

Abstract
Lipid metabolism, specifically fatty acid oxidation (FAO) mediated by carnitine palmitoyltransferase (CPT) 1A, has been described to be an important actor of ghrelin action in hypothalamus. However, it is not known whether CPT1A and FAO mediate the effect of ghrelin on the cortex. Here, we show that ghrelin produces a differential effect on CPT1 activity and γ-aminobutyric acid (GABA) metabolism in the hypothalamus and cortex of mice. In the hypothalamus, ghrelin enhances CPT1A activity while GABA transaminase (GABAT) activity, a key enzyme in GABA shunt metabolism, is unaltered. However, in cortex CPT1A activity and GABAT activity are reduced after ghrelin treatment. Furthermore, in primary cortical neurons, ghrelin reduces GABA release through a CPT1A reduction. By using CPT1A floxed mice, we have observed that genetic ablation of CPT1A recapitulates the effect of ghrelin on GABA release in cortical neurons, inducing reductions in mitochondrial oxygen consumption, cell content of citrate and α-ketoglutarate, and GABA shunt enzyme activity. Taken together, these observations indicate that ghrelin-induced changes in CPT1A activity modulate mitochondrial function, yielding changes in GABA metabolism. This evidence suggests that the action of ghrelin on GABA release is region specific within the brain, providing a basis for differential effects of ghrelin in the central nervous system.
Electronic supplementary material
The online version of this article (10.1007/s12035-018-0921-3) contains supplementary material, which is available to authorized users.
Keywords: Ghrelin, GABA, Fatty acid oxidation, CPT1A, Cortical neurons
Introduction
Acyl-ghrelin (hereafter referred to as ghrelin) is the only peripheral hormone with orexigenic effects described to date [1–3]. It is produced in stomach X/A-like cells where both ghrelin/obestatin preprohormone gene and ghrelin O-acyltransferase are expressed [4, 5]. Ghrelin’s main functions are related to the central control of energy homeostasis and its use. Specifically, ghrelin promotes food intake [6–9], increased body weight and adiposity [10–12], controls glucose homeostasis [13, 14] and growth hormone secretion [15], and enhances motivation for food intake [16–18].
Ghrelin functions in hypothalamus are mediated by growth hormone secretagogue receptor (GHSR) [19]. The molecular mechanisms involved under ghrelin activation of GHSR receptor are not completely understood, but in hypothalamus, AMP-dependent protein kinase (AMPK) has a key role in intracellular signal transduction [20–22]. AMPK activation due to ghrelin promotes acetyl-CoA carboxylase (ACC) inactivation, which reduces malonyl-CoA formation and consequently increases CPT1A activity [23]. CPT1A activation triggers a series of molecular events leading to increased expression of orexigenic agouti-related protein (AgRP) and neuropeptide Y (NPY) [24–27], which results in increased appetite.
Ghrelin has also been involved in GABAergic signaling. It has been reported that ghrelin inhibits firing of postsynaptic pro-opiomelanocortin (POMC)-expressing neurons by increasing presynaptic γ-aminobutyric acid (GABA) release [28]. This increase in GABA output in the orexigenic neurons is produced as a consequence of glutamic acid decarboxylase (GAD) expression [29]. Furthermore, CPT1A as an intermediate in ghrelin signaling in ventromedial hypothalamus has been found to increase the expression of vesicular GABA transporter (VGAT), which is considered the factor that controls GABA quantal size, in CPT1A activity-induced hyperphagic rats [30].
Three pathways control the cytoplasmic GABA content to be released: (1) GAD activity, which is the canonical pathway to generate GABA out of glutamate [31]; (2) GABA shunt, which is composed of two enzymatic reactions catalyzed in mitochondria by succinate semialdehyde dehydrogenase (SSADH) and GABA transaminase (GABAT) [32, 33]; and (3) GABA transport into small synaptic vesicles for its release, via VGAT.
Besides energy balance, other actions of ghrelin related to anxiety, cognition, stress, and sleep have been extensively studied [34–37]. For ghrelin to be involved in such processes, it must reach extra-hypothalamic areas. In fact, GHSR was first described in pituitary gland, hippocampus, ventral tegmental area, raphe nuclei, and hypothalamus [38]. However, its expression can be extended to neocortex, olfactory bulb, basal ganglia, and cerebellum [39]. Moreover, 76% of primary cortical neurons are GHSR-positive cells in in vitro culture [40, 41], which indicates that ghrelin may play an important role in these brain areas. However, little is known about the intracellular mediators involved in ghrelin action in cortical neurons.
In this study, we show that ghrelin’s action on cortical neurons involves CPT1A modulation that differs from that observed in hypothalamic neurons. In cortical neurons, ghrelin inhibits CPT1A activity and fatty acid oxidation (FAO), and reduces the levels of Krebs cycle intermediates such as citrate and α-ketoglutarate, GABA shunt enzymes, and GABA release under depolarization conditions. These data indicate that ghrelin modulates GABA in a region-specific fashion, which may account for the variation in ghrelin actions.
Materials and Methods
Animals and Treatments
The mice strains used in this project were C57BL/6J and CPT1A(loxP/loxP) mice obtained from HEPD0727_3_H09 clone from the European Conditional Mouse Mutagenesis (EUCOMM) programme. Once the karyotype had been studied, HEPD0727_3_H09 clone was chosen to be microinjected in blastocysts to obtain chimeric mice. Eventually, from the seven chimera that were obtained, we selected one male with 80% chimerism to obtain offspring. This mouse was crossed with C57BL/6J to obtain CPT1A(+/frt-loxP) mice. These mice were genotyped by analyzing the number of lacz-containing sequences by digital droplet PCR (ddPCR), as described below. These potentially conditional CPT1A mice were crossbred with C57BL6/J FLP recombinase-expressing mice to eliminate the lacz cassette and obtain CPT1A(+/loxP) mice (Supplemental Fig. 1). CPT1A(+/loxP) mice were crossbred to obtain homozygous CPT1A(loxP/loxP) mice.
In order to analyze the effects of ghrelin on several cortical and hypothalamic parameters, we injected a dose of 10 μg ghrelin (or the equivalent volume of phosphate-buffered solution, i.e. 300 μL) intraperitoneally (IP) at 0 min and another dose at 30 min in mice that had been food-deprived for 2 h after the dark period [42]. All the mice received the same amount of ghrelin, as their body weights were similar (25.74 ± 0.52 g for ghrelin-treated mice and 25.52 ± 061 g for the control group). We monitored eating time and food intake for 1 h after the first injection. One hour after the treatment, mice were sacrificed by cervical dislocation and their tissues collected. All the procedures with mice were approved by the Animal Experimentation Ethics Committee of the University of Barcelona (CEEA-UB). DAAM Permit #8173 and colony management Permit C-0020 were obtained from the Government of Catalonia, according to European Directive 2010/63/EU.
Cell Cultures and Treatments
Primary cortical neurons were directly obtained from fresh cortex as described by Solà et al. [43]. Cerebral cortices from C57BL/6J and CPT1A(loxP/loxP) mice were harvested from unborn pups on embryonic days from 15 to 17. No isolation with less than 60% viability was used. Then, 8 × 105 living cells per well (in 6-well plates) were seeded. Each litter yielded around 6 to 8 plates, which were precoated with 4 °C overnight incubation with 0.005% poly-l-lysine, prepared from 2× solution (Sigma-Aldrich, ref. P4707). Three hours after seeding, cells were attached and the medium was replaced with Neurobasal Medium (GIBCO, ref. 21103-049) supplemented at 1× with B27 (GIBCO, ref. 17504-044) and GlutaMax (GIBCO, ref. 35050-038), as well as 1% penicillin/streptomycin (100 U/mL final concentration) (GIBCO, ref. 15140122). Two days after culturing the cells, 2 μM cytosine β-d-arabinofuranoside (Sigma-Aldrich, ref. C1768) was added to avoid proliferation of non-neuronal cells to the medium. Every 2 days, the medium had to be changed by removing half of the previous medium and replacing it with fresh medium. Cells were prevented from drying out without any medium on them.
To delete CPT1A in the cortical neuron cells, we used CRE recombinase- and GFP-expressing adenoviral vectors (Ad-CRE-GFP) (Vector Biolabs, ref. 1700) with a titration of 4.22 × 109 pfu/mL and Ad-GFP (3.32 × 109 PFU/mL) as a control. We infected the cells with 100 PFU of Ad-CRE-GFP per cell. The medium was changed the next day. The medium used was the same as in the regular culture. Ghrelin treatment was conducted in both primary cortical neurons and GT1-7. Hypothalamic GT1-7 cells were pretreated for 3 h with 5 mM glucose pyruvate- and glutamine-enriched Dulbecco’s modified Eagle medium (DMEM). Primary cortical neurons were pretreated for 3 h with 5 mM glucose Neurobasal®-A medium (GIBCO, ref. 10888022). Then, cells were treated with 100 nM ghrelin (Sigma-Aldrich, ref. G8903) in pretreatment medium for 30 min. To pharmacologically inhibit CPT1A, we added 40 μg/mL etomoxir in the 30-min ghrelin treatment. To block the tricarboxylic cycle by inhibiting isocitrate dehydrogenase, we added 700 μM 2-hydroxyglutarate.
Metabolic Extracellular Flux XF Analysis
Cortical neuron cells were cultured in customized Seahorse 24-well plates. Before the measurement, cells were treated for 3 h with 5 mM glucose medium. In the last 30 min, the ghrelin treatment was carried out as previously explained. Then, cells were assayed for 1 h in XF Assay Medium (Seahorse Bioscience) plus 5 mM glucose. During the assay, we injected the following at the final concentrations shown: 2 μg/mL oligomycin, 0.16 μM FCCP, and 2 μM antimycin A (Sigma-Aldrich). Oxygen consumption rate (OCR) was calculated by plotting the oxygen tension of media as a function of time (pmol/min), and data were normalized by the protein concentration measured in each well. The results were quantified as the average of 8–10 wells ± SEM per time point in at least three independent experiments.
Amino Acid Neurotransmitters Release Assay
We carried out the experiments with cultured primary neurons on the 8th day of in vitro (8 DIV) culture, when they were mature enough. If an infection had to be made with Ad-CRE, the cells were infected on 6 DIV, to obtain maximum expression on 8 DIV. On that day, any pretreatment (5 mM glucose reduction for 3 h) and ghrelin treatment with 100 nM ghrelin was completed just before the amino acid neurotransmitter release assay. Two buffers are needed for this assay: Basal 5 mM KCl Hank’s buffer (K5) and depolarizing 90 mM KCl Hank’s buffer (K90). After treatment, the wells were washed with pre-warmed Hank’s Buffer (K5). Then, 1 mL K5 buffer was added per well and incubated for 10 min at 37 °C, so that neurons could stabilize in the buffer. The buffer was removed and cells were incubated with 1 mL K5 for 2 min and the supernatant was kept in a microcentrifuge tube. Then, cells were incubated with 1 mL K90 for 2 min and the supernatant was kept in a microcentrifuge tube. This process was repeated up to six times, alternating between K5 and K90 solutions and keeping the neurotransmitters released into the Hank’s solution for posterior analysis. As a control of non-vesicular neurotransmitter efflux, incubation for 2 min in 1 mL K90 was carried out without Ca2+ and 3 mM EGTA, and the supernatant was kept for analysis. All the samples were stored at − 20 °C until analysis and the cells in the plate were lysed with 0.2 mL NaOH 0.2 N to quantify the protein content for normalization. Amino acid neurotransmitter content was then measured from the samples by HPLC-MS/MS at the Institute of Biomedical Research of Barcelona (IIBB-CSIC).
Digital Droplet PCR
To check if CPT1A(+/frt-loxP) contained the correct integration cassette and the right number of copies in the genome, we genotyped the allele-specific CPT1A(+/frt-loxP) mice previous to generating CPT1A(+/loxP) mice, using a QX100 Droplet Digital PCR System (BioRad, ref. QX100) that had been adapted for the use of QX200 ddPCR EvaGreen Supermix dsDNA binding dye (BioRad, ref. 186-4035). Genomic DNA was obtained from the tail of CPT1A(+/frt-loxP) mice by proteinase K digestion and phenol-chloroform purification. One microgram of genomic DNA was digested for 1 h with 10 U of the restriction enzyme EcoRI. To perform ddPCR, the primers used were LACZfor GCTGGAGTGACGGCAGTTAT and LACZrev TACCCGTAGGTAGTCACGCA, and for the control TERTfor CCTCTGTGTCCGCTAGTTACA and TERTrev TCTTTGTACCTCGAGATGGCA. The amplicon sizes were 137 bp for TERT and 197 bp for LACZ. Due to specific characteristics of ddPCR, a temperature gradient was performed to optimize the annealing of the primers. The annealing temperatures tested were 58.9, 60.1, 61.0, and 61.6 °C. The selected temperature was 60.1 °C. The ddPCR mixture for each gene amplification contained the following: 4.4 ng of DNA sample, primer concentration at 0.5 μM each, and 11 μL of EvaGreen® supermix (BioRad) in a 22-μL final volume. A total of 20 μL of reaction mixture and 70 μL of Droplet Generation Oil were placed in the pertinent well of the DG8 cartridge (BioRad, ref. 186-3006). The bubbles were removed from the system, since they can interfere with emulsification. The cartridge was then placed in the QX100 droplet generator (BioRad, ref. 186-3002) for droplet generation. The droplets were transferred to a 96-well PCR plate. The program for amplification started with 5 min at 95 °C, followed by 39 cycles of 30 s at 95 °C and 1 min at 59 °C; finally, the temperature decreased to 4 °C for 5 min, was increased to 90 °C for 5 min, and the reaction was kept at 25 °C. The analysis by the QX100 Droplet Reader (BioRad, ref. 186-3003) and QuantaSoft Software showed that the tert/lacz ratio is 2.09, which suggest that only one copy of the construct is present in heterozygous CPT1A(+/frt-loxP) mice for each two copies of tert reference gene (Supplemental Fig. 1A and B). All the equipment was available from the Laboratory of Luminescence and Biomolecular Spectroscopy (LLEB), Scientific and Technical Services, Autonomous University of Barcelona (UAB).
mRNA Expression Analysis
Tissues were excised from six mice from each group, frozen, and stored at − 80 °C. Total RNA was extracted from frozen brain tissue using the RNeasy Lipid Tissue Mini kit (QIAGEN, ref. 25-0500-71), following the manufacturer’s indications with minor modifications. Total RNA was extracted from cultured cells using the Illustra Mini RNAspin kit (GE Lifesciences, ref. 25-0500-71), following the manufacturer’s indications with minor modifications. cDNA was obtained using TaqMan Reverse Transcription Kit (Applied Biosystems, ref. N8080234), from 1 μg total RNA from MBH or 400 μg from cell cultures, because of the varying RNA extraction yields. The manufacturer’s protocol was used with hexamer primers to obtain cDNA from all the mRNA. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using Power SYBR Green PCR Master Mix adapted for LightCycler 480 (Applied Biosystems, ref. 4367659), according to the manufacturer’s indications in the LightCycler 480 Instrument II (Roche, ref. 05015243001). The primers of the genes that were analyzed are described in Supplemental Table 1.
Analysis of Protein Levels
Protein expression analysis was obtained from four mice from the control group and 4 mice from the ghrelin-treated group. Frozen hypothalamus and cortex were homogenized in protein extraction buffer (30 mM Hepes, pH 7.4, 150 mM NaCl, 10% glycerol, 0.5% sodium deoxycholate [DOC], 1% Triton X-100 with phosphatase and protease inhibitors). Fifty micrograms of protein were analyzed on 10% SDS-PAGE gels and then transferred onto PVDF membranes (Millipore). The following primary antibodies were used: GAD65/GAD2 (1/1000; Cell Signaling ref. 5843), VGLUT2 (1/1000; Cell Signaling ref. 71555), and β-actin (1/50,000; Sigma-Aldrich). Blots were incubated with the appropriate IgG-HRP-conjugated secondary antibody. Protein bands were visualized using the ECL immunoblotting detection system (GE Healthcare) and developed on an ImageQuant LAS4000 mini Fuji luminescence imagining system. The bands were quantified by densitometry using ImageJ analysis software.
GABA Transaminase Activity
GABA transaminase Assay Kit (BMRService, ref. E134) was used according to the manufacturer’s indications. This kit is based on the sequential GABA transamination reaction and glutamate oxidation, which couples the reduction of iodonitrotetrazolium (INT) into INT-formazan (ε = 18 mM−1 cm−1 at 492 nm).
Determination of Tissue Acylcarnitine Content
Tissues for analysis were removed quickly, frozen in liquid nitrogen, and stored at − 80 °C prior to quantification. Acylcarnitines were analyzed using an Acquity UPLC-TOF system (Waters) with a BEH C8 column (1.7 μm particle size, 100 mm × 2.1 mm, Waters). The two mobile phases were 1 mM ammonium formate in methanol (phase A) and 2 mM ammonium formate in H2O (phase B), both phases with 0.05 mM formic acid. The following gradient was programmed: 0 min, 65% A; 10 min, 90% A; 15 min, 99% A; 17 min, 99% A; 20 min, 65% A, and a flow rate of 0.3 mL min−1. Quantification was carried out using the extracted ion chromatogram of each compound, with 50-mDa windows. The linear dynamic range was determined by injecting standard mixtures. Positive identification of compounds was based on the accurate mass measurement with an error < 5 ppm and their LC retention time was compared to that of a standard (± 2%).
Neuronal Tricarboxylic Acid Cycle Intermediates Analysis
Analysis of tricarboxylic acids was carried out by gas chromatography-mass spectrometry (GC-MS) detection, with a method adapted from the literature [44–46]. Experiments were performed on cortical cell cultures (2.5 × 105/well). At the end of the incubations, cells were washed with PBS, and the cell pellet was resuspended in 500 μl of Milli-Q water and frozen at − 20 °C until assayed (a separate fraction was set aside for protein quantification). For the preparation of extracts, the 500-μl samples were taken to a volume of 2 ml with water, and 1 ml of 8 M NaOH and 1 ml of 25 mg/ml hydroxylamine was added. The sample was then heated at 60 °C for 30 min, and the pH was adjusted by adding 1 ml of 6 N HCl. Sequential extractions were carried out as described [44] with the exception that samples were extracted twice with 2 ml of diethyl ether and twice with 2 ml of ethyl acetate. A total of 6 μl of 5 mM undecanedioic acid was added to the collection tube to serve as an internal standard of the derivatization procedure. Once completely evaporated with nitrogen gas, the final dry residue was resuspended in 75 μl of trimethylsilyl, incubated at 60 °C for 30 min and kept at − 20 °C until injected. A total of 2-μl samples were injected into a GC-MS (Agilent Technologies ref. 7890A-5975C), with an HP-5MS 60 × 0.25 × 0.25 capillary column, using a splitless method and pressure ramp, and the results were analyzed using ChemStation GC/MSD software. The ratio between the areas was normalized by the protein concentration of the sample (μg/μl).
Statistical Analysis
Data are expressed as mean ± SEM. Statistical significance was determined by two-way ANOVA and the Student’s t test, using Microsoft Excel and GraphPad Prism 6 software. A p value of < 0.05 was considered significant.
Results
Ghrelin Modulates CPT1A Activity Differentially in Cortex and Hypothalamus
To address the differential effects on CPT1A activity in the cortex and hypothalamus, we measured the CPT1A mRNA levels and total acylcarnitine content in both regions in ghrelin-treated and saline-injected control mice. As expected, ghrelin-treated mice showed a 2-fold increased food intake (Fig. 1a) and spent more time eating a chow diet (Fig. 1b). Cortical CPT1A mRNA levels were reduced up to 1.3-fold in ghrelin-treated mice and showed a slight increase in hypothalamus (Fig. 1c). Next, we measured the acylcarnitine content as an indicator of CPT1 activity in both tissues. Cortex and hypothalamus had similar acylcarnitine basal levels (4.83 ± 0.15 and 5.30 ± 0.3 pmol/μg, respectively), but in ghrelin-treated mice, cortical acylcarnitine content dropped to 48% while the hypothalamic pool increased 3-fold (Fig. 1d).
Fig. 1.

Differential effect of ghrelin in cortex and hypothalamus. a Changes in food intake. b Eating time. c CPT1A mRNA levels measured by qPCR (the cortex control group is the reference group). d Acylcarnitine levels measured by HPLC-MS/MS in mice after two ip ghrelin injections. Results are represented as mean ± SEM. n = 6; *p < 0.05; **p < 0.01, ***p < 0.001
Ghrelin Modulates GABA Shunt Enzymes Differentially in Cortex and Hypothalamus
To determine if this change in CPT1 activity correlates with changes in the expression of genes related to glutamate and GABA, we determined the mRNA levels of different genes involved in glutamate and GABA metabolism, taking as a reference the cortex control group. Vesicular glutamate transporters (VGLUT) 1, 2, and 3 showed different patterns: VGLUT1 slightly decreased in the hypothalamus and remained unaltered in cortex (Fig. 2a); while generally expressed VGLUT2 increased 5-fold in cortex (Fig. 2b) and GABAergic neuron-associated VGLUT3 decreased up to 0.6-fold in cortex (Fig. 2c), while both remain unaltered in hypothalamus.
Fig. 2.
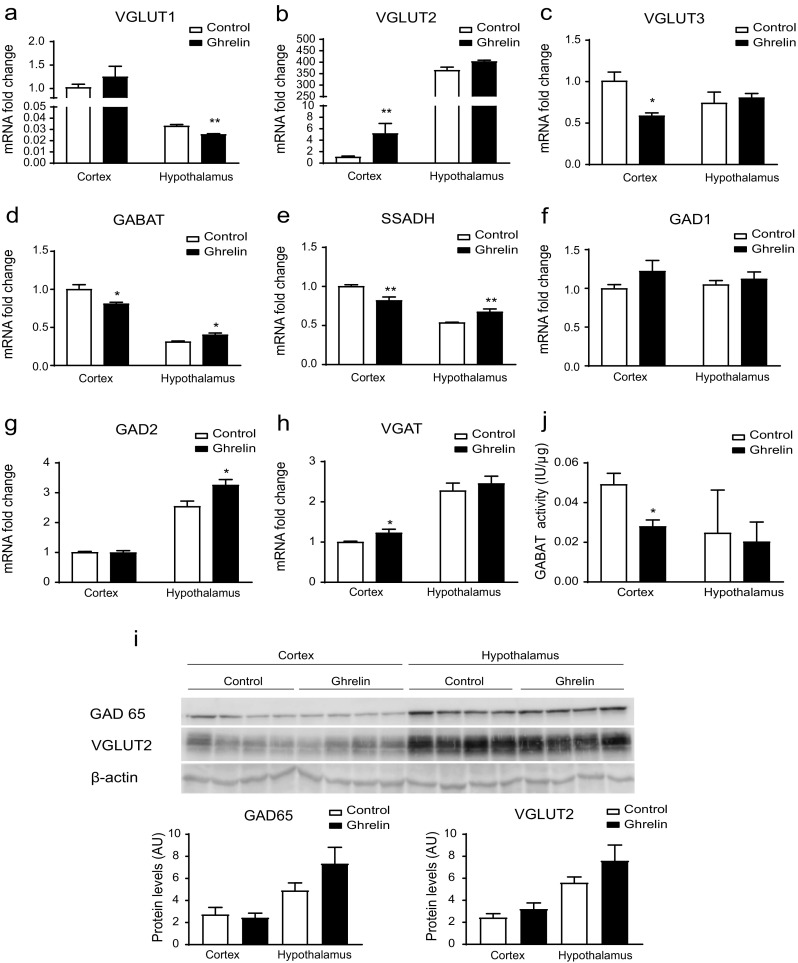
Ghrelin modifies GABA metabolism in cortex. Analysis of the following in the cortex and hypothalamus of mice after two ip ghrelin injections. a–c Relative mRNA levels of Vglut1, Vglut2, Vglut3 genes analyzed by qPCR. The cortex control group is the reference group. d–h Relative mRNA levels of GABA metabolism genes. The cortex control group is the reference group n = 6. j Levels of GABAT activity n = 4. i Representative Western Blot of the protein levels of GAD65, VGLUT2, and β-actin n = 4. Results are represented as mean + SEM.; *p < 0.05, **p < 0.01
We also assessed the modulation of GABA metabolism by ghrelin. mRNA levels of the GABA shunt genes (GABAT and SSADH) were higher in cortex than in hypothalamus (Fig. 2d, e). In ghrelin-treated mice, cortical levels were reduced, while hypothalamic levels increased. Thus, intraperitoneal ghrelin produced differential effects in cortex and hypothalamus in terms of GABA shunt genes. However, canonical GABA generators such as GAD did not follow this pattern. GAD1 seemed to be unaffected (Fig. 2f) and GAD2 increased in the hypothalamus of ghrelin-treated mice, while it remained unaltered in cortex (Fig. 2g). Vesicular GABA transporter (VGAT) mRNA levels slightly increased in cortex, and no changes were observed in the hypothalamus (Fig. 2h). Altogether, this indicates that GABA metabolism is altered in the cortex after ghrelin’s injection.
Next, we assessed changes in protein levels of GAD65/GAD2 and VGLUT2 in the cortex in ghrelin-treated mice. Neither cortical GAD65/GAD2 nor VGLUT2 protein levels changed after ghrelin treatment (Fig. 2i). However, cortical GABAT activity drops to 58% in ghrelin-treated mice, while hypothalamic GABAT remained unaltered (Fig. 2j). These results suggest that GABA metabolism is reduced in the cortex after ghrelin’s administration.
Ghrelin Reduces GABA Release and FAO in Primary Cortical Neurons
Given that the expression of GABA shunt enzymes is reduced by ghrelin in mouse cortex, we wanted to assess the direct effect of ghrelin on the GABA release of cultured primary neurons. We observed that released GABA at depolarizing 90 mM KCl concentration was significantly reduced up to 55% compared to 5 mM KCl in ghrelin-treated neurons at glycorrhachia-like glucose levels (5 mM glucose) [47] and in glucose-deprived medium (Fig. 3a). Furthermore, basal depolarized levels of released GABA were 4-fold higher at 5 mM glucose, compared to those at 0 and 25 mM glucose. Since both GABAergic and glutamatergic neurons can be found in primary cortical cultures, we assessed the effect on glutamate release as well. In the assay conditions, glutamate release was not affected at glycorrhachia-like glucose levels, but it was reduced in glucose-deprived cortical neurons (Fig. 3b). Since CPT1A expression and activity was reduced in the cortex by intraperitoneal ghrelin, we wanted to assess the effect of ghrelin on FAO. To do so, we performed metabolic extracellular flux (XF) analysis in a palmitate-, carnitine-, and glucose-enriched medium. Basal mitochondrial OCR in ghrelin-treated neurons was reduced by 80% (0.35 to 0.07 pmol/min/μg, p < 0.05) (Fig. 3c), which is in agreement with the reduction of CPT1 activity.
Fig. 3.

Ghrelin reduces GABA release and fatty acid oxidation in primary cortical neuronal cultures. a GABA release at different glucose concentrations. b Glutamate release at different glucose concentrations. c Analysis of oxygen consumption rate (OCR) after incubation with exogenous palmitate (Seahorse XF Analyser). Results are represented as mean + SEM. n = 4 in triplicates; *p < 0.05. ***p < 0.001
CPT1A Ablation Mimics Ghrelin’s Effect on GABA Release in Primary Cortical Neurons
Since ghrelin treatment in cortical neurons reduced CPT1A expression, FAO, and GABA release, we wanted to assess whether the ablation of CPT1A would affect GABA release in the same direction as ghrelin. Firstly, we generated a potentially conditional CPT1A knockout mouse by taking advantage of two heterozygous stem cell clones from the cell repository of the European Conditional Mouse Mutagenesis (EUCOMM) Program. CPT1A(loxP/loxP) mice were obtained from one of these clones (Supplemental Fig. 1). To assess the integrity of the loxP sequences, primary hepatocytes obtained from CPT1A(loxP/loxP) mice were infected with CRE-expressing adenovirus (CRE). The infection successfully removed the loxP-flanked region containing the CPT1A exon 4, since gDNA amplicon from both sides of the homologous region dropped to 219 bp compared to control (GFP) cells 1030-bp amplicon (Fig. 4a, b). Regarding CPT1A mRNA expression, CRE-infected primary cortical neurons showed a reduction in wt CPT1A and exon4-deleted CPT1A mRNA levels (Supplemental Fig. 2d). The amplicon from exon 3 to 5 from CRE-infected cDNA with an expected 116-bp length was barely detectable after 24- and 48-h infections, which indicates great instability of the deleted CPT1A mRNA product.
Fig. 4.
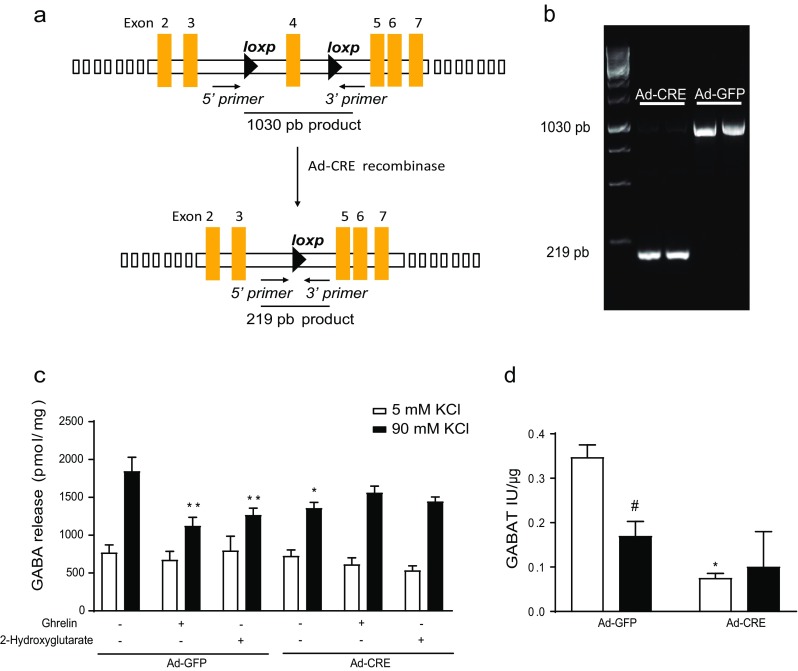
Effect of CPT1A deletion on GABA release in primary cortical neuronal cultures. Cortical neuron culture from CPT1A(loxP/loxP) embryo mice were obtained and incubated with Ad-Cre-GFP or Ad-GFP (100 PFU/cell). a Scheme of Cre recombination product. b Electrophoretic analysis of the gDNA amplicon after Cre recombinase effect. c Levels of GABA release after ghrelin and/or 2-hydroxyglutarate incubation. d Levels of GABAT activity in the presence of ghrelin. n = 4 in triplicate. Two-way ANOVA *p < 0.05, **p < 0.01 and t test #p < 0.05
Once we had generated the neuronal model, we assessed the effect of CPT1A deletion on the release of amino acid neurotransmitters from primary cortical neurons. We observed a 35% reduction in released GABA in depolarizing conditions, due to ghrelin or CPT1A deletion (Fig. 4b). Interestingly, when we blocked the tricarboxylic acid (TCA) cycle by inhibiting isocitrate dehydrogenase with 2-hydroxyglutarate at basal conditions, GABA release dropped with a similar trend to ghrelin’s effect. Analysis of mRNA levels of the GABA shunt genes did not show any change under ghrelin treatment. However, recombinase-induced CPT1A deletion significantly reduced mRNA levels of GABAT, SSADH (Supplemental Fig. 3 a, b). In addition, when we analyzed GABAT activity, CPT1A deletion reduced GABAT activity as it did ghrelin incubation (Fig. 4c). Furthermore, CPT1A deletion also reduced the mRNA levels of GAD1 involved in the canonical pathway to generate GABA out of glutamate and the mRNA levels of the GABA transporter VGAT as it did ghrelin treatment (Supplemental Fig. 3 c, e). All these results suggest that CPT1A ablation recapitulates the ghrelin’s effect on GABA metabolism and release, which indicates that CPT1A is a mediator of ghrelin’s action.
CPT1A Deletion and Ghrelin Treatment Reduce Intracellular Citrate and α-Ketoglutarate in Primary Cortical Neurons
Since TCA blockage produced similar effects on GABA release to those observed due to ghrelin and CPT1A deletion, we assessed the effect of both on TCA cycle intermediates. Both ghrelin and CPT1A deletion promoted a significant reduction in citrate, the main TCA cycle intermediate (Fig. 5a), and an 80% reduction of α-ketoglutarate (Fig. 5b). The other intermediates that were assessed (succinate, fumarate, and malate) remained unchanged (Fig. 5c–e).
Fig. 5.

Effect of the CPT1A deletion on the Krebs cycle metabolites in primary cortical neuron culture. The levels of the components of the Krebs cycle were measured in cortical neurons from CPT1A(loxP/loxP) mice incubated with Ad-Cre-GFP or Ad-GFP (100 PFU/cell) and ghrelin: a citrate, b α-cetoglutarate, c succinate, d fumarate, e malate. f Scheme of the ghrelin action in neuronal cortical cells. OAA oxaloacetate, CIT citrate, α-KG α-ketoglutarate, SUC succinate, SSA semialdehide succinate, GLU glutamate, ssv small single vesicle. Results are represented as mean ± SEM. n = 4 in duplicate; **p < 0.01, ***p < 0.001
Discussion
The orexigenic effect of ghrelin in hypothalamus has been extensively studied [9, 48, 49]. It involves several pathways, including AMPK activation and ACC phosphorylation [50, 51], leading to reduced production of malonyl-CoA and disinhibition of CPT1A [23, 24, 27, 52–54]. This results in the production and release of orexigenic neuropeptides AgRP and NPY, and GABA neurotransmitter. Our results confirm this effect: ghrelin intraperitoneal administration induces food intake in lean mice and increases the hypothalamic acylcarnitine pool, which indicates that CPT1A is activated. Besides its orexigenic effect in hypothalamus, ghrelin may enhance motivation for food intake, since it mediates in the rewarding effect of palatable food [16–18, 55]. Systemic administration of ghrelin causes dopamine release in the nucleus accumbens, which leads to a hedonic feeling of reward that is also needed for addiction development [17, 56]. This ghrelin effect involves several brain regions like the amygdala, hippocampus, and prefrontal cortex activated by neuronal projections from the nucleus accumbens, which suggests that ghrelin has an indirect effect in these brain regions [57]. This indirect ghrelin effect could explain our results in cortical tissue where, contrary to hypothalamus, ghrelin clearly reduces CPT1A activity. However, our studies on primary culture of cortical neurons show that ghrelin has a direct effect on these neurons, reducing CPT1A and FAO. Mechanistically, GHSR1a seems to mediate such effects, as it is expressed in cortex [58]. Although not demonstrated yet in the cortex, but increasingly observed in other brain regions, the potential heterodimerization of ghrelin receptor GHSR1a with other GPCR receptors may potentially modulate specific signal transduction in discrete sets of neurons in the brain (reviewed in [59, 60]). GHSR1a heterodimerizes with at least five different GPCRs: serotonin 2C receptors attenuating orexigenic ghrelin signaling [61], dopamine D1 and D2 receptors altering dopamine signaling [62, 63] and, at peripheral level, with melanocortin 3 receptors modulating ghrelin signaling [64]. Furthermore, another important player has emerged recently: the truncated ghrelin receptor lacking transmembrane domains 6 and 7, GHSR1b. This receptor is widely expressed in many tissues where it co-localizes with the GHSR1a receptor. It has been observed that GHSR1b modulates both internalization of the active GHSR1a receptor causing the subsequent ghrelin signaling attenuation, and the ability of GHSR1a to form oligomeric complexes with other receptors, inducing changes in ghrelin-induced signaling [65]. The cellular effects of these receptor-receptor interactions remain elusive in the major regions of brain, and further investigation will show a versatile system that can transduce signals through various signaling cascades, probably depending on the cellular microenvironment that will explain the pleiotropic actions of ghrelin in different brain areas.
Cortical neurons use a wide range of neurotransmitters. We assessed the effect of ghrelin on amino acid neurotransmitters, since most cortical neurons are either GABAergic (15%) or glutamatergic (85%) [66–68]. Here, we show that ghrelin reduces GABA release at glycorrhachia-like glucose levels (5 mM) [47], while glutamate release remains unaffected. An important part of GABA production comes from TCA cycle anaplerotic pathways [69, 70]. One of the TCA anaplerotic pathways is GABA shunt. GABA shunt is highly conserved through evolution from plants to vertebrates, with varying functions among the different species [71–73]. It activates itself in pathological events such as Alzheimer’s disease [74], epileptic episodes [75, 76], and after brain ischemia [77, 78]. Physiologically, GAD activity changes have been observed during fasting in hypothalamus. Other researchers have observed changes in GABA shunt activities related to modulation of food intake: three hyperphagic rat models show increased GAD activity in VMH and two of them have increased GABA shunt activities as well [79]. These observations, together with the previous statement regarding GAD [29] and VGAT [30], indicate that GABA metabolism modulation in hypothalamus depends on the nutritional state. This modulation might be extensible to other brain areas, since caloric restriction can modulate GAD isoenzyme expression in cerebellum, superior colliculus, temporal cortex [80], and visual cortex [81]. In our study (Fig. 5f), we show that either ghrelin or a FAO reduction due to CPT1A ablation can reduce GABAergic output from cortical neurons. Ghrelin reduction of GABAergic output in cortex could explain some of the central extra-hypothalamic effects of this gastric hormone.
The functional significance of a reduction in inhibitory neurotransmitters, such as GABA, in cortical neurons under ghrelin action suggests the excitatory/inhibitory balance is adjusted within neuronal networks to function properly, and could explain animal behavior related with foraging. The anxiogenic and alertness effect needed to complement hypothalamic effects for foraging in animals [82] and to block sleep [83] are evident in a paradigm in which inhibitory neurotransmitters, such as GABA, have reduced output. In addition, the reduction in FAO and mitochondrial respiration observed in our results could be related with a neuroprotective effect associated with ghrelin and fasting [35, 84, 85]. Ghrelin’s neuroprotective effect with enhanced memory and spatial learning in mice may be closely related to mitochondrial metabolism modulation in rodents [86, 87]. At molecular level, a reduction in FAO can contribute to a reduction in the production of reactive oxygen species, protecting the cell from oxidative stress and reducing apoptosis [88, 89]. CPT1A acts as part of the mechanism by which ghrelin can modulate mitochondrial processes, since the deletion of its gene promotes deep changes in the metabolic responsiveness of the neuron to ghrelin. Consequently, CPT1A and FAO may play a role in other important ghrelin functions, such as stimulating synapsis and modulating electrical activity, which could increase cortical networks to enhance memory and cognition [41]. Further studies are needed to clarify the role of CPT1A and FAO in the molecular mechanisms involved in the various ghrelin actions.
To sum up, our data demonstrate that ghrelin produces a differential reduction in cortical GABA output, compared to hypothalamus. This reduction is produced by a drop in FAO, which produces a subsequent drop in GABA metabolism and in TCA intermediates involved in GABA production, which would explain the reduction in GABA release. This evidence suggests that the action of ghrelin on GABA is region specific within the brain, providing a basis for ghrelin differential effects in the central nervous system.
Electronic Supplementary Material
(PDF 676 kb)
Acknowledgements
We acknowledge A. Zorzano and D. Sebastián (IRB, Barcelona, Spain) for the assistance with the Seahorse analysis.
Abbreviations
- AMPK
AMP-dependent protein kinase
- CPT
Carnitine palmitoyltransferase
- FAO
Fatty acid oxidation
- GABA
γ-Aminobutyric acid
- GABAT
GABA transaminase
- GHSR
Growth hormone secretagogue receptor
- GAD
Glutamic acid decarboxylase
- SSADH
Succinate semialdehyde dehydrogenase
- TCA
Tricarboxylic acid
- VGAT
Vesicular GABA transporter
- VGLUT
Vesicular glutamate transporter
Author Contributions
DS, LH and JFM conceived the experiments, JFM, SZ, MW and AG conducted the experiments in mice and cortex cell line, MPL and CS determined GABA levels, JGV and AR determined the levels of TCA metabolites, GF and JC determined acylcarnitine levels, DS, JFM, LH, ML and NC analyzed the results. DS wrote the manuscript. All authors reviewed the manuscript.
Funding Information
This work was supported by the Ministry of Spain (MINECO) (SAF2015-71026-R and BFU2015-70454-REDT/Adipoplast to ML, SAF2013-45887-R to LH, SAF2014-52223-C2-1-R to DS, and SAF2014-52223-C2-2-R to NC, cofunded by the European Regional Development Fund [ERDF]), the Centro de Investigación Biomédica en Red de Fisiopatología de la Obesidad y la Nutrición (CIBEROBN) (Grant CB06/03/0001 to DS), the Government of Catalonia (2014SGR465 to DS), the Fundació La Marató de TV3 (201627-30 to DS), the Xunta de Galicia (2015-CP079 to ML) and the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 281854 – the ObERStress project to ML.
Compliance with Ethical Standards
All the procedures with mice were approved by the Animal Experimentation Ethics Committee of the University of Barcelona (CEEA-UB). DAAM Permit #8173 and colony management Permit C-0020 were obtained from the Government of Catalonia, according to European Directive 2010/63/EU.
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Kojima M, Hosoda H, Date Y, et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 2.Nakazato M, Murakami N, Date Y, et al. A role for ghrelin in the central regulation of feeding. Nature. 2001;409(6817):194–198. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- 3.Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86(12):5992–5995. doi: 10.1210/jcem.86.12.8111. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, Witcher DR, Luo S, Onyia JE, Hale JE. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci U S A. 2008;105(17):6320–6325. doi: 10.1073/pnas.0800708105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell. 2008;132(3):387–396. doi: 10.1016/j.cell.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 6.Hotta M, Ohwada R, Akamizu T, et al. Ghrelin increases hunger and food intake in patients with restricting-type anorexia nervosa: a pilot study. Endocr J. 2009;56(9):1119–1128. doi: 10.1507/endocrj.K09E-168. [DOI] [PubMed] [Google Scholar]
- 7.Mericq V, Cassorla F, Bowers CY, et al. Changes in appetite and body weight in response to long-term oral administration of the ghrelin agonist GHRP-2 in growth hormone deficient children. J Pediatr Endocrinol Metab. 2003;16:981–985. doi: 10.1515/JPEM.2003.16.7.981. [DOI] [PubMed] [Google Scholar]
- 8.Druce MR, Wren AM, Park AJ, Milton JE, Patterson M, Frost G, Ghatei MA, Small C, Bloom SR. Ghrelin increases food intake in obese as well as lean subjects. Int J Obes. 2005;29(9):1130–1136. doi: 10.1038/sj.ijo.0803001. [DOI] [PubMed] [Google Scholar]
- 9.Al Massadi O, López M, Tschop M, et al. Current understanding of the hypothalamic ghrelin pathways inducing appetite and adiposity. Trends Neurosci. 2017;40(3):167–180. doi: 10.1016/j.tins.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407(6806):908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 11.Nass R, Pezzoli SS, Oliveri MC, Patrie JT, Harrell FE, Jr, Clasey JL, Heymsfield SB, Bach MA, Vance ML, Thorner MO. Effects of an oral ghrelin mimetic on body composition and clinical outcomes in healthy older adults: a randomized trial. Ann Intern Med. 2008;149(9):601–611. doi: 10.7326/0003-4819-149-9-200811040-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui H, López M, Rahmouni K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat Rev Endocrinol. 2017;13(6):338–351. doi: 10.1038/nrendo.2016.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egido EM, Rodríguez-Gallardo J, Silvestre RA, Marco J. Inhibitory effect of ghrelin on insulin and pancreatic somatostatin secretion. Eur J Endocrinol. 2002;146(2):241–244. doi: 10.1530/eje.0.1460241. [DOI] [PubMed] [Google Scholar]
- 14.Tassone F, Broglio F, Destefanis S, et al. Neuroendocrine and metabolic effects of acute ghrelin administration in human obesity. J Clin Endocrinol Metab. 2003;88(11):5478–5483. doi: 10.1210/jc.2003-030564. [DOI] [PubMed] [Google Scholar]
- 15.Takaya K, Ariyasu H, Kanamoto N, et al. Ghrelin strongly stimulates growth hormone (GH) release in humans. J Clin Endocrinol Metab. 2000;85(12):4908–4911. doi: 10.1210/jcem.85.12.7167. [DOI] [PubMed] [Google Scholar]
- 16.Egecioglu E, Jerlhag E, Salomé N, et al. Ghrelin increases intake of rewarding food in rodents. Addict Biol. 2010;15(3):304–311. doi: 10.1111/j.1369-1600.2010.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jerlhag E, Egecioglu E, Dickson SL, et al. Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol. 2006;11(1):45–54. doi: 10.1111/j.1369-1600.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- 18.Perello M, Sakata I, Birnbaum S, et al. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry. 2010;67(9):880–886. doi: 10.1016/j.biopsych.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kojima M, Hosoda H, Matsuo H, Kangawa K. Ghrelin: discovery of the natural endogenous ligand for the growth hormone secretagogue receptor. Trends Endocrinol Metab. 2001;12(3):118–122. doi: 10.1016/S1043-2760(00)00362-3. [DOI] [PubMed] [Google Scholar]
- 20.Andersson U, Filipsson K, Abbott CR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279(13):12005–12008. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- 21.Vázquez MJ, González CR, Varela L, Lage R, Tovar S, Sangiao-Alvarellos S, Williams LM, Vidal-Puig A, Nogueiras R, López M, Diéguez C. Central resistin regulates hypothalamic and peripheral lipid metabolism in a nutritional-dependent fashion. Endocrinology. 2008;149(9):4534–4543. doi: 10.1210/en.2007-1708. [DOI] [PubMed] [Google Scholar]
- 22.López M, Nogueiras R, Tena-Sempere M, Diéguez C. Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat Rev Endocrinol. 2016;12(7):421–432. doi: 10.1038/nrendo.2016.67. [DOI] [PubMed] [Google Scholar]
- 23.López M, Lage R, Saha AK, et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 2008;7(5):389–399. doi: 10.1016/j.cmet.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Andrews ZB, Liu Z-W, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454(7206):846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamegai J, Tamura H, Shimizu T, et al. Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and agouti-related protein mRNA levels and body weight in rats. Diabetes. 2001;50:2438–2443. doi: 10.2337/diabetes.50.11.2438. [DOI] [PubMed] [Google Scholar]
- 26.Lage R, Vázquez MJ, Varela L, Saha AK, Vidal-Puig A, Nogueiras R, Diéguez C, López M. Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J. 2010;24(8):2670–2679. doi: 10.1096/fj.09-150672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramírez S, Martins L, Jacas J, et al. Hypothalamic ceramide levels regulated by CPT1C mediate the orexigenic effect of ghrelin. Diabetes. 2013;62(7):2329–2337. doi: 10.2337/db12-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong Q, Ye C-P, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11(9):998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dicken MS, Hughes AR, Hentges ST. Gad1 mRNA as a reliable indicator of altered GABA release from orexigenic neurons in the hypothalamus. Eur J Neurosci. 2015;42(9):2644–2653. doi: 10.1111/ejn.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mera P, Mir JF, Fabriàs G, Casas J, Costa ASH, Malandrino MI, Fernández-López JA, Remesar X, Gao S, Chohnan S, Rodríguez-Peña MS, Petry H, Asins G, Hegardt FG, Herrero L, Serra D. Long-term increased carnitine palmitoyltransferase 1A expression in ventromedial hypotalamus causes hyperphagia and alters the hypothalamic lipidomic profile. PLoS One. 2014;9(5):e97195. doi: 10.1371/journal.pone.0097195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walls AB, Waagepetersen HS, Bak LK, Schousboe A, Sonnewald U. The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res. 2015;40(2):402–409. doi: 10.1007/s11064-014-1473-1. [DOI] [PubMed] [Google Scholar]
- 32.Hearl WG, Churchich JE. Interactions between 4-aminobutyrate aminotransferase and succinic semialdehyde dehydrogenase, two mitochondrial enzymes. J Biol Chem. 1984;259(18):11459–11463. [PubMed] [Google Scholar]
- 33.Schousboe A, Hertz L, Svenneby G. Uptake and metabolism of GABA in astrocytes cultured from dissociated mouse brain hemispheres. Neurochem Res. 1977;2(2):217–229. doi: 10.1007/BF00964098. [DOI] [PubMed] [Google Scholar]
- 34.Chuang JC, Perello M, Sakata I, et al. Ghrelin mediates stress-induced food-reward behavior in mice. J Clin Invest. 2011;121(7):2684–2692. doi: 10.1172/JCI57660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews ZB. The extra-hypothalamic actions of ghrelin on neuronal function. Trends Neurosci. 2011;34(1):31–40. doi: 10.1016/j.tins.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Lutter M, Sakata I, Osborne-Lawrence S, et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008;11(7):752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tolle V, Bassant MH, Zizzari P, Poindessous-Jazat F, Tomasetto C, Epelbaum J, Bluet-Pajot MT. Ultradian rhythmicity of ghrelin secretion in relation with gh, feeding behavior, and sleep-wake patterns in rats. Endocrinology. 2002;143(4):1353–1361. doi: 10.1210/endo.143.4.8712. [DOI] [PubMed] [Google Scholar]
- 38.Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494(3):528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mani BK, Walker AK, Lopez Soto EJ, Raingo J, Lee CE, Perelló M, Andrews ZB, Zigman JM. Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J Comp Neurol. 2014;522(16):3644–3666. doi: 10.1002/cne.23627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoyanova II. Ghrelin: a link between aging, metabolism and neurodegenerative disorders. Neurobiol Dis. 2014;72:72–83. doi: 10.1016/j.nbd.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 41.Stoyanova II, le Feber J. Ghrelin accelerates synapse formation and activity development in cultured cortical networks. BMC Neurosci. 2014;15(1):49. doi: 10.1186/1471-2202-15-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004;101(13):4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solà C, Cristòfol R, Suñol C, Sanfeli C. Primary cultures for neurotoxicity testing. In: Aschner M, editor. Cell Cult. Tech. New York: Springer; 2011. pp. 281–296. [Google Scholar]
- 44.Ribes A, Riudor E, Briones P, et al. Significance of bound glutarate in the diagnosis of glutaric aciduria type I. J Inherit Metab Dis. 1992;15:367–370. doi: 10.1007/BF02435978. [DOI] [PubMed] [Google Scholar]
- 45.Serra-Pérez A, Planas AM, Núñez-O’Mara A, et al. Extended ischemia prevents HIF1alpha degradation at reoxygenation by impairing prolyl-hydroxylation: role of Krebs cycle metabolites. J Biol Chem. 2010;285(24):18217–18224. doi: 10.1074/jbc.M110.101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka K, West-Dull A, Hine DG, Lynn TB, Lowe T. Gas-chromatographic method of analysis for urinary organic acids. II. Description of the procedure, and its application to diagnosis of patients with organic acidurias. Clin Chem. 1980;26(13):1847–1853. [PubMed] [Google Scholar]
- 47.Leen WG, Willemsen MA, Wevers RA, Verbeek MM. Cerebrospinal fluid glucose and lactate: age-specific reference values and implications for clinical practice. PLoS One. 2012;7(8):e42745. doi: 10.1371/journal.pone.0042745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrews ZB. Central mechanisms involved in the orexigenic actions of ghrelin. Peptides. 2011;32(11):2248–2255. doi: 10.1016/j.peptides.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 49.Müller TD, Nogueiras R, Andermann ML, et al. Ghrelin. Mol Metab. 2015;4(6):437–460. doi: 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.López M, Lelliott CJ, Vidal-Puig A. Hypothalamic fatty acid metabolism: a housekeeping pathway that regulates food intake. BioEssays. 2007;29(3):248–261. doi: 10.1002/bies.20539. [DOI] [PubMed] [Google Scholar]
- 51.Kohno D, Sone H, Minokoshi Y, Yada T. Ghrelin raises [Ca2+]i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem Biophys Res Commun. 2008;366(2):388–392. doi: 10.1016/j.bbrc.2007.11.166. [DOI] [PubMed] [Google Scholar]
- 52.Gao S, Serra D, Keung W, et al. Important role of ventromedial hypothalamic carnitine palmitoyltransferase-1a in the control of food intake. Am J Physiol Endocrinol Metab. 2013;305(3):E336–E347. doi: 10.1152/ajpendo.00168.2013. [DOI] [PubMed] [Google Scholar]
- 53.Obici S, Feng Z, Arduini A, et al. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med. 2003;9:756–761. doi: 10.1038/nm873. [DOI] [PubMed] [Google Scholar]
- 54.Serra D, Mera P, Malandrino MI, Mir JF, Herrero L. Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal. 2012;19(3):269–284. doi: 10.1089/ars.2012.4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skibicka KP, Hansson C, Alvarez-Crespo M, et al. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience. 2011;180:129–137. doi: 10.1016/j.neuroscience.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 56.Ross S, Peselow E. The neurobiology of addictive disorders. Clin Neuropharmacol. 2009;32(5):269–276. doi: 10.1097/WNF.0b013e3181a9163c. [DOI] [PubMed] [Google Scholar]
- 57.Goldstone AP, Prechtl CG, Scholtz S, Miras AD, Chhina N, Durighel G, Deliran SS, Beckmann C, Ghatei MA, Ashby DR, Waldman AD, Gaylinn BD, Thorner MO, Frost GS, Bloom SR, Bell JD. Ghrelin mimics fasting to enhance human hedonic, orbitofrontal cortex, and hippocampal responses to food. Am J Clin Nutr. 2014;99(6):1319–1330. doi: 10.3945/ajcn.113.075291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jerlhag E, Janson AC, Waters S, Engel JA. Concomitant release of ventral tegmental acetylcholine and accumbal dopamine by ghrelin in rats. PLoS One. 2012;7(11):e49557. doi: 10.1371/journal.pone.0049557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edwards A, Abizaid A (2017) Clarifying the ghrelin system’s ability to regulate feeding behaviours despite enigmatic spatial separation of the GHSR and its endogenous ligand. Int J Mol Sci 18(4). 10.3390/ijms18040859 [DOI] [PMC free article] [PubMed]
- 60.Howick K, Griffin BT, Cryan JF, Schellekens H (2017) From belly to brain: targeting the ghrelin receptor in appetite and food intake regulation. Int J Mol Sci 18(2). 10.3390/ijms18020273 [DOI] [PMC free article] [PubMed]
- 61.Schellekens H, Van Oeffelen WEPA, Dinan TG, Cryan JF. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. J Biol Chem. 2013;288(1):181–191. doi: 10.1074/jbc.M112.382473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kern A, Mavrikaki M, Ullrich C, et al. Hippocampal dopamine/DRD1 signaling dependent on the ghrelin receptor. Cell. 2015;163(5):1176–1190. doi: 10.1016/j.cell.2015.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kern A, Albarran-zeckler R, Walsh HE, Smith RG. NIH Public Access. 2013;73:317–332. [Google Scholar]
- 64.Rediger A, Piechowski CL, Yi CX, Tarnow P, Strotmann R, Grüters A, Krude H, Schöneberg T, Tschöp MH, Kleinau G, Biebermann H. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem. 2011;286(45):39623–39631. doi: 10.1074/jbc.M111.287607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Navarro G, Aguinaga D, Angelats E, et al. A significant role of the truncated ghrelin receptor GHS-R1b in ghrelin-induced signaling in neurons. J Biol Chem. 2016;291(25):13048–13062. doi: 10.1074/jbc.M116.715144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21(10):1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 67.Braitenberg V. Corticonics: neural circuits of the cerebral cortex. Trends Neurosci. 1992;15(4):156–157. doi: 10.1016/0166-2236(92)90361-B. [DOI] [Google Scholar]
- 68.Niciu MJ, Kelmendi B, Sanacora G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol Biochem Behav. 2012;100(4):656–664. doi: 10.1016/j.pbb.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sonnewald U. Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J Neurochem. 2014;131(4):399–406. doi: 10.1111/jnc.12812. [DOI] [PubMed] [Google Scholar]
- 70.Hassel B Carboxylation and anaplerosis in neurons and glia. Mol Neurobiol 22:21–40. 10.1385/MN:22:1-3:021 [DOI] [PubMed]
- 71.Fait A, Fromm H, Walter D, et al. Highway or byway: the metabolic role of the GABA shunt in plants. Trends Plant Sci. 2008;13(1):14–19. doi: 10.1016/j.tplants.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 72.Michaeli S, Fait A, Lagor K, et al. A mitochondrial GABA permease connects the GABA shunt and the TCA cycle, and is essential for normal carbon metabolism. Plant J. 2011;67(3):485–498. doi: 10.1111/j.1365-313X.2011.04612.x. [DOI] [PubMed] [Google Scholar]
- 73.Mamelak M. Sporadic Alzheimer’s disease: the starving brain. J Alzheimers Dis. 2012;31(3):459–474. doi: 10.3233/JAD-2012-120370. [DOI] [PubMed] [Google Scholar]
- 74.Lanctôt KL, Herrmann N, Mazzotta P, et al. GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can J Psychiatr. 2004;49:439–453. doi: 10.1177/070674370404900705. [DOI] [PubMed] [Google Scholar]
- 75.Kang T-C, Park S-K, Hwang IK, An SJ, Choi SY, Kwon OS, Baek NI, Lee HY, Won MH. The altered expression of GABA shunt enzymes in the gerbil hippocampus before and after seizure generation. Neurochem Int. 2003;42(3):239–249. doi: 10.1016/S0197-0186(02)00079-7. [DOI] [PubMed] [Google Scholar]
- 76.Yogeeswari P, Sriram D, Vaigundaragavendran J. The GABA shunt: an attractive and potential therapeutic target in the treatment of epileptic disorders. Curr Drug Metab. 2005;6(2):127–139. doi: 10.2174/1389200053586073. [DOI] [PubMed] [Google Scholar]
- 77.Kang T-C, Park S-K, Hwang I-K, An SJ, Choi SY, Cho SW, Won MH. Spatial and temporal alterations in the GABA shunt in the gerbil hippocampus following transient ischemia. Brain Res. 2002;944(1-2):10–18. doi: 10.1016/S0006-8993(02)02596-9. [DOI] [PubMed] [Google Scholar]
- 78.Seo JY, Lee CH, Cho JH, Choi JH, Yoo KY, Kim DW, Park OK, Li H, Choi SY, Hwang IK, Won MH. Neuroprotection of ebselen against ischemia/reperfusion injury involves GABA shunt enzymes. J Neurol Sci. 2009;285(1-2):88–94. doi: 10.1016/j.jns.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 79.Beverly JL, Martin RJ. Increased GABA shunt activity in VMN of three hyperphagic rat models. Am J Phys. 1989;256:R1225–R1231. doi: 10.1152/ajpregu.1989.256.6.R1225. [DOI] [PubMed] [Google Scholar]
- 80.Cheng CM, Hicks K, Wang J, et al. Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J Neurosci Res. 2004;77(2):270–276. doi: 10.1002/jnr.20144. [DOI] [PubMed] [Google Scholar]
- 81.Spolidoro M, Baroncelli L, Putignano E, et al. Food restriction enhances visual cortex plasticity in adulthood. Nat Commun. 2011;2:320. doi: 10.1038/ncomms1323. [DOI] [PubMed] [Google Scholar]
- 82.Thomas MA, Ryu V, Bartness TJ. Central ghrelin increases food foraging/hoarding that is blocked by GHSR antagonism and attenuates hypothalamic paraventricular nucleus neuronal activation. Am J Physiol Regul Integr Comp Physiol. 2016;310(3):R275–R285. doi: 10.1152/ajpregu.00216.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Szentirmai E, Hajdu I, Obal F, Krueger JM. Ghrelin-induced sleep responses in ad libitum fed and food-restricted rats. Brain Res. 2006;1088(1):131–140. doi: 10.1016/j.brainres.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 84.Gahete MD, Córdoba-Chacón J, Kineman RD, et al. Role of ghrelin system in neuroprotection and cognitive functions: implications in Alzheimer’s disease. Peptides. 2011;32(11):2225–2228. doi: 10.1016/j.peptides.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bayliss JA, Lemus MB, Stark R, Santos VV, Thompson A, Rees DJ, Galic S, Elsworth JD, Kemp BE, Davies JS, Andrews ZB. Ghrelin-AMPK signaling mediates the neuroprotective effects of calorie restriction in Parkinson’s disease. J Neurosci. 2016;36(10):3049–3063. doi: 10.1523/JNEUROSCI.4373-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin RS, Xu L, Yamada KA, Sleeman MW, Tschöp MH, Horvath TL. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9(3):381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 87.Davis JF, Choi DL, Clegg DJ, Benoit SC. Signaling through the ghrelin receptor modulates hippocampal function and meal anticipation in mice. Physiol Behav. 2011;103(1):39–43. doi: 10.1016/j.physbeh.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andrews ZB, Erion D, Beiler R, et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29(45):14057–14065. doi: 10.1523/JNEUROSCI.3890-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chung H, Seo S, Moon M, Park S. Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3β and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J Endocrinol. 2008;198(3):511–521. doi: 10.1677/JOE-08-0160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 676 kb)