

The nonstructural membrane protein encoded by open reading frame K15 of Kaposi's sarcoma-associated herpesvirus (KSHV) (HHV8) activates several intracellular signaling pathways that contribute to the angiogenic properties of KSHV in endothelial cells and to its reactivation from latency. A detailed understanding of how pK15 activates these intracellular signaling pathways is a prerequisite for targeting these processes specifically in KSHV-infected cells. By identifying pK15-associated cellular proteins using a combination of immunoprecipitation and mass spectrometry, we provide evidence that pK15-dependent signaling may occur from intracellular vesicles and rely on the endocytotic machinery. Specifically, a class II PI3K, PI3K-C2α, is recruited by pK15 and involved in pK15-dependent intracellular signaling and viral reactivation from latency. These findings are of importance for future intervention strategies that aim to disrupt the activation of intracellular signaling by pK15 in order to antagonize KSHV productive replication and tumorigenesis.
KEYWORDS: KSHV-K15, PI3K-C2α, lytic replication
ABSTRACT
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV)/human herpesvirus 8 (HHV-8) causes the angiogenic tumor KS and two B-cell malignancies. The KSHV nonstructural membrane protein encoded by the open reading frame (ORF) K15 recruits and activates several cellular proteins, including phospholipase Cγ1 (PLCγ1), components of the NF-κB pathway, as well as members of the Src family of nonreceptor tyrosine kinases, and thereby plays an important role in the activation of angiogenic and inflammatory pathways that contribute to the pathogenesis of KS as well as KSHV productive (lytic) replication. In order to identify novel cellular components involved in the biology of pK15, we immunoprecipitated pK15 from KSHV-infected endothelial cells and identified associated proteins by label-free quantitative mass spectrometry. Cellular proteins interacting with pK15 point to previously unappreciated cellular processes, such as the endocytic pathway, that could be involved in the function of pK15. We found that the class II phosphatidylinositol 3-kinase (PI3K) PI3K-C2α, which is involved in the endocytosis of activated receptor tyrosine kinases and their signaling from intracellular organelles, interacts and colocalizes with pK15 in vesicular structures abundant in the perinuclear area. Further functional analysis revealed that PI3K-C2α contributes to the pK15-dependent phosphorylation of PLCγ1 and Erk1/2. PI3K-C2α also plays a role in KSHV lytic replication, as evidenced by the reduced expression of the viral lytic genes K-bZIP and ORF45 as well as the reduced release of infectious virus in PI3K-C2α-depleted KSHV-infected endothelial cells. Taken together, our results suggest a role of the cellular PI3K-C2α protein in the functional properties of the KSHV pK15 protein.
IMPORTANCE The nonstructural membrane protein encoded by open reading frame K15 of Kaposi's sarcoma-associated herpesvirus (KSHV) (HHV8) activates several intracellular signaling pathways that contribute to the angiogenic properties of KSHV in endothelial cells and to its reactivation from latency. A detailed understanding of how pK15 activates these intracellular signaling pathways is a prerequisite for targeting these processes specifically in KSHV-infected cells. By identifying pK15-associated cellular proteins using a combination of immunoprecipitation and mass spectrometry, we provide evidence that pK15-dependent signaling may occur from intracellular vesicles and rely on the endocytotic machinery. Specifically, a class II PI3K, PI3K-C2α, is recruited by pK15 and involved in pK15-dependent intracellular signaling and viral reactivation from latency. These findings are of importance for future intervention strategies that aim to disrupt the activation of intracellular signaling by pK15 in order to antagonize KSHV productive replication and tumorigenesis.
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) was first discovered in 1994 by Yuan Chang and Patrick S. Moore in KS, a highly vascularized tumor of endothelial origin (1). It also plays an important role in the pathogenesis of two types of malignant B-cell-associated disorders known as primary effusion lymphoma (PEL) (2) and the plasmablastic variant of multicentric Castleman's disease (MCD) (3). The tumorigenic endothelial spindle cells in KS lesions as well as the malignant B cells in the two lymphoproliferative diseases are infected with KSHV (4–10). Similar to all other herpesviruses, latency is the default pathway after KSHV infection. Productive (lytic) replication can also occur spontaneously in a subpopulation of KSHV-infected tumor cells (6, 7, 11, 12). The KSHV genome contains several genes coding for proteins that are capable of eliciting cellular signaling pathways. Among these are the viral G-protein-coupled receptor (vGPCR), K1, viral interleukin-6 (vIL-6), homologues of cellular CC chemokines, and K15. Cellular signaling subsequently leads to the expression of inflammatory and angiogenic molecules that are known to play an important role in KSHV lytic replication and disease progression (13–20).
The K15 gene is located between open reading frame 75 (ORF75) and the terminal repeats at the end of the KSHV long unique region (LUR). It contains eight alternatively spliced exons encoding multiply spliced variants of the K15 transcript, which can be translated into K15 proteins featuring a variable number of transmembrane domains with attached short amino (N)-terminal and long carboxyl (C)-terminal cytoplasmic domains (16, 17, 21, 22). There are three highly divergent alleles of the K15 gene known so far in different KSHV genomes, predominant (P), minor (M), and N, which display low levels (less than 33%) of amino acid sequence homology (16, 17, 23). However, the C-terminal cytoplasmic domains of the proteins encoded by all three K15 alleles possess conserved signaling motifs, including a tumor necrosis factor receptor-associated factor (TRAF)-binding site, two Src homology (SH2)-binding sites (YASIL and YEEVL), and an SH3-binding site (PPLP), among others (16, 17).
The expression of K15 mRNA and protein can be detected in latently infected (unstimulated) cells, while their expression increases upon the induction of the KSHV lytic cycle (16, 17, 19, 21, 24–26). Consistent with its classical description as an early lytic gene, the promoter of the K15 gene can be trans-activated by regulator of transcription activation (RTA) in the PEL cell line BCBL-1 (27), and it has been shown to be expressed predominantly during the KSHV lytic cycle in PEL cell lines (28–30). However, both in cultured KSHV-infected cells and in KS tissue samples, the K15 locus in the KSHV genome displays activating epigenetic marks similar to the promoter region of the constitutively active latent genes (31, 32), and the K15 mRNA is expressed in a fashion similar to that of genes in the latency locus in endothelial cells of both blood and lymphatic origins, representing restricted and relaxed KSHV transcription profiles, respectively (33). Moreover, K15 mRNA is strongly expressed in KS lesions obtained from treatment-naive HIV-positive patients displaying both relaxed and restricted KSHV transcription profiles (34). These findings suggest that K15 is an inducible gene that can be expressed during latency or a “relaxed” form of latency. By using a monoclonal antibody (mAb) to the K15 C-terminal cytoplasmic tail, pK15 protein expression has been immunohistochemically detected in latently infected PEL cells and in KSHV latency-associated nuclear antigen (LANA)-positive plasmablasts in MCD (35). Similarly, we have recently demonstrated the expression of the K15 protein in KS tumors obtained from AIDS-KS patients (19).
Although K15 is a membrane protein, it primarily localizes within the cytoplasm as punctate structures that are abundant in the perinuclear area as well as in the cell periphery in medium to large patches both during overexpression and in the context of infection (17, 19, 21, 25, 26, 35–37). Colocalization experiments using markers of intracellular membrane-bound organelles showed the association of K15 with the Golgi apparatus, the endoplasmic reticulum, the late endosome, and, partially, with the lipid raft marker caveolin (19, 35, 38).
The K15 protein is capable of binding several cellular proteins with SH2 and/or SH3 domains by means of two SH2- and/or one SH3-binding site in its C-terminal cytoplasmic tail. A chimera of the K15 C-terminal cytoplasmic tail placed instead of the cytoplasmic domain of the CD8α molecule is constitutively tyrosine phosphorylated at the distal SH2-binding site (Y481EEVL) (21). This tyrosine residue can be bound and phosphorylated by members of the Src family of protein tyrosine kinases (22). The recruitment of phospholipase Cγ1 (PLCγ1) to this same phosphorylated tyrosine residue (Y481EEVL) in the K15 cytoplasmic tail induces the constitutive activation of the PLCγ1-calcineurin-NFAT (nuclear factor of activated T cells) pathway and the expression of downstream proangiogenic genes such as Dscr-1 (24–26, 36, 39). K15 also binds to the TRAF family of proteins, TRAF-1, -2, and -3, which are involved in the activation of NF-κB-mediated gene expression and c-Jun N-terminal kinase (JNK1) signaling (17, 22). In line with this, K15 expression induces the activation of the two mitogen-activated protein (MAP) kinases Erk2 and JNK as well as the NF-κB and AP-1 transcription factors, which was abolished with a point mutation (Y481F) in the K15 second-SH2-binding site, suggesting that the phosphorylation of this residue is an important step in K15-induced signaling (22, 39). In addition to the tyrosine residue at Y481EEVL, K15-mediated NF-κB activation also depends on the recruitment of the NF-κB-inducing kinase (NIK) and the IκB kinase (IKK) complex (IKKα and -β) to its membrane-proximal region containing the amino acid sequence RQRRRR (40). Consistent with its ability to activate the PLCγ1, MAP kinase, and NF-κB pathways, K15 induces the expression of a plethora of proangiogenic and proinflammatory factors, including Dscr-1, interleukin-8 (IL-8), IL-6, IL-1α/β, CCL20, CCL2, CXCl3, and Cox-2, mostly dependent on the phosphorylation of its Y481 residue (24, 25). K15 also contributes to KSHV-mediated endothelial spindle cell formation, angiogenesis, and invasiveness in infected endothelial cells as well as KSHV lytic replication (19, 24, 26).
The K15 SH3-binding site also recruits components of the endocytic machinery, including the SH3-C-domain-containing adaptor protein intersectin 2 (ITSN2), an indication that K15 might be involved in endocytic trafficking (36). In agreement with this notion, the overexpression of K15 in B cells can inhibit B-cell receptor (BCR) signaling activation (21, 41) and increase the rate of BCR internalization after anti-IgM antibody treatment (36). However, in BCR-positive (BCR+) B cells that had been immortalized with a recombinant Epstein-Barr virus (EBV) genome containing the K15 gene in place of the latent membrane protein 2A (LMP2A) gene, K15 inhibited neither the phosphorylation levels of BCR-associated signaling proteins, such as Syk and PLCγ2, nor the mobilization of intracellular calcium ions (Ca2+) upon BCR cross-linking, suggesting that K15 might rather play a positive role in the activation of BCR signaling (42).
pK15 has also been shown to interact with the antiapoptotic protein HAX-1 (HS1-associated protein X-1) through its other SH2-binding site (YASIL) (35). The functional consequence of this interaction is not well defined; however, when placed instead of the LMP2A gene in the EBV genome, K15 provides survival and proliferative advantages for infected BCR-negative B cells, which are otherwise proapoptotic because of the lack of survival signals from the BCR (42). In support of a role in host cell survival, pK15 also induces the expression of a number of antiapoptotic genes, including birc2, brc3, bf, A20, and bcl2a1 (25, 35, 42).
Taken together, the above-described observations show that KSHV pK15 can recruit several cellular proteins that play an important role in the activation of angiogenic and inflammatory pathways and that might provide a survival advantage to the infected cell (19, 22, 24–26, 40, 41). In this study, we have used immunoprecipitation followed by mass spectrometry (MS) in order to identify additional pK15-interacting partners and elucidate their functions. We found that the α-isoform of the class II phosphatidylinositol 3-kinase (PI3K) PI3K-C2α, which is involved in receptor endocytosis and signaling activation following the binding of cognate ligands, is a novel pK15-interacting partner. We show that PI3K-C2α colocalizes with pK15 in intracellular vesicles. Furthermore, similar to its role in receptor-mediated signaling, PI3K-C2α plays a role in the activation of pK15-mediated signaling as well as KSHV lytic reactivation in infected endothelial cells. Taken together, our results here demonstrate the importance of PI3K-C2α in pK15-mediated signaling and point to a role for this nonstructural viral membrane protein in mediating signaling from intracellular membrane vesicles as a key step in virus reactivation.
RESULTS
Identification of K15-interacting proteins by label-free quantitative proteomics.
Endothelial cells are among the targets of KSHV infection in vivo (4, 7–9) that play an important role in the pathogenesis of KS. The KSHV K15 protein, which we recently showed to be expressed in KS lesions (19), contributes to the invasiveness and angiogenic properties of infected endothelial cells in culture and to the formation of endothelial spindle cells as well as KSHV lytic replication (19, 24, 26). In order to identify new interacting partners of the pK15 protein, a conditionally immortalized endothelial cell line (HuARLT2), stably infected with a recombinant KSHV (HuARLT2-rKSHV), was used (see Materials and Methods). A rat monoclonal antibody to pK15, 6E7, directed against the PPLP SH3-binding motif in the cytoplasmic domain of pK15, was covalently cross-linked to protein A/G beads by using the pierce cross-link immunoprecipitation (IP) kit, which enables efficient antigen immunoprecipitation with less IgG contamination from the antibody. 6E7-coated beads were used to immunoprecipitate endogenous pK15 from HuARLT2-rKSHV cells, which were either left untreated or treated with a cocktail of a baculovirus encoding KSHV RTA and sodium butyrate (SB) to induce the lytic replication cycle (Fig. 1A). Cellular lysates were also prepared from uninfected parental HuARLT2 cells that had been treated similarly and used in parallel as a control. The efficiency of the IP was assessed by Western blot analysis of a small aliquot from each preparation (Fig. 1B). After scaling up the experiment to 5 × 108 KSHV-infected and uninfected HuARLT2 cells for each immunoprecipitation, the immunoprecipitated proteins were resolved on SDS-PAGE gels and visualized by staining with Coomassie brilliant blue dye.
FIG 1.

Experimental workflow and sample preparation. (A) Detailed scheme of the interaction proteomics workflow. The KSHV lytic cycle was induced by using RTA and SB in latently infected HuARLT2 cells or uninfected control parental cells. After 48 h, endogenous K15 protein was precipitated from both induced and uninduced samples by using a rat anti-K15 mAb (clone 6E7). (B) Western blot (WB) analysis, using antibody 10A6 to pK15, of a small aliquot of the IP preparation used for MS/MS analysis. Experiments were performed in two independent biological replicates.
Entire lanes of the Coomassie blue-stained SDS-PAGE gel were then cut into pieces, subjected to tryptic digestion, and analyzed by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS). Data were analyzed with MaxQuant software, and the resulting files were searched against the human and KSHV/human herpesvirus 8 (HHV-8) complete proteome sequences obtained from the UniProt database (https://www.uniprot.org/) and a set of commonly observed contaminants. Typical for an affinity-based enrichment (43), a total of 800 cellular proteins were identified. Potential nonspecific binders were excluded during subsequent data processing. pK15 was the only viral protein identified.
To identify specific pK15-interacting partners, the peptide label-free quantification (LFQ) intensity ratio of HuARLT2-rKSHV cells with or without induction of the KSHV lytic cycle to the respective uninfected control HuARLT2 cells was calculated. A total of 19 (Fig. 2A) and 154 (Fig. 2B) proteins were identified in uninduced and induced samples, respectively, from two independent experiments, with a cutoff set at 4-fold enrichment. The resulting list of candidate proteins from both induced and uninduced samples was then further filtered by removing known contaminants, including ribosomal and cytoskeletal proteins as well as metabolic enzymes, from the CRAPome (contaminant repository for affinity purification) database (http://www.crapome.org/). The cleared lists of pK15-interacting proteins identified from HuARLT2-rKSHV cells are shown in Table 1 (9 uninduced proteins) and Table 2 (75 proteins after lytic induction;). Only 3 proteins (labeled in blue in Fig. 2A) were common between the induced and uninduced samples (Fig. 2A and B).
FIG 2.

Distribution of proteins identified after MS/MS analysis. (A and B) Scatter plot showing the ratio of the log2 LFQ intensity of proteins from Hu-rKSHV cells to that of proteins from HuARLT2 cells, where experiment 1 is plotted against experiment 2, in uninduced cells (latent) (A) and after lytic induction (B). The names of proteins identified in both latent and lytic samples are shown in blue. The second dotted lines in panel B mark proteins enriched more than 64-fold for both experiments. The list of the pK15-interacting proteins identified in cells undergoing lytic KSHV replication (Table 2) was uploaded to the DAVID bioinformatics tool. (C and D) Gene Ontology (GO) annotations for enriched cellular compartments (C) and molecular functions (D), with a cutoff (P < 0.006) based on their fold enrichment, are shown.
TABLE 1.
List of 9 K15-interacting proteins identified by LC-MS/MS from latent infectiona
| Gene | Protein | Log2 FE, expt 1 | Log2 FE, expt 2 |
|---|---|---|---|
| CLTA | Clathrin light chain A | 2.7 | 2.8 |
| PSMC4 | 26S protease regulatory subunit 6B | 3.9 | 2.6 |
| SCCPDH | Saccharopine dehydrogenase-like oxidoreductase | 3.6 | 2.6 |
| PIK3C2A | Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit alpha | 2.5 | 2.4 |
| PCMTD1 | Protein-l-isoaspartate O-methyltransferase domain-containing protein 1 | 2.1 | 2.3 |
| SGCG | Zeta-sarcoglycan | 3.7 | 2.3 |
| ASS1 | Argininosuccinate synthase | 2.9 | 2.2 |
| PSMD6 | 26S proteasome non-ATPase regulatory subunit 6 | 3.4 | 2.2 |
| ADRM1 | Proteasomal ubiquitin receptor ADRM1 | 2.7 | 2.2 |
The list of pK15-interacting proteins that were identified in two independent experiments with more than 4-fold enrichment from unstimulated cells was cleared of potential nonspecific binders found in the CRAPome database (http://crapome.org/), and the resulting list of proteins is shown. FE, fold enrichment.
TABLE 2.
List of 75 K15-interacting proteins identified by LC-MS/MS after lytic reactivationa
| Gene(s) | Protein(s) | Log2 FE, expt 1 | Log2 FE, expt 2 |
|---|---|---|---|
| PSMD11 | 26S proteasome non-ATPase regulatory subunit 11 | 7.7 | 2.5 |
| PSMC1 | 26S protease regulatory subunit 4 | 2.1 | 2.6 |
| SEPT2 | Septin-2 | 4.0 | 3.0 |
| DBN1 | Drebrin | 6.6 | 5.0 |
| SEPT9 | Septin-9 | 4.9 | 3.4 |
| LIMA1 | LIM domain and actin-binding protein 1 | 5.2 | 6.6 |
| CTTN | Src substrate cortactin | 7.5 | 8.6 |
| HADHA | Trifunctional enzyme subunit alpha, mitochondrial; long-chain enoyl-CoA hydratase; long chain 3-hydroxyacyl-CoA dehydrogenase | 2.4 | 2.9 |
| MYO1B | Unconventional myosin-Ib | 5.4 | 5.6 |
| ARF4, ARF5 | ADP-ribosylation factor 4, ADP-ribosylation factor 5 | 2.9 | 3.2 |
| MYO1C | Unconventional myosin-Ic | 3.4 | 4.6 |
| TMOD3 | Tropomodulin-3 | 3.1 | 5.3 |
| BASP1 | Brain acid soluble protein 1 | 7.1 | 5.8 |
| KPRP | Keratinocyte proline-rich protein | 3.0 | 4.3 |
| CLTA | Clathrin light chain A | 6.5 | 9.0 |
| G3BP2 | Ras GTPase-activating protein-binding protein 2 | 3.8 | 4.1 |
| GNB1, GNB2,GNB4 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1, guanine nucleotide-binding protein subunit beta-2, guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-4 | 7.3 | 3.8 |
| CKAP4 | Cytoskeleton-associated protein 4 | 3.9 | 5.4 |
| DLST | Dihydrolipoyllysine residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial | 5.3 | 4.5 |
| PPP1R12A | Protein phosphatase 1 regulatory subunit 12A | 2.9 | 7.2 |
| AP2B1 | AP-2 complex subunit beta | 8.0 | 4.3 |
| RBM14 | RNA-binding protein 14 | 4.5 | 2.4 |
| DNAJB11 | DnaJ homologue subfamily B member 11 | 2.9 | 2.1 |
| ACTR2 | Actin-related protein 2 | 7.3 | 8.1 |
| PDHA1 | Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial | 3.3 | 4.7 |
| HADHB | Trifunctional enzyme subunit beta, mitochondrial, 3-ketoacyl-CoA thiolase | 2.3 | 4.1 |
| FXR1, FXR2 | Fragile X mental retardation syndrome-related protein 1, fragile X mental retardation syndrome-related protein 2 | 4.1 | 3.2 |
| ACTR3 | Actin-related protein 3 | 7.6 | 5.0 |
| ARPC4 | Actin-related protein 2/3 complex subunit 4 | 7.2 | 8.7 |
| RAI14 | Ankycorbin | 6.1 | 6.0 |
| VAT1 | Synaptic vesicle membrane protein VAT-1 homologue | 3.2 | 3.0 |
| MYO1D | Unconventional myosin-Id | 5.7 | 4.8 |
| H1F0 | Histone H1.0, N-terminally processed | 7.7 | 4.7 |
| MRPS27 | 28S ribosomal protein S27, mitochondrial | 2.1 | 2.8 |
| PAWR | PRKC apoptosis WT1 regulator protein | 6.1 | 6.0 |
| GSN | Gelsolin | 5.9 | 4.7 |
| SVIL | Supervillin | 5.0 | 4.2 |
| AP2A1 | AP-2 complex subunit alpha-1 | 5.5 | 3.9 |
| AP2A2 | AP-2 complex subunit alpha-2 | 6.4 | 6.1 |
| PIK3C2A | Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit alpha | 6.9 | 6.6 |
| PICALM | Phosphatidylinositol-binding clathrin assembly protein | 8.2 | 5.0 |
| ACOT9 | Acyl-coenzyme A thioesterase 9, mitochondrial | 6.5 | 5.7 |
| FLOT1 | Flotillin-1 | 6.3 | 4.6 |
| ARPC3 | Actin-related protein 2/3 complex subunit 3 | 3.7 | 7.6 |
| DECR1 | 2,4-Dienoyl-CoA reductase, mitochondrial | 6.8 | 4.2 |
| HSDL2 | Hydroxysteroid dehydrogenase-like protein 2 | 3.8 | 3.4 |
| FHL2 | Four-and-a-half LIM domains protein 2 | 7.1 | 5.4 |
| CYR61 | Protein CYR61 | 5.0 | 5.3 |
| REPS1 | RalBP1-associated Eps domain-containing protein 1 | 5.0 | 3.6 |
| RABEP1 | Rab GTPase-binding effector protein 1 | 5.0 | 7.7 |
| DAB2 | Disabled homologue 2 | 7.9 | 5.9 |
| LEPRE1 | Prolyl 3-hydroxylase 1 | 2.0 | 2.3 |
| SNX9 | Sorting nexin, sorting nexin-9 | 5.7 | 4.5 |
| FRYL | Protein furry homologue-like | 3.9 | 3.2 |
| ARPC5 | Actin-related protein 2/3 complex subunit 5 | 5.2 | 3.0 |
| STOM | Erythrocyte band 7 integral membrane protein | 2.7 | 4.4 |
| CD59 | CD59 glycoprotein | 2.8 | 5.6 |
| MYL3, MYL1 | Myosin light chain 3; myosin light chain 1/3, skeletal muscle isoform | 6.3 | 3.4 |
| IKBIP | Inhibitor of nuclear factor κB kinase-interacting protein | 2.6 | 3.3 |
| PBDC1 | Protein PBDC1 | 3.5 | 2.8 |
| GIPC1 | PDZ domain-containing protein GIPC1 | 3.1 | 6.3 |
| SCCPDH | Saccharopine dehydrogenase-like oxidoreductase | 4.5 | 4.4 |
| NAA50 | N-Alpha-acetyltransferase 50 | 2.5 | 3.8 |
| TGM2 | Protein-glutamine gamma-glutamyltransferase 2 | 5.0 | 3.8 |
| FGF2 | Fibroblast growth factor, fibroblast growth factor 2 | 2.5 | 7.5 |
| FECH | Ferrochelatase, mitochondrial | 6.5 | 6.0 |
| ISG15 | Ubiquitin-like protein ISG15 | 6.4 | 4.8 |
| ARPC5L | Actin-related protein 2/3 complex subunit 5-like protein | 5.1 | 2.5 |
| MME | Neprilysin | 4.2 | 5.7 |
| CD109 | CD109 antigen | 5.6 | 5.2 |
| SQRDL | Sulfide:quinone oxidoreductase, mitochondrial | 4.2 | 4.0 |
| FCHO2 | F-BAR domain-only protein 2 | 2.7 | 2.7 |
| MX2 | Interferon-induced GTP-binding protein Mx2 | 6.1 | 5.3 |
| SERPINE1 | Plasminogen activator inhibitor 1 | 7.1 | 4.4 |
| ANGPT2 | Angiopoietin-2 | 3.4 | 5.4 |
The pK15-interacting proteins identified from cells treated with a reactivation cocktail (RTA plus SB) was processed similarly to those in Table 1, and the resulting list of proteins is shown here. Proteins annotated to be involved in the endocytic pathway by using the DAVID bioinformatics tool are in boldface type. FE, fold enrichment.
In order to investigate whether specific cellular compartments and molecular functions were enriched in our list of pK15-interacting proteins, we performed Gene Ontology (GO) and pathway analyses of the hits by using the DAVID bioinformatics resource (https://david.ncifcrf.gov/) (44). The pK15 interactome (Table 1) from latent infection (without lytic induction) shows an enrichment of proteins from the proteasome complex and the proteasome KEGG pathway. In contrast, a similar analysis of the pK15 interactome (Table 2) upon induction of the KSHV lytic cycle revealed the enrichment of biological processes involved in endocytosis and intracellular transport as well as different signaling pathways, angiogenesis, and actin cytoskeleton reorganization. Based on the GO annotation (Fig. 2C), the most enriched cellular compartment was the Arp2/3 protein complex (fold enrichment [FE] = 133), followed by the endolysosome membrane (FE = 76) and the AP-2 adaptor complex (FE = 70). There were also different cellular compartments related to endocytosis and vesicular transport, such as clathrin-coated pit (FE = 21), clathrin-coated vesicle (FE = 20), vesicle (FE = 11), and cytoplasmic vesicle (FE = 7). Consistent with the cellular compartment analysis, the molecular function annotation (Fig. 2D) shows a strong enrichment for proteins with clathrin adaptor activity (FE = 111), followed by actin-dependent ATPase activity (FE = 50) and microfilament motor activity (FE = 33), among others. As pK15 is involved in the induction of angiogenesis and invasiveness in KSHV-infected endothelial cells (24, 26), a strong representation of proteins involved in biological processes related to the cellular cytoskeleton is not a surprise. It is, however, curious to see a strong representation of proteins involved in endocytosis and intracellular/vesicular transport. We also performed a KEGG pathway enrichment analysis. In agreement with the cellular compartment annotation (Fig. 2C), endocytosis was among the most significantly enriched KEGG pathways (data not shown). Taken together, the GO and pathway analyses revealed a strong association of pK15 with cellular proteins involved in the endocytic pathway and with cellular cytoskeleton rearrangement.
Signaling from the KSHV K15 protein resembles that of a constitutively active receptor tyrosine kinase (RTK). Recent evidence suggests that signaling from membrane proteins such as RTKs occurs not only at the cell membrane but also from intracellular compartments (45). The identification of a number of pK15-interacting cellular proteins (Table 2) involved in vesicular transport and endocytosis and the fact that pK15, a membrane protein, is localized intracellularly in LAMP1-positive vesicles and at the Golgi apparatus (19) support the notion that endocytosis might play a role in the biology of K15. Alternatively, K15 itself might also be involved in the regulation of endocytosis and intracellular trafficking. In support of this hypothesis, a previous study that analyzed B-cell proteins that bind the K15 SH3-binding domain (PPLP) by mass spectrometry also demonstrated the enrichment of components of the endocytic machinery, such as the endocytic regulator intersectin 2, Pacsin 2, the trafficking protein Eps15R, and clathrin (36). However, we did not identify these proteins in our analysis, possibly as a result of our antibody binding to the PPLP site and thereby interfering with their interaction with pK15 or as a result of the different KSHV-infected cells used in our study. Based on the above-mentioned premises, proteins involved in endocytosis and intracellular/vesicular transport were candidates for follow-up and further functional analysis.
K15 interacts with PI3K-C2α.
To investigate the role of individual pK15-interacting partners, we selected the alpha isoform of the class II PI3K PI3K-C2α (Tables 1 and 2), which was one of the few proteins enriched more than 64-fold after lytic induction and also enriched more than 2-fold in latently infected (uninduced) samples (Fig. 2A and B and 3A). PI3K-C2α is known to be involved in several diverse biological processes, including membrane receptor internalization and signaling, endocytosis, intracellular trafficking, and exocytosis (46, 47).
FIG 3.

pK15 interacts with PI3K-C2α through its C-terminal cytoplasmic tail. (A) Log2-fold enrichment of PI3K-C2α using the LFQ intensity obtained from the induced and uninduced samples in the experiments in Fig. 1 and 2. (B to D) HEK-293T cells were cotransfected with plasmid vectors expressing GFP-tagged PI3K-C2α and Flag-tagged pK15. After 48 h, cellular lysates were collected, pulldown assays were performed by using anti-Flag M2 control IgG-conjugated beads, and coprecipitation of PI3K-C2α was analyzed by Western blotting (B and C), or pulldown assays were performed by using GFP-Trap or control beads, and coprecipitation of K15 was analyzed by Western blotting (D). The expression levels of the individual proteins in the cellular lysate are shown as input. (E) Endogenous PI3K-C2α protein from a HEK 293T cellular lysate was pulled down by using a purified GST fusion protein of the full-length K15 C-terminal cytoplasmic tail or a GST control. Results are representative of data from at least 3 experiments.
To confirm the interaction between pK15 and PI3K-C2α (Fig. 3A), HEK-293T cells were cotransfected with vectors expressing green fluorescent protein (GFP)-tagged PI3K-C2α (PI3K-C2α-GFP) and Flag-tagged pK15 (Flag-K15), and a coimmunoprecipitation (co-IP) assay was performed. First, Flag-pK15 was precipitated by using an anti-Flag M2 affinity gel or beads conjugated with control IgG. The presence of PI3K-C2α was checked by Western blot analysis. Vice versa, GFP-tagged PI3K-C2α was pulled down using GFP-Trap or control beads to see the coprecipitation of pK15; the expression levels of the two proteins are shown in parallel as input. The results in Fig. 3B to D show that PI3K-C2α specifically interacts with pK15, confirming the MS results. This was also further verified by pulling down endogenous PI3K-C2α from HEK-293T cells with the purified C-terminal cytoplasmic tail of the pK15 protein fused to glutathione S-transferase (GST) (GST-K15) (Fig. 3E).
pK15 and PI3K-C2α colocalize in vesicular structures in the perinuclear area.
PI3K-C2α catalyzes the production of phosphatidylinositol 3 phosphate [PtdIns(3)P] at endosomal membranes and of the less-abundant PtdIns(3,4)P2 at the plasma membrane. The protein localizes not only to the plasma membrane but also intracellularly to different endosomal compartments, including vesicles of the recycling compartment, the trans-Golgi network, and clathrin-coated vesicles (48–51). Similarly, pK15 also localizes to vesicular structures in the perinuclear area that are positive for markers of the Golgi apparatus and the lysosomal marker LAMP1 (19, 26). In order to visualize pK15/PI3K-C2α complexes in infected cells and to validate data from our MS analysis with an independent method, the intracellular localization of endogenous pK15 and PI3K-C2α proteins in stably infected HuARLT2-rKSHV cells was analyzed by immunofluorescence assay (IFA) using antibodies specific to each protein. The result shows that in pK15-expressing HuARLT2-rKSHV cells, the two proteins colocalize in vesicle-like structures that are abundant in the perinuclear area (Fig. 4A). We also analyzed the localization of the two proteins after cotransfection of plasmid vectors expressing GFP-fused PI3K-C2α or pK15 in HeLa-CNX cells and a subsequent staining of pK15 with the rat anti-pK15 monoclonal antibody, while PI3K-C2α was visualized by means of its GFP tag. Consistent with the localization of the endogenously expressed proteins (Fig. 4A), the two proteins associate with each other in similar vesicular structures in the perinuclear area of transfected cells (Fig. 4B).
FIG 4.

pK15 colocalizes with PI3K-C2α on vesicular structures in the perinuclear area. (A) Stably infected HuARLT2-rKSHV cells were stained with rat mAb 18E5 to pK15, followed by Cy3-conjugated anti-rat secondary antibody (red), with subsequent staining using a mouse anti-PI3K-C2α mAb and a FITC-conjugated anti-mouse secondary antibody (green). (B) HeLa-CNX cells cotransfected with vectors expressing GFP-tagged PI3K-C2α (green) and pK15 were stained with rat mAb 18E5 to pK15, followed by a Cy3-conjugated anti-rat secondary antibody (red). Images were acquired using a Leica confocal microscope. DIC, differential interference contrast.
PI3K-C2α binds to a defined region in the cytoplasmic domain of pK15.
In order to define the region in the K15 protein (pK15) involved in the association with PI3K-C2α, we used the K15 C-terminal cytoplasmic tail of both the K15 alleles K15P and K15M with either the wild-type (wt) sequence or a sequence carrying mutations in the SH3-binding site (ΔSH3), the SH2-binding site YEEVL (YF), or a combination of the two mutations as GST fusion proteins to pull down GFP-tagged PI3K-C2α transfected into HEK-293 cells. The structural organization of the K15 protein and its signaling motifs are shown in a cartoon representation in Fig. 5A. The result shows that PI3K-C2α binds to both the wild-type and the mutant GST-K15 proteins of both P and M types (Fig. 5B), indicating that neither the SH3-binding site (PPLP) nor the YEEVL SH2-binding site is involved in the recruitment of PI3K-C2α to K15.
FIG 5.

Mapping the K15 region necessary for PI3K-C2α binding. (A) Schematic representation of the full-length K15 protein and its signaling motifs. (B) Purified GST fusion proteins of the full-length cytoplasmic tails of wild-type (wt) K15P and K15M or with mutations at either the SH3-binding site (PPxPP387–391 to AAxAA387–391/ΔSH3), the SH2-binding site (Y481 to F481 [YF] for K15P and Y489F for K15M), or a combination of both (ΔSH3 YF) as well as a GST control were used to pull down GFP-tagged PI3K-C2α that was transfected into HEK-293T cells. (C) Schematic representation of C-terminal or N-terminal deletion constructs of the pK15 cytoplasmic domain fused to GST. (D) GST-K15 fusion constructs in panel C were used to pull down GFP-tagged PI3K-C2α transfected into HEK-293T cells. (E) GST pulldown of endogenous PI3K-C2α protein from HEK-293T cells using a series of triple-alanine scanning mutants derived from the last C-terminal deletion mutant (C4 in panel C). (F) Pulldown of transiently expressed GFP-tagged PI3K-C2α using either the wild type or two triple-alanine scanning mutants in the full-length K15 C-terminal cytoplasmic tail fused to GST. Results are representative of data from at least three independent experiments.
Next, we performed similar GST pulldown assays using N-terminally or C-terminally truncated fragments of the pK15 C-terminal cytoplasmic tail as GST fusion proteins, as presented schematically in Fig. 5C. As shown in Fig. 5D, GFP-tagged PI3K-C2α binds to the wild type and all C-terminally truncated mutants but not the N-terminally truncated mutants of the pK15 cytoplasmic tail, indicating the requirement of the membrane-proximal region of the pK15 C-terminal cytoplasmic tail for the binding of PI3K-C2α. To further narrow down the interaction region, a series of triple-alanine scanning mutants in the shortest C-terminal deletion mutant (K15 amino acid sequence between residues 355 and 374) (40) was used to pull down endogenous PI3K-C2α protein from HEK-293 cells. As shown in Fig. 5E, the first two alanine scanning mutants (359 RQR/AAA and 362 RRR/AAA) were no longer able to bind to PI3K-C2α; this was also true in the context of the full-length pK15 cytoplasmic tail fused to GST (Fig. 5F). Taken together, the results so far suggest that the membrane-proximal region of the pK15 C-terminal cytoplasmic tail located between amino acids 359 and 364 is involved in the interaction with PI3K-C2α.
PI3K-C2α is involved in the reactivation of KSHV from latency and pK15-dependent activation of PLCγ1.
Recruitment of PI3K-C2α to activated receptor tyrosine kinases such as vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2) (51), transforming growth factor β (TGFβ) receptor (TGFβR) (52), epidermal growth factor receptor (EGFR) (53), and insulin receptor (IR) (54, 55) as well as G-protein-coupled receptors such as S1P1 (56) and CXCR2 (57) after ligand binding is required for their internalization and formation of signaling endosomes and, subsequently, cellular responses to the stimulus.
Knockdown of PI3K-C2α in endothelial cells reduces PtdIns(3)P-enriched VEGFR2- and S1P1-positive endosomes, VEGF- and S1P-induced endosomal signaling, and, consequently, endothelial cell migration, proliferation, tube formation, and barrier integrity (51, 56). Similarly, the depletion of PI3K-C2α in endothelial cells and the subsequent reduction in PtdIns(3,4)P2 at the plasma membrane also impair TGFβR internalization and its association with smad anchor for receptor activation (SARA) on early endosomes in response to TGFβ1 stimulation; this in turn abolishes TGFβ1-induced Smad2/3 phosphorylation and nuclear localization, which is important for the expression of VEGF-A and its effect on endothelial cell migration and capillary tube formation (52). Consequently, global or endothelial-specific knockout of PI3K-C2α in mice results in embryonic lethality due to defective vasculogenesis and primary cilium organization.
We have previously shown that pK15 and its activation of cellular signaling pathways play an important role in KSHV lytic reactivation (19). To investigate whether PI3K-C2α could play a similar role in the activation of K15-induced signaling, we transfected HeLa-CNX cells first with either a control (scrambled [Scr]) small interfering RNA (siRNA) or a pool of four siRNAs against PI3K-C2α and then with a plasmid vector expressing pK15 or an empty vector control. In the Scr siRNA-transfected cells (Fig. 6), pK15 expression induces PLCγ1 Y783 and Erk1/2 T202/Y204 phosphorylation, whereas depleting PI3K-C2α mRNA from these cells reduced the pK15-dependent phosphorylation of PLCγ1 and Erk1/2, suggesting that PI3K-C2α may be involved in the pK15-dependent activation of these signaling pathways. PI3K-C2α knockdown also seems to reduce pK15 expression or stability, which might contribute to the reduction in the phosphorylation of PLCγ1 and Erk1/2.
FIG 6.
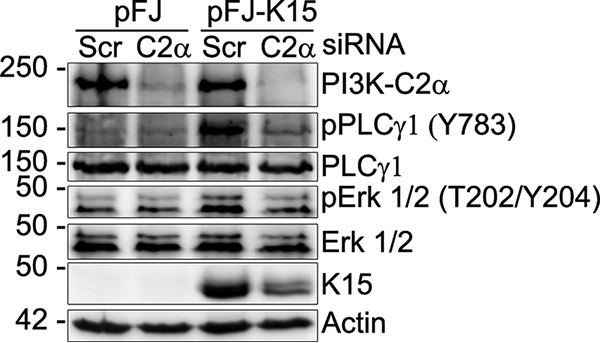
PI3K-C2α is required for the pK15-induced phosphorylation of PLCγ1 and ERK1/2. HeLa-CNX cells were transfected with either a scrambled (Scr) siRNA as a control or a pool of siRNAs against PI3K-C2α. After 6 h, cells were transfected with a plasmid vector expressing wild-type K15P or an empty vector control. After 48 h, cells were collected, and the phosphorylation level of the indicated proteins was assessed by Western blotting using specific antibodies.
To explore the involvement of PI3K-C2α in KSHV lytic reactivation, we depleted PI3K-C2α from stably KSHV-infected endothelial cells (HuARLT2-rKSHV) by using 4 individual siRNAs or a pool of these individual siRNAs, and an siRNA against K15 was used in parallel as a positive control; the KSHV lytic cycle was induced by using a reactivation cocktail of KSHV-RTA and sodium butyrate as described previously (19). The rKSHV.219 virus expresses GFP under the control of a constitutive promoter, which serves as a marker of infection, and red fluorescent protein (RFP) under the control of the viral lytic gene PAN promoter, which serves as a marker of lytic reactivation (58). The results (Fig. 7A and B) show that in cells transfected with the control (Scr) siRNA, the induction of the KSHV lytic cycle increased the number of RFP-expressing cells, while the depletion of PI3K-C2α from these cells with either the individual or pooled siRNAs reduced the number of RFP-positive cells upon lytic reactivation to a greater extent than with the depletion of K15 itself. Similarly, analysis of the expression of the KSHV lytic genes K-bZIP and ORF45 by Western blotting (Fig. 7C) also revealed that silencing of PI3K-C2α leads to the reduced expression of both viral lytic genes compared to cells transfected with the control (Scr) siRNA, suggesting the involvement of PI3K-C2α early in the virus life cycle.
FIG 7.

Depletion of PI3K-C2α from infected endothelial cells leads to reduced KSHV lytic replication. HuARLT2-rKSHV cells were microporated with either a control siRNA (Scr), four individual siRNAs or a pool of these siRNAs against PI3K-C2α, or a single siRNA against K15, and the KSHV lytic cycle was induced 24 h later using a cocktail of RTA and SB. (A to C) Forty-eight hours after lytic induction, images for GFP and RFP expression were acquired (A), and the number of RFP-positive cells from five or more fields was quantified; cells were then lysed, and the expression level of the indicated viral proteins was assessed by Western blotting (C). Results are representative of data from two independent experiments. The scatter plot (B) represents the means ± standard deviations (SD) for five or more fields. Ordinary one-way analysis of variance (ANOVA) was used to determine P values. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (D and E) Primary lymphatic endothelial cells infected with KSHV for 2 weeks (LEC-rKSHV) were transfected with either the pooled PI3K-C2α siRNA or a scrambled control. After 72 h, cells and the culture supernatant were collected. The expression levels of the indicated proteins were analyzed by Western blotting (D), and the amount of released infectious virus was assayed by titrating the culture supernatant on HEK-293 cells (E). A Mann-Whitney test was used to determine P values. **, P < 0.01.
In stably infected primary lymphatic endothelial (LEC-rKSHV) cells, which are known to support KSHV lytic replication without stimulation (19, 33), siRNA-mediated knockdown of PI3K-C2α also reduced the expression of K-bZIP and ORF45 as well as the amount of infectious virus released compared to control (Scr) siRNA-transfected cells (Fig. 7D and E), indicating that PI3K-C2α is also involved in KSHV spontaneous lytic replication in the absence of external stimuli. The decreased production of extracellular virus after PI3K-C2α depletion (Fig. 7E) could result from the effect of PI3K-C2α on viral early gene expression (Fig. 7D); however, a possible additional effect of PI3K-C2α on the later steps of the viral life cycle, i.e., envelopment and release, cannot be ruled out at this point. Taken together, the results so far suggest that PI3K-C2α might be involved in the early stage of KSHV lytic replication, presumably through its interaction with pK15 and its activation of cellular signaling pathways. Alternatively, PI3K-C2α might be required for the formation of pK15-containing vesicles that could serve as signaling platforms and could maintain pK15 stability.
DISCUSSION
The KSHV K15 protein interacts with several cellular proteins and can activate several signaling pathways, such as PLCγ-calcineurin-NFAT, MAP kinase/extracellular signal-regulated kinase (ERK), and NF-κB (22, 24, 36, 40). The activation of these pathways induces the expression of a plethora of proangiogenic and proinflammatory molecules that are involved in KSHV-induced invasiveness and angiogenic tube formation in infected endothelial cells as well as endothelial spindle cell formation and viral lytic replication (19, 24–26), which are all important in KSHV-mediated pathogenesis (Fig. 8).
FIG 8.
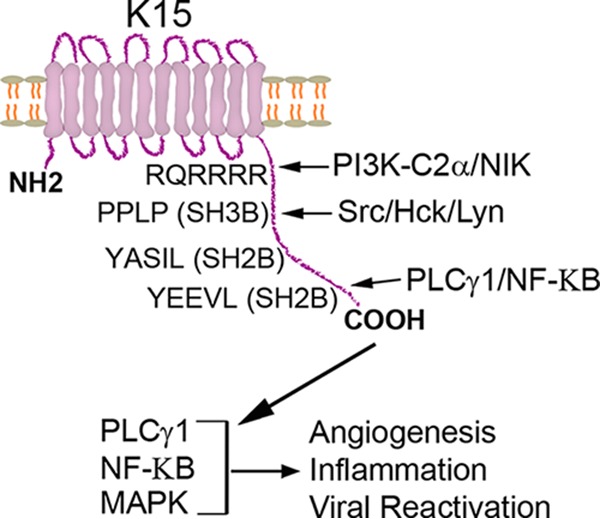
Schematic representation of pK15-mediated signaling. The pK15 protein can recruit PLCγ1 and NF-κB as well as members of the Src family of tyrosine kinases, activating the PLCγ1, MAP kinase (MAPK), and NF-κB signaling pathways. The activation of these cellular signaling pathways in turn leads to the expression of angiogenic and inflammatory molecules, which are important for the pathogenesis of KSHV-mediated diseases and virus lytic replication. Additionally, the interaction of pK15 with the class II PI3K PI3K-C2α, which also contributes to KSHV lytic replication, might play a role in its intracellular localization (by mediating its internalization); it is possible that pK15 signals from intracellular membrane-bound organelles.
In this study, we have used coimmunoprecipitation together with label-free quantitative mass spectrometry (43, 59) in order to identify novel proteins that have the capacity to interact with pK15 in an unbiased manner (Fig. 1). This allowed a definition of the pK15 interactome in the context of KSHV latent and lytic infection (Tables 1 and 2 and Fig. 2). Gene Ontology categorization of the 75 newly identified potential pK15-interacting proteins from reactivated samples revealed the enrichment of cellular processes, such as endocytosis, whose role in the biology of KSHV pK15 had not previously been appreciated, as well as those related to the previously reported functions of pK15, such as cytoskeleton reorganization and signaling pathways (Fig. 2C and D).
Endocytosis regulates signaling induced by signaling receptors and other integral membrane proteins and their downstream effects after ligand binding, and conversely, the endocytic machinery is also affected by receptor signaling (45). Since pK15 resembles a constitutively active signaling receptor, components of the endocytic pathway identified in our MS analysis were of special interest in order to obtain new insights into pK15-mediated signaling and downstream effects in this regard. We focused on one of the highly enriched candidate proteins, phosphatidylinositol 4-phosphate 3-kinase (PI3K) C2 domain-containing subunit alpha (PI3K-C2α) (Fig. 3A). PI3K-C2α belongs to the class II PI3Ks together with the β and γ isoforms that are known to be refractory to the classical PI3K inhibitors wortmannin and LY294002 (60, 61). PI3K-C2α is ubiquitously expressed in a wide variety of mammalian tissues (60, 62) and generates PtdIns(3)P at endosomal membranes and the less-abundant PtdIns(3,4)P2 at the plasma membrane. It is involved in diverse cellular processes, including signal transduction, endocytosis, vesicular trafficking, and exocytosis (47, 63). PI3K-C2α is implicated in both dynamin-dependent and -independent endocytic pathways (49, 64, 65).
We identified a novel interaction between PI3K-C2α and pK15 in KSHV-infected endothelial cells (Fig. 1 to 4) and found that PI3K-C2α is involved in pK15-mediated signaling activation as well as in KSHV lytic replication in infected endothelial cells (Fig. 6 and 7). GST pulldown assays using wild-type and mutant versions of the cytoplasmic domains of both K15 alleles K15P and K15M showed that the interaction with PI3K-C2α involves the cytoplasmic domain of pK15 (Fig. 3E and 5B) but is independent of the SH2- and SH3-binding sites (Fig. 5B) that are known to be required for the recruitment of other cellular signaling components (21, 22, 24–26, 41).
Endocytosis has already been shown to be critical for the activation of KSHV K1-mediated cellular signaling (66). Similarly, pK15 has also been implicated in the recruitment of components of the endocytic machinery (ITSN2, Eps15, and Pacsin 2) through its SH3-binding site and in the increase of the rate of internalization of the B-cell receptor and its signaling activation after ligand binding (36, 42). In keeping with these observations, we found pK15 to recruit PI3K-C2α (Fig. 1 to 4), which plays a crucial role in the internalization of several activated cell surface signaling receptors and the subsequent activation of their downstream signaling targets and cellular response (51–57). The pK15 interactome contained additional cellular proteins involved in endocytosis and vesicular transport, such as the AP-2 complex, sorting Nexin-9 (SNX9), RalBP1-associated Eps domain-containing protein 1 (REPS1), and the phosphatidylinositol-binding clathrin assembly protein (PICALM), among others (Table 2).
We have confirmed the binding of PI3K-C2α to pK15 (Fig. 3) and show that the tyrosine residue in the SH2-binding site Y481EEVL of pK15, which is responsible for the activation of both the PLCγ1 and Erk1/2 signaling pathways and the expression of the majority of K15-induced genes (21, 22, 24–26), is not involved in the interaction with PI3K-C2α (Fig. 5B). Similarly, an SH3-binding site in the pK15 cytoplasmic domain does not appear to contribute to this interaction (Fig. 5B). In contrast, the K15 mutants 359–361 RQR/AAA and 362–364 RRR/AAA are defective in PI3K-C2α binding (Fig. 5E and F). In both KSHV-infected and K15-transfected cells, pK15 associates with PI3K-C2α in intracellular vesicular structures that are abundant in the perinuclear area (Fig. 4), and the depletion of PI3K-C2α reduces K15-mediated PLCγ1 and Erk1/2 activation (Fig. 6).
We recently showed the involvement of pK15 and of signaling pathways activated by pK15 in KSHV lytic replication (19). Here, we show that the depletion of PI3K-C2α in KSHV-infected endothelial cells by siRNA also leads to reduced viral lytic replication, as demonstrated by the reduced number of RFP-positive cells and the decreased expression of the viral early lytic genes K-bZIP and ORF45 (Fig. 7A to C), suggesting an effect of PI3K-C2α on the early steps of the viral lytic cycle. PI3K-C2α depletion in spontaneously lytic KSHV-infected lymphatic endothelial cells (LECs) also resulted in reduced viral early lytic gene expression and release of infectious virus (Fig. 7D and E). These results, together with the fact that K15 is strongly associated with components of the endocytic machinery, suggest a role for PI3K-C2α-mediated endocytosis in pK15-mediated signaling, KSHV reactivation, and lytic replication. PI3K-C2α knockdown also reduced the pK15 protein level in transfected as well as infected cells (Fig. 6 and 7C and D). It is therefore possible that the binding of PI3K-C2α could play a role in the maintenance of pK15 in vesicular structures, which can in turn contribute to protein stability; i.e., pK15 will dissociate from vesicular membranes in the absence of PI3K-C2α, thereby targeting it for degradation. In addition to its role in endocytic trafficking through PtdIns(3,4)P2 production and endosomal sorting through PtdIns(3)P, PI3K-C2α is also involved in the late steps of exocytosis of neurotransmitters and insulin granules (67, 68). Furthermore, PI3K-C2α has recently been reported to be involved in the secondary envelopment and release of human cytomegalovirus (HCMV) virions (69). We cannot exclude the possibility that the reduced production of infectious KSHV particles in PI3K-C2α-depleted lymphatic endothelial cells (Fig. 7E) might also imply an additional role of PI3K-C2α during the later steps of the viral life cycle, such as virion maturation and release. The strong association of pK15 with components of the extracellular exosome and secreted proteins (Fig. 2C and Table 2) as well as its colocalization with the Golgi network (19), which is known to be the site of herpesvirus secondary envelopment and maturation, could also argue in favor of this hypothesis. However, even if PI3K-C2α were to also be involved during virion maturation, our observation that the depletion of PI3K-C2α from KSHV-infected endothelial cells reduces the expression of viral early lytic proteins, whose expression is dependent on pK15-mediated signaling (19), clearly indicates an important role during the early stages of lytic cycle activation. Distinguishing the effects of both pK15 and PI3K-C2α on early lytic viral gene expression from a possible role during the later stages of the viral life cycle will require further detailed investigation. We also note that a previous report on the Epstein-Barr virus LMP2 protein, which is encoded in the same region of the EBV genome as its positional homologue in KSHV, K15, showed that it may be involved in signaling from intracellular vesicular compartments (70).
Taken together, in this study, we have identified novel pK15-interacting cellular proteins using affinity enrichment-based quantitative mass spectrometry. We have shown that one of the identified proteins, PI3K-C2α, is involved in the activation of cellular signaling pathways by pK15 and of KSHV lytic replication. The known involvement of PI3K-C2α and its catalytic products PtdIns(3,4)P2 and PtdIns(3)P in endocytic trafficking and endosomal sorting, respectively, together with the composition of the pK15 interactome reported here suggest that the formation or trafficking of endocytic vesicles may be required for pK15-dependent signaling and its role in KSHV lytic replication.
MATERIALS AND METHODS
Plasmids and constructs.
Eukaryotic expression plasmids pFJ-EA (71) and pFJ-K15P clone 35 (21) were kind gifts from Jae U. Jung (University of Southern California, Los Angeles, CA, USA), pFJ-K15P clone 35 RQR359-361/AAA and pFJ-K15P clone 35 RRR362-364/AAA were reported previously (40), and a PI3K-C2α-GFP plasmid containing a GFP tag at the C terminus was kindly provided by Marco Falasca (Queen Mary University of London, London, UK). GST fusion constructs pGEX-6P-1 (catalogue number 28-9546-48) and pGEX-4T-1 (catalogue number 28-9545-49) were purchased from Amersham Pharmacia; pGEX-6P-1 sK15P347-489 was purchased from GeneArt GmbH; pGEX-3X K15P 355-489 (wild type [wt]), pGEX-3X K15P 355-472 (CΔ1), pGEX-3X K15P 355-438 (CΔ2), and pGEX-3X K15P 355-374 (CΔ4) were reported previously (17); pGEX 6P-1 K15P 365-489 (NΔ1) and pGEX 6P-1 K15P 410-489 (NΔ2) were generated by deleting the N-terminal 18 and 63 amino acids, respectively, of the K15 cytoplasmic tail from pGEX-6P-1 K15 347-489; pGEX-4T-1 K15P 355-489 359-361 RQR/AAA, pGEX-4T-1 K15P 355-489 362-364 RRR/AAA, pGEX-3X K15P CΔ4 359-361 RQR/AAA, pGEX-3X K15P CΔ4 362-364 RRR/AAA, pGEX-3X K15P CΔ4 365-367 IYT/AAA, pGEX-3X K15P CΔ4 368-370 RDQ/AAA, pGEX-3X K15P CΔ4 371-373 NLH/AAA, and pGEX-3X K15P CΔ4 374 H/A were reported previously (40); and pGEX-4T-1 K15P YF, pGEX-4T-1 K15P ΔSH3, and pGEX-4T-1 K15P YF ΔSH3 were reported previously (41).
Antibodies.
The production and epitope mapping of the rat anti-KSHV K15 monoclonal antibodies clone 10A6 (used for Western blotting) and clone 18E5 (used for immunofluorescence) were described previously (19, 26), and the rat K15 monoclonal antibody clone 6E7 (used for immunoprecipitation) was produced and epitope mapped (epitope LPPFFR at the SH3-binding site) similarly. The primary antibodies rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalogue number 14C10), rabbit anti-PLCγ1 (catalogue number 2822), rabbit anti-phospho-PLCγ1 (Tyr783; catalogue number 2821), mouse anti-phospho-MAP kinase p44/42 (Thr202/Tyr204; catalogue number 91062), mouse anti-MAP kinase p44/42 (catalogue number 9107), and rabbit anti-PI3K-C2α (catalogue number 12402) (used for Western blotting) were purchased from Cell Signaling Technology; mouse anti-KSHV ORF45 (catalogue number sc-53883) and mouse anti-KSHV K-bZIP (catalogue number sc-69797) were purchased from Santa Cruz Biotechnology Inc.; rat anti-KSHV ORF73 (LNA-1; catalogue number 13-210-100) was purchased from Advanced Biotechnology Inc.; mouse anti-PI3K-C2α (catalogue number 611046) (used for IFAs) was purchased from BD Transduction Laboratories; and mouse anti-β-actin (catalogue number A5441) was purchased from Sigma-Aldrich. The horseradish peroxidase (HRP)-conjugated secondary antibodies goat anti-rabbit IgG (catalogue number P0448) and rabbit anti-mouse IgG (catalogue number P0260) were purchased from Dako, and goat anti-rat IgG (catalogue number 3050-05) was purchased from Southern Biotech. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (catalogue number 115-095-146; Dianova) and Cy3-conjugated donkey anti-rat IgG (catalogue number 712-165-153; Jackson Immuno Research) were used for IFAs.
Cells and viruses.
HEK-293 (ATCC CRL-1573) and HeLa-CNX (DSMZ ACC 57) cells were maintained in Dulbecco's modified Eagle medium (DMEM; Gibco, Life Technologies, Paisley, UK) supplemented with 10% heat-inactivated fetal bovine serum (FBS; HyClone, Cramlington, UK). BJAB-rKSHV.219 cells, described previously (72), are BJAB cells (DSMZ ACC 757) stably infected with a recombinant virus, rKSHV.219. The virus is derived from JSC-1 cells and contains a puromycin resistance gene as a selectable marker; it can express red fluorescent protein (RFP) under the control of the KSHV lytic gene PAN promoter and the green fluorescent protein (GFP) from the cellular EF-1α promoter (58). These cells were grown and maintained in RPMI 1640 medium (Gibco, Life Technologies, Paisley, UK) supplemented with 20% heat-inactivated FBS in the presence of 4.2 μg/ml puromycin (catalogue number A2856; AppliChem GmbH, Darmstadt, Germany). Primary juvenile foreskin lymphatic endothelial cells (LECs; Lonza) were provided by Päivi M. Ojala (University of Helsinki, Helsinki, Finland) and were grown in EGM-2MV medium (Lonza, Walkersville, MD, USA). HuARLT2-rKSHV.219 cells, also described previously (73), are HuARLT2 cells (74) stably infected with rKSHV.219 and were grown and maintained in EGM-2MV medium in the presence of 1 μg/ml doxycycline and 5 μg/ml puromycin. SF-9 insect cells (DSMZ ACC 125) were grown and maintained in Grace's insect medium (Gibco, Life Technologies, Paisley, UK) supplemented with standard-quality 10% FBS (PAA Laboratories GmbH, Pasching, Austria) and 100 U/ml of penicillin-streptomycin (CytoGen GmbH, Sinn, Germany) at 28°C.
Recombinant virus production and infection.
The recombinant virus rKSHV.219 was produced as described previously (19, 26, 72). Briefly, 6 × 105 BJAB-rKSHV.219 cells/ml were grown in the presence of 2.5 μg/ml of anti-human IgM antibody (catalogue number 2020-01; Southern Biotech) for 4 to 5 days in Techne spinner flasks (Cole-Parmer GmbH, Wertheim, Germany) with agitation at 60 rpm. Cells and debris were cleared from the culture supernatant containing the virus by low-speed centrifugation. The cleared supernatant was then filtered through a 0.45-μm filter, virus particles were concentrated by ultracentrifugation at 15,000 rpm for 5 h in a type 19 rotor (Beckman Coulter Inc., Brea, CA, USA), and the pellet containing virus was resuspended in EBM-2MV medium. A total of 2.7 × 104 HEK-293 cells per well of a 96-well plate were plated and infected by serial dilution of the virus preparation on the next day in order to determine the infectious virus titer. After 3 days, the number of GFP-positive cells was counted, and the infectious virus titer was determined.
The KSHV regulator of transcription activation (RTA) cloned in a baculovirus expression vector was kindly provided by J. Vieira (University of Washington, Seattle WA, USA) and propagated in SF-9 insect cells. In stably infected cells, the KSHV lytic cycle was then induced as described previously (58) by using a reactivation cocktail of an SF-9 cell culture supernatant containing the baculovirus in combination with sodium butyrate (catalogue number B5887; Sigma-Aldrich, St. Louis, MO, USA).
Primary LECs between passages 2 and 5 were plated at a density of 5 × 105 cells per well of a 6-well plate and infected the next day with rKSHV.219 at a multiplicity of infection (MOI) of 1 by spinoculation (450 × g for 30 min at 30°C) in the presence of 10 μg/ml Polybrene (catalogue number H9268; Sigma-Aldrich, Milwaukee, WI, USA). At 2 days postinfection, the medium was changed to selection medium containing 0.25 μg/ml puromycin, and the stably infected cells (LEC-rKSHV.219) were maintained under selection for 2 weeks by refreshing the medium every 2 days.
Transfection of recombinant DNA.
HEK-293 cells at a density of 5 × 105 cells were plated into each well of a 6-well plate. On the next day, 1 μg of the individual expression vectors was transfected by using the Fugene 6 transfection reagent (Promega, Madison, WI, USA) according to the manufacturer's protocol. Forty-eight hours after transfection, cells were washed once and lysed in the respective buffers for either Western blot analysis or an immunoprecipitation assay.
Transfection of small interfering RNA.
Transient transfection of small interfering RNA (siRNA) in HeLa-CNX cells was performed by using the Lipofectamine 2000 reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA) according to the manufacturer's recommendations. Briefly, 2.5 × 105 cells were plated per well of a 12-well plate. After 24 h, 150 pmol of each individual siRNA or 5 μl of the transfection reagent was diluted in 250 μl of low-serum Opti-MEM medium (Gibco, Life Technologies, Paisley, UK), mixed, and incubated for 20 min at room temperature. The siRNA-Lipofectamine 2000 complex was then applied to cells dropwise. After 6 h, medium was changed, and cells were transfected with a K15 expression construct or an empty vector control. After 48 h of transfection, cells were collected for Western blot analysis.
HuARLT2-rKSHV cells were transiently transfected with siRNA by using the Neon transfection system (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Briefly, 1 × 105 HuARLT2-rKSHV cells were microporated with 150 pmol of each individual siRNA, and the KSHV lytic cycle was induced after 24 h by using a cocktail of KSHV RTA and sodium butyrate. Forty-eight hours after lytic induction, images for GFP and RFP expression were acquired, and subsequently, cells were lysed for the analysis of the expression levels of KSHV lytic proteins by Western blotting. The number of RFP-positive cells from a minimum of five fields was quantified by using CellProfiler software (75).
Transient transfection of LECs or LEC-rKSHV.219 cells with individual siRNAs complexed with Lipofectamine RNAiMAX reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA) was performed according to the manufacturer's recommendations. Briefly, 6 μl of Lipofectamine RNAiMAX reagent and 150 pmol of each individual siRNA were each diluted in 150 μl of low-serum Opti-MEM medium and combined. The siRNA-transfection reagent complex was then incubated for 20 min at room temperature and applied dropwise onto a semiconfluent monolayer of cells in a well of a 6-well plate. Seventy-two hours after transfection, the culture supernatant and cells were collected for virus titration and Western blot analysis, respectively. Control/nontargeting scrambled (Scr) siRNA pool 2 (catalogue number D-001206-14-20), siRNA 1 against KSHV K15 targeting exon 8 (CAACCACCUUGGCAAUAAU), and siGENOME human PI3K-C2α (5286) siRNA SMARTpool or a set of 4 individual siRNAs (AGGAAGUGCUGCAGAAUAA, GAAGAGAGAUCGACAGCAA, GGAUUUCAGCUACCAGUUA, and CAAGGAAGCUUACCUAUCU) were all purchased from Dharmacon, GE Health Care.
Western blot analysis.
Western blot analysis was performed as described previously (19). Cells were lysed directly on the plate by using 1× SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% [wt/vol] SDS, 10% [vol/vol] glycerol, and 0.01% [wt/vol] bromophenol blue) and centrifuged at 20,000 × g for 10 min at 4°C. The cleared total cellular lysate (not boiled) was then separated on SDS-PAGE gels and transferred onto 0.45-μm nitrocellulose membranes (Amersham, GE Health Care Europe GmbH, Freiburg, Germany). Membranes were then incubated with a blocking solution (5% [wt/vol] nonfat milk [Carl Roth GmbH+Co.KG, Karlsruhe, Germany] in phosphate-buffered saline [PBS] plus Tween [PBS-T] or IgG-free bovine serum albumin [BSA] [Carl Roth GmbH+Co.KG, Karlsruhe, Germany] in Tris-buffered saline plus Tween [TBS-T]) for 1 h at room temperature (RT) and probed with an appropriate primary antibody in blocking solution overnight at 4°C. (Note that for the analysis of phosphoproteins and corresponding total proteins, 5% BSA in TBS-T buffer was used throughout the experiment.) After washing them three times, membranes were then incubated with the corresponding HRP-conjugated secondary antibodies for an hour at RT. After another three washes, a standard enhanced chemiluminescence (ECL) kit (catalogue number 34096; Thermo Scientific, Rockford, IL, USA) was used to develop the signals. For the list of primary and secondary antibodies used for Western blotting, see the section on antibodies above.
Immunofluorescence assay.
Immunofluorescence staining of both overexpressed and endogenous proteins was performed as described previously (19). Briefly, 2 × 105 HeLa-CNX cells per well of a 6-well plate were plated onto sterile 20-by-20-mm coverslips and transfected the next day with 2 μg of each plasmid DNA using the Fugene 6 transfection reagent. After 2 days, cells were washed with 1× PBS and fixed with 4% paraformaldehyde (PFA) in PBS for 20 min at RT. Subsequently, PFA was quenched with 125 mM glycine for 10 min at RT, and cells were washed in 1× PBS and permeabilized with 0.2% Triton X-100 for 10 min at RT. Coverslips were then blocked for an hour at 37°C with 10% FBS in PBS and incubated with the respective primary antibody in blocking solution for another hour at 37°C. Cells were then washed three times and incubated with the corresponding fluorescently labeled secondary antibodies for 45 min at 37°C. For immunofluorescence labeling of endogenous proteins, 2 × 105 HuARLT2-rKSHV cells per well of a 6-well plate were plated onto coverslips. After 2 days, cells were washed in 1× PBS and fixed with 100% ice-cold methanol at −20°C. Coverslips were then washed thoroughly with 1× PBS, blocked, and stained with the respective primary and secondary antibodies. Images were acquired with a Leica TCS SP2 AOBS confocal microscope (Leica Microsystems, Wetzlar, Germany).
Immunoprecipitation assay.
Immunoprecipitation (IP) of endogenous pK15 from stably KSHV-infected endothelial cells was performed using a rat anti-KSHV K15 monoclonal antibody (clone 6E7 [IgG2b], described previously [19, 26]) and the Pierce cross-linking IP kit (catalogue number 26147; Thermo Fisher Scientific Inc., Rockford, IL, USA) according to the manufacturer's protocol. Immunoprecipitations were performed by using lysates from equal numbers (5 × 108) of HuARLT2-rKSHV cells with or without the induction of the KSHV lytic cycle; the parental uninfected HuARLT2 cells, treated similarly, were used in parallel as a control. The resulting immunoprecipitate eluted from both the sample and control preparations was then alkylated with acrylamide at a concentration of up to 2% and separated by SDS-PAGE. After staining with Coomassie brilliant blue dye, complete lanes from each sample were cut for mass spectrometric analysis.
LC-MS/MS analysis.
Each SDS-PAGE gel lane was cut into smaller gel pieces, further minced into 1-mm3 pieces, and further processed as described previously (76). Peptide samples were analyzed with a shotgun approach and data-dependent analysis with an LC-MS/MS system (RSLC, LTQ Orbitrap Velos; Thermo Fisher). Raw data were processed using MaxQuant (77) and human entries of the UniProt database containing common contaminants. Proteins were identified with settings yielding a false discovery rate of 0.01 on the protein and peptide levels. Bioinformatic analysis was done using Perseus software (version 1.5.2.6) (78) and the DAVID bioinformatics resource (https://david.ncifcrf.gov/).
Coimmunoprecipitation assay.
HEK-293T cells were plated at a density of 5 × 105 cells per well of a 6-well plate and transfected with 1 μg of each plasmid DNA the next day. Forty-eight hours after transfection, cells were washed once with 1× PBS and lysed in 300 μl/well of IP lysis/wash buffer (25 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% [vol/vol] NP-40, 5% [vol/vol] glycerol) in the presence of protease inhibitors (100 μM aprotinin, 10 μM leupeptin, 100 μM phenylmethylsulfonyl fluoride [PMSF], 1 mM benzamidine, 1.46 μM pepstatin A). A total of 250 μl of the cleared cellular lysate was then incubated with 10 μl prewashed EZ view Red anti-Flag M2 affinity agarose beads (catalogue number F2426; Sigma-Aldrich, St. Louis, MO, USA) or GFP-Trap_A or its control beads (ChromoTek GmbH, Munich, Germany) with gentle agitation, overnight, at 4°C. Beads were then washed 6 times in ice-cold IP wash buffer by centrifugation at 2,000 × g for 1 min at 4°C, and the bound proteins were eluted with 5× SDS loading buffer (300 mM Tris-HCl [pH 6.8], 50% [vol/vol] glycerol, 10% [wt/vol] SDS, 0.1% [wt/vol] bromophenol blue, 300 mM β-mercaptoethanol) and analyzed by Western blotting, together with the input lysates.
Production and purification of GST-tagged proteins.
Escherichia coli Rosetta cells transformed with expression plasmids for the respective GST fusion proteins were inoculated into 20 ml of LB medium supplemented with ampicillin and chloramphenicol and grown overnight at 37°C in an orbital shaker. The next morning, the culture grown overnight was diluted 1:10 and continued to grow at 37°C to an optical density at 600 nm (OD600) of ∼0.8, and the expression of the GST-fused protein was induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for 5 h at 30°C. Cells were harvested by centrifugation for 10 min at 5,000 × g and lysed by sonication (3 bursts of 30 s each) in 1 ml of ice-cold bacterial lysis buffer (PBS plus protease inhibitors) per 20 ml of bacterial culture, and 0.5% (vol/vol) NP-40 was subsequently added (Branson 250 sonifier). The bacterial lysate was then cleared by centrifugation at 20,000 × g for 10 min at 4°C. A total of 300 μl of a 50% glutathione-Sepharose bead slurry (Amersham Biosciences) was first washed 3 times and resuspended in 700 μl of bacterial lysis buffer supplemented with protease inhibitors. Seven hundred microliters of the washed glutathione-Sepharose bead suspension was added to 10 ml of the precleared lysate and incubated overnight at 4°C on a rolling platform. Beads were pelleted down, washed 3 times in bacterial lysis buffer, aliquoted, and stored at −80°C. The protein concentration was estimated by running a small aliquot on an SDS-PAGE gel together with known concentrations of BSA. The gel was subsequently stained with Coomassie blue.
GST pulldown assay.
HEK-293T cells, empty or transfected with an expression construct, were lysed in IP lysis/wash buffer supplemented with protease inhibitors. Equal amounts of washed glutathione-Sepharose beads bound to a GST-fused protein were then incubated with 300 μl of the cleared cellular lysate at 4°C, overnight, with gentle agitation. Beads were then settled by centrifugation at 2,000 × g for 1 min, washed 5 times with IP lysis/wash buffer, resuspended in 5× SDS loading buffer, and analyzed by SDS-PAGE and Western blotting.
ACKNOWLEDGMENTS
We are very thankful to the Hannover Medical School Confocal Laser Microscopy Facility and Rudolf Bauerfeind (Hannover Medical School) for providing expert advice in confocal microscopy and imaging. We acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG; SCHU-1668-3/1) to T.F.S. and the Deutsche Akademische Austauschdienst as well as the Hannover Biomedical Research School (HBRS) and Center for Infection Research (ZIB) to B.A. and N.S.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 4.Boshoff C, Schulz TF, Kennedy MM, Graham AK, Fisher C, Thomas A, McGee JO, Weiss RA, O'Leary JJ. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat Med 1:1274–1278. doi: 10.1038/nm1295-1274. [DOI] [PubMed] [Google Scholar]
- 5.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A 96:4546–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katano H, Sato Y, Kurata T, Mori S, Sata T. 2000. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi's sarcoma, and multicentric Castleman's disease. Virology 269:335–344. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- 7.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. Differential viral protein expression in Kaposi's sarcoma-associated herpesvirus-infected diseases: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. Am J Pathol 156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, Herrington CS, Moore PS, Schulz TF. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J Virol 71:5915–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71:715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li JJ, Huang YQ, Cockerell CJ, Friedman-Kien AE. 1996. Localization of human herpes-like virus type 8 in vascular endothelial cells and perivascular spindle-shaped cells of Kaposi's sarcoma lesions by in situ hybridization. Am J Pathol 148:1741–1748. [PMC free article] [PubMed] [Google Scholar]
- 11.Blasig C, Zietz C, Haar B, Neipel F, Esser S, Brockmeyer NH, Tschachler E, Colombini S, Ensoli B, Sturzl M. 1997. Monocytes in Kaposi's sarcoma lesions are productively infected by human herpesvirus 8. J Virol 71:7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiou CJ, Poole LJ, Kim PS, Ciufo DM, Cannon JS, ap Rhys CM, Alcendor DJ, Zong JC, Ambinder RF, Hayward GS. 2002. Patterns of gene expression and a transactivation function exhibited by the vGCR (ORF74) chemokine receptor protein of Kaposi's sarcoma-associated herpesvirus. J Virol 76:3421–3439. doi: 10.1128/JVI.76.7.3421-3439.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93:14862–14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neipel F, Albrecht JC, Fleckenstein B. 1997. Cell-homologous genes in the Kaposi's sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity? J Virol 71:4187–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neipel F, Albrecht JC, Ensser A, Huang YQ, Li JJ, Friedman-Kien AE, Fleckenstein B. 1997. Human herpesvirus 8 encodes a homolog of interleukin-6. J Virol 71:839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poole LJ, Zong JC, Ciufo DM, Alcendor DJ, Cannon JS, Ambinder R, Orenstein JM, Reitz MS, Hayward GS. 1999. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of Kaposi's sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J Virol 73:6646–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glenn M, Rainbow L, Aurade F, Davison A, Schulz TF. 1999. Identification of a spliced gene from Kaposi's sarcoma-associated herpesvirus encoding a protein with similarities to latent membrane proteins 1 and 2A of Epstein-Barr virus. J Virol 73:6953–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandford G, Choi YB, Nicholas J. 2009. Role of ORF74-encoded viral G protein-coupled receptor in human herpesvirus 8 lytic replication. J Virol 83:13009–13014. doi: 10.1128/JVI.01399-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abere B, Mamo TM, Hartmann S, Samarina N, Hage E, Ruckert J, Hotop SK, Busche G, Schulz TF. 2017. The Kaposi's sarcoma-associated herpesvirus (KSHV) non-structural membrane protein K15 is required for viral lytic replication and may represent a therapeutic target. PLoS Pathog 13:e1006639. doi: 10.1371/journal.ppat.1006639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Chen W, Sanders MK, Brulois KF, Dittmer DP, Damania B. 2016. The K1 protein of Kaposi's sarcoma-associated herpesvirus augments viral lytic replication. J Virol 90:7657–7666. doi: 10.1128/JVI.03102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi JK, Lee BS, Shim SN, Li M, Jung JU. 2000. Identification of the novel K15 gene at the rightmost end of the Kaposi's sarcoma-associated herpesvirus genome. J Virol 74:436–446. doi: 10.1128/JVI.74.1.436-446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, Schulz TF. 2003. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J Virol 77:9346–9358. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayward GS, Zong JC. 2007. Modern evolutionary history of the human KSHV genome. Curr Top Microbiol Immunol 312:1–42. [DOI] [PubMed] [Google Scholar]
- 24.Bala K, Bosco R, Gramolelli S, Haas DA, Kati S, Pietrek M, Havemeier A, Yakushko Y, Singh VV, Dittrich-Breiholz O, Kracht M, Schulz TF. 2012. Kaposi's sarcoma herpesvirus K15 protein contributes to virus-induced angiogenesis by recruiting PLCgamma1 and activating NFAT1-dependent RCAN1 expression. PLoS Pathog 8:e1002927. doi: 10.1371/journal.ppat.1002927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkmann MM, Pietrek M, Dittrich-Breiholz O, Kracht M, Schulz TF. 2007. Modulation of host gene expression by the K15 protein of Kaposi's sarcoma-associated herpesvirus. J Virol 81:42–58. doi: 10.1128/JVI.00648-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gramolelli S, Weidner-Glunde M, Abere B, Viejo-Borbolla A, Bala K, Ruckert J, Kremmer E, Schulz TF. 2015. Inhibiting the recruitment of PLCgamma1 to Kaposi's sarcoma herpesvirus K15 protein reduces the invasiveness and angiogenesis of infected endothelial cells. PLoS Pathog 11:e1005105. doi: 10.1371/journal.ppat.1005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong EL, Damania B. 2006. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus K15 gene. J Virol 80:1385–1392. doi: 10.1128/JVI.80.3.1385-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenner RG, Alba MM, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol 75:891–902. doi: 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulose-Murphy M, Ha NK, Xiang C, Chen Y, Gillim L, Yarchoan R, Meltzer P, Bittner M, Trent J, Zeichner S. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J Virol 75:4843–4853. doi: 10.1128/JVI.75.10.4843-4853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gunther T, Grundhoff A. 2010. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 6:e1000935. doi: 10.1371/journal.ppat.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun R, Tan X, Wang X, Wang X, Yang L, Robertson ES, Lan K. 2017. Epigenetic landscape of Kaposi's sarcoma-associated herpesvirus genome in classic Kaposi's sarcoma tissues. PLoS Pathog 13:e1006167. doi: 10.1371/journal.ppat.1006167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang HH, Ganem D. 2013. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 13:429–440. doi: 10.1016/j.chom.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosseinipour MC, Sweet KM, Xiong J, Namarika D, Mwafongo A, Nyirenda M, Chiwoko L, Kamwendo D, Hoffman I, Lee J, Phiri S, Vahrson W, Damania B, Dittmer DP. 2014. Viral profiling identifies multiple subtypes of Kaposi's sarcoma. mBio 5:e01633-14. doi: 10.1128/mBio.01633-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C. 2002. K15 protein of Kaposi's sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol 76:802–816. doi: 10.1128/JVI.76.2.802-816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim CS, Seet BT, Ingham RJ, Gish G, Matskova L, Winberg G, Ernberg I, Pawson T. 2007. The K15 protein of Kaposi's sarcoma-associated herpesvirus recruits the endocytic regulator intersectin 2 through a selective SH3 domain interaction. Biochemistry 46:9874–9885. doi: 10.1021/bi700357s. [DOI] [PubMed] [Google Scholar]
- 37.Sander G, Konrad A, Thurau M, Wies E, Leubert R, Kremmer E, Dinkel H, Schulz T, Neipel F, Sturzl M. 2008. Intracellular localization map of human herpesvirus 8 proteins. J Virol 82:1908–1922. doi: 10.1128/JVI.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith CG, Kharkwal H, Wilson DW. 2017. Expression and subcellular localization of the KSHV K15P protein during latency and lytic reactivation in primary effusion lymphoma cells. J Virol 91:e01370-17. doi: 10.1128/JVI.01370-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho NH, Choi YK, Choi JK. 2008. Multi-transmembrane protein K15 of Kaposi's sarcoma-associated herpesvirus targets Lyn kinase in the membrane raft and induces NFAT/AP1 activities. Exp Mol Med 40:565–573. doi: 10.3858/emm.2008.40.5.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Havemeier A, Gramolelli S, Pietrek M, Jochmann R, Sturzl M, Schulz TF. 2014. Activation of NF-kappaB by the Kaposi's sarcoma-associated herpesvirus K15 protein involves recruitment of the NF-kappaB-inducing kinase, IkappaB kinases, and phosphorylation of p65. J Virol 88:13161–13172. doi: 10.1128/JVI.01766-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pietrek M, Brinkmann MM, Glowacka I, Enlund A, Havemeier A, Dittrich-Breiholz O, Kracht M, Lewitzky M, Saksela K, Feller SM, Schulz TF. 2010. Role of the Kaposi's sarcoma-associated herpesvirus K15 SH3 binding site in inflammatory signaling and B-cell activation. J Virol 84:8231–8240. doi: 10.1128/JVI.01696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinbruck L, Gustems M, Medele S, Schulz TF, Lutter D, Hammerschmidt W. 2015. K1 and K15 of Kaposi's sarcoma-associated herpesvirus are partial functional homologues of latent membrane protein 2A of Epstein-Barr virus. J Virol 89:7248–7261. doi: 10.1128/JVI.00839-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keilhauer EC, Hein MY, Mann M. 2015. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol Cell Proteomics 14:120–135. doi: 10.1074/mcp.M114.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 45.Sorkin A, von Zastrow M. 2009. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol 10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazza S, Maffucci T. 2011. Class II phosphoinositide 3-kinase C2alpha: what we learned so far. Int J Biochem Mol Biol 2:168–182. [PMC free article] [PubMed] [Google Scholar]
- 47.Jean S, Kiger AA. 2014. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci 127:923–928. doi: 10.1242/jcs.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Domin J, Gaidarov I, Smith ME, Keen JH, Waterfield MD. 2000. The class II phosphoinositide 3-kinase PI3K-C2alpha is concentrated in the trans-Golgi network and present in clathrin-coated vesicles. J Biol Chem 275:11943–11950. doi: 10.1074/jbc.275.16.11943. [DOI] [PubMed] [Google Scholar]
- 49.Gaidarov I, Smith ME, Domin J, Keen JH. 2001. The class II phosphoinositide 3-kinase C2alpha is activated by clathrin and regulates clathrin-mediated membrane trafficking. Mol Cell 7:443–449. doi: 10.1016/S1097-2765(01)00191-5. [DOI] [PubMed] [Google Scholar]
- 50.Borner GH, Antrobus R, Hirst J, Bhumbra GS, Kozik P, Jackson LP, Sahlender DA, Robinson MS. 2012. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol 197:141–160. doi: 10.1083/jcb.201111049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshioka K, Yoshida K, Cui H, Wakayama T, Takuwa N, Okamoto Y, Du W, Qi X, Asanuma K, Sugihara K, Aki S, Miyazawa H, Biswas K, Nagakura C, Ueno M, Iseki S, Schwartz RJ, Okamoto H, Sasaki T, Matsui O, Asano M, Adams RH, Takakura N, Takuwa Y. 2012. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med 18:1560–1569. doi: 10.1038/nm.2928. [DOI] [PubMed] [Google Scholar]
- 52.Aki S, Yoshioka K, Okamoto Y, Takuwa N, Takuwa Y. 2015. Phosphatidylinositol 3-kinase class II alpha-isoform PI3K-C2alpha is required for transforming growth factor beta-induced Smad signaling in endothelial cells. J Biol Chem 290:6086–6105. doi: 10.1074/jbc.M114.601484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arcaro A, Zvelebil MJ, Wallasch C, Ullrich A, Waterfield MD, Domin J. 2000. Class II phosphoinositide 3-kinases are downstream targets of activated polypeptide growth factor receptors. Mol Cell Biol 20:3817–3830. doi: 10.1128/MCB.20.11.3817-3830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown RA, Domin J, Arcaro A, Waterfield MD, Shepherd PR. 1999. Insulin activates the alpha isoform of class II phosphoinositide 3-kinase. J Biol Chem 274:14529–14532. doi: 10.1074/jbc.274.21.14529. [DOI] [PubMed] [Google Scholar]
- 55.Leibiger B, Moede T, Uhles S, Barker CJ, Creveaux M, Domin J, Berggren PO, Leibiger IB. 2010. Insulin-feedback via PI3K-C2alpha activated PKBalpha/Akt1 is required for glucose-stimulated insulin secretion. FASEB J 24:1824–1837. doi: 10.1096/fj.09-148072. [DOI] [PubMed] [Google Scholar]
- 56.Biswas K, Yoshioka K, Asanuma K, Okamoto Y, Takuwa N, Sasaki T, Takuwa Y. 2013. Essential role of class II phosphatidylinositol-3-kinase-C2alpha in sphingosine 1-phosphate receptor-1-mediated signaling and migration in endothelial cells. J Biol Chem 288:2325–2339. doi: 10.1074/jbc.M112.409656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turner SJ, Domin J, Waterfield MD, Ward SG, Westwick J. 1998. The CC chemokine monocyte chemotactic peptide-1 activates both the class I p85/p110 phosphatidylinositol 3-kinase and the class II PI3K-C2alpha. J Biol Chem 273:25987–25995. doi: 10.1074/jbc.273.40.25987. [DOI] [PubMed] [Google Scholar]
- 58.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 59.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Domin J, Pages F, Volinia S, Rittenhouse SE, Zvelebil MJ, Stein RC, Waterfield MD. 1997. Cloning of a human phosphoinositide 3-kinase with a C2 domain that displays reduced sensitivity to the inhibitor wortmannin. Biochem J 326(Part 1):139–147. doi: 10.1042/bj3260139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Falasca M, Maffucci T. 2007. Role of class II phosphoinositide 3-kinase in cell signalling. Biochem Soc Trans 35:211–214. doi: 10.1042/BST0350211. [DOI] [PubMed] [Google Scholar]
- 62.El Sheikh SS, Domin J, Tomtitchong P, Abel P, Stamp G, Lalani EN. 2003. Topographical expression of class IA and class II phosphoinositide 3-kinase enzymes in normal human tissues is consistent with a role in differentiation. BMC Clin Pathol 3:4. doi: 10.1186/1472-6890-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campa CC, Franco I, Hirsch E. 2015. PI3K-C2alpha: one enzyme for two products coupling vesicle trafficking and signal transduction. FEBS Lett 589:1552–1558. doi: 10.1016/j.febslet.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, Muller R, Zarbakhsh S, Gulluni F, Hirsch E, Krauss M, Schultz C, Schmoranzer J, Noe F, Haucke V. 2013. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499:233–237. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 65.Krag C, Malmberg EK, Salcini AE. 2010. PI3KC2alpha, a class II PI3K, is required for dynamin-independent internalization pathways. J Cell Sci 123:4240–4250. doi: 10.1242/jcs.071712. [DOI] [PubMed] [Google Scholar]
- 66.Tomlinson CC, Damania B. 2008. Critical role for endocytosis in the regulation of signaling by the Kaposi's sarcoma-associated herpesvirus K1 protein. J Virol 82:6514–6523. doi: 10.1128/JVI.02637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meunier FA, Osborne SL, Hammond GR, Cooke FT, Parker PJ, Domin J, Schiavo G. 2005. Phosphatidylinositol 3-kinase C2alpha is essential for ATP-dependent priming of neurosecretory granule exocytosis. Mol Biol Cell 16:4841–4851. doi: 10.1091/mbc.e05-02-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dominguez V, Raimondi C, Somanath S, Bugliani M, Loder MK, Edling CE, Divecha N, da Silva-Xavier G, Marselli L, Persaud SJ, Turner MD, Rutter GA, Marchetti P, Falasca M, Maffucci T. 2011. Class II phosphoinositide 3-kinase regulates exocytosis of insulin granules in pancreatic beta cells. J Biol Chem 286:4216–4225. doi: 10.1074/jbc.M110.200295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polachek WS, Moshrif HF, Franti M, Coen DM, Sreenu VB, Strang BL. 2016. High-throughput small interfering RNA screening identifies phosphatidylinositol 3-kinase class II alpha as important for production of human cytomegalovirus virions. J Virol 90:8360–8371. doi: 10.1128/JVI.01134-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lynch DT, Zimmerman JS, Rowe DT. 2002. Epstein-Barr virus latent membrane protein 2B (LMP2B) co-localizes with LMP2A in perinuclear regions in transiently transfected cells. J Gen Virol 83:1025–1035. doi: 10.1099/0022-1317-83-5-1025. [DOI] [PubMed] [Google Scholar]
- 71.Takebe Y, Seiki M, Fujisawa J, Hoy P, Yokota K, Arai K, Yoshida M, Arai N. 1988. SR alpha promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol Cell Biol 8:466–472. doi: 10.1128/MCB.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kati S, Hage E, Mynarek M, Ganzenmueller T, Indenbirken D, Grundhoff A, Schulz TF. 2015. Generation of high-titre virus stocks using BrK.219, a B-cell line infected stably with recombinant Kaposi's sarcoma-associated herpesvirus. J Virol Methods 217:79–86. doi: 10.1016/j.jviromet.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 73.Alkharsah KR, Singh VV, Bosco R, Santag S, Grundhoff A, Konrad A, Sturzl M, Wirth D, Dittrich-Breiholz O, Kracht M, Schulz TF. 2011. Deletion of Kaposi's sarcoma-associated herpesvirus FLICE inhibitory protein, vFLIP, from the viral genome compromises the activation of STAT1-responsive cellular genes and spindle cell formation in endothelial cells. J Virol 85:10375–10388. doi: 10.1128/JVI.00226-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.May T, Butueva M, Bantner S, Markusic D, Seppen J, MacLeod RA, Weich H, Hauser H, Wirth D. 2010. Synthetic gene regulation circuits for control of cell expansion. Tissue Eng Part A 16:441–452. doi: 10.1089/ten.tea.2009.0184. [DOI] [PubMed] [Google Scholar]
- 75.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jochim N, Gerhard R, Just I, Pich A. 2014. Time-resolved cellular effects induced by TcdA from Clostridium difficile. Rapid Commun Mass Spectrom 28:1089–1100. doi: 10.1002/rcm.6882. [DOI] [PubMed] [Google Scholar]
- 77.Tyanova S, Temu T, Cox J. 2016. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 78.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. 2016. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]