome (Leica VT 1200S, Wetzlar, Germany). Loading of cells with Flou-4-AM allowed visualization of free intracellular calcium transients. Slices were incubated at 37°C for 30 min with 175 μL of 20% Pluronic® F-127 in DMSO (Life Technologies, Eugene, OR) dissolved in 4 mL of aCSF consisting of, in mM: NaCl 127; KCl 1.8; KH2PO4 1.2; CaCl2 2.4; MgSO4 1.3; NaHCO3 26; glucose 15, and 5 μM Fluo-4-AM (Life Technologies) with 95% O2/5% CO2 blown over the surface of the incubant. To fluorescently label astrocytes, the slices were incubated in sulfurorhodamine 101 (SR-101, 1μM; Molecular Probes, Eugene, OR) added to the incubant at 33°C for 30 min. The slices were incubated in fresh aCSF for 30 min at room temperature prior to imaging. Imaging was carried out at room temperature in a stage-mounted imaging chamber (volume = ~1.5 mL) perfused with oxygenated aCSF or drugs via peristaltic pump at a flow rate of 3 mL/min and transit time of 2 min from the reservoir to the chamber. Due to the flow rate into, and volume of the imaging chamber, the effective concentration of applied drugs were dependent on the time of application. A 10 s application (500 μL bolus) introduced into the perfusion line yielded an effective concentration in the imaging chamber equal to 1/3 the initial concentration, while a bolus > 30 s yielded effective concentrations equal to the initial concentration. Thus, 10 s applications of 100 μM and 1 mM glutamate yielded effective concentrations of 33 μM and 333μM respectively, while application of peptides (> 30 s) were effectively applied at the initial concentration. Peptides were dissolved in DI water at a concentration of 1 mM, and stored at −20°C until dilution to final concentration in aCSF on the day of imaging. Single plane scanning two-photon images of the dorsal horn were collected by 800 nm IR excitation at a depth of 30–60 μm from the surface of the slice at a frame rate of 0.5 Hz for the duration of the experiment using the same imaging equipment as above. After completion of the two-photon time-lapse protocol a confocal image was captured to visualize SR-101 positive cells (astrocytes).
Time-lapse image files were analyzed using Fiji. Following image stabilization [24] and application of a fire lookup table, cellular profiles were identified for analysis based on morphology and responsiveness to 1 mM glutamate. Profiles were outlined and marked as regions of interest (ROI) using Fiji’s ROI manager. The mean Fluo-4 fluorescence intensity of individual ROIs was calculated for the duration of the experiment (35–40 min). Intensity values for individual ROIs during the ten images in the time-lapse sequence (20 s) immediately preceding drug exposure were averaged to obtain resting values. A maximum search function was used to identify peak response intensity in the next 100 frames (200 s). Response amplitude was calculated by subtracting the resting intensity value from the peak intensity value. The fold change from the baseline glutamate response was calculated using the formula: [(amplitude #2) − (amplitude #1)] / (amplitude #1). Profiles displaying spontaneous activity during the calculation periods were excluded from analysis. Analyzed profiles were presumed to be neuronal because they were negative for the astrocyte marker SR-101 and exhibited calcium transients in response to glutamate of similar amplitude and kinetics as those described previously for neurons as opposed to microglia [10,11].
Spinal cord electrophysiology
Adult male Sprague Dawley rats were deeply anesthetized with urethane (Sigma; 2 g/kg i.p.), intubated with PE-240 tubing (Becton Dickenson, East Rutherford, NJ), and mounted in a fixed head and spinal stereotaxic frame with body temperature maintained by a homeothermic blanket control unit. The dorsal surface of the L3-L6 spinal cord was exposed by laminectomy and bathed in 0.9% saline. A 5–10 MΩ stainless steel microelectrode (0.005 inch; FHC, Bowdoin, ME) driven by a microstep motor and controller (Burleigh, Newton, NJ) was advanced into the dorsal horn in search of spinal neurons with mechanical receptive fields (RFs) in the left hind paw. Extracellular signals were amplified (DAM-80, WPI, Sarasota, FL) and filtered for noise (Hum Bug, Quest Scientific, North Vancouver, Canada) before being digitally recorded with Spike 2 data acquisition software (CED, Cambridge, England). Mechanical stimuli were delivered manually in the form of brush (paint brush stroked on skin), pressure (large arterial clip), pinch (small arterial clip) delivered for 10 s each to the RF in the left hind paw with a 10 s inter-stimulus interval. The pinch stimulus was judged by experimenters to be frankly painful when applied to their skin, and the larger arterial clip was described as moderately intense pressure, but not painful. Responses to mechanical stimulation were calculated as the difference between the sums of spikes occurring during the 10 s stimulus, less the sum of spikes occurring in the preceding 10 s inter-stimulus interval. To test thermal responses, a feedback-controlled Peltier device (Thermal Devices Inc., Golden Valley, MN, 49 mm2 contact area) delivered a series of thermal stimuli (10 s) from a resting temperature of 30°C down to 10°C, back to 30°C, then up to 50°C. Thermal-evoked responses were calculated as the sum of spikes occurring during the stimulus, less the sum of spikes occurring during a 10 s baseline period preceding the cold and heat ramp. To test cooling responses 20 μl of acetone were applied to the receptive field and allowed to evaporate, responses were calculated as above. Baseline responses were recorded to three sequential sets of graded mechanical stimuli followed by a single set of thermal stimuli prior to application of TLQP-62 (50 nM) or vehicle (saline) to the dorsal surface of the spinal cord for 15–18 min. Following wash with saline, the mechanical and thermal stimuli were re-applied every 5 and 10 min for the next 30 min, and then every 10 and 20 min, respectively, until the end of the recording. Electrolytic lesions (anodal current, 25 PA, 15–40 s) through the tip of recording electrodes were routinely made at the end of recording sessions for subsequent histological verification of recording sites in the dorsal horn. Recordings were analyzed in Spike 2 and identified single units were classified as wide dynamic range (WDR) or high threshold (HT) as previously reported [9]. Briefly, HT units were classified based on a response to brush <0.2 Hz and a pinch response larger than the response to pressure. Units were classified as WDR if they responded to brush, pressure and pinch in a graded fashion.
Statistics
Statistical analysis was performed in GraphPad Prism 7 and utilized α=0.05. All data analyzed by ANOVA or Student’s t-test passed a test for normality. Behavioral data were analyzed by two-way repeated measure ANOVA with Dunnett’s post test. Immunofluorescence: Population analysis of DRG neuron anti-AQEE immunofluorescence was compared between surgical groups by Kolmogorov-Smirnov testing of their cumulative frequency distributions. Percent DRG neurons positive for anti-AQEE immunofluorescence and the mean ratio of anti-AQEE immunofluorescence mean grey value in ipsilateral/contralateral dorsal horns were compared by unpaired Student’s t-test. Western blot: The densities of anti-AQEE labeled bands were normalized to actin bands, and treatments were compared by unpaired Student’s t-test. Population analysis for Ca2+ transient imaging and in vivo recording: For physiological testing two standard deviations (SD) was used as the criteria to identify neuronal profiles (Ca2+ transient imaging) and spinal neurons (in vivo electrophysiology) with significantly altered responses relative to controls. Because 2SD represents 95% of normally distributed data, our utilization of α=0.05 identifies fold changes >2SD as significant. Proportions of profiles and units were compared using Fisher’s exact test. Experimenters blinded to the treatment performed testing and or analyses. Mean responses for Ca2+ transient imaging: The mean fold change from 1st response was calculated by averaging fold change from 1st response across all glutamate-responsive, SR-101(−), profiles from all slices exposed to a given pharmacological treatment that were derived from the same animal. The number of animals was used as the value of n and a minimum of 3 animals were used per group. To account for varying numbers of slices analyzed per animal, the group mean and variance was weighted using the formulas: , and . Treatments were compared using unpaired Student’s t-test (mouse) or one-way ANOVA with Dunnett’s post test (rat).
Results
Localization of C-terminal VGF peptides in dorsal horn and identification of spinal TLQP-62
The neurosecretory protein VGF is proteolytically processed ([32], Fig. 1A). VGF-derived peptides containing the C-terminus, including TLQP-62, are recognized by our in-house generated antibody, anti-AQEE30, which was raised against the 30 C-terminal amino acids of VGF (Fig. 1A) and has been previously characterized [33,41].
Figure 1. Spinal VGF is derived from multiple sources and the C-terminal peptide TLQP-62 is present in spinal cord.

A) The neurosecretory protein VGF undergoes proteolytic processing to yield multiple bioactive peptides. The C-terminal VGF peptide antibody, anti-AQEE30, was generated against a peptide containing the final 30 amino acids in the VGF sequence (AQEE30), and binds all VGF fragments containing the C-terminus. B) Western blot analysis of spinal cord lysates with anti-AQEE30 demonstrates the presence of TLQP-62 in the spinal cord (Lane S, spinal cord lysate spiked with 0.125 pmol TLQP-62 peptide standard; Lanes 1–3, spinal cord lysates from 3 naïve mice). C) Localization of anti-AQEE30 immunoreactivity to central primary afferent processes by GFP co-immunolabeling in Pirt-GCamp3 mice. Scale bars = 50 μm. D) Zoomed in image of the area outlined by the white box in C illustrates extensive colocalization of anti-AQEE30 and GFP immunolabeling. Scale bar = 10 μm. E) Localization of anti-AQEE30 immunoreactivity to central primary afferent processes by co-labeling for CGRP and IB4. Scale bars = 50 μm. F) Zoomed in image of the area outlined by the white box in E. Scale bar = 10 μm.
The presence of endogenous bioactive C-terminal VGF peptides in spinal cord has not been shown. We employed Western blot analysis of lumbar spinal cord lysates from naïve mice to examine C-terminal VGF fragments using anti-AQEE30. Figure 1B shows full length VGF as well as bands corresponding to C-terminal proteolytic fragments of decreasing molecular weight. To determine if any of the bands correspond to the peptide TLQP-62, one of the samples was spiked with a peptide standard (lane S). Direct comparison of VGF fragments in spinal cord lysates from three mice (lanes 1–3) with the spiked sample identified the fragment running at a molecular weight of ~6 kDa (arrow, Fig. 1B) as TLQP-62, demonstrating bioavailability in the spinal cord.
Spinal VGF originates from both primary afferent and intrinsic spinal cord neurons [37,41]. To quantify these potential sources of C-terminal VGF peptides, we utilized Pirt-GCamp3 mice, in which a variant of GFP is expressed in >96% of all primary sensory neurons and can be visualized using GFP immunolabeling [27]. Double-labeling of spinal cord sections with anti-AQEE30 and anti-GFP indicated extensive colocalization (Fig. 1C & D), and quantitative analysis of the spinal colocalization showed that 70% (SEM = 0.6%, n = 6 animals) of anti-AQEE30 immunoreactivity in the dorsal horn was localized in GFP-positive spinal primary afferent processes while the remaining 30% (SEM = 0.6%, n = 6 animals) was associated with other structures. We also evaluated the relationship of anti-AQEE30 immunoreactivity to labeling for the established markers of spinal primary afferent processes CGRP and IB4 (Fig. 1E & F). Quantitative analysis demonstrated that 28% (SEM = 3%, n = 6 animals) of anti-AQEE30 immunoreactivity colocalized with the marker of nonpeptidergic small diameter primary afferent neurons IB4. Additionally, 23% (SEM = 1.2%, n = 6 animals) of anti-AQEE30 immunoreactivity was localized within peptidergic primary afferent terminals, based on colocalization with CGRP immunoreactivity.
Immunoneutralization of C-terminal VGF peptides attenuates the development of nerve injury-induced tactile hypersensitivity
The involvement of endogenous VGF C-terminal peptides in the development of tactile hypersensitivity following peripheral nerve injury has not been demonstrated. We used immunoneutralization to test the hypothesis that endogenous C-terminal VGF peptides participate in the initiation of nerve injury-induced hypersensitivity. This neuropeptide immunoneutralization method has been demonstrated to work effectively in the hippocampus using anti-AQEE30 [33]. In addition, we have shown that anti-AQEE30 dose-dependently attenuates the previously reported hyperalgesia induced by intrathecal TLQP-62 [13] in the warm water tail withdrawal test (See Figure, Supplemental Digital Content 1). This observation demonstrates that anti-AQEE30 is able to interfere with the functional effects of TLQP-62, presumably through immunoneutralization of the peptide.
We administered anti-AQEE30 intrathecally immediately prior to peripheral nerve damage (SNI model). Mechanical withdrawal thresholds were compared in untreated SNI mice and SNI mice that received intrathecal injection of anti-AQEE30 (1 μg) or a non-specific guinea pig IgG (1 μg). Mice injected with anti-AQEE30 showed significantly higher ipsilateral paw withdrawal threshold on day 1 post-surgery compared to controls, and this effect persisted on days 2, 3, and 7 post-surgery (P<0.001, n = 4 animals per group, Two-way ANOVA; Fig. 2A).
Figure 2. Immunoneutralization of C-terminal VGF peptides delays the progression of peripheral nerve injury-induced mechanical hypersensitivity.
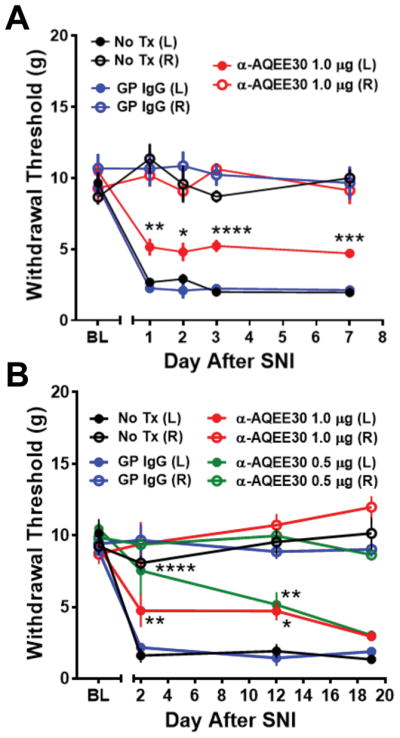
A) Spared Nerve Injury (SNI) mice treated with anti-AQEE30 at the time of injury show decreased mechanical hypersensitivity at 1, 2, 3 and 7 days post-injury compared to mice treated with control IgG (GP IgG) or untreated mice (No Tx). B) In a separate cohort of mice, mechanical hypersensitivity is reduced in SNI mice treated with anti-AQEE30 (0.5 or 1 μg) compared to controls for 12 days post injury but is fully manifest by 19 days post-surgery. Data are represented as mean +/− SEM. *, **, ***, **** = P<0.05, P<0.01, P<0.001, P<0.0001 compared to No Tx control by ipsilateral two-way ANOVA with Dunnett’s post test.
In an independent experiment a separate cohort of mice was tested over a prolonged time course to determine the duration of anti-AQEE30 action following a single intrathecal injection at the time of injury. Increased ipsilateral paw withdrawal thresholds persisted up to 12 days, but the effect was absent on day 19 post-surgery (P<0.001, n = 4 animals per group, two-way ANOVA with Dunnett’s post test; Fig. 2B). In this experiment, we also observed that a lower dose of anti-AQEE30 (0.5 μg) yielded similar attenuation of hypersensitivity, suggesting that both doses were within the range of maximally effective concentrations. Animals receiving intrathecal administration of non-specific IgG developed behavioral hypersensitivity indistinguishable from untreated controls (P>0.05, repeated measure two-way ANOVA with Dunnett’s post test). These results demonstrate for the first time that immunoneutralization of endogenous C-terminal VGF peptides at the time of injury delays the development of hypersensitivity.
Spinal TLQP-62 is elevated during the establishment of neuropathic pain
We next asked whether the level of spinal TLQP-62 increased following peripheral nerve damage. First, we quantified anti-AQEE30 immunoreactivity both in the ipsilateral L4 DRG as well as the dorsal horns of L3-L4 mouse spinal cord in sham- and SNI-operated mice 3 days post-surgery (Fig. 3A–D). Population analysis of anti-AQEE30 immunoreactivity in DRG was performed in 6 sham- and 6 SNI-operated mice. The representative confocal images in figure 3A show increased anti-AQEE30 immunoreactivity among DRG neurons of SNI mice relative to sham controls. This observation is quantified and displayed in figure 3B as a plot of cumulative frequency distributions of individual DRG neuron mean grey values. The rightward shift of the cumulative distribution of labeling intensity in DRG neurons from SNI mice (dashed line) relative to sham controls indicates increased anti-AQEE30 immunoreactivity after nerve injury (P<0.0001, Kolmogorov-Smirnov test). In addition to this population analysis, we found that the proportion of anti-AQEE30 labeled DRG neurons was higher in SNI- compared to sham-operated mice (sham, 27.1%, SEM = 4.0% n = 6 animals; SNI, 41.6%, SEM = 2.2% n = 6 animals, P<0.05, unpaired Student’s t-test). To quantify anti-AQEE30 immunoreactivity in the spinal cord dorsal horn, confocal images of both the ipsilateral and contralateral side were acquired (Fig. 3C). The ratio of the mean grey value of anti-AQEE30 immunoreactivity between the ipsilateral and contralateral dorsal horns was calculated and the mean across animals was compared between groups (Fig. 3D). This analysis demonstrated an increase in ipsilateral dorsal horn anti-AQEE30 immunoreactivity 3 days post SNI surgery relative to sham controls (P<0.01, n = 6 animals per group, unpaired Student’s t-test).
Figure 3. Spinal TLQP-62 is elevated during the establishment of neuropathic pain.

A) Representative confocal images of anti-AQEE30 immunoreactivity in ipsilateral DRG of mice 3 days post sham or SNI surgery. Scale bar: 100 μm. B) Cumulative frequency histogram of individual DRG neuron mean grey values (anti-AQEE30 immunoreactivity) identified in the L4 DRG of mice 3 days post sham (solid line, 6 mice) or SNI (dashed line, 6 mice) surgery. **** = P<0.0001, Kolmogorov-Smirnov test. C) Representative confocal images of anti-AQEE30 immunoreactivity in the contralateral and ipsilateral dorsal horn of mouse spinal cord at the level of L4 3 days post sham or SNI surgery. Scale bar: 50 μm. D) The average anti-AQEE30 immunoreactivity mean grey value ratio between the ipsilateral and contralateral L4 dorsal horn is elevated in SNI animals relative to sham controls 3 days post-surgery. ** = P<0.01, unpaired Student’s t-test. E) Representative Western blot with anti-AQEE30 shows C-terminal VGF fragments in ipsilateral spinal cord lysates from SNI and sham mice collected 3 days post-surgery. Full-length VGF is represented by a doublet of bands (arrowheads in Fig. 3E), where the lower molecular weight band is pro-VGF that is generated after the cleavage of the signal peptide. F) Densitometry analysis demonstrated that both full length VGF protein (left) and the C-terminal peptide TLQP-62 (right) are elevated in SNI animals compared to sham. Measurements for full-length VGF included both bands in the doublet and were performed using the low intensity image shown above the actin image.* = P<0.05, unpaired Student’s t-test. Data represented as mean +/− SEM.
We next performed Western blot densitometry analysis of C-terminal VGF fragments in ipsilateral lumbar spinal cord lysate from SNI- and sham-operated mice at day 3 post-surgery (Fig 3E and F). Images of anti-AQEE30-labeled membranes were collected at different intensity settings for optimal quantification of the high- and low-density bands, corresponding to full-length VGF and TLQP-62, respectively. Quantitative analysis of band intensity indicated that the bands corresponding to TLQP-62 (arrows, Fig. 3E) and full-length VGF (arrowhead, Fig. 3E) were significantly elevated in lysates from mice with SNI compared to sham controls (Fig. 3F, P<0.05, n = 6 animals per group, unpaired Student’s t-test).
TLQP-62 potentiates neuronal glutamate responses in the dorsal horn through a kinase-dependent mechanism
Having demonstrated a functional consequence of endogenous C-terminal VGF peptide immunoneutralization following peripheral nerve injury, as well as an injury-induced increase in spinal TLQP-62, we investigated potential mechanisms of action of TLQP-62 in enhancing nociceptive signaling in the spinal cord. Based on its modulation of excitatory neurotransmission in the hippocampus [1,4], we hypothesized that exposure to TLQP-62 potentiates glutamatergic responses in the superficial dorsal horn. To this end, we performed two-photon imaging of presumptive superficial dorsal horn neuronal profiles (see methods) loaded with the fluorescent Ca2+ indicator Fluo-4 in spinal cord slices from both mice and rats. A submaximal concentration of glutamate (100 μM, 10 s) was applied following a 2-min baseline period. After a subsequent 5-min washout period, the slice was incubated without (Fig. 4A) or with (Fig. 4B) TLQP-62 in the perfusate (aCSF). The response to a second application of submaximal glutamate was then recorded in order to quantify potentiation of glutamatergic responses. Exposure to 100 μM (10 s) glutamate yielded stable submaximal Ca2+ transients before and after the 15 min aCSF incubation period (Fig. 4A). In a significant proportion of Flou-4+/SR101− presumptive neuronal profiles, incubation with TLQP-62 resulted in increased amplitudes of glutamate-induced Ca2+ transients (Fig. 4B). Potentiation was defined as fold change from baseline that was > 2SD from the mean fold change observed in the control (aCSF) condition. Glutamate responses were potentiated in a significantly larger proportion of neuronal profiles from slices of mouse spinal cord exposed to 50 nM TLQP-62 compared to aCSF (aCSF: 9/147 profiles; 50 nM TLQP-62: 58/157 profiles; P<0.0001, Fisher’s exact test; Fig. 4C). In spinal cord slices from rat, incubation in 50 and 500 nM, but not 5 nM TLQP-62 significantly increased the proportion of Flou-4+ neuronal profiles with potentiated glutamate responses (Fisher’s exact test; Fig 4D). Proteolytic processing of TLQP-62 yields the bioactive peptides TLQP-21 and AQEE-30. Therefore, we tested whether these peptides alone or in combination reproduce the potentiating effects of TLQP-62. Incubation with 50 nM TLQP-21 (0/54 profiles), 50 nM AQEE-30 (2/58 profiles), or their combination (0/62 profiles) did not significantly increase the proportion of neuronal profiles with potentiated glutamatergic responses compared to aCSF (P>0.05, Fisher’s exact test).
Figure 4. TLQP-62 potentiates neuronal glutamate responses in the dorsal horn.

A & B) The ability of TLQP-62 to potentiate submaximal glutamate responses was tested by measuring the change in intensity of the Ca2+ indicator Fluo-4 following glutamate exposure before (1) and after (2) 15-min incubation with aCSF (A) or TLQP-62 (B). Responses to 100 μM glutamate were shown to be submaximal by subsequent application of 1 mM glutamate (3). Incubation in aCSF resulted in submaximal glutamate responses of similar amplitude (A), while incubation with TLQP-62 yielded a notable increase in the amplitude of the second glutamate response, relative to the first (B). Scale bars = 10 μm. C & D) Scatter plots of the fold change in submaximal glutamate response from all glutamate responsive, Fluo-4+ neuronal profiles relative to baseline. The mean and 2SD envelope of the aCSF group are depicted by dotted lines. The proportion of Fluo-4+ neuronal profiles exhibiting a fold change > 2SD (ratio indicated above scatter) is significantly elevated following incubation with TLQP-62 at ≥ 50 nM in neuronal profiles of the superficial dorsal horn in transverse spinal cord slices derived from both mice (C) and rats (D). This effect is eliminated in the presence of the kinase inhibitor K252a (D). Lines and bars represent mean +/− SD; **, **** = P<0.01, P<0.0001 compared to aCSF by Fisher’s Exact test. E & F) Mean fold change of fluorescent intensity averaged across all glutamate responsive profiles imaged per animal (# animals = n) for mice (E) and rats (F). Bars represent mean +/− SEM, means and variance weighted for the number of slices per animal; number below x-axis details the number of slices and (number of animals) used in each group; E, ** = P<0.01 unpaired Student’s t-test; F, **, *** = P<0.01, P<0.001 compared to aCSF by one-way ANOVA with Dunnett’s post test.
In the hippocampus, TLQP-62 induces neuroplasticity via a BDNF-dependent mechanism involving signaling through the TrkB receptor [1,4,33,48]. To test whether the potentiation of glutamate-induced Ca2+ transients by TLQP-62 was due to an active kinase-dependent event consistent with BDNF/TrkB signaling, we examined the effects of the classically used Trk inhibitor K252a [19,28,46]. K252a (200 nM), applied concomitantly with TLQP-62, prevented the peptide-induced increase in the proportion of presumptive neuronal profiles with potentiated glutamate responses (5/178 profiles, P>0.05, Fisher’s exact test compared to aCSF). Application of this broad kinase inhibitor (K252a 200 nM) alone did not alter the proportion of cells with potentiated glutamate induced Ca2+ transients compared to aCSF (2/121 profiles; P>0.05, Fisher’s exact test).
The raw data shown as a scatter plot in Figure 4C & D is recapitulated in Figure 4E & F as means averaged across all profiles within a slice and compared between groups utilizing the number of animals as n and weighted to account for the number of slices used per animal. This statistical analysis demonstrates the robustness of glutamate-induced Ca2+ responses by TLQP-62. We found a significant increase in the mean fold change of peak glutamate responses between 50 nM TLQP-62 (1.38 +/− 0.27, n = 5 animals) and aCSF (−0.05 +/− 0.04, n = 4 animals; P<0.01, unpaired Student’s t-test) in spinal cord slices from mice (Fig. 4E). Similarly, in rats we found a significant increase in the mean fold change of peak glutamate responses between 50 nM and 500 nM, but not 5 nM, TLQP-62 compared to aCSF (aCSF: 0.02 +/− 0.08 n = 6 animals; 5 nM TLQP-62: 0.05 +/− 0.15 n = 4 animals; 50nM TLQP-62: 1.18 +/− 0.29 n = 4 animals; 500 nM TLQP-62: 0.79 +/− 0.18 n = 3 animals, P<0.001, one-way ANOVA with Dunnett’s post test, Fig 4D). Further, there was no significant difference in mean fold change of peak glutamate responses between aCSF and 500 nM TLQP-62 + 200 nM K252a (0.14 +/− 0.14 n = 4 animals, P>0.05), nor 200 nM K252a alone (0.15 +/− 0.14 n= 5 animals, P>0.05). Taken together, these results demonstrate the ability of TLQP-62 to robustly induce kinase-dependent potentiation of glutamatergic signaling in the dorsal horn.
TLQP-62 increases responses of high threshold dorsal horn neurons to moderate mechanical stimulation in vivo
We next examined the effects of TLQP-62 on the response properties of dorsal horn neurons in vivo. We analyzed the responses of single units recorded from rat spinal dorsal horn along the experimental timeline outlined in Figure 5A. All histologically recovered recording sites were within the dorsal horn (examples shown in Fig. 5B). Fifteen high threshold (HT) and 23 wide dynamic range (WDR) units with low rates of spontaneous activity (<1 Hz) were analyzed. We assessed the effect of 50 nM TLQP-62 or control (saline) treatment on single-unit spiking frequency in response to mechanical stimuli. The baseline responses prior to application of saline or TLQP-62 were not significantly different between the two experimental groups among HT or WDR units (Fig 5C; P>0.05, repeated measures two-way ANOVA with Bonferroni-corrected post-hoc test).
Figure 5. TLQP-62 increases responses of high threshold dorsal horn neurons to moderate mechanical stimulation.

A) Experimental outline. Spinal single units recorded in vivo were tested for response to successive 10 s mechanical stimulation in the form of brush (B), pressure (Pr) and pinch (Pi) with a 10 s inter-stimulus-interval. Following three sets of baseline responses, TLQP-62 (50 nM) or vehicle was applied to the exposed spinal cord for 15 minutes, after which responses were assessed for up to 1 h. B) Histologically identified recording sites. C) Baseline responses of high threshold (HT) and wide dynamic range (WDR) units were not significantly different between TLQP-62 and vehicle exposed groups (P > 0.05; Fisher’s Exact test). The number of units of each type is shown in parentheses. D) The response of single units was deemed significantly altered if the fold change relative to baseline response was > 2SD of that observed in the vehicle group. A significantly larger proportion of HT but not WDR units exposed to TLQP-62 exhibited an increase in response to pressure, relative to controls; *=P<0.05 Fisher’s Exact test. E) Representative example of HT units exposed to saline (left) and TLQP-62 (right). Note the emergence of a pressure response following exposure to TLQP-62.
Units were categorized as significantly altered by the experimental treatment if the fold change from baseline was > 2SD from the mean change observed in the saline group at the equivalent time point (Fig 5D). This analysis indicated that HT units were significantly more responsive to moderate mechanical stimuli after exposure to 50 nM TLQP-62 relative to saline exposure (P<0.05, Fisher’s exact test), but not to pinch (P>0.05, Fisher’s exact test). In contrast to HT units, the proportions of WDR units with altered activity were not different between the 50nM TLQP-62 and saline-exposed groups for any of the modalities tested (P > 0.05, Fisher’s exact test). Figure 5E illustrates the enhanced response properties of HT units to a moderate mechanical stimulus following exposure to TLQP-62. These in vivo single unit recordings demonstrate the ability for TLQP-62 to enhance nociceptive information transfer in the dorsal horn.
With regard to thermal stimuli, of the 15 HT units recorded, at baseline three responded to acetone cooling alone, one responded to cold (10°C) alone and six responded to heat (50°C) alone. Whereas among the 23 WDRs recorded, at baseline four responded to acetone cooling alone, none responded to cold, six responded to both acetone cooling and heat, three responded to heat only and one responded to all three thermal stimuli. The proportion of units with significantly altered thermal responses did not vary significantly between 50 nM TLQP-62 exposed units and controls for any of the thermal responses tested among either HT or WDR units (P>0.05, Fisher’s exact test). Further, the proportion of units with significantly altered spontaneous activity (# of spikes 10 s prior to the first mechanical stimulus in the sequence) was not significantly different between 50 nM TLQP-62 exposed units and controls among either HT or WDR units (P>0.05 Fisher’s exact test; data not shown).
Discussion
VGF peptides are potential mediators of the transition from acute to chronic pain. This study demonstrates the contribution of endogenous C-terminal VGF peptides in the establishment of neuropathic pain and provides insight into the functions of the C-terminal peptide TLQP-62 in the dorsal horn. Consistent with previous reports of the involvement of TLQP-62 in hippocampal neuroplasticity [1,3,4,23,33,48,49], we show that TLQP-62 potentiates spinal glutamatergic signaling ex vivo. Further, in vivo recordings suggest that TLQP-62-induced neuroplasticity also potentiates responses of dorsal horn neurons to sensory stimuli.
C-terminal VGF peptides contribute to the establishment of neuropathic pain
This study demonstrates for the first time the presence of TLQP-62 in spinal cord as well as its increase after nerve injury. These findings provide evidence for the bioavailability of TLQP-62 during the development of neuropathic pain. We have shown that treatment with anti-AQEE30 at the time of nerve injury delays the development of mechanical allodynia, presumably through immunoneutralization of endogenous C-terminus VGF-derived peptides. Among the bioactive C-terminal VGF peptides (TLQP-62, AQEE-30, LQEQ-19), TLQP-62 is the only one currently known to modulate spinal neuroplasticity, and therefore its immunoneutralization likely contributes to the attenuation of mechanical allodynia. Our immunofluorescent analysis of Pirt-GCamp3 mice shows that spinal VGF is predominantly of primary afferent origin, supporting the idea that TLQP-62 may function as sensory neuron-derived mediator of the establishment of chronic pain after nerve injury. However, TLQP-62 produced by spinal neurons may also contribute to mechanisms of neuroplasticity since following nerve injury VGF mRNA is increased in both DRG and spinal cord [37]. The observation of nerve injury-induced increase in VGF levels in DRG and spinal cord using immunofluorescent analysis reported here is in agreement with previous reports [33,41].
Previously, we observed attenuation of nerve injury-induced hypersensitivity following immunoneutralization of the peptide TLQP-21 [14], which corresponds to the N-terminal portion of TLQP-62 [32]. The anti-TLQP21 antibody used in those experiments recognized the C-terminus of TLQP-21 and does not sequester full length TLQP-62. Taken together, our immunoneutralization studies suggest that multiple bioactive VGF fragments contribute to nerve injury-induced spinal neuroplasticity, although the relative spinal levels of different peptides are unknown.
The ability of a single intrathecal injection of anti-AQEE30 given at the time of injury to delay the progression of mechanical allodynia for at least 12 days is striking. We postulate that the site of action of the antibody is within the dorsal horn of spinal cord, consistent with evidence for penetration of intrathecally delivered IgG within the CNS parenchyma [40]. Moreover, recent pharmacokinetic analyses demonstrate that CNS parenchymal IgG clearance is orders of magnitude slower than clearance from other tissues, and a substantial fraction of the antibody remains within the interstitial space for at least 24 h [6,12]. Prolonged TLQP-62 sequestration may interfere with signaling pathways that participate in the establishment of persistent maladaptive neuroplasticity leading to central sensitization. TLQP-62 has been demonstrated to function in learning and memory by regulating BDNF expression and facilitating BDNF signaling in the hippocampus [32,33]. The neurotrophin-inducible vgf gene is similarly robustly upregulated by BDNF [1,15]. Thus, BDNF and VGF may function in a reinforcing manner to facilitate neuroplasticity in the spinal cord. Notably, continuous BDNF signaling maintains glutamatergic potentiation of spinal neurons following peripheral nerve injury [19]. BDNF is also thought to contribute to the failure of inhibitory gating mechanisms in the dorsal horn, resulting in the facilitation of HT neuron activity by Aβ-fiber activation [8,16,19,34,39]. Therefore, prolonged TLQP-62 sequestration may have lasting effects through disruption of persistent BDNF-dependent mechanisms of mechanical allodynia.
TLQP-62 potentiates glutamatergic signaling in the dorsal horn
Using two-photon microscopy to quantify changes in intracellular free Ca2+ in dorsal horn neurons, we observed potentiated glutamate signaling in 19% of neuronal profiles following TLQP-62 exposure. In addition, we provide evidence that the mechanism of action of TLQP-62 depends on kinase-mediated signaling. Originally characterized as an inhibitor of PKC [25], K252a was suggested to inhibit receptor tyrosine kinases selectively when administered at a concentration of 200 nM [20,28], and its use as a TrkB inhibitor has persisted [19]. The potential involvement of TrkB activation in the spinal potentiating effects of TLQP-62 is consistent with extensive evidence showing that TLQP-62 functions through BDNF/TrkB signaling in hippocampus [1,3,4,23,33,48]. A functional relationship between TLQP-62 and BDNF would represent an exciting potential mechanism for the involvement of TLQP-62 in the pathophysiology of neuropathic pain. Further work will be needed to elucidate the mechanism(s) of TLQP-62-induced potentiation of glutamate-induced Ca2+ transients and its dependence on BDNF/TrkB signaling.
Potentiation of glutamate-evoked Ca2+ transients in individual profiles may reflect both direct postsynaptic receptor activation as well as glutamate-induced input from other neurons in the network. Work in the hippocampus has recently identified BDNF-dependent effects of TLQP-62 on modifications to the glutamatergic postsynaptic compartment [23], suggesting that a similar mechanism may operate in the dorsal horn. The observed TLQP-62-induced increase in glutamate responsiveness may also be the result of increases in presynaptic glutamate release, or possibly, a reduction in synaptic inhibition either directly onto the profile or elsewhere in the circuit. Importantly, all of these potential mechanisms have been shown to occur following peripheral nerve damage [2,19,47]. Taken together, elevated spinal levels after nerve injury and the ability to potentiate glutamate-evoked Ca2+ transients in dorsal horn neurons position TLQP-62 as a potential mediator of the reported nerve injury-induced increase in Ca2+ responses to glutamate [7]. Under physiological conditions the TLQP-62-induced potentiation of glutamatergic signaling could contribute to the transfer of sensory information that may otherwise be sub-threshold.
TLQP-62 potentiates responses of dorsal horn neurons in vivo
Our in vivo single-unit experiments support the hypothesis that TLQP-62 mediates changes in the spinal processing of sensory inputs. Relative to control treatment, TLQP-62 selectively increased the responsiveness of nociceptive-specific HT neurons to moderate mechanical stimulation. In contrast to a previous study, which analyzed WDR units [37], we did not observe TLQP-62-induced changes in spontaneous activity, brush responses and acetone responses of WDR units relative to control treatment. Methodological differences could account for the disparate outcomes of the two studies. The thermal responsiveness of HT units with significantly increased response to tactile stimuli following exposure to TLQP-62 was heterogeneous: three units were activated by heat, and two were unresponsive to the thermal stimuli tested. The only HT unit that was not sensitized to moderate mechanical stimulation by TLQP-62 was responsive only to the acetone cooling stimulus. Analysis of a larger sample size of HT units is required to determine if the effects of TLQP-62 are restricted to a functional subset of HT neurons.
Our observation of a TLQP-62-induced increase in the responsiveness of HT units suggests that the peptide potentiates nociceptive signaling in the dorsal horn. Our experiments did not distinguish between projection neurons and local interneurons, both of which can be identified functionally as HT [7,45]. TLQP-62 may affect the responsiveness of HT neurons through direct action on the recorded neuron or through a network level effect that results in increased excitatory or decreased inhibitory synaptic input to HT neurons. Indeed, accumulating evidence indicates that modifications within dorsal horn circuits lead to failure of inhibitory gating mechanisms on excitatory interneurons and mechanical allodynia following peripheral nerve injury [31,34,36,38]. The activation of TLQP-62 exposed HT neurons by stimuli that did not elicit a response in the baseline state is consistent with a potential function of the peptide in the development of mechanical allodynia. A caveat in this interpretation is that the mechanical stimuli used in our recordings (brush, pressure, pinch) are distinct from stimuli used to establish mechanical allodynia behaviorally (i.e. von Frey filaments). Therefore, it remains to be determined in future in vivo recordings whether TLQP-62 induces increased responsiveness of HT neurons to stimuli that correspond to lowered mechanical withdrawal thresholds.
Conclusion
We show here for the first time that endogenous spinal TLQP-62 is increased following peripheral nerve damage and functions in the development of mechanical hypersensitivity. Further, we demonstrate the peptide’s ability to potentiate spinal glutamatergic signaling and enhance nociceptive transmission via increased responsiveness of HT spinal neurons. Thus, TLQP-62 may function as a mediator of neuroplasticity during the establishment of chronic pain, contributing to the development of mechanical allodynia. Elucidation of the precise neuroanatomical substrate underlying the ability of TLQP-62, and other injury induced neuropeptides, to facilitate transfer of sensory input along nociceptive pathways may offer mechanistic insight into the pathophysiology of neuropathic pain.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. George Wilcox for use of equipment and facilities, Dr. Guillermo Marques for guidance in multiphoton confocal imaging, and Rebecca Speltz and Galina Kalyuzhnaya for technical assistance. Supported by PHS grant R01 DE021996 (LV, SRS), R01 NS088518 (LV), T32 DA007097 (AS), F32 NS100438 (AS).
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, Black IB. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J Neurosci Off J Soc Neurosci. 2003;23:10800–10808. doi: 10.1523/JNEUROSCI.23-34-10800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardoni R. Role of Presynaptic Glutamate Receptors in Pain Transmission at the Spinal Cord Level. Curr Neuropharmacol. 2013;11:477–483. doi: 10.2174/1570159X11311050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behnke J, Cheedalla A, Bhatt V, Bhat M, Teng S, Palmieri A, Windon CC, Thakker-Varia S, Alder J. Neuropeptide VGF Promotes Maturation of Hippocampal Dendrites That Is Reduced by Single Nucleotide Polymorphisms. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18030612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bozdagi O, Rich E, Tronel S, Sadahiro M, Patterson K, Shapiro ML, Alberini CM, Huntley GW, Salton SRJ. The neurotrophin-inducible gene Vgf regulates hippocampal function and behavior through a brain-derived neurotrophic factor-dependent mechanism. J Neurosci Off J Soc Neurosci. 2008;28:9857–9869. doi: 10.1523/JNEUROSCI.3145-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridges D, Thompson SW, Rice AS. Mechanisms of neuropathic pain. Br J Anaesth. 2001;87:12–26. doi: 10.1093/bja/87.1.12. [DOI] [PubMed] [Google Scholar]
- 6.Cooper PR, Ciambrone GJ, Kliwinski CM, Maze E, Johnson L, Li Q, Feng Y, Hornby PJ. Efflux of monoclonal antibodies from rat brain by neonatal Fc receptor, FcRn. Brain Res. 2013;1534:13–21. doi: 10.1016/j.brainres.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 7.Cordero-Erausquin M, Inquimbert P, Schlichter R, Hugel S. Neuronal networks and nociceptive processing in the dorsal horn of the spinal cord. Neuroscience. 2016;338:230–247. doi: 10.1016/j.neuroscience.2016.08.048. [DOI] [PubMed] [Google Scholar]
- 8.Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, Koninck YD. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 9.Dado RJ, Katter JT, Giesler GJ. Spinothalamic and spinohypothalamic tract neurons in the cervical enlargement of rats. II. Responses to innocuous and noxious mechanical and thermal stimuli. J Neurophysiol. 1994;71:981–1002. doi: 10.1152/jn.1994.71.3.981. [DOI] [PubMed] [Google Scholar]
- 10.Doolen S, Blake CB, Smith BN, Taylor BK. Peripheral nerve injury increases glutamate-evoked calcium mobilization in adult spinal cord neurons. Mol Pain. 2012;8:56. doi: 10.1186/1744-8069-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doolen S, Cook J, Riedl M, Kitto K, Kohsaka S, Honda CN, Fairbanks CA, Taylor BK, Vulchanova L. Complement 3a receptor in dorsal horn microglia mediates pronociceptive neuropeptide signaling. Glia. 2017 doi: 10.1002/glia.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eigenmann MJ, Fronton L, Grimm HP, Otteneder MB, Krippendorff B-F. Quantification of IgG monoclonal antibody clearance in tissues. mAbs. 2017;9:1007–1015. doi: 10.1080/19420862.2017.1337619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fairbanks CA. Spinal delivery of analgesics in experimental models of pain and analgesia. Adv Drug Deliv Rev. 2003;55:1007–1041. doi: 10.1016/s0169-409x(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 14.Fairbanks CA, Peterson CD, Speltz RH, Riedl MS, Kitto KF, Dykstra JA, Braun PD, Sadahiro M, Salton SR, Vulchanova L. The VGF-derived peptide TLQP-21 contributes to inflammatory and nerve injury-induced hypersensitivity. Pain. 2014;155:1229–1237. doi: 10.1016/j.pain.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferri G-L, Possenti R. vgf A neurotrophin-inducible gene expressed in neuroendocrine tissues. Trends Endocrinol Metab. 1996;7:233–239. doi: 10.1016/s1043-2760(96)00123-3. [DOI] [PubMed] [Google Scholar]
- 16.Ferrini F, De Koninck Y. Microglia Control Neuronal Network Excitability via BDNF Signalling. Neural Plast. 2013 doi: 10.1155/2013/429815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaskell H, Moore RA, Derry S, Stannard C. Oxycodone for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2014:CD010692. doi: 10.1002/14651858.CD010692.pub2. [DOI] [PubMed] [Google Scholar]
- 18.Gilron I, Baron R, Jensen T. Neuropathic pain: principles of diagnosis and treatment. Mayo Clin Proc. 2015;90:532–545. doi: 10.1016/j.mayocp.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 19.Hildebrand ME, Xu J, Dedek A, Li Y, Sengar AS, Beggs S, Lombroso PJ, Salter MW. Potentiation of Synaptic GluN2B NMDAR Currents by Fyn Kinase Is Gated through BDNF-Mediated Disinhibition in Spinal Pain Processing. Cell Rep. 2016;17:2753–2765. doi: 10.1016/j.celrep.2016.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Horch HW, Krüttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–364. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- 21.Hylden JL, Wilcox GL. Intrathecal substance P elicits a caudally-directed biting and scratching behavior in mice. Brain Res. 1981;217:212–215. doi: 10.1016/0006-8993(81)90203-1. [DOI] [PubMed] [Google Scholar]
- 22.Jaken RJP, Joosten EAJ, Knüwer M, Miller R, van der Meulen I, Marcus MAE, Deumens R. Synaptic plasticity in the substantia gelatinosa in a model of chronic neuropathic pain. Neurosci Lett. 2010;469:30–33. doi: 10.1016/j.neulet.2009.11.038. [DOI] [PubMed] [Google Scholar]
- 23.Jiang C, Lin W-J, Sadahiro M, Labonté B, Menard C, Pfau ML, Tamminga CA, Turecki G, Nestler EJ, Russo SJ, Salton SR. VGF function in depression and antidepressant efficacy. Mol Psychiatry. 2017 doi: 10.1038/mp.2017.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. [Accessed 10 Apr 2018];Kang Li @ CMU - Image Stabilizer Plugin for ImageJ. n.d Available: http://www.cs.cmu.edu/~kangli/code/Image_Stabilizer.html.
- 25.Kase H, Iwahashi K, Matsuda Y. K-252a, a potent inhibitor of protein kinase C from microbial origin. J Antibiot (Tokyo) 1986;39:1059–1065. doi: 10.7164/antibiotics.39.1059. [DOI] [PubMed] [Google Scholar]
- 26.Khan N, Smith MT. Neurotrophins and Neuropathic Pain: Role in Pathobiology. Mol Basel Switz. 2015;20:10657–10688. doi: 10.3390/molecules200610657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YS, Chu Y, Han L, Li M, Li Z, LaVinka PC, Sun S, Tang Z, Park K, Caterina MJ, Ren K, Dubner R, Wei F, Dong X. Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron. 2014;81:873–887. doi: 10.1016/j.neuron.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koizumi S, Contreras ML, Matsuda Y, Hama T, Lazarovici P, Guroff G. K-252a: a specific inhibitor of the action of nerve growth factor on PC 12 cells. J Neurosci. 1988;8:715–721. doi: 10.1523/JNEUROSCI.08-02-00715.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuner R, Flor H. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci. 2016;18:20–30. doi: 10.1038/nrn.2016.162. [DOI] [PubMed] [Google Scholar]
- 30.LaCroix-Fralish ML, Austin J-S, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152:1888–1898. doi: 10.1016/j.pain.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 31.Leitner J, Westerholz S, Heinke B, Forsthuber L, Wunderbaldinger G, Jäger T, Gruber-Schoffnegger D, Braun K, Sandkühler J. Impaired excitatory drive to spinal GABAergic neurons of neuropathic mice. PloS One. 2013;8:e73370. doi: 10.1371/journal.pone.0073370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levi A, Ferri G-L, Watson E, Possenti R, Salton SRJ. Processing, distribution, and function of VGF, a neuronal and endocrine peptide precursor. Cell Mol Neurobiol. 2004;24:517–533. doi: 10.1023/B:CEMN.0000023627.79947.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin W-J, Jiang C, Sadahiro M, Bozdagi O, Vulchanova L, Alberini CM, Salton SR. VGF and Its C-Terminal Peptide TLQP-62 Regulate Memory Formation in Hippocampus via a BDNF-TrkB-Dependent Mechanism. J Neurosci. 2015;35:10343–10356. doi: 10.1523/JNEUROSCI.0584-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Dong H, Gao Y, Gong Y, Ren Y, Gu N, Zhou S, Xia N, Sun Y-Y, Ji R-R, Xiong L. A feed-forward spinal cord glycinergic neural circuit gates mechanical allodynia. J Clin Invest. 2013;123:4050–4062. doi: 10.1172/JCI70026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo C, Kuner T, Kuner R. Synaptic plasticity in pathological pain. Trends Neurosci. 2014;37:343–355. doi: 10.1016/j.tins.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Miraucourt LS, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through PKCgamma interneurons. PloS One. 2007;2:e1116. doi: 10.1371/journal.pone.0001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moss A, Ingram R, Koch S, Theodorou A, Low L, Baccei M, Hathway GJ, Costigan M, Salton SR, Fitzgerald M. Origins, actions and dynamic expression patterns of the neuropeptide VGF in rat peripheral and central sensory neurones following peripheral nerve injury. Mol Pain. 2008;4:62. doi: 10.1186/1744-8069-4-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peirs C, Williams S-PG, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, Seal RP. Dorsal Horn Circuits for Persistent Mechanical Pain. Neuron. 2015;87:797–812. doi: 10.1016/j.neuron.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petitjean H, Pawlowski SA, Fraine SL, Sharif B, Hamad D, Fatima T, Berg J, Brown CM, Jan L-Y, Ribeiro-da-Silva A, Braz JM, Basbaum AI, Sharif-Naeini R. Dorsal Horn Parvalbumin Neurons Are Gate-Keepers of Touch-Evoked Pain after Nerve Injury. Cell Rep. 2015;13:1246–1257. doi: 10.1016/j.celrep.2015.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pizzo ME, Wolak DJ, Kumar NN, Brunette E, Brunnquell CL, Hannocks M-J, Abbott NJ, Meyerand ME, Sorokin L, Stanimirovic DB, Thorne RG. Intrathecal antibody distribution in the rat brain: surface diffusion, perivascular transport and osmotic enhancement of delivery. J Physiol. 2017 doi: 10.1113/JP275105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riedl MS, Braun PD, Kitto KF, Roiko SA, Anderson LB, Honda CN, Fairbanks CA, Vulchanova L. Proteomic analysis uncovers novel actions of the neurosecretory protein VGF in nociceptive processing. J Neurosci Off J Soc Neurosci. 2009;29:13377–13388. doi: 10.1523/JNEUROSCI.1127-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shields SD, Eckert WA, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain Off J Am Pain Soc. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- 44.Siniscalco D, Giordano C, Rossi F, Maione S, de Novellis V. Role of Neurotrophins in Neuropathic Pain. Curr Neuropharmacol. 2011;9:523–529. doi: 10.2174/157015911798376208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takazawa T, MacDermott AB. Synaptic pathways and inhibitory gates in the spinal cord dorsal horn. Ann N Y Acad Sci. 2010;1198:153–158. doi: 10.1111/j.1749-6632.2010.05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- 47.Taylor BK. Spinal inhibitory neurotransmission in neuropathic pain. Curr Pain Headache Rep. 2009;13:208–214. doi: 10.1007/s11916-009-0035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thakker-Varia S, Behnke J, Doobin D, Dalal V, Thakkar K, Khadim F, Wilson E, Palmieri A, Antila H, Rantamaki T, Alder J. VGF (TLQP-62)-induced neurogenesis targets early phase neural progenitor cells in the adult hippocampus and requires glutamate and BDNF signaling. Stem Cell Res. 2014;12:762–777. doi: 10.1016/j.scr.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thakker-Varia S, Krol JJ, Nettleton J, Bilimoria PM, Bangasser DA, Shors TJ, Black IB, Alder J. The neuropeptide VGF produces antidepressant-like behavioral effects and enhances proliferation in the hippocampus. J Neurosci Off J Soc Neurosci. 2007;27:12156–12167. doi: 10.1523/JNEUROSCI.1898-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West SJ, Bannister K, Dickenson AH, Bennett DL. Circuitry and plasticity of the dorsal horn--toward a better understanding of neuropathic pain. Neuroscience. 2015;300:254–275. doi: 10.1016/j.neuroscience.2015.05.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.