

Abstract
Purpose
Adoptive T cell immunotherapy (ACT) has emerged as a viable therapeutic for peripheral and central nervous system (CNS) tumors. In peripheral cancers, optimal efficacy of ACT is reliant on dendritic cells (DCs) in the tumor microenvironment. However, the CNS is largely devoid of resident migratory DCs to function as antigen-presenting cells during immunotherapy. Herein, we demonstrate that cellular interactions between adoptively-transferred tumor-reactive T cells and bone marrow-derived HSPCs lead to the generation of potent intratumoral DCs within the CNS compartment.
Experimental Design
We evaluated HSPC differentiation during ACT in vivo in glioma-bearing hosts and HSPC proliferation and differentiation in vitro using a T cell co-culture system. We utilized FACS, ELISAs, and gene expression profiling to study the phenotype and function of HSPC-derived cells ex vivo and in vivo. To demonstrate the impact of HSPC differentiation and function on anti-tumor efficacy, we performed survival experiments.
Results
Transfer of HSPCs with concomitant ACT led to the production of activated CD86+CD11c+MHCII+ cells consistent with DC phenotype and function within the brain tumor microenvironment. These intratumoral DCs largely supplanted abundant host myeloid-derived suppressor cells. We determined that during ACT, HSPC-derived cells in gliomas rely on T cell-released IFN-γ to differentiate into DCs, activate T cells, and reject intracranial tumors.
Conclusions
Our data supports the use of HSPCs as a novel cellular therapy. While DC vaccines induce robust immune responses in the periphery, our data demonstrates that HSPC transfer uniquely generates intratumoral DCs that potentiate T cell responses and promote glioma rejection in situ.
Keywords: Hematopoietic stem and progenitor cell (HSPC), Adoptive cellular therapy (ACT), Glioblastoma (GBM), Dendritic Cell (DC), Immunotherapy
Introduction
Median survival for malignant gliomas is near 15-20 months with chemotherapy, radiation, tumor-treating fields, and surgical resection (1, 2). To overcome the dearth of curative therapies, immunotherapeutic strategies including adoptive T cell immunotherapy (ACT) and dendritic cell (DC) vaccination have emerged that synergize with standard of care and promote tumor rejection (3-11). Recently, it has been demonstrated that the success of ACT in peripheral cancers is dependent on intratumoral DCs (12). However, few DCs have been found in the homeostatic brain as well as the brain tumor microenvironment (13-15). On the contrary, there is significant infiltration of pro-tumor myeloid-derived suppressor cells (MDSCs) in brain tumors that diminish the success of therapies (16, 17). Our prior studies have demonstrated that intravenously administered hematopoietic stem and progenitor cells (HSPCs) traffic to sites of malignant glioma growth and significantly enhance the efficacy of ACT, at least in part, through the chemotactic attraction of tumor-specific lymphocytes into the tumor microenvironment (5). In this report, we demonstrate that HSPCs interface with tumor-reactive lymphocytes and differentiate into potent antigen-presenting cells (APCs) of a DC phenotype, supplant intratumoral MDSCs, present captured tumor antigens leading to enhancement of intratumoral T cell activation, and promote the immunologic rejection of invasive gliomas.
HSPCs have recently become characterized as significant immunomodulatory cells during infection, autoimmunity, and cancer (5, 6, 18-20). In the context of peripheral solid tumor-bearing hosts, several studies have characterized HSPCs as immunosuppressive, tumor-promoting, and as seeds for metastasis (17, 21-27). On the contrary, few studies have described HSPC-mediated expansion and recruitment of T cells as a novel method of potentiating cytotoxic responses (5, 6). While there are conflicting reports on the immunologic role of HSPCs in peripheral cancers, HSPCs selectively migrate to malignant gliomas through the SDF-1-CXCR4 axis (28) and refrain from migration to organs including liver, lung, muscle, and spleen (29). Additionally, HSPC migration to gliomas can be enhanced with standard therapies such as irradiation and temozolomide (30). However, the differentiation of HSPCs and the functions of their progeny in the brain tumor microenvironment remains unclear. Our goal here was to determine the role of HSPC progeny in glioma immunity in situ during adoptive cell transfer. To accomplish this, we studied HSPC differentiation in the brain tumor microenvironment, the function of HSPC-derived cells, and mechanisms of synergy between HSPCs and tumor-reactive T cells.
We therefore investigated HSPC differentiation and function in brain tumor-bearing hosts during ACT and host conditioning (4-11). Here we demonstrate that HSPCs in the brain tumor microenvironment supplant host MDSCs and differentiate into CD86+CD11c+MHCII+ activated DCs. This differentiation occurs through tumor-reactive T cell-released cytokines including interferon-γ (IFN-γ) and its signaling through IFN-γ receptor (IFN-γR) on HSPCs. While activated DC vaccines are capable of induction of peripheral immune responses (3, 31), our data demonstrates that HSPC transfer uniquely leads to accumulation of intratumoral DCs in malignant gliomas and supplants immunosuppressive MDSCs within the tumor microenvironment. These findings have significant implications for ACT in the treatment of refractory brain tumors.
Methods
Mice
Five- to eight-week-old female C57BL/6 mice (Jackson, 000664), transgenic DsRed mice (Jackson, 006051), transgenic GREAT mice (Jackson, 017580), and IFN-γR−/− mice (Jackson, 003288) were used for experiments. All investigators adhered to the “Guide for the Care and Use of Laboratory Animals” and the University of Florida Animal Care Services are fully accredited by the American Association for Accreditation of Laboratory Animal Care. All studies were approved by the Institutional Animal Care and Use Committee and are covered under protocol number 201607966.
RNA isolation
Total tumor RNA (ttRNA) isolation from tumor cell lines was performed with RNeasy mini kit (Qiagen, 74104) per the manufacturer’s protocol.
Tumor-reactive T cells
Tumor-reactive T cells were generated as previously described through ex-vivo expansion with bone marrow-derived DCs (BMDCs) (5).
Tumor models
Tumor-bearing experiments were performed in syngeneic sex-matched C57BL/6 mice. The KR158B-luc glioma line (provided by Dr. Karlyne M. Reilly) has been verified histologically as high-grade glioma and gene expression analysis by RNA Seq demonstrated appropriate haplotype background and expression of astrocytoma-associated genes. KR158B-luc cells (104) were implanted into the caudate nucleus by injecting 2mm lateral to midline at the bregma suture and 3mm deep (5, 32). NSC tumor cells were generated through previously described in vitro culture of sorted granule neuron precursor cells (33). NSC medulloblastoma cells (1×103) were implanted into the cerebellum 1mm lateral to the midline and 3mm deep (33, 34). K2 brain stem glioma cells (provided by Dr. Oren Becher) were developed through previously described methods including an induced H3.3K27M mutation in the progenitor cells of the brainstem (35). K2 cells (1×105) were implanted into the brain stem of mice 1mm caudal to the lambda suture on the midline and 3.5 mm deep. Tumors were injected with a stereotactic frame (Stoelting, 53311) and a 250μL syringe (Hamilton, 81120) with a 25-gauge needle. All lines tested negative for mycoplasma contamination (IDEXX, 9/26/2017) and if passaged in vitro, were passaged less than 10 times after thawing.
HSPC Transfer
Hematopoietic stem and progenitor cells were freshly derived from bone marrow of naïve C57BL/6 mice. For in vivo tracking experiments, HSPCs were harvested from naïve DsRed mice. After red blood cell lysis, bone marrow was prepared for lineage depletion by MACS multistand with lineage depletion kit and LS columns (Miltenyi Biotec, 130-090-858, 130-042-401, and 130-042-303).
Adoptive Cellular Therapy
Treatment of tumor-bearing mice began with 5Gy non-myeloablative (NMA) lymphodepletion or 9Gy myeloablation (MA) by total body irradiation (TBI) with X-rays (X-RAD 320, Precision X-ray) 4 days post-intracranial injection. On day 5 post-intracranial tumor injection, mice received a single intravenous (IV) injection of 107 autologous ex vivo-expanded ttRNA T cells +/− the inclusion of 3.5×105 lin− HSPCs (Miltenyi Biotec, 130-090-858). Beginning day 6 post-tumor injection, 2.5×105 ttRNA-pulsed BMDC vaccines were injected intradermally posterior to the pinna weekly for three vaccines.
Brain Tumor Processing
Tumor resection extended to gross borders of tumor mass near the site of injection. Tumors were dissociated mechanically with a sterilized razor blade and chemically with papain (Worthington, NC9809987) for 30 minutes. Tumors were filtered with a 70μm cell strainer (BD Biosciences, 08-771-2) prior to antibody incubation.
Flow cytometry
Flow cytometry was performed on the BD Biosciences FACS Canto-II. IFN-γ release by T cells from transgenic GREAT mice were detected at FL-1 for yellow fluorescent protein (YFP) that is expressed under the IFN-γ promoter. Cells from transgenic DsRed mice were detected at FL-2. FACS-sorting was completed using the BD Biosciences FACS Aria II. Cells were prepared ex vivo as described above and suspended in 2% FBS (Seradigm, 97068-091) in PBS (Gibco, 10010-049). Antibodies were applied per manufacturer’s recommendation with isotype controls (Supplementary Table 1). Analysis and flow plots were generated with FlowJo version 10 (Tree Star) after omission of doublets and debris and were gated on size and granularity.
T cell function assays and supernatant transfer system
In vitro experiments utilized restimulation assays including effector cells (T cells) and targets (pulsed DCs or tumor cell lines) that are co-cultured in a 10:1 ratio in 96-well U-bottom plates in triplicate as a measure of T cell activity. IFN-γ Platinum ELISAs (Affymetrix, BMS606) were performed on acellular media that was harvested and frozen from the supernatants after 48 hours. The supernatant transfer system utilized the 10:1 ratio of T cells and DCs to generate supernatants. Anti-GM-CSF (Affymetrix, 16-7331-85), anti-IFN-γ (Affymetrix, 16-7311-85), and recombinant IFN-γ (Fisher, 50-925-7) were used for HSPC differentiation cultures.
PCR Array
Brain tumors were dissociated, RNA isolated with the RNeasy Kit, and analyzed with the RT2 Profiler Array Cancer Inflammation and Immunity Crosstalk PCRarray (Qiagen, PAMM-181ZD-12).
Statistical analysis
All experiments were analyzed in Prism 7 (GraphPad). The median survival for tumor-bearing animals is 25-42 days. Tumor-bearing survival experiments in this manuscript are n≥7 and analyzed with the Mantel-Cox log-rank test. An unpaired, Mann-Whitney rank sum test or unpaired, Student’s t-test was applied for two-group comparisons for in vivo experiments or in vitro experiments, respectively. Significance is determined as p<0.05. Animal studies were powered to include n=5 randomized mice per group unless otherwise noted. The authors pre-established that no animals or samples were to be excluded from analysis.
Results
We have previously demonstrated that the success of HSPCs and ACT in malignant gliomas is reliant on cellular interactions between DCs, T cells, and HSPCs (5). While HSPCs promote T cell recruitment to the brain tumor microenvironment, it remained unclear how T cells and HSPCs were interacting within malignant gliomas, and what the cellular fates were of HSPC-derived progeny. Given previous reports of HSPC-mediated expansion of cytotoxic T cells (6), and the dependence of ACT on intratumoral DCs in peripheral malignancies (12), we hypothesized that HSPC-derived cells that migrate to brain tumors directly promote anti-tumor cytotoxic immunity in situ. We therefore studied the intratumoral differentiation of HSPCs and functional capacity of progeny to determine mechanisms of how HSPCs promote antitumor immunity.
We evaluated these interactions during ACT that uses total tumor RNA (ttRNA)-pulsed BMDCs to expand tumor reactive lymphocytes for several orthotopic brain tumor models. Intracranially-implanted Nf1+/− p53+/− (KR158B-luc) malignant gliomas, CMYC-driven (NSC) medulloblastomas, and H3.3K27M mutant (K2) brainstem gliomas were all responsive to the combination of adoptively-transferred tumor-reactive lymphocytes, intradermal BMDC vaccination, and IV-administered lin− HSPC transfer after MA 9Gy TBI. This HSPCs+ACT treatment platform extended median overall survival and promoted long-term cures in all aggressive brain tumor models (p≤.0001; Fig. 1). To determine the mechanism of action, we studied KR158B-luc tumor-bearing hosts after 5Gy NMA conditioning and ACT with and without syngeneic HSPC transfer from bone marrow of DsRed transgenic animals to track the fate of HSPC progeny. Similar to the adoptive therapies utilized in the melanoma setting (6, 7, 9), the anti-tumor efficacy of ACT in our model is greatest in the MA 9Gy setting. However, we studied the NMA 5Gy setting to offer the benefits of lymphodepletion while removing the requirement for bone marrow HSPC rescue when doing experiments with or without the use of wild-type (WT) HSPC transfer. This allowed for continuous investigation of a treatment platform with experimental flexibility.
Figure 1. ACT and HSPC transplant in malignant glioma, brainstem glioma, and medulloblastoma tumor-bearing mice.
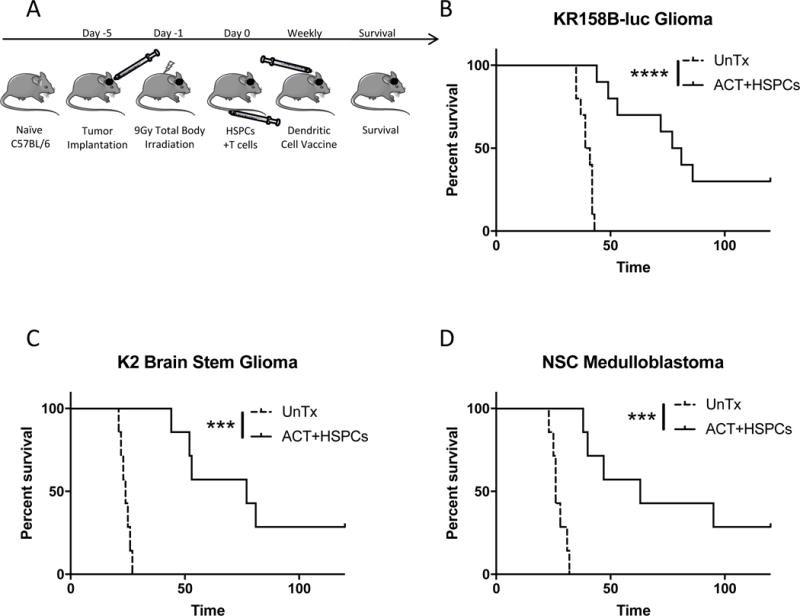
A). Experimental layout of treatment platform for tumor-bearing mice. Treatment includes HSPCs and ACT (tumor-reactive T cells and DC vaccine) after MA 9Gy TBI host conditioning. B). Survival of KR158B-luc glioma-bearing mice untreated or treated with HSPC transfer and ACT. A previous report demonstrated the requirement for all components of HSPCs, ACT, and host conditioning for greatest efficacy of therapy. Control groups with monotherapies can be found therein. C). Survival of K2 brain stem glioma tumor-bearing mice untreated or treated with HSPCs and ACT. K2 resembles diffuse intrinsic pontine glioma (DIPG). D). Survival of NSC medulloblastoma tumor-bearing mice untreated or treated with HSPCs and ACT. *P<.05, **P<.01, ***P<.001, ****P<.0001, by Mantel-Cox Log Rank Test for survival experiments (n≥7).
We determined that IV-administered HSPCs migrate to brain tumors, engraft, and persist within the tumor microenvironment 7 days post-transfer (Fig. 2A-B). During an initial phenotype of HSPC-derived cells in brain tumors, we discovered that a fraction of HSPC-derived cells upregulated the CD11c DC marker (HSPC-derived CD11c+: 16.7%, Fig. 2B). Given the tendency for brain tumors to be resistant to DC infiltration, we investigated the relative migration capacity of HSPCs or ex vivo-expanded BMDCs from DsRed animals after systemic injection. BMDCs were largely incapable of migrating into the tumor microenvironment as we found that IV-administered HSPCs are 192-fold better at engrafting in brain tumors (p=0.0079; Fig. 2B). These results demonstrated the unique capacity for HSPC tropism to malignant brain tumors and propensity to differentiate into intratumoral CD11c+ cells. Previous reports have attributed HSPC migration to gliomas to the SDF-1-CXCR4 chemotactic axis (28) and this has been corroborated in our model systems (not shown). To determine whether HSPCs migrate to gliomas as multipotent or lineage-restricted cells, we FACS-sorted DsRed+ HSPC-derived cells from brain tumors and used the isolated cells to rescue non-tumor bearing animals treated with MA TBI (Fig. 2C). HSPC-derived cells isolated from brain tumor 3 hours after infusion were capable of rescuing 60% of myeloablated hosts, demonstrating that pluripotent stem cells within the HSPC transfer reach the brain tumor microenvironment shortly after injection and retain immunologic reconstitution capacity (WT HSPCs: 100% survival, 3hrs. p.t. HSPCs: 60% survival; not significant; Fig. 2D). However, only 20% of mice could be rescued from myeloablative therapy using HSPC-derived cells isolated from the tumor microenvironment 24 hours after transplant, demonstrating the significant onset of lineage-restriction (WT HSPCs: 100% survival, 24hrs. p.t. HSPCs: 20% survival; p=.0133; Fig. 2D).
Figure 2. HSPC engraftment in gliomas.
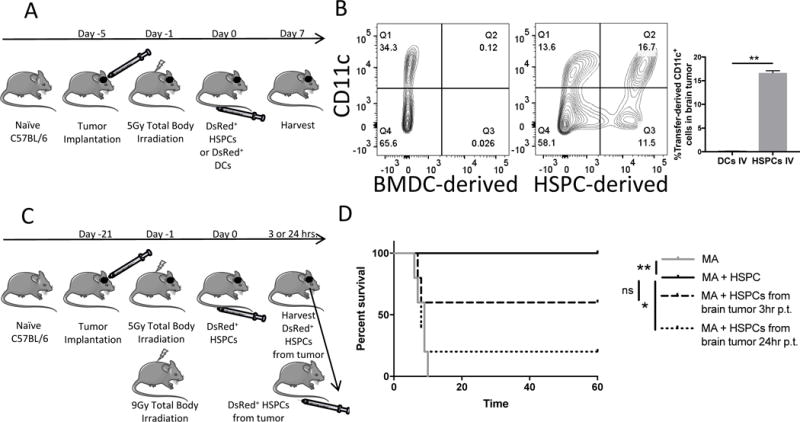
A). Experimental layout of experiment in Figure 2D. HSPCs or DCs were administered IV after 5Gy irradiation with no T cell transfer. Experimental structure utilized to inquire into the ability of HSPCs to migrate to brain tumors in comparison to BMDCs, a differentiated cell type. B). Representative FACS plots of brain tumors of animals treated according to Figure. 2A with HSPCs or DC IV. CD11c is a global marker for DCs. C). Experimental layout of experiment performed with HSPC-derived cells isolated from brain tumor shortly after HSPC transplant to test multipotency in a separate lethally-irradiated host. D). Survival curve of 9Gy myeloablated (MA) non-tumor-bearing animals after various IV transplants. MA+HSPCs group demonstrates successful bone marrow rescue of MA animals. Meanwhile, DsRed+ HSPC-derived cells were FACS-sorted from brain tumors of animals treated with 5Gy and HSPCs either 3 hrs. or 24 hrs. post-transfer (p.t.). Then HSPC-derived cells were transplanted into MA animals to determined capability to rescue MA hosts. All data represent the mean +/−SD. *P<.05, **P<.01, ***P<.001, ****P<.0001, by Mann-Whitney t test for in-vivo studies (n=5) and by Mantel-Cox Log Rank Test for survival experiment (n=5).
We next investigated HSPC-derived cells in brain tumors during ACT (Fig. 3A). HSPC-derived cells persisted in brain tumors long after transfer, including near moribund endpoint 31 days after injection (Fig. 3B, Supplementary Fig. S1A). During engraftment in tumor, HSPCs supplanted host cells in the brain tumor microenvironment and this displacement was facilitated by immunotherapeutic treatment. Specifically, when HSPCs are administered alone after 5Gy, HSPCs drive a 57.5% decrease in host DsRed− Ly-6G/6C+ MDSCs (-HSPCs: 13.92%; +HSPCs: 5.92%, p<.0001; Fig. 3C, Supplementary Fig. S1B). When administered during ACT, HSPCs drive an 87.7% decrease in host MDSCs indicating that ACT facilitates this process (ACT alone: 24.18%; ACT+HSPCs: 2.98%, p<.0001; Fig. 3C, Supplementary Fig. S1C). To ensure that the reduction in intratumoral host MDSCs represented a true displacement and not altered expansion of other cellular components, we evaluated the absolute counts of MDSCs within the tumor microenvironment. We also discovered that nearly all Ly-6G/6C+ cells expressed CD11b+ and restricted the MDSC definition to CD11b+Ly-6G/6C+ (36-39). HSPCs drove a 60% decrease in total host CD11b+Ly-6G/6C+ MDSCs while ACT+HSPCs drove a 77% decrease in total host CD11b+Ly-6G/6C+ MDSCs (HSPCs, p=.036, ACT+HSPCs, p=.0015; Fig. 3D). To further define the intratumoral CD11b+Ly-6G/6C+ population as an MDSC population (38), we analyzed their functional capacity to suppress T cells. To perform this, we evaluated FACS-sorted tumor-infiltrating host MDSCs in a T cell stimulation assay measuring release of IFN-γ by activated tumor-specific T cells cultured with ttRNA-pulsed BMDCs. When tumor-derived host CD11b+Ly-6G/6C+ cells were included in this assay, they caused a 3-fold suppression of IFN-γ release by tumor-specific T cells (p<.0001; Fig. 3E). To characterize the MDSC population with a functionally suppressive molecule of import in the tumor microenvironment (40), we investigated the expression of PD-L1 on host MDSCs. We determined that 85% of Ly-6G/6C+ cells express PD-L1 and that HSPC transfer supplants PD-L1+ MDSCs as well (5Gy: 6.3%, 5Gy+HSPC+ACT: 1.5%, p=0.0079; Fig. 3, F-G). These results demonstrate that tumor-infiltrating MDSCs are immunosuppressive and that HSPC transfer can supplant these populations in brain tumors. This highlights an important pathway to reprogram the suppressive signature of the brain tumor microenvironment.
Figure 3. Host immunity in brain tumors during HSPC engraftment.
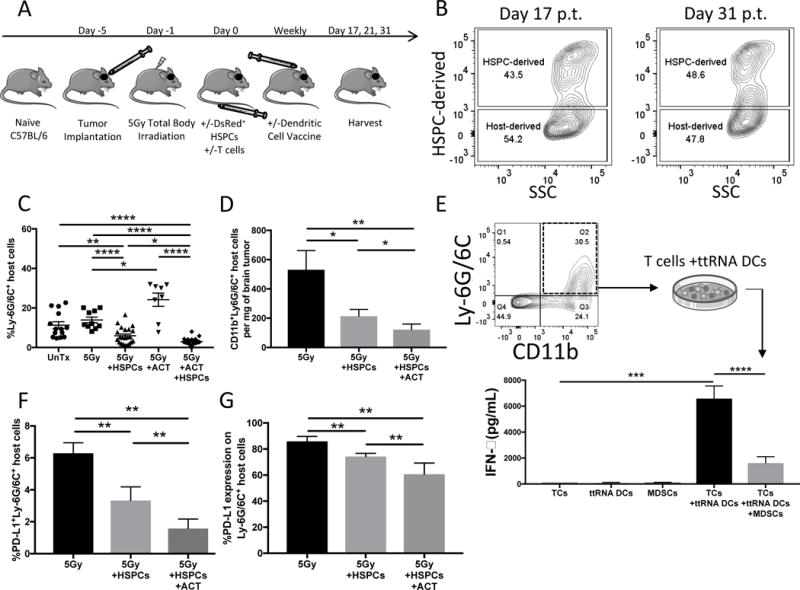
A). Experimental layout for remainder of in-vivo experiments including +/−HSPC transfer and +/−ACT (TCs with BMDC vaccine) following 5Gy non-myeloablative total body irradiation. Experimental plan similar to plan in Figure 1A but includes 5Gy irradiation instead of 9Gy to remove the requirement for HSPC transplant and DsRed+ HSPCs are administered to allow tracking of HSPC-derived cells (DsRed+) or Host-derived cells (DsRed−). B). Representative flow cytometry of brain tumors of animals harvested 17d and 31d post-ACT and 350,000 HSPCs. Experiment performed twice. C). Bar graphs represents flow cytometry of brain tumors of animals 21d post-treatment. Groups include untreated (n=14), 5Gy alone (n=10), 5Gy+HSPC transfer (n=22), 5Gy+ACT (n=8), or 5Gy+ACT+HSPC (n=24) transfer. Gated on all DsRed− host-derived cells. Data is pooled from >2 experiments and represents mean +/− SEM. Data from individual experiments is separated in supplemental fig. S1. D). Bar graph represents absolute cell count flow cytometry of brain tumors of animals 21d post-treatment with 5Gy alone (n=7), 5Gy+HSPCs (n=12), or 5Gy+HSPCs+ACT (n=15). Data is pooled from 2 experiments and represents mean +/− SEM. Gated on all DsRed− host-derived cells. Myeloid-derived suppressor cell (MDSC) marker was narrowed from Ly-6G/6C+ in Panel C to CD11b+Ly-6G/6C+ double-positive MDSCs. E). FACS-sorting of host-derived CD11b+Ly-6G/6C+ cells from brain tumors of mice 21d post-treatment with ACT without HSPCs. 40,000 FACS-sorted host-derived MDSCs were introduced into a co-culture restimulation assay with 40,000 ttRNA-pulsed DCs and 400,000 tumor-reactive T cells. 48h later an IFN-γ ELISA was performed on the supernatants to determined the degree to which host-derived MDSCs suppressed T cell activation by DCs. Experiment performed twice and data represents mean +/− SD. F). Bar graph represents flow cytometry of brain tumors 21d post-treatment with 5Gy, 5Gy+HSPCs or 5Gy+HSPCs+ACT. Data represents mean +/− SD of PD-L1+Ly-6G/6C+ host MDSCs. PD-L1 is a suppressive molecule found on tumor-infiltrating MDSCs. G). Further phenotypic characterization of host MDSCs. Bar graph represents flow cytometry percent expression of PD-L1 on the host Ly-6G/6C+ MDSC population in brain tumor. Data represents mean +/− SD. *P<.05, **P<.01, ***P<.001, ****P<.0001, by unpaired students t test for in-vitro studies (n=3) and by Mann-Whitney t test for in-vivo studies (n≥5).
We next evaluated the phenotype of HSPC-derived cells in brain tumors of ACT-treated animals. When HSPCs are administered in conjunction with ACT (3, 5), the HSPCs differentiate into CD11c+CD11b+MHCII+ DCs in the tumor microenvironment with high expression of co-stimulatory activation marker CD86 and low expression of CD80 (HSPC-derived cells 21d post-transfer-CD11c+:83.7%, CD11b+:84.5%, MHC II:79.9%, CD86+:76.2%, CD80+:14.8%; Fig. 4, A-B, Supplementary Fig. S1D) (41). Detailed profiling indicates that HSPCs display little differentiation into CD3+ lymphocytes, c-kit+ stem cells, F4/80+ macrophages, PD-L1+ APCs, or Ly-6G/6C+ MDSCs and that HSPC-derived CD11c+ cells are largely negative for DC subtype markers CD8 and CD103 (Fig. 4, A-B, Supplementary Fig. S1D). We also studied the durability of HSPC-derived DCs in brain tumors and determined they persist and outnumber the HSPC-derived MDSCs even until Day 31 post-transfer when suboptimally treated animals are nearing moribund endpoint (Fig. 4C). Interestingly, the small population of HSPCs that do differentiate into MDSCs are less suppressive in a T cell stimulation assay than host-derived MDSCs (HSPC-derived: 3108.6 pg/mL, Host-derived: 1619.7 pg/mL IFN-γ, p=.0159; Supplementary Fig. S1E). Given the lack of HSPC differentiation into MDSCs, during HSPC transfer with ACT there remains a significant net loss of total MDSCs in the tumor microenvironment while HSPC-derived DCs engraft (Supplementary Fig. S1F).
Figure 4. HSPC differentiation in brain tumors during adoptive T cell immunotherapy.
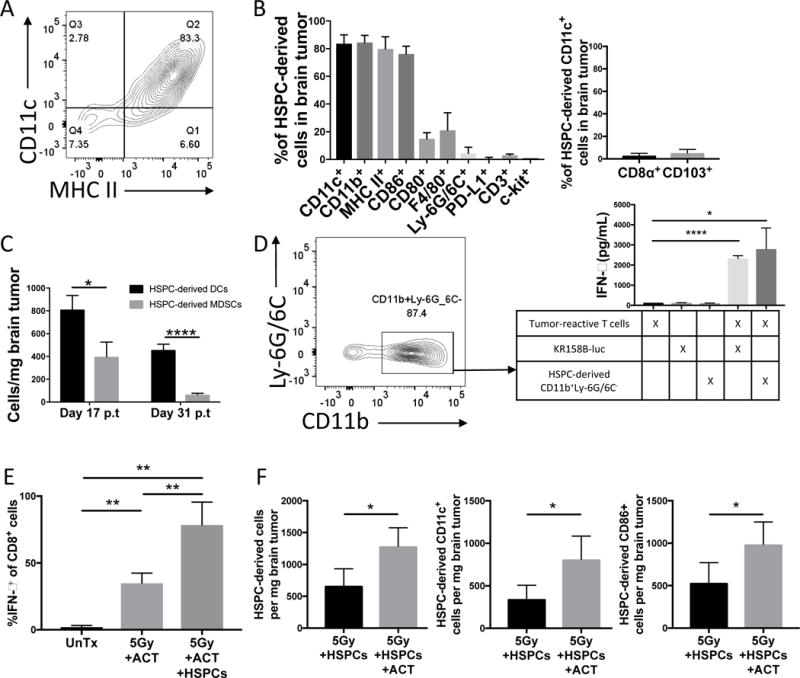
A). Representative flow cytometry of brain tumor of animal 21 days post-treatment with ACT and HSPCs. Gated on DsRed+ HSPC-derived cells. B). Graph demonstrates percent expression of various markers on HSPC-derived cells isolated from brain tumors of animals treated with 5Gy+ACT+HSPCs 21d post-transfer. C). Graph demonstrates absolute counts of surface expression of CD11c+MHCII+ (HSPC-derived DCs) compared to surface expression of CD11b+Ly-6G/6C+ (HSPC-derived MDSCs) over time course after treatment with 5Gy+ACT+HSPCs. D). FACS-sorting of brain tumors of animals treated with 5Gy+ACT+HSPCs 21d post-transfer. Gated on DsRed+(PE+) HSPC-derived cells. 40,000 CD11b+Ly-6G/6− myeloid cells were introduced into a co-culture restimulation assay with 400,000 tumor-reactive T cells to identify antigen-presentation capacity of HSPC-derived cells. After 48h, IFN-γ ELISA was performed on supernatants to determine T cell activation level. Experiment performed twice. E). Bar graph represents flow cytometry of brain tumors of mice either untreated, treated with ACT, or treated with ACT+HSPCs. ACT T cells were derived from GREAT mice that express YFP under the IFN-γ promoter to identify IFN-γ-expression in vivo on CD8+ cells. MFI included in supplemental figures. Experiment performed twice. F). Absolute counts of flow cytometry of brain tumors of mice treated with 5Gy+HSPCs +/− ACT 21d post-transfer. Percent expression panel is in supplemental fig. S2C. Experiment performed twice. All data represent the mean +/−SD. *P<.05, **P<.01, ***P<.001, ****P<.0001, by unpaired students t test for in-vitro studies (n=3) and by Mann-Whitney t test for in-vivo studies (n≥5).
To evaluate the function of HSPC-derived DCs, we FACS-sorted HSPC-derived CD11b+Ly-6G/6C− cells, a population of non-MDSC CD11b+ HSPC-derived cells (87.4% of HSPC-derived cells), from brain tumors of mice treated with ACT and DsRed+ HSPC transfer. We then co-cultured HSPC-derived DCs with tumor-specific T cells in a T cell restimulation assay and identified significant activation of polyclonal tumor-specific T cells with an 18-fold increase in IFN-γ (p=.0117; Fig. 4D). This level of T cell activation was comparable to direct recognition of KR158B-luc glioma cells by tumor reactive lymphocytes in vitro, demonstrating efficient antigen capture and presentation by HSPC-derived cells isolated from brain tumors. To demonstrate the same function completely in vivo, we treated tumor-bearing animals with ACT alone or ACT with HSPCs. Tumor-reactive lymphocytes were generated from GREAT mice, whose cells express yellow fluorescent protein (YFP) under the control of the IFN-γ promoter. Therefore, we could track the antigen-specific activation of tumor-reactive T cells by IFN-γ reporter expression in vivo. While ACT led to significant IFN-γ expression in the brain tumor microenvironment, the inclusion of HSPCs doubled the amount of IFN-γ-expressing tumor-reactive T cells and the amount of IFN-γ expression by tumor-reactive T cells in the brain tumor compared to ACT alone (YFP+ of CD8+-UnTx: 1.95%; ACT: 34.89%, p=.0079 over UnTx; ACT+HSPCs: 78.48%, p=.0079 over UnTx, p=.0079 over ACT; Fig. 4E, Supplementary Fig. S1G). This experiment demonstrated that HSPC-derived DCs induce significant intratumoral T cell activation. This falls in stark contrast to the immunosuppressive function of the intratumoral host MDSCs that are supplanted by HSPC transfer.
Since ACT alone induced a significant expression of IFN-γ in the brain tumor microenvironment, we next hypothesized that intratumoral cytokines could impact HSPC differentiation into DCs. Therefore, we determined by flow cytometry that ACT T cells infiltrate brain tumor and by whole brain tumor PCRarray that ACT induces a genetic upregulation of IFN-γ and other cytotoxic T cell genes (Fold upregulation-Ifng: 4-fold, Gzma: 18-fold, Gzmb: 3.2-fold, Fasl: 5.2-fold, ccl5: 4.5-fold; Supplementary Fig. S2, A-B). To gain an understanding of the impact of ACT on host and HSPC-derived cells in brain tumors, we compared cells that express CD11c+ or MHC II+ with and without treatment with ACT or HSPCs. We discovered that while host populations never expressed greater than 40% of either CD11c or MHC II, the HSPC-derived population expresses 84% CD11c and 80% MHC II only during ACT (Supplementary Fig. S2C). Flow cytometry also showed that ACT increased the percentage of HSPC-derived CD11c+ cells from 15% to 84% and MHC II+ cells from 5% to 80% (p=0.0079; Supplementary Fig. S2C). When we performed absolute cell counts to verify this effect, ACT doubled the absolute number of total HSPC-derived cells, CD11c+, and CD86+ HSPC-derived cells in brain tumors (Fig. 4F). To further assess the impact of ACT on HSPCs, we determined by PCRarray that when ACT is combined with HSPCs, there is an upregulation of IFN-γ signaling, interferon-responsive genes, and APC-released cytokines including Il12a and Il12b (Fold upregulation-Ifng: 2.6-fold, Irf1: 2.05-fold, Myd88: 2.05-fold, Tnfsf10: 1.66-fold, Il12a: 5.51-fold, Il12b: 3.42-fold, Il6: 2.13-fold, Cxcl9: 1.65-fold, Ccl5: 2.44-fold, Cxcl10: 1.88-fold; Supplementary Fig. S2, D-E). This suggests a feedback loop in which activated T cells influence HSPC differentiation into DCs, while HSPC-derived DCs in turn promote T cell activation.
Given the impact ACT had on HSPC engraftment and gene signatures, and the appearance that IFN-γ was critical, we sought to verify the role of activated T cell-released cytokines that drive HSPC differentiation (42, 43). Therefore, we performed supernatant transfer onto HSPCs to determine the effects of soluble factors released by tumor-reactive T cells when cultured with BMDCs presenting cognate antigens (Fig. 5A). Supernatant transfers onto freshly-isolated HSPCs included conditioned media containing acellular supernatants from 2 day co-cultures of ttRNA T cells with ttRNA-pulsed BMDCs (hereafter referred to as TC+DC supernatants). After 3 day culture of HSPCs in the conditioned media, soluble factors caused a >100-fold increase in HSPC proliferation by CellTrace Violet (p=.0021; Fig. 5, B-C, Supplementary Fig. S3). Importantly, TC+DC supernatants polarized HSPC differentiation in the proliferated populations towards DCs and away from MDSCs (MHC II+: 81.2%, CD11b+Ly-6G/6C+: 27.3%; Fig. 5, B-C). Moreover, the largest CellTrace Violet peaks significantly correlated with decreased expression of MDSC markers and the increased expression of activated DC markers including MHC II co-expressed with CD11c and CD86 on HSPC-derived cells (Fig. 5, B-C, Supplementary Fig. S3). On the contrary, supernatants from T cells that had not engaged their cognate antigens expressed by BMDC targets (T cells alone) polarized HSPC differentiation towards MDSCs (83%) and away from DCs (9%). This demonstrates that HSPCs differentiate into DCs in the presence of soluble factors released by activated T cells.
Figure 5. Impact of T cell-released soluble factors on HSPC proliferation and differentiation.
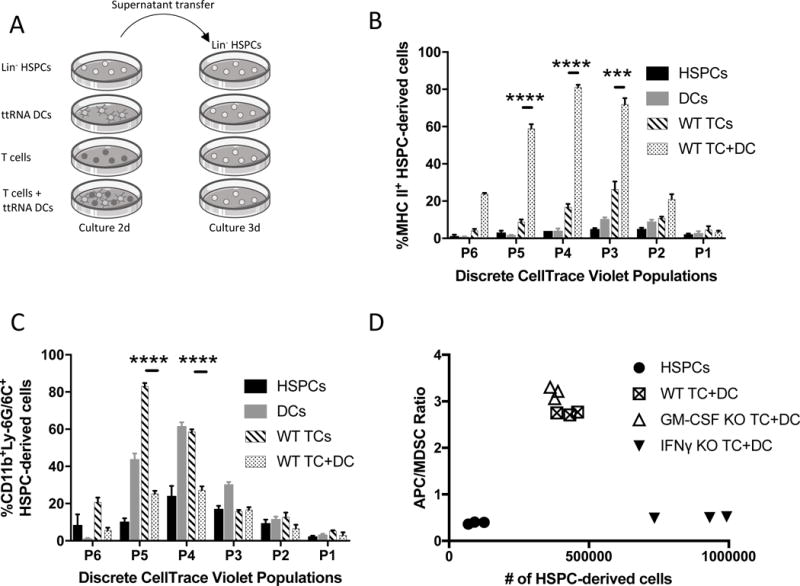
A). Experimental outline for supernatant transfer system to determine to role of T cell-released soluble factors. Supernatants were collected after centrifugation at 500rcf and filtered through a 0.2 μm syringe filter. Supernatant generation: T cells-2 million/mL, DCs-200,000/mL. Recipient culture included 200,000 HSPCs. Conditioned media was comprised of 1mL RPMI (with 10%FBS, 1% penicillin/streptomycin) and 1mL supernatant transfer media. B). Bar graph represents flow cytometry percent of HSPC-derived MHCII+ APCs after 3d of supernatant transfer conditioned media. Bar patterns indicate different supernatant transfer condition; all cells analyzed are the phenotype of only HSPC-derived cells. Gated on CellTrace Violet (CTV) proliferation peaks; P0-no proliferation, P6-most proliferative population. Experiment performed twice. C). Bar graph represents flow cytometry percent of HSPC-derived CD11b+Ly-6G/6C+ MDSCs after 3d of supernatant transfer conditioned media. Gated on CTV proliferation peaks. Experiment performed twice. D). Graph represents flow cytometry data from supernatant transfer experiment performed with wild-type T cells, GM-CSF knockout (KO) T cells, or IFN-γ KO T cells with ttRNA DC target cells. Y axis represents ratio of HSPC-derived APCs (MHC II+) to HSPC-derived MDSCs (CD11b+Ly-6G/6C+). Higher ratio is an increase in HSPC differentiation into APCs instead of MDSCs, lower ratio if the opposite. X axis represents the number of HSPC-derived cells per well. Experiment performed twice. All data represent the mean +/−SD. *P<.05, **P<.01, ***P<.001, ****P<.0001, by unpaired students t test for in-vitro studies (n=3).
We then compared the phenotype and functional capacity of HSPC-derived cells generated by 3-day culture to BMDCs generated as previously described through a 9-day selection culture. This determined that HSPC-derived cells largely expressed a similar phenotype to BMDCs including high CD11c, CD11b, MHC II, and CD86 expression (Supplementary Fig. S4A). To confirm that these HSPC-derived cells were functionally DCs, we tested the capacity of these HSPC-derived cells to prime naïve T cells. To perform this, we harvested HSPC-derived DCs or BMDCs from culture, pulsed them with ttRNA, and used them to prime GREAT mice. We then measured a significant and nearly equivalent increase in IFN-γ-producing CD3+ cells and IFN-γ release by CD3+ T cells in spleens of HSPC-derived DC and BMDC-primed hosts (%: p=0.0079, MFI: p=.0317; Supplementary Fig. S4B). This indicated that while HSPC-derived DCs from brain tumors could activate tumor-reactive T cells, in vitro HSPC-derived DCs can also prime naïve T cells in the periphery at a level similar to traditional BMDCs.
To determine if HSPC differentiation into DCs was dependent on T cells recognizing cognate antigen, supernatants were collected from either co-cultures of KR158B-luc-specific T cells+B16F10OVA cells (irrelevant target) or KR158B-luc T cells+KR158B-luc ttRNA-pulsed BMDCs (relevant target expressing tumor-derived antigens). We then performed the supernatant transfer onto HSPCs and determined that antigen-matched TC+DC supernatants were significantly better at upregulating MHC II on HSPCs compared to the antigen-mismatched supernatants (B16F10OVA: 28% vs. ttRNA DC: 70%; p=.0002; Supplementary Fig. S5A). We also determined that T cell recognition of KR158B-luc glioma cells generated soluble factors that promoted HSPC differentiation into CD11c+MHCII+CD86+ DCs (Supplementary Fig. S5B). Overall, this indicates that T cell-released cytokines significantly shift after T cell activation with antigen-specific target cells to promote HSPC differentiation into DCs. Additionally, this indicated that tumor-reactive T cells should likely release these cytokines into the tumor microenvironment in vivo.
We hypothesized that IFN-γ would likely be the key T cell-released cytokine in this system given the preponderance of IFN-γ upregulation in ACT-treated brain tumors (Supplementary Fig. S2B) and the described role for IFN-γ in myelopoiesis, APC activation, and broad anti-cancer immunity (44-48). Therefore, we performed a series of in vitro experiments with selective inclusion or depletion of T cell-released cytokines. We evaluated GM-CSF in parallel with IFN-γ given its similarly important role in HSPC differentiation. We first demonstrated that T cells release IFN-γ and GM-CSF after recognition of antigen-specific targets (p<.0001 for IFN-γ and GM-CSF; Supplementary Fig. S6). We next performed GM-CSF and IFN-γ antibody blockade during supernatant transfer. Antibody blockade caused a significant decrease in HSPC-derived DCs during supernatant transfer with 25μg/mL anti-IFN-γ (57% decrease) or 25μg/mL anti-GM-CSF (77% decrease; Supplementary Fig. S7A). To confirm these results, we evaluated supernatant transfer with T cells from GM-CSF and IFN-γ cytokine knockout animals. This generated functional T cells that produced a milieu of activated T cell cytokines after antigenic stimulation, including TNF-alpha, without the contribution of either IFN-γ or GM-CSF (Supplementary Fig. S7B). When analyzing HSPCs cultured in IFN-γ-deficient TC+DC supernatants, HSPCs proliferated more and significantly increased their differentiation into MDSCs (90% increase, p<0.0001) and decreased differentiation into DCs (65% decrease, p<0.0001) when compared to WT TC+DC supernatants (Fig. 5D, Supplementary Fig. S8). On the contrary, GM-CSF-deficient supernatant transfer mildly impaired HSPC proliferation and differentiation. This suggests a role for T cell-released IFN-γ in arresting HSPC proliferation for the selective differentiation into a DC instead of an MDSC. We then tested the impact of 1μg of IFN-γ on 3-day in vitro culture of HSPCs from WT or IFN-γ receptor knockout (IFN-γR−/−) mice. This demonstrated that HSPC-derived cells express 5-fold more MHC II when treated with 125 pg/mL IFN-γ (p<0.0001) and that this upregulation is abrogated when the HSPCs are from IFN-γR−/− mice (p<0.0001; Fig. 6A).
Figure 6. Impact of IFN-γ on anti-tumor efficacy of HSPCs and ACT.
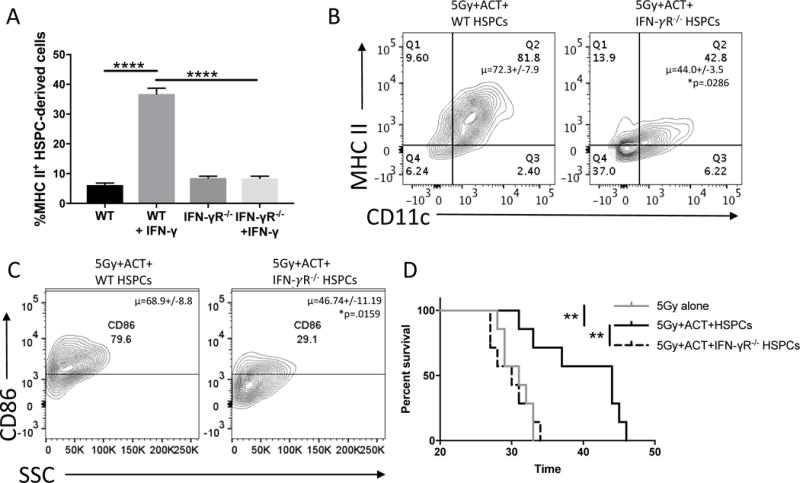
A). Bar graph represents flow cytometry of 3d. in-vitro culture of HSPCs either UnTx or with 125pg/mL IFN-γ. Groups on the left were performed with HSPCs from WT mice, groups on right were performed with HSPCs from IFN-γR−/− mice. Experiment performed twice. B). Representative flow cytometry of brain tumors of mice 21d post-treatment with 5Gy, ACT, and WT HSPCs or IFN-γR−/− HSPCs. Without IFN-γR, HSPC-derived cells have decreased CD11+MHCII+ co-expression indicating decreased DC phenotype. In this experiment, DsRed mice were the tumor-bearing hosts while WT mice or IFN-γR−/− mice were the source of HSPCs. This allowed tracking of HSPC-derived populations in the DsRed− compartment. Experiment performed twice. C). Representative flow cytometry of brain tumors of mice 21d post-treatment with 5Gy, ACT, and WT HSPCs or IFN-γR−/− HSPCs. Flow plots are analyzing CD86 expression on previously gated CD11c+MHCII+ HSPC-derived cells. Previous reports have indicated these co-stimulatory-expressing DCs are critical to brain immune responses. Experiment performed twice. D). Survival curve of brain tumor-bearing mice treated with 5Gy, 5Gy+ACT+HSPCs, or 5Gy+ACT+HSPCs from an IFN-γR−/− mouse. All data represent the mean +/−SD. *P<.05, **P<.01, ***P<.001, ****P<.0001, by unpaired students t test for in-vitro studies (n=3), by Mann-Whitney t test for in-vivo studies (n≥4) and Mantel-Cox Log Rank Test for survival experiments (n=7).
Having demonstrated the capacity for T cell derived IFN-γ to drive HSPC-differentiation in vitro in an antigen-dependent manner, and the effectiveness of ACT in increasing intratumoral IFN-γ, we evaluated whether IFN-γ signaling was critically involved in intratumoral HSPC differentiation. To do so, we analyzed the intratumoral differentiation of WT or IFN-γR−/− HSPCs in mice treated with ACT. This demonstrated that IFN-γR−/− HSPCs have a significantly impaired ability to differentiate into CD11c+MHCII+ DCs (WT: 71.98%; IFN-γR−/−: 44.27%; p=.0357; Fig. 6B, Supplementary Fig. S9). Additionally, IFN-γR−/− HSPC-derived CD11c+MHCII+ cells have an impaired ability to upregulate the CD86 co-stimulatory activation marker (32% decrease; WT: 68.9%; IFN-γR−/−: 46.74%; p=.0159; Fig. 6C). Lastly, IFN-γR−/− HSPCs differentiated into CD11b+Ly-6G/6C+ MDSCs 131% more than WT HSPCs (WT HSPCs: 3.6% MDSCs, IFN-γR−/− HSPCs: 8.3% MDSCs; p=.0161; Supplementary Fig. S9).
Since removing IFN-γR from HSPCs impaired the ability to generate intratumoral DCs, we hypothesized that IFN-γR−/− HSPCs would have an impaired ability to prolong survival in conjunction with ACT in tumor-bearing animals. We utilized a 5Gy model system since IFN-γR−/− HSPCs are poor rescuers of 9Gy-treated host bone marrow (<40% survival; p=.013). We therefore treated mice with 5Gy alone, 5Gy+ACT+WT HSPCs, or 5Gy+ACT+IFN-γR−/− HSPCs. This experiment demonstrated that the overall anti-tumor efficacy of the adoptive cellular therapy platform is completely abrogated when HSPCs do not possess IFN-γR (WT HSPCs+ACT: 32% median survival benefit, IFN-γR−/− HSPCs+ACT: -3% median survival benefit compared to 5Gy alone, p=0.0053; Fig. 6D). These results demonstrate that the survival benefit generated by HSPCs+ACT is reliant on IFN-γ signaling within transferred HSPCs and highlights a critical role of these cells in mediating anti-tumor efficacy.
In summary, HSPCs interact coordinately with T cells during ACT to promote the immunologic rejection of malignant brain tumors through a reliance upon an IFN-γ-driven feedback loop between HSPCs and tumor-reactive T cells. In this synergy, HSPCs rely upon T cell-released IFN-γ to drive the generation of potent, activated HSPC-derived DCs in the brain tumor microenvironment. In turn, HSPC-derived DCs function as in situ activators of tumor-reactive T cells. Through capitalizing on these mechanistic understandings, HSPC transfer and ACT hold significant potential as effective cellular immunotherapies for malignant brain tumors and potentially other solid malignancies.
Discussion
While HSPCs possess the ability to differentiate into any hematopoietic cell, many reports demonstrate that HSPCs in solid cancer-bearing hosts are immunosuppressive (21-27). Specifically, they are polarized to differentiate into MDSCs and can home to the bone marrow, spleen, tumor site, and the peripheral blood to impair immune responses and even promote metastases. Our study demonstrates that the combination of ACT and HSPC transfer can reprogram the typical immunosuppressive differentiation and function of HSPCs in solid cancers. While we focused on the brain tumor microenvironment because HSPCs are tropic for this region, it will be interesting to investigate the cellular interplay in peripheral lymphoid organs and to see if these findings can be translated to peripheral solid cancers to supplant host MDSCs, promote the differentiation of DCs, and thereby promote cytotoxic rejection of tumors.
Dogma indicates that the brain is immunoprivileged to protect neurons from immunologic targeting and manage critical skull pressures (15, 49); however, recent studies have demonstrated considerable immune activity in the brain, including the cytotoxic rejection of central nervous system malignancies (4). Despite this success, the CNS has a lack of activated DCs in the brain at homeostasis (13-15). This diminishes sampling of tissues that are immune-protected and can delay or impair the stimulation of an inflammatory response in the brain. While certain APCs can be activated in the brain during autoimmunity and infection, in the context of brain malignancies, brain immunoprivilege works in unison with the already immunosuppressive microenvironment of brain tumors to doubly impair APC activation and infiltration (17). This can manifest with a downregulation of MHC class I and II as well as co-stimulatory molecules including CD86 (14, 17).
Importantly, recent reports have indicated the success of ACT for peripheral cancers is reliant on intratumoral activated DCs (12). Our study provides a mechanism for counteracting the lack of activated DC infiltration in brain tumors. Through HSPC transfers and the simultaneous infusion of ACT, we can generate a significant population of DCs that promote cytotoxic responses. These HSPC-derived cells are particularly potent because of the CD86 co-stimulatory marker that enables the generation of a strong T cell response (14, 50). Moreover, given the upregulation of IL-12 in brain tumors treated with HSPCs and ACT, it will be interesting to investigate the role of HSPC-derived DC-released cytokines that promote immune cell development and anti-tumor immunity.
Our study suggests that during HSPC+ACT therapy there is an intricate network of functions including chemotaxis, proliferation, differentiation, T cell activation, and cytotoxic tumor destruction that requires further investigation. This future investigation will include investigation of other cell types including NK cells, other cytokines including type-1 interferons (12), and attention to immunosuppressive gene signatures. Given the highly pliable nature of the microenvironment, we anticipate that there will be a collaborative set of alterations in the brain tumor microenvironment that extends beyond our current findings to yield an impairment of immunosuppression in favor of immune rejection. Additionally, while we have demonstrated that HSPCs act as their own immunologic agent in promoting anti-tumor immunity, there is already a keen interest in using HSPCs as a carrier to intracranial tumors. It will be interesting to see if HSPCs can be loaded as an immunomodulatory vehicle to further reprogram the brain tumor microenvironment.
Potential limitations to ACT and HSPC therapy could include HSPC differentiation into suppressive cells in the periphery, an exhaustion of HSPC-derived DCs near endpoint, tumor escape from immunoediting, or the potential for HSPC-derived DCs and T cells to have checkpoint blockade molecule interactions. In future studies it will be important to test the timing of tumor implantation and treatment. Ongoing studies evaluating mobilized HSPCs, synergism with checkpoint blockade, host conditioning with chemotherapy, overcoming tumor escape from T cell immunotherapy, and endogenous tumor models will be instrumental to continue to improve our translation to the clinic.
While brain tumors are largely resistant to DC infiltration and activation (13, 14, 49), we have demonstrated a unique mechanism of supplanting MDSCs while generating intragliomal DCs with potent immunologic functions. Importantly, this synergy combines a minimization of immune suppression and concomitant immune activation. Taken together, this data implies that HSPCs are more than just stem or progenitor cells capable of rescuing bone marrow, but cells capable of synergistically enhancing in situ anti-tumor immunity in the immunosuppressive brain tumor microenvironment.
Supplementary Material
Translational Relevance.
Malignant gliomas are notoriously recalcitrant and often overcome any attempts at engendering effective anti-tumor immunity. We have previously demonstrated hematopoietic stem and progenitor cells (HSPCs) play a novel and synergistic role in enhancing the effectiveness of adoptive T cell therapy targeting invasive malignant gliomas. Herein, we demonstrate that adoptive cellular therapy incorporating HSPC transfer is effective against multiple preclinical brain tumor models (glioblastoma, diffuse intrinsic brain stem glioma, and Group 3 medulloblastoma) and that HSPCs are driven by T cell-released IFN-γ to differentiate into potent intratumoral dendritic cells (DCs) in situ within the tumor microenvironment. These HSPC-derived DCs additionally supplant intratumoral host myeloid-derived suppressor cells, potentiate T cell activation in situ, and drive intracranial tumor rejection. These findings unravel the mechanistic cross-talk between tumor-specific T cells and HSPCs within malignant gliomas during adoptive cellular therapy and point to the novel utilization of HSPCs in cellular immunotherapy.
Acknowledgments
This research was supported by the University of Florida Health Cancer Center Predoctoral Award (T. Wildes); National Cancer Institute R01 CA195563 (D. Mitchell); American Brain Tumor Association Research Collaboration Grant (C. Flores); Alex’s Lemonade Stand Young Investigator Grant (C. Flores); Florida Center for Brain Tumor Research Grant (C. Flores); the Preston A. Wells, Jr. Endowment at the University of Florida; and University of Florida Clinical and Translational Sciences Award (5UL1TR001427-03).
Footnotes
Conflict of interest: CTF and DAM have patents related to material disclosed in this publication that have been licensed to iOncologi, Inc. CTF and DAM hold interests in iOncologi, Inc., a biotechnology company focused on immuno-oncology. TJW, AG, KAD, BMW, DJD, and RSA declare no conflicts of interest.
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: A clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Taillibert S, Kanner AA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. JAMA. 2015;314:2535–2543. doi: 10.1001/jama.2015.16669. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell DA, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–369. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong JJ, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–4898. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores C, et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology. 2015;4:e994374. doi: 10.4161/2162402X.2014.994374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrzesinski C, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wrzesinski C, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. Journal of immunotherapy (Hagerstown, Md: 1997) 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batich KA, et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clinical Cancer Research. 2017;23:1898–1909. doi: 10.1158/1078-0432.CCR-16-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gururangan S, et al. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. Journal of Clinical Oncology. 1998;16:2486–2493. doi: 10.1200/JCO.1998.16.7.2486. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg SA, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–723.e714. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends in Immunology. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Fabry Z, Schreiber HA, Harris MG, Sandor M. Sensing the microenvironment of the central nervous system: immune cells in the central nervous system and their pharmacological manipulation. Current Opinion in Pharmacology. 2008;8:496–507. doi: 10.1016/j.coph.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 16.Dejaegher J, Van Gool S, De Vleeschouwer S. Dendritic cell vaccination for glioblastoma multiforme: review with focus on predictive factors for treatment response. Immunotargets and Therapy. 2014;3:55–66. doi: 10.2147/ITT.S40121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol. 2011;11:685–692. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao JL, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell stem cell. 2014;14:445–459. doi: 10.1016/j.stem.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 21.Wu WC, et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci U S A. 2014;111:4221–4226. doi: 10.1073/pnas.1320753111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sio A, et al. Dysregulated hematopoiesis caused by mammary cancer is associated with epigenetic changes and hox gene expression in hematopoietic cells. Cancer Res. 2013;73:5892–5904. doi: 10.1158/0008-5472.CAN-13-0842. [DOI] [PubMed] [Google Scholar]
- 23.Pu S, et al. Identification of early myeloid progenitors as immunosuppressive cells. Scientific reports. 2016;6:23115. doi: 10.1038/srep23115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang WW, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giles AJ, et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre-metastatic Niche. Cancer Res. 2016;76:1335–1347. doi: 10.1158/0008-5472.CAN-15-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casbon AJ, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112:E566–575. doi: 10.1073/pnas.1424927112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabatabai G, et al. Lessons from the bone marrow: how malignant glioma cells attract adult haematopoietic progenitor cells. Brain: a journal of neurology. 2005;128:2200–2211. doi: 10.1093/brain/awh563. [DOI] [PubMed] [Google Scholar]
- 29.Hasenbach K, et al. Monitoring the glioma tropism of bone marrow-derived progenitor cells by 2-photon laser scanning microscopy and positron emission tomography. Neuro Oncol. 2012;14:471–481. doi: 10.1093/neuonc/nor228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabatabai G, Frank B, Mohle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-beta-dependent HIF-1alpha-mediated induction of CXCL12. Brain: a journal of neurology. 2006;129:2426–2435. doi: 10.1093/brain/awl173. [DOI] [PubMed] [Google Scholar]
- 31.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 32.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 33.Pham CD, et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin Cancer Res. 2016;22:582–595. doi: 10.1158/1078-0432.CCR-15-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York, NY) 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 35.Cordero FJ, et al. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Molecular cancer research: MCR. 2017;15:1243–1254. doi: 10.1158/1541-7786.MCR-16-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-Specific Inhibition of CD8+ T Cell Response by Immature Myeloid Cells in Cancer Is Mediated by Reactive Oxygen Species. The Journal of Immunology. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 38.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nature Immunology. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 40.Lu C, Savage N, Singh N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. The Journal of Immunology. 2017;198:124.129–124.129. doi: 10.1080/2162402X.2016.1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mildner A, Jung S. Development and Function of Dendritic Cell Subsets. Immunity. 2014;40:642–656. doi: 10.1016/j.immuni.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 42.Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell stem cell. 2014;14:460–472. doi: 10.1016/j.stem.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 44.de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood. 2014;124:2479–2486. doi: 10.1182/blood-2014-04-568451. [DOI] [PubMed] [Google Scholar]
- 45.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. Journal of leukocyte biology. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 46.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 47.Giermasz AS, et al. Type-1 polarized dendritic cells primed for high IL-12 production show enhanced activity as cancer vaccines. Cancer immunology, immunotherapy: CII. 2009;58:1329–1336. doi: 10.1007/s00262-008-0648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaliński P, Schuitemaker JHN, Hilkens CMU, Wierenga EA, Kapsenberg ML. Final Maturation of Dendritic Cells Is Associated with Impaired Responsiveness to IFN-γ and to Bacterial IL-12 Inducers: Decreased Ability of Mature Dendritic Cells to Produce IL-12 During the Interaction with Th Cells. The Journal of Immunology. 1999;162:3231–3236. [PubMed] [Google Scholar]
- 49.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunological reviews. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.