

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Development of thrombocytopenia in HIT is modulated by the (re)distribution of PF4 among hematopoietic and endothelial cell surfaces.
Redistribution of PF4 from platelets to other hematopoietic cells may limit thrombocytopenia but promote prothrombotic processes in HIT.
Abstract
Heparin-induced thrombocytopenia (HIT) is a prothrombotic disorder initiated by antibodies to platelet factor 4 (PF4)/heparin complexes. PF4 released from platelets binds to surface glycosaminoglycans on hematopoietic and vascular cells that are heterogenous in composition and differ in affinity for PF4. PF4 binds to monocytes with higher affinity than to platelets, and depletion of monocytes exacerbates thrombocytopenia in a murine HIT model. Here we show that the expression of PF4 on platelets and development of thrombocytopenia are modulated by the (re)distribution of PF4 among hematopoietic and endothelial cell surfaces. Binding of PF4 to platelets in whole blood in vitro varies inversely with the white cell count, likely because of the greater affinity of monocytes for PF4. In mice, monocyte depletion increased binding of PF4 to platelets by two- to three-fold. Induction of HIT in mice caused a transient >80-fold increase in binding of HIT antibody to monocytes vs 3.5-fold increase to platelets and rapid transient monocytopenia. Normalization of monocyte counts preceded the return in platelet counts. Exposure of blood to endothelial cells also depletes PF4 from platelet surfaces. These studies demonstrate a dynamic interchange of surface-bound PF4 among hematopoetic and vascular cells that may limit thrombocytopenia at the expense of promoting prothrombotic processes in HIT.
Visual Abstract
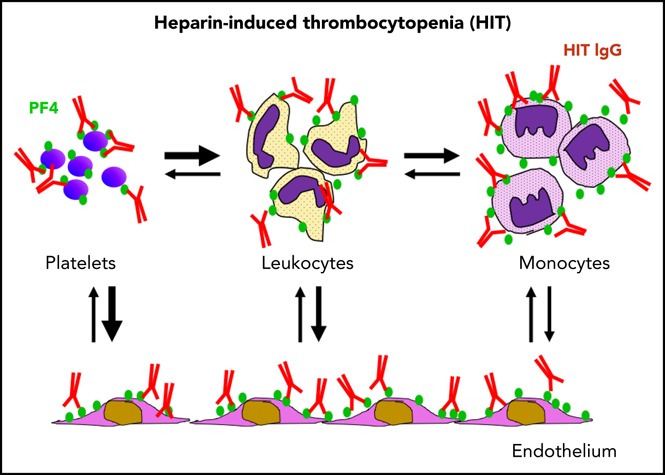
Introduction
Heparin-induced thrombocytopenia (HIT) is an iatrogenic disorder characterized by thrombocytopenia and a high risk of limb- or lifethreatening thrombosis.1-3 Pathogenic HIT antibodies react with a multimolecular complex of platelet factor 4 (PF4) and polyanions such as heparin or cell-surface glycosaminoglycans.4,5 Immuglobulin G–containing immune complexes involving PF4 bound to membrane chondroitin sulfate (CS) activate platelets through FcγIIA receptors,5,6 contributing to the risk of thrombosis. However, the intense generation of thrombin in HIT devolves from immune complexes that also form on the surface of monocytes,7 neutrophils,8 and endothelial cells9 that express glycosaminoglycans with higher affinity than platelet CS, inducing expression of tissue factor, release of microparticles and formation of neutrophil extracellular traps, and initiating diverse procoagulant pathways.10-15
The way in which this diversity of PF4 binding sites affects immune complex formation on platelets, the development of and recovery from thrombocytopenia, and the propensity for thrombosis is only partially understood. Depletion of peripheral monocytes using clodronate liposomes or inhibition of monocyte function using gadolinium chloride markedly attenuated thrombus development in the passive immunization murine model of HIT. However, unexpectedly, monocyte depletion also exacerbated the severity of thrombocytopenia in the same mice.7 This argues against platelet activation as the sole cause of thrombocytopenia in this model. Here we tested an alternative explanation for these findings involving the dynamic partitioning of PF4, and thereby HIT antigen, among hematopoetic and vascular cells, including platelets. We find that redistribution of PF4 away from platelets to monocytes and endothelium may mitigate the severity of thrombocytopenia at the cost of leaving sufficient platelets in the circulation to participate in the prothrombotic pathways activated on diverse cell types in the circulation. The clinical implications of these findings are discussed.
Materials and methods
In vitro distribution of PF4-FITC in whole blood
Human blood from healthy, aspirin-free volunteers was collected in 0.129 M of sodium citrate (v/v, 10:1) supplemented with 300 nM of carbacyclin. Mouse blood was drawn from the inferior vena cava into a syringe containing 1:7 (v/v) citrate-dextrose solution (ACD; Sigma-Aldrich) and immediately diluted 1:1 (v/v) in modified Tyrode’s buffer (134 mM of NaCl, 3 mM of KCl, 0.3 mM of NaH2PO4, 2 mM of MgCl2, 5 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 5 mM of glucose, 12 mM of NaHCO3, and 0.1% bovine serum albumin; Sigma A7030) containing carbacyclin (300-nM final concentration). Samples of human or Cxcl4−/− (PF4null) mouse16 whole blood (100 µL) were incubated with PF4–fluorescein isothiocyanate (FITC) at the indicated concentrations in the presence of species-specific anti-CD41 or anti-CD45 monoclonal antibodies (mAbs), and anti-hCD14 or anti-mCD115 mAbs to identify cell subsets, for 45 minutes at room temperature. The results of initial studies showed that binding of PF4 to platelets and red blood cells (RBCs) in whole blood was affected by incubation time but not by fixation. Thereafter, cells were fixed in 2% paraformaldehyde fixation buffer (BD Bioscience), and surface-bound PF4-FITC was analyzed by flow cytometry (LSRFortessa; BD Bioscience). Individual cell types were identified based on binding the population-specific marker and forward scatter (platelets CD41+CD45−, monocytes CD45brightCD115+CD41−, lymphocytes CD45bright, CD115−CD41−, neutrophils CD45dimCD115−CD41−, RBCs CD45−CD115−CD41−FSChigh). In some experiments, the DNA dye Hoechst 33342 (10-µg/mL final concentration; Thermo Scientific) was used to gate nucleated cells. The total binding of PF4 was expressed as the geometric mean fluorescence intensity (MFI) of FITC in the presence of a specific amount of added PF4 after subtracting the background fluorescence of each cell type measured in the absence of PF4. To normalize the binding of PF4 to each cell type with different background fluorescence, we used the distribution resolution metric (Rd), which was defined as the difference in the geometric means of 2 populations, MFIPF4x and MFIctrl (MFI in the presence of PF4 at concentration x and control), divided by the sum of the robust standard deviations of the distributions (rSDPF4x + rSDctrl) as follows: Rd = (MFIPF4x − MFIctrl)/(rSDPF4x + rSDctrl). The distribution resolution metric measures the degree of separation between distribution curves of fluorescence intensities, incorporates measurement noise, and is independent of the type of detector being used or its relative gain settings.17 It is more relevant for comparison of populations with different background fluorescences and fluorescent variabilities (spread of the intensities) than the ratio of MFIPF4x/MFIctrl and was used for comparison of cell populations of different sizes.
In some experiments, peripheral blood monocytes were depleted in PF4null mice using clodronate liposomes7 before blood draw. Total white blood cells (WBCs) were isolated from PF4null mice by positive selection using biotin-labeled anti-mCD45 antibody and CELLection Biotin Binder Kit (ThermoScientific). Platelet-rich plasma (PRP) was prepared from PF4null mice as described,14 various numbers of WBCs were added to the PRP, and the final WBC and platelet numbers were calculated using Hemavet 950 (Drew Scientific).
Redistribution of endogenous PF4 released from activated platelet in vitro
Human blood was collected from healthy, aspirin-free volunteers in 0.129 M of sodium citrate (v/v, 10:1). After blocking Fc receptors with mouse serum (v/l, 10:1), CD41a-phycoerythrin, CD14–Alexa 647, and Hoechst 33342 were added and samples were activated with the protease-activated receptor-1 selective agonist (TFLLR-NH2; Sigma) at a final concentration of 10 µM for 15 minutes at 37°C. Released PF4 bound to the surface of individual cells was detected by Alexa 488–labeled polyclonal anti–heparin PF4 (hPF4), Alexa 488–labeled RTO (10 µg/mL), or Alexa 488–labeled KKO (10 µg/mL). In some experiments, heparin was added at the indicated concentrations at the end of 15 minutes of incubation.
Murine studies
Passive immunization studies to simulate HIT were carried out using hPF4- and FcγRIIA-double transgenic mice as described.9 Mice were injected IV with 1 mg/kg of Alexa 488–labeled KKO or TRA at the 0 time point. Retroorbital blood samples drawn at the indicated time points were stained with anti-mCD45, anti-mCD115, and anti-mCD41. KKO binding to peripheral blood cells was estimated by flow cytometry. Individual cell types were identified based on binding the population-specific markers and forward scatter (platelets CD41+CD45−, monocytes CD45brightCD115+CD41−, lymphocytes CD45bright, CD115−CD41−, neutrophil CD45dimCD115−CD41−, and RBC CD45−CD115−CD41−FSChigh). KKO binding to each subpopulation was estimated as the increase in geometric MFI of Alexa 488–KKO over the matched Alexa 488–TRA sample. Absolute platelet and WBC counts were obtained using the Hemavet 950, and absolute numbers of monocytes, lymphocytes, and neutrophils were calculated based on the proportion of these populations of CD45+ events estimated by flow cytometry.
Endothelialized microfluidic channel
A well-plate microfluidic device (Fluxion Biosciences) was used to analyze surface distribution of PF4 on various blood cell types under flow in endothelial-lined channels. Human umbilical vein endothelial cells (HUVECs; Lonza) at passage 3-4, at a concentration of 106 cells/mL, were seeded onto fibronectin-coated (1 µg/mL; Sigma) channels of a low shear stress 48-well Bioflux plate at a shear stress of 2 dyn/cm2, allowed to adhere, and cultured at 37°C at 5% CO2 in endothelial cell growth medium (EGM; BulletKit; Lonza) until confluence throughout the channel was obtained.9
To saturate the endothelial cells with PF4, some of the HUVEC-coated channels were preincubated with hPF4 resuspended in EGM flowed through the channel at a concentration of either 0 or 100 µg/mL at 2 dyn/cm2 for 1 hour at 37°C. Unbound PF4 was washed out from the channels with Hank’s Balanced Salt Solution (Gibco) for 2 minutes at 37°C at a shear stress of 3 dyn/cm2 just before perfusion with blood samples. PF4null blood samples were then perfused through the channels at a shear stress of 7 dyn/cm2 for 10 minutes.
To elicit a targeted photochemical injury to the HUVECs within the microfluidic channels, hematoporphyrin dihydrochloride (50 µg/mL in EGM; Santa Cruz Biotechnology), which generates reactive oxygen species upon exposure to blue light (metal halide fluorescence light source through the FITC filter set irradiation; 50-microsecond exposure at 4 spots evenly distributed along the microfluidic channel), was perfused over the endothelial cells. Uninjured channels exposed to the light exposured to EGM without hematoporhyrin served as controls. Whole blood samples from PF4null mice containing 10 or 100 µg of hPF4/mL were then perfused through the channels at 7 dyn/cm2 for 10 minutes at 37°C. The channels were then washed with EGM at 7 dyn/cm2 for 40 seconds, and staining for cell-bound PF4 was measured with Alexa 488–KKO and for human von Willebrand factor (VWF) with rabbit anti-human VWF antibody (DAKO) and Alexa 647–labeled goat anti-rabbit immunoglobulin G (Life Technologies).
Statistical analysis
Differences between 2 groups were compared using a 2-sided Student t test or a Mann-Whitney U test. Differences between >2 groups were determined using a 2-way analysis of variance and Bonferroni post hoc multiple comparison tests. Statistical analyses were performed using Microsoft Excel 2011 and GraphPad Prism 7.0 (GraphPad Software). Differences were considered statistically significant when P values were ≤.05.
Institutional approval
Samples were obtained under a protocol approved by the institutional review board for studies involving human subjects at the Children’s Hospital of Philadelphia. All animal protocols were approved by the Institutional Animal Care and Use Committee at Children’s Hospital of Philadelphia.
Results
Partitioning of exogenous hPF4 in whole blood in vitro
We and others hypothesized previously that preferential binding of PF4, and the resultant formation of HIT antigen, on monocytes and neutrophils initiates prothrombotic processes while sparing sufficient platelets to participate in thrombosis.8,10,13,14 To test this concept, we first measured the partitioning of FITC-labeled hPF4 to each cell type in whole blood using flow cytometry (Figure 1). PF4null murine blood (Figure 1A,C) was used to avoid PF4 release from platelets that might compete with binding of hPF4. Monocytes bound the most PF4 per cell with a steep increase in binding at an external concentration of 2 µg/mL (Figure 1A). Binding of PF4 to neutrophils and lymphocytes followed a similar dose-dependent pattern, but maximal surface binding was ∼3-fold less than that observed on monocytes. Platelets bound ∼100-fold less PF4 than monocytes at low PF4 concentrations and up to 50-fold less at saturation. More PF4 bound to monocytes than to other cell types when the increase of fluorescent intensity at increasing PF4 concentration was corrected for cell size and background fluorescence as described in “Materials and methods” (Figure 1C). Similar results were seen in human whole blood, a setting in which it is likely that unlabeled hPF4 was being released concurrently from the platelets (Figure 1B,D).
Figure 1.

In vitro distribution of cell-bound PF4 in whole mouse or human blood. (A-D) The ordinate depicts the MFI generated by FITC-PF4 bound to the indicated cell types. (A,C) PF4null murine blood. (B,D) Human blood. (A-B) MFI per cell after background subtraction. (C-D) Similar to panels A and B, but results are expressed as the increase in fluorescence intensity using Rd. Mean ± 1 standard deviation is shown in each. N = 5. Monocytes (mono), green squares; neutrophils (neu), blue triangles; lymphocytes (lymph), inverted purple triangles; RBCs, blue Xs; and platelets (plt), red circles.
Surprisingly, when calculated per total surface area of all cells present in the sample, the RBC pool contained the highest amount of bound PF4 at all concentrations tested, although the surface-bound PF4 level per cell was quite low (supplemental Figure 1, available on the Blood Web site). This suggests that RBCs may serve as an important, low-affinity reservoir for PF4 in the circulation. Platelets comprised the next-largest overall pool of bound PF4. The overall amount of PF4 bound to the platelet pool increased at a high concentration of added PF4 (150 µg/mL), compared with a lower PF4 concentration (2 µg/mL). Similar results were seen when human blood was studied in place of murine blood (supplemental Figure 1). This increased binding to platelets at high PF4 is consistent with the known higher affinity of PF4 for the more complex mixture of GAG sidechains expressed on leukocytes vs lower-affinity binding to platelet CS, likely leading to saturation of the leukocyte compartment at high external concentrations of ligand.
Partitioning of endogenous hPF4 released from platelets in whole blood in vitro
We next asked whether a similar pattern of binding is observed when PF4 is released from activated platelets. PF4 released from platelets activated by the selective PAR-1 agonist TFLLR-NH2 binds predominantly to monocytes (Figure 2A). This was followed by binding to neutrophils and lymphocytes, with the least binding to platelets, results consistent with our observations on the distribution of exogenous PF4 (Figure 1D). Relative total surface PF4 was measured using the anti-PF4 mAb RTO, binding of which is not influenced by heparin or GAGs.18,19 Although RTO recognizes the epitope on PF4 monomer in solution,19 it does bind to PF4 complexes on platelet surfaces.20 In our experiments, the binding of RTO tracked with that seen with polyclonal anti-PF4 antibody (supplemental Figure 2). We therefore used RTO as a marker for total PF4 binding. Surface HIT antigen was detected by the binding of mAb KKO, as shown previously.5 Binding of KKO to the various cell types followed a relative distribution similar to that of surface total PF4. Monocytes bound the most KKO and platelets the least (Figure 2A, right), although the latter bound more PF4 in total (Figure 2A, left), indicating less effective HIT antigen assembly on platelet membranes. A similar distribution was seen when directly labeled PF4 was examined. We tracked KKO binding in subsequent studies to assess the impact of intercellular redistribution of HIT antigenicity.
Figure 2.

In vitro distribution of endogenous hPF4 released from activated platelets in whole human blood in the absence and presence of heparin. TFLLR-NH2 was added to whole human blood. Total cell-surface–bound PF4 on RBCs, platelets (plts), monocytes (mono), neutrophils (neu), and lymphocytes (lymph) was detected using RTO and HIT antigenicity using KKO. (B) Effect of increasing amounts of heparin (0, 0.1, 0.4, and 100 U/mL) on RTO and KKO binding. (A,B) Data are expressed as increase of MFI after stimulation compared with unstimulated samples using distribution resolution metric (RD), where value 0 means complete overlap and value ≥1 means complete separation of the distribution curves; values >0 mean increase in binding. Mean ± 1 standard deviation of the change in surface-bound PF4 against a hypothetical value of 0 for no change is shown. (A) N = 6. (B) N = 3. Distinct experiments performed in duplicate. *P < .05, ***P < .0005, ****P < .0001 using 2-way analysis of variance with Dunnett’s multiple comparisons test.
Influence of heparin on intercellular distribution of PF4
We then examined the effect of heparin on the intercellular distribution of PF4 at various levels of heparin, including at a clinically relevant concentration (0.4 U/mL).21 Heparin decreased binding of PF4 to all cell surfaces in a dose-dependent manner (Figure 2B, left). Low concentrations of heparin (0.1 U/mL) increased binding of KKO to all cell types, as previously reported22 (Figure 2B, right), consistent with a redistribution of cell-surface PF4 into oligomeric complexes,7 whereas higher concentrations (0.4 and 100 U/mL) depleted cell-surface antigen expression. However, even at the highest heparin concentration tested (100 U/mL), appreciable PF4 remained bound onto monocytes, neutrophils, and platelets (Figure 2B, left).
Influence of WBCs on platelet-surface PF4 levels
We then postulated that the effect of WBCs was similar to adding heparin to PF4-coated platelets (ie, the more WBCs present, the fewer PF4 and consequently fewer HIT antigenic complexes formed on the surface of platelets). Conversely, the lower the WBC count, the more PF4 and HIT antibody bound to the residual platelets, and the more rapid the resultant clearance of the most heavily sensitized platelets from the circulation. To test this hypothesis, we examined whether at higher WBC/platelet ratios, fewer PF4 and consequently fewer HIT antigenic complexes form on the surface of the platelets. To simulate this situation, we isolated PRP from PF4null mice to which increasing numbers of WBCs were added above and below the average of ∼3.2 WBCs to 103 platelets observed in vivo (Figure 3A). The amount of HIT antigen bound per platelet varied inversely with the WBC/platelet ratio. As an independent approach, whole blood from PF4null mice that had or had not been pretreated with clodronate-containing liposomes to eliminate circulating monocytes7 was exposed to PF4. Platelets in monocyte-depleted samples expressed more surface-bound HIT antigen than in samples containing monocytes (Figure 3B), an effect that was especially evident at lower concentrations of added PF4. This difference decreased at high PF4 concentrations that likely saturate the monocyte surface. These data provide insight into our published observation that monocyte-depleted mice develop more severe thrombocytopenia after induction of HIT than control mice7; in the absence of monocytes, their platelets would be expected to express more PF4 and therefore HIT antigen. Consequently, more HIT antibodies can bind, enhancing platelet clearance.
Figure 3.

Effect of the WBC/platelet ratio on cell-surface–bound PF4 and HIT antigenicity in vitro. (A) PRP was prepared from PF4null mouse blood. Recombinant hPF4 (100 µg/mL) and varying numbers of isolated total WBCs were added. PF4 bound to the surface of platelets was measured using KKO. Mean ± 1 standard deviation is shown. N = 3 per arm, each in duplicate; P < .05 compared with baseline value (no WBC cells added, dashed line) using Wilcoxon matched-pairs test. (B) Monocytes were depleted in PF4null mice using clodronate liposomes. The indicated amounts of PF4 were added to monocyte-depleted or control nondepleted PF4null blood, and platelet-bound PF4 was measured based on binding of KKO to platelets by flow cytometry. Relative binding in the monocyte-depleted blood compared with the nondepleted blood is shown at the indicated PF4 concentrations with 1 = no difference (dashed line). Mean ± 1 standard deviation is shown. N = 3 mice per arm. P < .05 compared with baseline value (nondepleted blood) using Wilcoxon matched-pairs test.
PF4 binds preferentially to monocytes in vivo
We then asked whether similar changes in the distribution of HIT antigen develop in vivo using the passive immunization murine model of HIT.5,23 Within 15 minutes of infusing KKO, monocytes developed a >80-fold increase in the surface binding of KKO over baseline (Figure 4A). By 4 hours, this increase dropped to ∼50% of the peak value and by 48 hours to ∼25% (Figure 4A). This contrasts with a ∼3.5-fold increase in peak KKO binding to platelets at 15 minutes that slowly disipated over the ensuing 48 hours. PF4 binding to neutrophils peaked at 1 hour and showed an intermediate ∼12-fold increase that remained almost stable over the next 2 days. Monocytopenia to 25% of baseline by 2 hours postinfusion and a more modest thrombocytopenia to 50% of baseline developed concurrently (Figure 4B). Monocytes returned to normal followed by monocytosis by 24 to 48 hours, replenishing competing binding sites for PF4 followed by a gradual return of platelet counts to normal.
Figure 4.

Changes in circulating hematopoetic cells in the passive immunization murine model of HIT. HIT was induced in hPF4+/FcγRIIA+ mice by IV injecting 1 mg/kg of KKO. Pre refers to blood levels drawn before KKO infusion for that cell line. (A) Surface-bound KKO was detected by flow cytometry and then normalized to preinfusion level. (B) Same as panel A, but for blood cell counts. Dashed horizontal line indicates preinfusion level for comparison. Basal pre-KKO absolute platelet count was 948 ± 180 × 103/µL, and absolute monocyte count was 0.92 ± 0.24 × 103/µL. Mean ± 1 standard deviation is shown. N = 3 (24-48 hours) to 6 (0-4 hour time points). *P < .01, **P < .001, ***P < .0001 compared with baseline pre value using analysis of variance.
Redistribution of PF4 between hematopoetic and vascular endothelial cells
PF4 also binds to vascular endothelial cells, which express a complex surface of densely packed glycosaminoglycans within the glycocalyx.24 Binding of PF4 and HIT-associated antibodies to the endothelium leads to cell activation25,26 and a prothrombotic state.9 Therefore, we analyzed how endothelial cells affect PF4 binding to circulating platelets and WBCs under flow conditions. PF4null murine blood supplemented with hPF4 (0-100 µg/mL) was perfused through microfluidic channels coated with HUVECs that had or had not been preincubated with a near-saturating concentration of hPF4 (100 µg/mL; Figure 5). When whole blood was perfused through PF4-free endothelial-lined channels, PF4 was displaced more effectively from platelets than from WBCs to the endothelium (Figure 5A). When the endothelial surface was preexposed to 100 µg/mL of PF4, both platelets and WBCs in PF4-free whole blood acquired surface PF4 as they transited through the channels, whereas platelets, and to a lesser extent WBCs, in blood preexposed to PF4 lost PF4 to the endothelium (Figure 5B). These data show that the endothelium participates in a back-and-forth interchange of PF4 with circulating hematopoetic cells even within the short (∼20 mm) length of channels and ∼8-second transit time.
Figure 5.

Effect of endothelial cells on binding of PF4 to platelets and WBCs. Whole blood from PF4null mice was flowed through a microfluidic channel coated with HUVECs that had been either previously unexposed to PF4 (A) or previously incubated with 100 µg/mL of hPF4 (B). (A-B) The infused whole blood was also preincubated with 0, 10, or 100 µg/mL of PF4, as indicated. Shown is relative cell-surface HIT antigenicity detected using KKO on platelets (red circles) or WBCs (yellow squares) after traversing the channel compared with results before entering the channel. N = 6 separate studies per arm, each in duplicate. *P < .01, **P < .001 with values determined using analysis of variance of calculated values against a hypothetical value 1 = no change (dashed horizontal line).
Most patients who develop HIT have underlying vascular disorder and/or evidence of inflammation.27 Therefore, we asked whether endothelial injury would alter the observations in Figure 5. HUVECs were subjected to hematoporphyrin photochemical injury, which affects the glycocalyx and leads to VWF release.9 Endothelial injury within the microfluidic channels was affirmed by release of VWF (Figure 6A). When channels lined by endothelial cells were perfused with whole blood preexposed to PF4, significantly higher levels of KKO bound to injured HUVECs than to resting HUVECs (Figure 6B), suggesting that the distribution of PF4 on cell surfaces may involve the vascular lining and its state of activation as well as its binding to intravascular cells.
Figure 6.

Endothelial activation enhances PF4 binding. HUVECs lining microfluidic wells were injured with hematoporphyrin and perfused with PF4null murine blood supplemented with 10 or 100 µg/mL of hPF4 as indicated. (A) VWF exposure on the surface of endothelial cells showing the effects of the photochemical injury using binding of a labeled anti-VWF antibody. —, no injury; +++, photochemical injury. N = 4 separate experiments, each performed in duplicate; each experimental result is shown as well as the mean. P values were determined using analysis of variance comparing MFI of injured with noninjured endothelium. (B) Same as panel A, but for PF4 binding to endothelial cells detected by labeled KKO.
Discussion
With the discovery of the key role of PF4/heparin complexes as the antigenic target in HIT by Amiral et al4 25 years ago, it was thought that the pathogenesis of HIT was largely understood. However, this important observation did not fully explain why this often mild thrombocytopenic disorder was associated with such a marked increase in prothrombotic tendency or why present-day therapeutic interventions do not fully ablate the progression of thrombotic complications in all patients. We proposed previously that the major role of soluble PF4/heparin complexes in HIT was triggering the immune response, but that a key target was PF4 bound to surface glycosaminoglycans on platelets5 as well as on leukocytes7,8 and vascular endothelial cells.9 However, the way in which this distribution of PF4 and the resultant HIT antigen among these cell types affect the development of thrombocytopenia and thrombosis has not been established.
In this report, we show that formation and stability of the HIT antigen constitute a dynamic process that reflects a balance between binding of PF4 to diverse hematopoietic and vascular endothelial cell surfaces and depends on variation in cell counts and changes in the composition of cell-surface glycosaminoglycans, especially in response to inflammation and injury. Monocytes bind more exogenous PF4 or endogenously released PF4 than do platelets, even accounting for vast differences in estimated surface area, likely because of their expression of higher-affinity glycosaminoglycans, as we have shown previously.7 Because of this affinity, a relatively small number of leukocytes, and in particular monocytes, influence platelet PF4 levels. Raising the leukocyte/platelet ratio or exposing whole blood to endothelium elicits a loss of PF4 from the platelet and a redistribution onto other cell surfaces. It is likely that alteration of GAGs caused by inflammation28-30 would further shift this redistribution of PF4 from platelets to monocytes and endothelium, thereby enhancing their susceptibility to antibody-induced activation. Thus, our studies suggest that the protection afforded platelets by reducing surface HIT antigenicity through redistribution of PF4 may mitigate the severity of thrombocytopenia, but at the expense of leaving sufficient activated platelets in the circulation to participate in thrombus development. The potential contribution of the previously unreported reservoir of RBC-associated PF4 to the perpetuation of HIT may be important because of the large number of RBCs in circulation, but this needs further investigation.
In the passive antibody-mediated murine model of HIT, binding of PF4 and HIT antibody to monocytes exceeds binding to platelets, confirming in vitro observations. Monocytopenia develops rapidly, perhaps preceding the development of thrombocytopenia. We propose that PF4-targeted, activated monocytes either adhered to the vessel wall31 or were incorporated into thrombi. The monocytosis that followed was likely a result of replacement by cells mobilized from other pools32 that initially were inaccessible to circulating PF4. Additional studies are needed to determine if the monocytopenia we observed in the murine model develops in patients with HIT, but its brevity and rapid replacement may hinder detection. Morever, the effect on monocytes will also have to take into account the contribution of endothelial binding of PF4 that may be enhanced by underlying changes in the vasculature in affected patients.33
Our findings involving dynamic partitioning of PF4 in blood in HIT provide a deeper understanding of the complexity and dynamic nature of events in HIT and may have several clinical implications. The binding of PF4 to platelets vs monocytes, other leukocytes, and endothelium, as well as levels of circulating heparin, can each affect which cells are targeted and activated and/or cleared and whether thrombosis will develop. Binding of PF4 to glycosaminoglycans on monocytes, neutrophils, and endothelium, leading to formation of immune complexes and induction of tissue factor7,10-12 and initiation of other procoagulant pathways,13,14 may contribute to the intense prothrombotic phenotype and the perpetuation of HIT once heparin has dissipated. Delayed-onset HIT, which develops days after heparin discontinuation,34 may in part be related to such shifting dynamics and cell targeting. Recent observations that addition of PF4 to donor platelets increases detection of HIT antibody in vitro35 could reflect that saturation of leukocyte binding sites and high PF4 concentrations are necessary for sensitization of platelets in vivo.36,37 In vitro assays to detect monocyte or neutrophil activation in whole blood may enhance diagnostic specificity. In the proposed therapeutic use of nonanticoagulant heparins38 to displace PF4 from platelets, such heparins would have to be administered in amounts sufficient to eliminate HIT antigen on higher-affinity cell surfaces to be effective. Dynamic distribution of surface-bound PF4 among hematopoetic cells may explain successful high-dose IV immunoglobulin therapy in HIT.39 Raising platelet counts while concurrently inhibiting FcγRIIA-mediated platelet activation40 could provide a sink for PF4 and help attenuate the risk of thrombosis as well. Other efforts to inhibit monocyte, neutrophil, and perhaps endothelial cell activation41,42 may provide novel adjunctive approaches to intense systemic anticoagulation in this at-risk population.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants P01HL110860 (L.R., D.M., V.H., G.M.A., D.B.C., M.P.), R01HL139448 and HL136512 (L.R., D.B.C., and M.P.), R01-HL-142122 (G.M.A.), R01HL142122 (D.B.C., M.P.); American Heart Association grant 17GRNT33460445 (L.R.); the Shanghai Jiao Tong University K. C. Wong Medical Fellowship Fund (J.D.); and National Natural Science Foundation of China grant 81770131 (J.D.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.D., D.M., and L.R. carried out most of the studies shown and wrote the manuscript; V.H., V.T., and H.S.A. assisted in several studies; G.M.A. and D.B.C. helped in manuscript preparation and data interpretation; and L.R. and M.P. designed the studies and helped in data interpretation and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lubica Rauova, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, ARC, Room 316, Philadelphia, PA 19130; e-mail: lubica@email.chop.edu.
References
- 1.Greinacher A, Farner B, Kroll H, Kohlmann T, Warkentin TE, Eichler P. Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis. A retrospective analysis of 408 patients. Thromb Haemost. 2005;94(1):132-135. [DOI] [PubMed] [Google Scholar]
- 2.Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med. 1996;101(5):502-507. [DOI] [PubMed] [Google Scholar]
- 3.Elalamy I, Tardy-Poncet B, Mulot A, et al. ; GEHT HIT Study Group. Risk factors for unfavorable clinical outcome in patients with documented heparin-induced thrombocytopenia. Thromb Res. 2009;124(5):554-559. [DOI] [PubMed] [Google Scholar]
- 4.Amiral J, Bridey F, Dreyfus M, et al. . Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost. 1992;68(1):95-96. [PubMed] [Google Scholar]
- 5.Rauova L, Zhai L, Kowalska MA, Arepally GM, Cines DB, Poncz M. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: diagnostic and therapeutic implications. Blood. 2006;107(6):2346-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelton JG, Sheridan D, Santos A, et al. . Heparin-induced thrombocytopenia: laboratory studies. Blood. 1988;72(3):925-930. [PubMed] [Google Scholar]
- 7.Rauova L, Hirsch JD, Greene TK, et al. . Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood. 2010;116(23):5021-5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao Z, Visentin GP, Dayananda KM, Neelamegham S. Immune complexes formed following the binding of anti-platelet factor 4 (CXCL4) antibodies to CXCL4 stimulate human neutrophil activation and cell adhesion. Blood. 2008;112(4):1091-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes V, Johnston I, Arepally GM, et al. . Endothelial antigen assembly leads to thrombotic complications in heparin-induced thrombocytopenia. J Clin Invest. 2017;127(3):1090-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arepally GM, Mayer IM. Antibodies from patients with heparin-induced thrombocytopenia stimulate monocytic cells to express tissue factor and secrete interleukin-8. Blood. 2001;98(4):1252-1254. [DOI] [PubMed] [Google Scholar]
- 11.Kasthuri RS, Glover SL, Jonas W, et al. . PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcγRI. Blood. 2012;119(22):5285-5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khairy M, Lasne D, Amelot A, et al. . Polymorphonuclear leukocyte and monocyte activation induced by plasma from patients with heparin-induced thrombocytopenia in whole blood. Thromb Haemost. 2004;92(6):1411-1419. [DOI] [PubMed] [Google Scholar]
- 13.Pouplard C, Iochmann S, Renard B, et al. . Induction of monocyte tissue factor expression by antibodies to heparin-platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood. 2001;97(10):3300-3302. [DOI] [PubMed] [Google Scholar]
- 14.Tutwiler V, Madeeva D, Ahn HS, et al. . Platelet transactivation by monocytes promotes thrombosis in heparin-induced thrombocytopenia. Blood. 2016;127(4):464-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warkentin TE. Heparin-induced thrombocytopenia: IgG-mediated platelet activation, platelet microparticle generation, and altered procoagulant/anticoagulant balance in the pathogenesis of thrombosis and venous limb gangrene complicating heparin-induced thrombocytopenia. Transfus Med Rev. 1996;10(4):249-258. [DOI] [PubMed] [Google Scholar]
- 16.Eslin DE, Zhang C, Samuels KJ, et al. . Transgenic mice studies demonstrate a role for platelet factor 4 in thrombosis: dissociation between anticoagulant and antithrombotic effect of heparin. Blood. 2004;104(10):3173-3180. [DOI] [PubMed] [Google Scholar]
- 17.Ortyn WE, Hall BE, George TC, et al. . Sensitivity measurement and compensation in spectral imaging. Cytometry A. 2006;69(8):852-862. [DOI] [PubMed] [Google Scholar]
- 18.Arepally GM, Kamei S, Park KS, et al. . Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood. 2000;95(5):1533-1540. [PubMed] [Google Scholar]
- 19.Cai Z, Yarovoi SV, Zhu Z, et al. . Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat Commun. 2015;6:8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen TH, Greinacher A. Platelet factor 4/heparin complexes present epitopes differently on solid-phase vs platelet surfaces. Blood. 2017;129(26):3498-3501. [DOI] [PubMed] [Google Scholar]
- 21.Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med. 1993;119(2):104-109. [DOI] [PubMed] [Google Scholar]
- 22.Sheridan D, Carter C, Kelton JG. A diagnostic test for heparin-induced thrombocytopenia. Blood. 1986;67(1):27-30. [PubMed] [Google Scholar]
- 23.Reilly MP, Taylor SM, Hartman NK, et al. . Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcgammaRIIA. Blood. 2001;98(8):2442-2447. [DOI] [PubMed] [Google Scholar]
- 24.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cines DB, Tomaski A, Tannenbaum S. Immune endothelial-cell injury in heparin-associated thrombocytopenia. N Engl J Med. 1987;316(10):581-589. [DOI] [PubMed] [Google Scholar]
- 26.Visentin GP, Ford SE, Scott JP, Aster RH. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specific for platelet factor 4 complexed with heparin or bound to endothelial cells. J Clin Invest. 1994;93(1):81-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lubenow N, Hinz P, Thomaschewski S, et al. . The severity of trauma determines the immune response to PF4/heparin and the frequency of heparin-induced thrombocytopenia. Blood. 2010;115(9):1797-1803. [DOI] [PubMed] [Google Scholar]
- 28.Petricevich VL, Michelacci YM. Proteoglycans synthesized in vitro by nude and normal mouse peritoneal macrophages. Biochim Biophys Acta. 1990;1053(2-3):135-143. [DOI] [PubMed] [Google Scholar]
- 29.Uhlin-Hansen L, Eskeland T, Kolset SO. Modulation of the expression of chondroitin sulfate proteoglycan in stimulated human monocytes. J Biol Chem. 1989;264(25):14916-14922. [PubMed] [Google Scholar]
- 30.Uhlin-Hansen L, Wik T, Kjellén L, Berg E, Forsdahl F, Kolset SO. Proteoglycan metabolism in normal and inflammatory human macrophages. Blood. 1993;82(9):2880-2889. [PubMed] [Google Scholar]
- 31.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107(3):321-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sunderkötter C, Nikolic T, Dillon MJ, et al. . Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172(7):4410-4417. [DOI] [PubMed] [Google Scholar]
- 33.Pitsilos S, Hunt J, Mohler ER, et al. . Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemost. 2003;90(6):1112-1120. [DOI] [PubMed] [Google Scholar]
- 34.Warkentin TE, Kelton JG. Delayed-onset heparin-induced thrombocytopenia and thrombosis. Ann Intern Med. 2001;135(7):502-506. [DOI] [PubMed] [Google Scholar]
- 35.Vayne C, Guery EA, Kizlik-Masson C, et al. . Beneficial effect of exogenous platelet factor 4 for detecting pathogenic heparin-induced thrombocytopenia antibodies. Br J Haematol. 2017;179(5):811-819. [DOI] [PubMed] [Google Scholar]
- 36.Nazi I, Arnold DM, Warkentin TE, Smith JW, Staibano P, Kelton JG. Distinguishing between anti-platelet factor 4/heparin antibodies that can and cannot cause heparin-induced thrombocytopenia. J Thromb Haemost. 2015;13(10):1900-1907. [DOI] [PubMed] [Google Scholar]
- 37.Padmanabhan A, Jones CG, Curtis BR, et al. . A novel PF4-dependent platelet activation assay identifies patients likely to have heparin-induced thrombocytopenia/thrombosis. Chest. 2016;150(3):506-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joglekar MV, Quintana Diez PM, Marcus S, et al. . Disruption of PF4/H multimolecular complex formation with a minimally anticoagulant heparin (ODSH). Thromb Haemost. 2012;107(4):717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azimov MB, Slater ED. Persistent heparin-induced thrombocytopenia treated with IVIg. Chest. 2017;152(3):679-680. [DOI] [PubMed] [Google Scholar]
- 40.Padmanabhan A, Jones CG, Pechauer SM, et al. . IVIg for treatment of severe refractory heparin-induced thrombocytopenia. Chest. 2017;152(3):478-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lhermusier T, van Rottem J, Garcia C, et al. . The Syk-kinase inhibitor R406 impairs platelet activation and monocyte tissue factor expression triggered by heparin-PF4 complex directed antibodies. J Thromb Haemost. 2011;9(10):2067-2076. [DOI] [PubMed] [Google Scholar]
- 42.Reilly MP, Sinha U, André P, et al. . PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood. 2011;117(7):2241-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.