

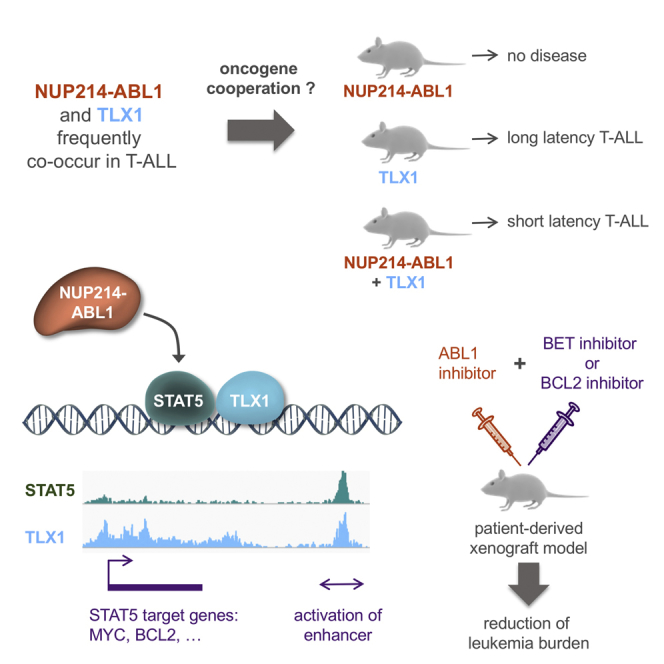
Summary
The NUP214-ABL1 fusion is a constitutively activated tyrosine kinase that is significantly associated with overexpression of the TLX1 and TLX3 transcription factors in T cell acute lymphoblastic leukemia (T-ALL). Here we show that NUP214-ABL1 cooperates with TLX1 in driving T-ALL development using a transgenic mouse model and human T-ALL cells. Using integrated ChIP-sequencing, ATAC-sequencing, and RNA-sequencing data, we demonstrate that TLX1 and STAT5, the downstream effector of NUP214-ABL1, co-bind poised enhancer regions, and cooperatively activate the expression of key proto-oncogenes such as MYC and BCL2. Inhibition of STAT5, downregulation of TLX1 or MYC, or interference with enhancer function through BET-inhibitor treatment leads to reduction of target gene expression and induction of leukemia cell death.
Keywords: oncogenes, leukemia, signaling, transcriptional regulation, cooperation, mouse model, cancer
Graphical Abstract
Highlights
-
•
TLX1 and STAT5 signaling cooperate in the development of T-ALL
-
•
TLX1 and STAT5 co-bind and activate oncogenic enhancers
-
•
A feedforward loop leads to MYC activation and recruitment to TLX1/STAT5 enhancers
-
•
Synergy between ABL1 inhibitors and BET/BCL2 inhibitors improves therapy response
Vanden Bempt et al. show that NUP214-ABL1 cooperates with TLX1 to drive T cell acute lymphoblastic leukemia (T-ALL) development and that STAT5, the downstream effector of NUP214-ABL1, and TLX1 cooperatively activate the expression of MYC and BCL2. Inhibition of STAT5, TLX1, MYC, or BCL2 induces T-ALL cell death.
Significance
Unraveling the molecular mechanisms underlying the cooperation between TLX1 and NUP214-ABL1/STAT5 signaling improves our understanding of oncogene cooperation, and contributes to the development of effective targeted therapies. Our data show that TLX1 and STAT5 directly cooperate at the transcriptional level through activation of enhancer regions of key proto-oncogenes such as MYC and BCL2. Based on these findings, we demonstrate synergy between ABL1 kinase inhibitors and BET or BCL2 inhibitors to reduce the growth of NUP214-ABL1/TLX-positive T-ALL cells in vitro and in vivo.
Introduction
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological disease, which arises from the malignant transformation of developing T cell progenitors due to the accumulation of oncogenic aberrations (Belver and Ferrando, 2016, Girardi et al., 2017). Although survival rates are currently close to 90% in children, adult cases still have a poor prognosis (Bassan and Hoelzer, 2011, Pui et al., 2015). Moreover, current therapy is associated with both short-term and long-term side effects, especially devastating for children (Haddy et al., 2009). Therefore, there remains an urgent need to uncover the molecular mechanisms underlying T-ALL in order to identify therapeutic targets and to develop personalized targeted therapies.
T-ALL can be subdivided into clinically relevant subgroups, based on the ectopic and mutually exclusive expression of transcription factors, such as TAL1/2, LMO1/2, TLX1/3, HOXA9/10, or NKX2-1. Each subgroup presents with different immunophenotypes and gene expression patterns that reflect developmental arrest at different stages of T cell maturation (Homminga et al., 2011, Van Vlierberghe et al., 2012). Furthermore, next-generation sequencing has contributed to the discovery of >100 genes that are recurrently mutated in T-ALL (De Keersmaecker et al., 2013, Li et al., 2016, Liu et al., 2017, Vicente et al., 2015).
TLX1 and TLX3 are not expressed in the hematopoietic system but are ectopically expressed in approximately 30% of T-ALL cases (Belver and Ferrando, 2016, Girardi et al., 2017). This is the result of chromosomal translocations that place TLX1 or TLX3 under control of regulatory elements of the T cell receptor locus or of the BCL11B locus, respectively. Overexpression of TLX1 in mouse T cells results in the development of T cell leukemia/lymphoma with long latency, illustrating the oncogenic potential of TLX1. These leukemias/lymphomas frequently harbor inactivating mutations in Bcl11b, and were typically aneuploid as a result of TLX1-driven defects in mitotic checkpoint activation (De Keersmaecker et al., 2010). Additional studies have shown that TLX1 target genes show significant overlap with NOTCH1 target genes (Durinck et al., 2015), and that TLX1 binds to the T cell receptor alpha locus (TRA), thereby inhibiting its recombination and blocking differentiation of the T cells (Dadi et al., 2012). Together, these data provide first hints as to how ectopic TLX1 expression can contribute to T-ALL development by disturbing NOTCH1 signaling and T cell differentiation. The role of TLX3 in T-ALL is less well studied, but Ferrando and coworkers have shown that TLX1- or TLX3-expressing tumors are characterized by a highly related gene expression signature and that TLX1 and TLX3 share 75% of their direct target genes as determined by chromatin immunoprecipitation (ChIP)-chip (Della Gatta et al., 2012).
Besides aberrant transcription factor expression, T-ALL cases harbor mutations that lead to constitutive activation of signaling pathways such as the NOTCH1, PI3K-AKT, RAS-MAPK, and IL7R-JAK-STAT pathways (Canté-Barrett et al., 2016, Degryse et al., 2014, Mullighan et al., 2009, Weng et al., 2004, Zenatti et al., 2011). Mutations in the PI3K-AKT pathway are most frequent in TAL1-positive T-ALL cases, while activating mutations in JAK1, JAK3 or IL7R, leading to constitutive STAT5 phosphorylation, are most often found in immature and TLX/HOXA-positive T-ALL (de Bock et al., 2018, Liu et al., 2017). In addition to IL7R-JAK mutations, ABL1 activation, through the generation of fusion genes with NUP214, EML1, or ETV6, results in aberrant STAT5 activation (De Keersmaecker et al., 2008a). The NUP214-ABL1 fusion is the most frequent ABL1 fusion in T-ALL, which is generated by episomal amplification (Graux et al., 2004). The NUP214-ABL1 kinase is able to confer cytokine-independent growth to Ba/F3 cells and to drive T cell leukemia when overexpressed from viral vectors in mouse bone marrow cells in vivo (De Keersmaecker et al., 2008a). Compared with BCR-ABL1, NUP214-ABL1 is a relatively weak oncoprotein with lower kinase activity, and a 2- to 3-fold higher sensitivity to the kinase inhibitor imatinib (De Keersmaecker et al., 2008b). Patients with NUP214-ABL1-positive T-ALL have been treated with imatinib, albeit with variable success (Clarke et al., 2011, Crombet et al., 2012, Deenik et al., 2009, Koschmieder et al., 2014, Stergianou et al., 2005).
From numerous sequencing studies, it has become evident that NUP214-ABL1+ T-ALL cases are almost exclusively found within TLX1/3-positive cases (Graux et al., 2009, Kleppe et al., 2010, Liu et al., 2017). This association between NUP214-ABL1 and TLX1/3 expression suggests a possible synergism between these two oncogenic aberrations in driving the development, progression, and/or maintenance of T-ALL, which we have investigated in the current study.
Results
The NUP214-ABL1 Fusion and TLX1/TLX3 Expression Co-occur in T-ALL Patients and Cooperate in the Development of T-ALL in a Transgenic Mouse Model
The NUP214-ABL1 kinase is a known driver of proliferation in T-ALL (Graux et al., 2004, Kleppe et al., 2010). Examination of publicly available clinical T-ALL sequencing data (Burmeister et al., 2006, Graux et al., 2004, Graux et al., 2009, Homminga et al., 2011, Liu et al., 2017, Soulier et al., 2005) confirmed that within the NUP214-ABL1+ cases, almost all cases are TLX1/TLX3 positive, while in a general T-ALL cohort only 32% of the cases are TLX1/TLX3 positive (p < 0.0001) (Figure 1A and Table S1). This significant co-occurrence between NUP214-ABL1 and TLX1/3 in T-ALL patients suggested that these lesions might cooperate in the initiation, development, and/or maintenance of T-ALL.
Figure 1.

Expression of NUP214-ABL1 and TLX1 Is Required to Induce T-ALL in a Transgenic Mouse Model
(A) Pie chart representing the percentage of T-ALL (left) or NUP214-ABL1-positive T-ALL (right) with TLX1 or TLX3 expression.
(B) Schematic overview of the transgenic mouse models used in this study. Red triangles represent LoxP sites. A conditional loxP-STOP-loxP NUP214-ABL1 knockin mouse model (abbreviated as LSL-NA) was generated. NUP214-ABL1 expression was initiated by crossing LSL-NA mice with CD4-Cre mice. Co-expression of NUP214-ABL1 and TLX1 was achieved by crossing NA mice with Tg(Lck-TLX1) mice, resulting in Tg(CD4 Cre; NUP214-ABL1; Lck TLX1) mice (abbreviated as NA + TLX1).
(C) Kaplan-Meier overall survival curve comparing NA + TLX1, TLX1, and NA mice.
(D) Representative fluorescence-activated cell sorting (FACS) analysis of GFP expression in NA + TLX1 mice at end-stage disease compared with wild-type (WT) cells for spleen, thymus, peripheral blood (PB), and bone marrow (BM).
(E–G) Peripheral white blood cell count (WBC) (E), spleen weight (F), and thymus weight (G) at end-stage disease for NA + TLX1 mice compared with NA and LSL-NA mice (end stage for NA and LSL-NA defined as >360 days). Star indicates NA + TLX1 mouse that presented with an elevated WBC, but did not present with an enlarged spleen or thymus at end stage. Statistical significance was calculated using a Mann-Whitney test. Data are presented as mean ± SD. N.s., not significant.
(H) Representative FACS analysis for CD4 and CD8 expression in GFP-positive NA + TLX1 leukemic cells from the peripheral blood compared with NA and LSL-NA peripheral blood cells.
(I) H&E and immunohistochemical staining for CD3 and Cre in spleen cells from LSL-NA, NA, and NA + TLX1 mice. Scale bars represent 100 μm.
(J) Kaplan-Meier overall survival curve of secondary (using cells from three different primary NA + TLX1 mice) and tertiary transplants.
(K) Growth curve of ex vivo primary immature pro T cells expressing EML1-ABL1, TLX1 or both. Data are presented as mean ± SD.
(L) Kaplan-Meier overall survival curve of mice transplanted with hematopoietic stem/progenitor cells expressing EML1-ABL1, TLX1 or EML1-ABL1+TLX1.
See also Figures S1–S4 and Table S1.
To investigate the potential cooperation of NUP214-ABL1 with TLX1, we generated a conditional transgenic mouse model Tg(NUP214-ABL1), in which the expression of NUP214-ABL1-IRES-GFP is blocked by a LoxP stop cassette (hereafter designated LSL-NA, Figures 1B and S1A). These mice were subsequently crossed with Tg(CD4-Cre) mice for targeted expression of NUP214-ABL1 within developing T cells beginning from the CD4+CD8+ double-positive stage (hereafter designated NA mice, Figures 1B and S1A). CD4-Cre-driven expression of NUP214-ABL1 alone was insufficient to cause T-ALL development in the NA mouse model over a 400-day observation period, and there were no profound T cell developmental defects (Figures 1C and S1B–S1G). Similarly, crossing the LSL-NA mice with CD2-Cre or CD19-Cre drivers, to activate NUP214-ABL1 expression in the common lymphoid progenitor or B cell progenitor stages, did not result in strong lymphoid abnormalities or disease development (Figure S2). Together, these data show that the expression of a single copy of NUP214-ABL1 within lymphoid progenitors was insufficient to drive leukemia development.
We next sought to determine whether co-expression of TLX1 with NUP214-ABL1 could drive T-ALL development. To this end, NA mice were crossed with Tg(Lck-TLX1) mice (designated TLX1) (Figure 1B), expressing TLX1 under control of the T cell-specific Lck promoter (De Keersmaecker et al., 2010), and this resulted in mice in which both NUP214-ABL1 and TLX1 were expressed in developing T cells (designated NA + TLX1) (Figures 1B, S3A, and S3B). In this instance, NA + TLX1 mice developed an aggressive T cell leukemia with a significantly shorter latency (median overall survival = 217 days) compared with TLX1 mice (median overall survival = 385 days) and NA mice (no leukemia) (p < 0.001). At end-stage disease, all NA + TLX1 mice had leukemic cell infiltration into the spleen, thymus, and bone marrow (Figure 1D), and the leukemic cells showed strong phosphorylation of STAT5, a downstream effector of NUP214-ABL1 (Figures S3C and S3D). Leukemic mice had increased white blood cell counts, with the majority also presenting with splenomegaly and enlarged thymi (Figures 1E–1G). Phenotypic analysis revealed that the major leukemic clone in the NA + TLX1 mice was an immature CD4+CD8+ T cell population (Figure 1H). Histopathological analysis further confirmed a T cell leukemia with severe expansion of the white pulp and infiltration of the red pulp by atypical CD3+ lymphoid cells, resulting in the loss of splenic tissue architecture in NA + TLX1 mice (Figure 1I). The disease was transplantable to secondary and tertiary transplants, confirming the disease to be an acute leukemia (Figure 1J).
To further extend these findings, we tested whether TLX1 could cooperate with EML1-ABL1, another ABL1 fusion detected in TLX1-positive T-ALL (Vanden Bempt et al., 2018, De Keersmaecker et al., 2005). First, we tested co-expression of EML1-ABL1 and TLX1 in ex vivo cultured primary mouse pro T cells (Bornschein et al., 2018, Gehre et al., 2015). Cells expressing EML1-ABL1 and TLX1 proliferated in the absence of interleukin-7 (IL-7) and outcompeted cells expressing EML1-ABL1 or TLX1 alone (Figure 1K). Next, to confirm these findings in vivo, we transplanted murine hematopoietic stem/progenitor cells transduced with retroviral vectors containing EML1-ABL1, TLX1, or EML1-ABL1 + TLX1 into irradiated recipient mice. After 100 days, 5 of 8 (63%) of the EML1-ABL1 + TLX1 mice had developed a fatal leukemia, whereas EML1-ABL1 or TLX1 alone was not able to induce leukemia during the observation period (Figures 1L and S4). Taken together, these data show that both NUP214-ABL1 and EML1-ABL1 can cooperate with TLX1 to transform T cells ex vivo and induce leukemia in vivo.
NUP214-ABL1/TLX1-Driven Leukemia Cells Are Characterized by a STAT5 Gene Signature
To elucidate the underlying transcriptional programs and identify key transcription factors driving the leukemia development in NA + TLX1 mice, we performed a global gene expression analysis using RNA sequencing (RNA-seq) in conjunction with an in silico analysis of regulatory sequences using i-CisTarget (Herrmann et al., 2012, Verfaillie et al., 2015). Genes that were most significantly up- or downregulated in NA + TLX1 CD4+CD8+ leukemia cells compared with normal CD4+CD8+ T cells were enriched for STAT5, SPI1, and ETS/RUNX binding sites in their regulatory regions (Figures 2A and 2B). In addition, we found significant upregulation of the JAK-STAT pathway genes in NA + TLX1 leukemic cells (Figures 2C and S5A). STAT5 is indeed known to be phosphorylated and activated by NUP214-ABL1 (De Keersmaecker et al., 2008a). To specifically determine the contribution of STAT5 target genes, we performed ChIP sequencing (ChIP-seq) with anti-STAT5 antibodies. We observed a strong enrichment of the genes bound by STAT5 (as defined by ChIP-seq) in the upregulated transcripts in the NA + TLX1 leukemic cells compared with wild-type (WT) CD4+CD8+ T cells (Figures 2D and S5B). Many of the upregulated genes, including Myc and Bcl2, are known as canonical STAT5 target genes (Figures 2E and S5C). In addition, activation of STAT5 and expression of TLX1 together led to increased expression of Myc and Bcl2 compared with STAT5 or TLX1 alone in ex vivo cultured pro T cells (Figure 2F).
Figure 2.

NUP214-ABL1 and TLX1 Upregulate the JAK-STAT Pathway
(A) Transcription factor binding motifs of the top transcription factors identified by i-CisTarget to regulate NA + TLX1 gene expression patterns. NES, normalized enrichment score.
(B) i-CisTarget transcriptional network showing genes regulated by STAT5.
(C) Heatmap representing the differential gene expression of canonical JAK-STAT signaling pathway genes in WT, NA, TLX1, and NA + TLX1 mice.
(D) Gene set enrichment analysis (GSEA) showing enrichment of STAT5 target genes (as defined by ChIP-seq) in the differentially expressed genes in NA + TLX1 mice compared with WT. NES, normalized enrichment score.
(E) qRT-PCR analysis of Myc and Bcl2 mRNA in the different transgenic mouse models. Statistical significance calculated using unpaired two-tailed t test with equal variance. Data are presented as mean ± SD.
(F) qRT-PCR analysis of Myc and Bcl2 in ex vivo immature pro T cells expressing EML1-ABL1, TLX1 or both. Statistical significance calculated using unpaired two-tailed t test with equal variance. Data are presented as mean ± SD.
(G) qRT-PCR in ALL-SIL cells after a 2-day antisense oligo-mediated knockdown of STAT5A or STAT5B. Data are presented as mean ± SD.
(H) GSEA to show enrichment of TLX1 target genes (genes with a TLX1 ChIP peak and downregulated after TLX1 gapmer treatment) in differentially expressed genes after imatinib treatment. NES, normalized enrichment score.
(I–K) Volcano plot (I) showing up- and downregulated genes (top) and GSEA to show enrichment of STAT5 target genes in differentially expressed genes (bottom) after imatinib treatment (500 nM imatinib or DMSO for 3 hr) in leukemic cells harvested from NA + TLX1 mice (n = 3; experiment was performed with cells harvested from three separate mice). (J) Volcano plot showing up- and downregulated genes (top) and GSEA to show enrichment of STAT5 target genes in differentially expressed genes (bottom) after imatinib treatment (500 nM imatinib or DMSO for 3 hr) in ALL-SIL cells (n = 3; experiment was performed as three independent repeats). (K) Venn diagram showing overlap between the upregulated genes in NA + TLX1 versus WT mice and genes that are downregulated after imatinib treatment (p = 8.7 × 10−159).
(L) Cell number of NA + TLX1 spleen cells expressing STAT5 N642H or empty vector, treated for 48 hr with 500 nM imatinib or DMSO. Data are presented as mean ± SD.
See also Figure S5.
These data indicate that NA + TLX1 leukemia cells are largely characterized by activation of STAT5 target genes. To further determine the contribution of STAT5 and TLX1 in transcriptional regulation, we knocked down STAT5A/STAT5B or TLX1 in the human T-ALL cell line ALL-SIL (expressing NUP214-ABL1 and TLX1), and found that STAT5 target genes were in both cases downregulated (Figures 2G, 2H, S5D, and S5E). Moreover, inhibition of STAT5 activation by treatment of the cells with the ABL1 kinase inhibitor imatinib led to downregulation of the STAT5 target genes (Figures 2I, 2J, and S5F–S5H), and these genes significantly overlapped with the set of genes specific for the mouse NA + TLX1 leukemia cells (p < 0.001) (Figure 2K). In addition, expression of a constitutive active STAT5B(N642H) mutant (Ariyoshi et al., 2000, Bandapalli et al., 2014) in mouse NA + TLX1 leukemia cells could rescue the effect of imatinib treatment on cell viability (Figure 2L), confirming that STAT5 is one of the major factors driving the proliferation and survival of the NA + TLX1 leukemia cells. Taken together, these gene expression data indicate a strong STAT5 signature in the NA + TLX1 leukemia cells, with marked upregulation of STAT5 target genes in the presence of TLX1.
TLX1 and STAT5 Have Overlapping Binding Sites Genome-wide and Are Preferentially Associated with Enhancer Regions
To gain further insight into how the STAT5 and TLX1 transcription factors might cooperate in the transcriptional regulation of NA + TLX1-driven leukemia, we mapped the binding sites of STAT5 and TLX1 genome-wide. ChIP-seq analysis of murine NA + TLX1 leukemic cells revealed that STAT5 and TLX1 binding sites overlap significantly throughout the genome (p < 0.001) and that binding was enriched in imatinib-responsive genes. Motif analysis showed that RUNX motifs were highly enriched in the STAT5/TLX1 binding sites. Indeed, RUNX1 also co-binds with STAT5 and TLX1 in the leukemic cells as confirmed by ChIP-seq (Figures 3A and 3B). STAT5 and TLX1 predominantly co-occupy enhancer regions, as shown by the presence of enhancer marks p300, BRD4, and H3K27ac. These regions were already bound by RUNX1 in normal CD4+CD8+ T cells, where they were marked with H3K4me1 rather than H3K27ac, indicating that these regions are in a poised state in WT cells (Figures 3A and 3C). Given the strong co-occupancy at enhancers, we next ranked the H3K27ac signals from regions bound by TLX1 to identify important regulatory regions, also referred to as super-enhancers. Many TLX1-bound regions were found to contain high-intensity H3K27ac signals (Figure 3D). These enhancer regions included numerous STAT5 responsive genes with strong H3K27ac signals localized either intragenically (e.g., Bcl2) or downstream (e.g., Socs1 and Pim1) and were marked by p300 and BRD4 enhancer marks, but not by the promoter-associated H3K4me3 mark (Figures 3E–3G). These data show that STAT5 and TLX1 cooperate on a genome-wide level mainly by co-occupying regulatory enhancer regions.
Figure 3.

STAT5 and TLX1 Co-bind Regulatory Regions throughout the Genome
(A) Centered read density heatmaps of ChIPmentation ChIP-seq signals for the binding of TLX1, STAT5, p300, BRD4, ETS1, RUNX1, and the histone marks H3K27ac and H3K4me1. Heatmaps centered and ranked on TLX1 signal strength in leukemic NA + TLX1 and WT mouse cells.
(B) GSEA comparing genes bound by both STAT5 and TLX1 and differentially expressed genes after treatment with imatinib in ALL-SIL cells.
(C) Venn diagram showing the total amount of TLX1 and STAT5 peaks that fall within the 90,804 H3K27ac peaks for NA + TLX1 leukemic cells (left). Pie chart (right) showing ChIP-seq peak co-occurrence of STAT5 and TLX1 in mouse NA + TLX1 leukemic cells in relation to promoter regions (H3K4me3+/H3K27ac+) and enhancer regions (H3K27ac+/BRD4+/p300+/H3K4me3−).
(D) Enhancer regions ranked on H3K27ac signal for NA + TLX1 leukemic cells and ALL-SIL. Only H3K27ac clusters with overlapping TLX1 peaks are shown. Gene labels are distance-based if not intragenic.
(E–G) Representative ChIP-seq tracks for canonical STAT5 target genes Bcl2 (E), Pim1 (F), and Socs1 (G) showing STAT5, TLX1, H3K4me3, p300, BRD4, and H3K27ac binding in NA + TLX1 leukemic mouse cells.
TLX1 Binding Is Correlated with Open Enhancer Regions of STAT5 Response Genes
Given the strong correlation between STAT5 and TLX1 binding in active enhancer regions, global chromatin architecture was further investigated using assay for transposase-accessible chromatin using sequencing (ATAC-seq). Comparing ATAC-seq profiles of CD4+CD8+ NA + TLX1 leukemic cells with CD4+CD8+ T cells from WT mice revealed that many STAT5 target genes had increased chromatin accessibility in NA + TLX1 cells. An in silico analysis using i-CisTarget (Herrmann et al., 2012, Verfaillie et al., 2015) identified STAT5, ETS1, and RUNX1 binding motifs in the more accessible chromatin regions (Figure 4A). These regions were found to be associated with genes that are differentially expressed in NA + TLX1 mice (Figure 4B). Only 16% of all chromatin regions with appearing ATAC peaks were located in promoter regions, while 42% were located in enhancer regions (Figure 4C), indicating that the gene expression changes in NA + TLX1 cells compared with NA or TLX1 cells were mainly due to enhancer-mediated effects. Indeed, we found that the more accessible regions in the NA + TLX1 leukemic cells were mostly marked by the H3K27ac enhancer mark, and only rarely by the H3K4me3 promoter mark (Figure 4D). A global comparison of ATAC peaks between non-leukemic WT, NA, and TLX1 CD4+CD8+ cells and leukemic NA + TLX1 cells showed that regions that were previously inaccessible were now accessible, bound by STAT5, TLX1, p300, and BRD4, and marked by H3K27ac. These regions were not accessible in WT, NA, or TLX1 cells, nor was any H3K27ac detected. However, these regions were already marked by H3K4me1, showing that these enhancers are already in a poised state in normal CD4+CD8+ thymocytes (Figure 4D). Among these more accessible chromatin regions were the internal Bcl2 enhancer, the Pim1 downstream enhancer, and the Notch-dependent Myc enhancer (N-ME) (Herranz et al., 2014, Yashiro-Ohtani et al., 2014), while the promoter regions of these genes showed only minimal changes in chromatin structure (Figures 4E and S6).
Figure 4.

TLX1 and STAT5 Bind in Newly Accessible Enhancer Regions
(A) Volcano plot of differential ATAC-seq peak height in CD4+CD8+ NA + TLX1 leukemic cells versus CD4+CD8+ WT cells. Peaks in STAT5 response genes are shown as blue dots. Transcription factor binding motifs enriched through i-CisTarget analysis in the appearing peaks are shown on the right. NES, normalized enrichment score.
(B) GSEA of appearing ATAC peaks in relation to differentially expressed genes in CD4+CD8+ NA + TLX1 leukemic cells versus CD4+CD8+ WT cells.
(C) Pie chart representing the genomic locations of appearing ATAC-seq peaks in CD4+CD8+ NA + TLX1 cells versus CD4+CD8+ WT cells. A promoter region is defined as a region with combined H3K4me3 and H3K27ac marks. An enhancer region is defined as a region with H3K27ac signal or BRD4 binding or p300 binding, but no H3K4me3.
(D) Read density heatmaps of ATAC-seq and ChIP-seq signals (STAT5, TLX1, p300, BRD4, H3K27ac, H3K4me1, H3K4me3) in CD4+CD8+ WT, NA, TLX1, and NA + TLX1 cells, centered around the top 2,000 appearing and disappearing ATAC-seq peaks.
(E) ATAC-seq tracks (performed in CD4+CD8+ WT, NA, TLX1, and NA + TLX1 cells) and ChIP-seq tracks (H3K4me1, H3K27ac in WT cells and STAT5, TLX1, H3K27ac in NA + TLX1 cells) at the Bcl2 locus, the Pim1 locus, and the Myc locus.
See also Figure S6.
TLX1 and STAT5 Activate a Feedforward Loop Engaging MYC in TLX1/STAT5 Complexes
To determine whether TLX1 and STAT5 co-bound regulatory regions were enriched for additional transcription factor binding sites, we integrated ChIP-seq and RNA-seq data from mouse and human leukemia cells and performed an in silico analysis using i-CisTarget (Herrmann et al., 2012, Verfaillie et al., 2015). This analysis revealed that MYC and MAX binding motifs were significantly enriched specifically in genes positively regulated by STAT5 (i.e., downregulated after imatinib treatment) (Figure 5A). To verify MYC binding to these regions, we performed ChIP-seq experiments with MYC antibodies to characterize genome-wide binding sites of MYC in human ALL-SIL cells. Strikingly, the binding pattern of MYC overlapped significantly with STAT5 and TLX1 (p < 0.001) (Figures 5B and 5C). Moreover, MYC was bound together with STAT5 and TLX1 in regulatory regions of STAT5 target genes, e.g. OSM, PIM1, BCL2, and MYC (Figures 5D–5G). To determine whether MYC was actively involved in the regulation of these genes, we treated the cells with MYC small interfering RNA (siRNA) or with the BET inhibitor JQ1, which indirectly causes MYC downregulation. Knockdown of MYC led to a reduction of STAT5 target gene expression, supporting an active role for MYC in the expression of STAT5 target genes in the leukemia cells expressing NUP214-ABL1 and TLX1 (Figures 5H and 5I). A more detailed analysis of the MYC locus revealed that STAT5 and TLX1 were bound to the promoter region (Figure 5G) as well as to regions within the 1.4-Mb downstream super-enhancer region. This downstream super-enhancer region has a well-characterized Notch-dependent enhancer region (N-ME) (Herranz et al., 2014, Yashiro-Ohtani et al., 2014) and also previously characterized STAT5 binding regions (Pinz et al., 2016, Shi et al., 2013). These regions were characterized by a strong H3K27ac signal and distinct regions co-bound by BRD4, STAT5, TLX1, and MYC itself (Figure 5J), suggesting a feedforward loop in the regulation of MYC expression.
Figure 5.

MYC Co-binds and Co-regulates STAT5 and TLX1 Target Genes
(A) In silico i-CisTarget analysis for enriched transcription factor motifs found within in regulatory regions of genes that are positively or negatively regulated by STAT5.
(B) Read density heatmaps of ChIP-seq signals on TLX1 binding locations for different transcription factors or epigenetic marks ranked on TLX1 signal strength (top) or STAT5 signal strength (bottom) in ALL-SIL cells.
(C) Venn diagram showing the total amount of TLX1 and STAT5 peaks that fall within the 41,229 H3K27ac peaks (left) and the total amount of STAT5 + TLX1 peaks that overlap with MYC peaks (right) in ALL-SIL cells.
(D–G) ChIP-seq tracks (performed on ALL-SIL cells) of STAT5, TLX1, MYC, p300, BRD4, and H3K27ac at canonical STAT5 regulated genes OSM (D), PIM1 (E), BCL2 (F), and MYC (G) loci.
(H) qRT-PCR in ALL-SIL cells treated with MYC siRNAs for 48 hr. Data are presented as mean ± SD. Statistical significance calculated using unpaired two-tailed t test with equal variance.
(I) qRT-PCR of STAT5 target genes in ALL-SIL cells treated with 500 nM JQ1 for 6 hr. Data are presented as mean ± SD. Statistical significance calculated using unpaired two-tailed t test with equal variance.
(J) ChIP-seq tracks (performed on ALL-SIL cells) of STAT5, TLX1, MYC, p300, BRD4, and H3K27ac at the MYC enhancer locus, 1.4 Mb downstream of the MYC gene.
Taken together, these data show that MYC upregulation in NUP214-ABL1/TLX1 leukemic cells is a direct effect of STAT5 and TLX1 binding to regulatory regions, and that MYC reinforces the transcriptional program initiated by these transcription factors.
Downstream Effectors of NUP214-ABL1 and TLX1 Can Be Targeted to Improve Treatment Strategies
Having established a role for STAT5, MYC, and TLX1 in the expression of key regulatory genes controlling cell survival and proliferation through enhancer activation, we sought to exploit these findings using a targeted therapeutic approach within NUP214-ABL1-positive patient T-ALL samples. Given that NUP214-ABL1 and TLX1 activate enhancer regions, we tested the therapeutic efficacy of BET inhibitors, which are known to disrupt active enhancers (Lovén et al., 2013). In addition, we selected to inhibit BCL2 as a downstream target of NUP214-ABL1 and TLX1, since a small-molecule inhibitor is already available for this target (Souers et al., 2013).
Treatment of ALL-SIL cells or T-ALL patient-derived xenograft (PDX) cells (X12 and XD82, Figures S7A and S7B) showed reduced viability upon treatment with imatinib (ABL1 inhibitor), JQ1 (BET inhibitor), and ABT-199 (BCL2 inhibitor, Venetoclax). Treatment with a combination of these inhibitors was even more potent (Figures 6A–6C). To explore the synergistic properties of the different inhibitors in more detail, we combined imatinib with a BET inhibitor (JQ1, iBET-762 or CPI0610) (Figures 6D, 6E, and S7C–S7H) or a BCL2-inhibitor (ABT-199) (Figures 6F, 6G, and S7I–S7L). We calculated synergy using the zero interaction potential (ZIP) method, and observed a clear average synergistic effect for all combinations. Moreover, we showed that the maximum ZIP synergy score for each combination exceeds 10, which means a 10% additional cell inhibition due to the synergy, compared with a solely additive effect of the two drugs (Ianevski et al., 2017). Combining imatinib with JQ1 resulted in a stronger reduction of BCL2 expression compared with the single agents (Figure 6H), which was in line with the more drastic inhibitory effect of the inhibitor combination on cell viability.
Figure 6.

Downstream Effectors of NUP214-ABL1 and TLX1 Can Be Targeted to Improve Treatment Strategies
(A) Viability of ALL-SIL cells after 48 hr treatment with 500 nM imatinib, 500 nM JQ1, 500 nM ABT-199, or a combination of these inhibitors (500 nM + 500 nM). (n = 3; experiment was performed as three independent repeats, and data are presented as mean ± SD).
(B) Viability of NUP214-ABL1+TLX3+ X12 PDX cells after 48 hr treatment with 500 nM imatinib, 500 nM JQ1, 500 nM ABT-199, or a combination of these inhibitors (500 nM + 500 nM). Statistical significance calculated using unpaired two-tailed t test with equal variance (n = 2; the experiment was performed using cells harvested from two different xenograft mice, and data are presented as mean ± SD). Statistical significance calculated using unpaired two-tailed t test with equal variance.
(C) Viability of NUP214-ABL1+TLX3+ XD82 PDX cells after 48 hr treatment with 500 nM imatinib, 500 nM JQ1, 500 nM ABT-199, or a combination of these inhibitors (500 nM + 500 nM) (n = 2; the experiment was performed using cells harvested from two different xenograft mice, and data are presented as mean ± SD). Statistical significance calculated using unpaired two-tailed t test with equal variance.
(D) Growth of X12 PDX cells after 48 hr treatment with imatinib with or without JQ1 (1 μM). Data are presented as mean ± SD.
(E) Synergy matrix plot showing δ-scores for X12 PDX cells treated with imatinib + JQ1 (average ZIP synergy score = the average δ-score for the whole range of concentrations shown in the synergy matrix; max ZIP synergy score = maximal score for a specific dose combination).
(F) Growth of X12 PDX cells after 48 hr treatment with imatinib with or without ABT-199 (0.5 μM). Data are presented as mean ± SD.
(G) Synergy matrix plot showing δ-scores for X12 PDX cells treated with imatinib + ABT-199 (average ZIP synergy score = the average δ-score for the whole range of concentrations shown in the synergy matrix; max ZIP synergy score = maximal score for a specific dose combination).
(H) qRT-PCR analysis of BCL2 and MYC mRNA expression levels in ALL-SIL cells after 6 hr of treatment with 500 nM imatinib, 500 nM JQ1, or in combination. Data are presented as mean ± SD. Statistical significance calculated using unpaired two-tailed t test with equal variance.
(I and J) Percentage of human CD45 cells detected by flow cytometry in peripheral blood samples (I) or spleen samples (J) of mice treated for 10 days with JQ1 (50 mg/kg/day), imatinib (100 mg/kg/day), or a combination of imatinib + JQ1. Data are presented as mean ± SD.
(K) Spleen weight of mice treated with JQ1, imatinib, or a combination of imatinib + JQ1. Data are presented as mean ± SD.
(L) Percentage of human CD45 detected by flow cytometry in peripheral blood samples of mice treated with ABT-199 (20 mg/kg/day), imatinib (100 mg/kg/day), or a combination of imatinib + ABT-199. Gray bar indicates treatment period. imatinib versus imatinib + ABT-199: p = 0.0048 (unpaired t test). ABT-199 versus imatinib + ABT-199: p = 0.0027 (unpaired t test). Data are presented as mean ± SD.
(M) Expression levels of MYC (left), BCL2 (middle), and PIM1 (right) in patients from different T-ALL subgroups. Patients harboring the NUP214-ABL1 fusion are represented by an orange star. Statistical significance was calculated using a Mann-Whitney test. Data are presented as mean ± SD.
See also Figure S7.
To test the combination treatment in vivo, we injected NSG mice with X12 PDX cells, and treated these mice for 10 consecutive days with imatinib, JQ1, ABT-199, or a combination of imatinib with JQ1 or ABT-199. Combined treatment with imatinib and JQ1 or ABT-199 reduced leukemia burden significantly more than single agents (Figures 6I–6L, S7M, and S7N). Investigation of expression data from NUP214-ABL1+ T-ALL cases (Liu et al., 2017) confirmed that MYC and BCL2 levels are consistently high in these cases, providing evidence that this subgroup could indeed benefit from combined treatment with imatinib and BET or BCL2 inhibitors (Figure 6M).
In summary, we have shown that STAT5, MYC, and TLX1 cooperatively activate key target genes implicated in proliferation and survival. Inhibition of these downstream targets of STAT5, MYC, and TLX1 together with inhibition of NUP214-ABL1 greatly improves therapy response in relevant T-ALL patient samples and could be a viable treatment option for NUP214-ABL1+ patients (Figure 7).
Figure 7.

Schematic Overview of the Mechanism of Cooperation between STAT5 and TLX1
Discussion
Recent next-generation sequencing studies encompassing large cohorts of T-ALL patient samples have led to the identification of a large number of mutations across multiple genes, indicating that on average ten or more potential oncogenic lesions are found in the leukemia cells at diagnosis (De Keersmaecker et al., 2013, Liu et al., 2017, Seki et al., 2017, Vicente et al., 2015). Based on these large datasets, we are now able to observe specific and recurrent associations between genetic aberrations, implying a degree of reciprocal dependency and potential cooperation. One of the challenges that remains is to functionally assess whether these associations are indeed cooperative and whether they are essential oncogenic drivers of the disease. Subsequent to this confirmation, it is important to determine whether these cooperating mutations drive leukemia development either by initiating separate but complementary changes in the cell or by directly cooperating with each other at the signaling or transcriptional level.
In this study, we reanalyzed genetic data from 457 T-ALL patients (Burmeister et al., 2006, Graux et al., 2004, Graux et al., 2009, Homminga et al., 2011, Liu et al., 2017, Soulier et al., 2005) and confirmed that the NUP214-ABL1-positive subgroup of T-ALL is significantly and almost exclusively associated with ectopic TLX1 or TLX3 expression, suggesting potential cooperation between NUP214-ABL1 signaling and TLX-driven transcriptional deregulation. Previous studies have shown that NUP214-ABL1 or TLX1 expression alone can lead to the development of T-ALL in mouse models, albeit with a long latency. Using a transgenic mouse model, we show that NUP214-ABL1 and TLX1 co-expression indeed accelerates T-ALL development. Both genetic and functional data described here demonstrate that STAT5 is a key player in the cooperation of NUP214-ABL1 with TLX1/TLX3. STAT5 is phosphorylated and activated by NUP214-ABL1, and we show that, in the leukemic context, STAT5 and TLX1 co-bind regulatory regions genome-wide and regulate target gene expression mainly through the activation of otherwise silent/poised and inaccessible enhancers.
Our finding that STAT5 and TLX1 regulate the expression of their target genes mainly through binding of more accessible enhancer regions indicates that leukemia development in this model is mainly driven by cell-identity-specific genes (Hnisz et al., 2013, Zeng et al., 2016). TLX1- and STAT5-occupied regions were bound by typical enhancer marks BRD4 and p300, both of which are necessary for transcriptional elongation, and were also marked by H3K27 acetylation.
MYC is a key oncogenic driver known as a general transcriptional activator with a large number of target genes, affecting many different cellular processes, including cell growth and genomic instability (Adhikary and Eilers, 2005). We document co-binding of STAT5 and TLX1 at the Notch-dependent MYC enhancer (N-ME) 1.4 Mb downstream of the MYC promoter (Herranz et al., 2014, Yashiro-Ohtani et al., 2014). No STAT5-TLX1 binding was detected at the blood enhancer cluster, an enhancer region 1.7 Mb downstream of the MYC promoter important for expression in B cells and granulocytes (Bahr et al., 2018, Shi et al., 2013). Notably, in the current study MYC was found to co-bind enhancer regions with STAT5 and TLX1 across the genome, including the N-ME. Binding of MYC occurred specifically to genes that were also sensitive to imatinib treatment, and depletion of MYC resulted in downregulation of these imatinib-sensitive genes. Taken together, these findings indicate a critical MYC-driven feedforward loop in the upregulation of STAT5/TLX1 target genes implicated in leukemia development. This mechanism of enhancer activation, combined with previously studied effects of TLX1 on NOTCH1 signaling and TCR recombination (Dadi et al., 2012, Durinck et al., 2015, Della Gatta et al., 2012, De Keersmaecker et al., 2010), can begin to explain how NUP214-ABL1 and TLX1 drive the development of T-ALL.
Our findings on the cooperation of NUP214-ABL1 (through STAT5) with TLX1 and MYC also provide opportunities for improved treatment of NUP214-ABL1-positive T-ALL cases. Imatinib treatment to inhibit NUP214-ABL1 kinase activity and STAT5 activation has been tested in several T-ALL patients, with variable success (Clarke et al., 2011, Crombet et al., 2012, Deenik et al., 2009, Koschmieder et al., 2014, Stergianou et al., 2005). Now that we understand that STAT5 and TLX1 occupy enhancer regions and that MYC is an important component of the STAT5/TLX1 complex, inhibition of enhancer activity and/or MYC expression may provide improved therapeutic opportunities for these cases. Treatment of the NUP214-ABL1/TLX1-expressing cell line ALL-SIL and human NUP214-ABL1/TLX3-expressing T-ALL xenografts with the BET inhibitor JQ1 in combination with imatinib showed synergistic anti-leukemia effects. Thus, this combination may be a viable treatment option for NUP214-ABL1-positive T-ALL patients. The use of BET inhibitors could serve a double purpose in these cases by disrupting activated enhancers in general and more specifically MYC expression, since MYC expression depends strongly on the activity of the downstream super-enhancer (Lovén et al., 2013). Clinical trials are currently ongoing for multiple BET inhibitors (Amorim et al., 2016, Berthon et al., 2016, Whitfield et al., 2017). Additionally, combining imatinib with the BCL2-inhibitor ABT-199 could also enhance therapy response.
In conclusion, we demonstrate that the oncogenic kinase NUP214-ABL1, through its downstream effector STAT5, directly cooperates with TLX1 at the transcriptional level. STAT5 and TLX1 selectively increase the accessibility of enhancer regions and drive a feedforward loop at the MYC enhancer, thereby reinforcing the activation of their oncogenic targets. Our data identify MYC and BCL2 as novel targets for therapy in NUP214-ABL1-positive T-ALL.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CD45 (clone 2D1) (APC) | Thermo Fischer Scientific | Cat# 17-9459-42, RRID:AB_10718532 |
| Rat monoclonal anti-CD4 (PE vio770) | Miltenyi Biotec | Cat# 130-102-784 RRID:AB_2659911 |
| Rat monoclonal anti-CD8a (Vioblue) | Miltenyi Biotec | Cat# 130-102-431 RRID:AB_2659889 |
| Rat monoclonal Anti-Gr1 (Vioblue) | Miltenyi Biotec | Cat# 130-102-233, RRID:AB_2659865 |
| Rat monoclonal Anti-Cd11b (APC-vio770) | Miltenyi Biotec | Cat# 130-096-834, RRID:AB_2660135 |
| Rat monoclonal anti-CD4 (clone GK1.5) (PE Cy7) | Thermo Fischer Scientific | Cat# 25-0041-81, RRID:AB_469575 |
| Rat monoclonal anti-CD8a (clone 53-6.7) (APC Cy7) | Thermo Fisher Scientific | Cat# 47-0081-82, RRID:AB_1272185 |
| Armenian hamster monoclonal anti-TCR-b (clone H57-597) (PE) | Thermo Fischer Scientific | Cat# 12-5961-82 RRID:AB_466066 |
| Mouse monoclonal anti-phospho-Stat5 (pY694) (PerCPCy5.5) | BD Biosciences | Cat# 560118, RRID:AB_1645551 |
| Rabbit monoclonal anti-Phospho-Stat5 (Tyr694) (C11C5) | Cell Signaling Technology | Cat# 9359S, RRID:AB_823649 |
| Rabbit polyclonal anti-Stat5 | Cell Signaling Technology | Cat# 9363, RRID:AB_2196923 |
| Mouse monoclonal Anti-β–Actin (Clone AC-15) | Merck | Cat# A5441, RRID:AB_476744 |
| Rabbit polyclonal anti-Stat5 (C17) | Santa Cruz Biotechnology | Cat# sc-835 RRID:AB_632446 |
| Mouse monoclonal anti-Stat5 (A-9) | Santa Cruz Biotechnology | Cat# sc-74442 RRID:AB_1129711 |
| Rabbit monoclonal anti-Stat5 (L-20) | Santa Cruz Biotechnology | Cat# sc-1081 RRID:AB_632448 |
| Rabbit polyclonal anti-HOX11 (C18) | Santa Cruz Biotechnology | Cat# sc-880 RRID:AB_2203789 |
| Rabbit polyclonal anti-MYC | Santa Cruz Biotechnology | Cat# sc-764 RRID:AB_631276 |
| Rabbit anti-EP300 | Santa Cruz Biotechnology | Cat# SC-585X RRID:AB_2616339 |
| Rabbit polyclonal anti-BRD4 | Bethyl | Cat# A301-985A50 RRID:AB_2631449 |
| Mouse monoclonal anti-ETS1 (8A8) | Thermo Fischer Scientific | Cat# MA5-15609, RRID:AB_10978854 |
| Rabbit polyclonal anti-RUNX1 | Active Motif | Cat# 39000 |
| Rabbit anti-H3K4me1 | Active motif | Cat# 39297, RRID:AB_2615075 |
| Rabbit monoclonal anti-H3K4me3 (clone C42D8) | Cell Signaling Technology | Cat# 9751, RRID:AB_2616028 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat# ab4729 RRID:AB_2118291 |
| Rabbit polyclonal Anti-H3K27me3 | Millipore | Cat# 07-449, RRID:AB_310624 |
| Rabbit monoclonal anti- Cre Recombinase (D7L7L) XP® | Cell Signaling Technology | Cat# 15036 |
| Goat polyclonal anti-CD3 epsilon | Santa Cruz Biotechnology | Cat# Sc-1127; RRID:AB_631128 |
| Biological Samples | ||
| Patient-derived xenografts (PDX) | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imatinib (Gleevec) | Selleck Chemicals | Cat# S2475; CAS ID:152459-95-5 |
| JQ1 | Selleck Chemicals | Cat# S7110; CID ID: 1268524-71-5 |
| ABT-199 | MedChem Express | Cat# HY-15531; CAS ID: 1257044-40-8 |
| iBET-762 | Merck | Cat# SML1272; CAS ID: 1260907-17-2 |
| CPI0610 | Axon Med Chem | Cat# 2594; CAS ID: 1380087-89-7 |
| DMSO | Millipore | Cat# 102950; CAS ID: 67-68-5 |
| Polybrene Infection / Transfection Reagent | Merck | Cat# TR-1003-G |
| Recombinant murine IL7 | Peprotech | Cat# 217-17 |
| Recombinant murine IL6 | Peprotech | Cat# 216-16 |
| Recombinant murine IL3 | Peprotech | Cat# 213-13 |
| Recombinant murine SCF | Peprotech | Cat# 250-03 |
| DLL4-Fc | Bornschein et al., 2018 | |
| Mouse monoclonal anti-Human IgG1 Fc | Abcam | Cat# ab1927; RRID:AB_956021 |
| 1M Tris-HCI, pH 8.0 | Thermo Fischer Scientific | Cat# 15568025 |
| Sodium dodecyl sulphate | VWR Chemicals | Cat# 444464T; Cas ID 151-21-3 |
| Sodium deoxycholate | Merck | Cat# D6750; Cas ID: 302-95-4 |
| Glycerol | Thermo Fischer Scientific | Cat# 15514-011 |
| IGEPAL® CA-630 | Merck | Cat# I8896; CAS ID: 9016-45-9 |
| Triton™ X-100 | Merck | Cat# T8787 CAS ID: 9002-93-1 |
| Tween 20 | MP Biomedicals | Cat# 11TWEEN201, CAS ID: 9005-64-5 |
| Tween 40 | Merck | Cat# P1504; CAS ID: 9005-66-7 |
| EDTA | Millipore | Cat# 108421; CAS ID: 6381-92-6 |
| EGTA | Merck | Cat# E3889; CAS ID 67-42-5 |
| NaCl | Fischer Scientific | Cat# S/3160; CAS ID: 7647-14-5 |
| MgCl2 | Merck | Cat# M8266; CAS ID: 7786-30-3 |
| LiCl | Merck | Cat# L4408; CAS ID: 7447-41-8 |
| N-Lauroylsarcosine sodium salt solution 20% | Merck | Cat# L7414; CAS ID: 137-16-6 |
| Formaldehyde | Merck | Cat# 25,254-9; CAS ID: 50-00-0 |
| cOmplete™ Protease Inhibitor Cocktail, EDTA-free | Merck | Cat# COEDTAF-RO |
| Magna ChIP™ Protein A+G Magnetic Beads | Millipore | Cat# 16-663 |
| Glycine Solution (10X) | Cell Signalling Technology | Cat# 7005S |
| ChIP Elution Buffer (2X) | Cell Signalling Technology | Cat# 7009S |
| 5 M NaCl | Cell Signalling Technology | Cat# 7010S |
| Proteinase K | Thermo Fischer Scientific | Cat# EO0491 |
| RNAse A | Thermo Fischer Scientific | Cat# EN0531 |
| Tagmentation enzyme | Illumina | Cat# 15027916 |
| Tagment buffer | Illumina | |
| KAPA HiFi HotStart Ready Mix | KAPA Biosystems | Cat# KK2602 |
| Agencourt AMPure XP beads | Fischer Scientific | Cat# A63881 |
| Resuspension buffer | Illumina | Cat# 15026770 |
| Ethanol absolute | VWR | Cat# 20821.296, CAS ID: 64-17-5 |
| Cell lysis buffer 10x | Cell signaling technology | Cat# 9803 |
| Sodium Orthovanadate | Merck | Cat# 450243, CAS ID: 13721-39-6 |
| cOmplete™ Protease Inhibitor Cocktail | Merck | Cat# CO-RO |
| Digitonin | Merck | Cat# D141; CAS ID: 11024-24-1 |
| Formalin, 10% | Sigma-aldrich | Cat# F5554 |
| Critical Commercial Assays | ||
| GoScript™ Reverse Transcriptase kit | Promega | Cat# A5001 |
| Maxwell® 16 LEV simplyRNA Purification Kit | Promega | Cat# AS1270 |
| GoTaq® qPCR Master Mix | Promega | Cat# A6001 |
| MinElute PCR purification kit | Qiagen | Cat# 28006 |
| ATPlite luminescence system | Perkin Elmer | Cat# 6016949 |
| Easysep Mouse hematopoietic progenitor cell isolation kit | Stem Cell Technologies | Cat# 19856 |
| Western lighting chemiluminescence reagent plus | Perkin Elmer | Cat# NEL105 |
| ImmPRESS HRP Anti-Goat IgG (Peroxidase) Polymer Detection Kit, made in Horse | Maravai Life Sciences | Cat# MP-7405 |
| EnVision+ System | Agilent | Cat# K400011-2 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE102209 |
| Human reference genome GRCh37/hg19 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| Mouse reference genome GRCm38/mm10 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| Experimental Models: Cell Lines | ||
| ALL-SIL | DSMZ | Cat# ACC-511, RRID:CVCL_1805 |
| PEER | DSMZ | Cat# ACC 6, RRID:CVCL_1913 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson lab | Cat# 000664 |
| Mouse: C57BL/6 LSL-NA | GenOway | N/A |
| Mouse: C57BL/6 NA | This paper | N/A |
| Mouse: C57BL/6 NA+TLX1 | This paper | N/A |
| Mouse: C57BL/6 TLX1 | De Keersmaecker et al., 2010 | |
| Mouse: C57/BL/6 CD4-Cre | The Jackson lab | Cat# 017336 |
| Mouse: C57/BL/6 CD2-Cre | Vacchio et al., 2014, The Jackson lab | Cat# 027406 |
| Mouse: C57/BL/6 CD19-Cre | The Jackson lab | Cat# 018958 |
| Oligonucleotides | ||
| Primers for genotyping | This paper | See Table S2 |
| Primers for qRT-PCR | This paper | See Table S3 |
| Negative control antisense LNA gapmer | Qiagen | Cat# LG00000002 |
| TLX1 antisense LNA gapmer | Qiagen | Cat# 339511 |
| STAT5A antisense LNA gapmer | Qiagen | Cat# 339511 |
| STAT5A antisense LNA gapmer | Qiagen | Cat# 339511 |
| STAT5B antisense LNA gapmer | Qiagen | Cat# 339511 |
| STAT5B antisense LNA gapmer | Qiagen | Cat# 339511 |
| Silencer™ Select Negative Control No. 1 siRNA | Thermo Fischer Scientific | Cat# 4390843 |
| Silencer™ Select s9129 MYC siRNA | Thermo Fischer Scientific | Cat# 4392420 |
| MYC siRNA sc-29226 | Santa Cruz Biotechnology | Cat# sc-29226 |
| Recombinant DNA | ||
| MSCV-EML1-ABL1-IRES-GFP | De Keersmaecker et al., 2005 | |
| MSCV-STAT5 N642H-IRES-GFP | Bandapalli et al., 2014 | |
| MSCV-TLX1-IRES-mCherry | This paper | |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | www.flowjo.com |
| GraphPad Prism | Graphpad software | www.graphpad.com |
| qBase+ | Biogazelle | www.qbaseplus.com |
| CLC Main workbench | Qiagen Bioinformatics | www.qiagenbioinformatics.com/products/clc-main-workbench/ |
| Integrative Genomics Viewer (IGV) | Broad Institute | software.broadinstitute.org/software/igv/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| deeptools | Ramírez et al., 2016 | http://deeptools.readthedocs.io/en/latest/ |
| RSAT peak-motifs | Thomas-Chollier et al., 2012 | http://pedagogix-tagc.univ-mrs.fr/rsat/RSAT_portal.html |
| fastq-mcf | Ea-utils | https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md |
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Tophat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTSeq | Anders et al., 2015 | http://htseq.readthedocs.io/en/release_0.9.1/ |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| SynergyFinder | Ianevski et al., 2017 | https://synergyfinder.fimm.fi |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jan Cools (jan.cools@kuleuven.vib.be).
Tg(Lck TLX1) mice were provided by the lab of Adolfo Ferrando, af2196@columbia.edu.
Experimental Model and Subject Details
In Vivo Animal Studies
NSG mice were bred in-house. Tg(Lck TLX1), Tg(CD4-Cre), Tg(CD2-Cre) and Tg(CD19-Cre) mice have been described elsewhere (De Keersmaecker et al., 2010, Lee et al., 2001, Rickert et al., 1997, Sawada et al., 1994, Vacchio et al., 2014). Tg(NUP214-ABL1) mice were developed in collaboration with genOway, France. A LoxP-Stop-LoxP NUP214-ABL1 IRES GFP expression cassette was inserted into the Rosa26 locus via homologous recombination in embryonic stem cells, which were injected into blastocysts (Figure S1A). Crossing the offspring with wild-type females resulted in the creation of a Tg(NUP214-ABL1) mice strain. Tg(NUP214-ABL1) mice were crossed with Tg(CD4 Cre) mice to generate the Tg (CD4 Cre;NUP214-ABL1) mouse model. Tg (CD4 Cre;NUP214-ABL1) mice were crossed with Tg(Lck TLX1) mice to generate the Tg(CD4 Cre;NUP214-ABL1;Lck TLX1) mouse model. Primers for genotyping are listed in Table S2. Leukemia development was monitored by biweekly peripheral blood withdrawal. Secondary and tertiary transplants were performed using 6-8 weeks old in-house bred BL/6 recipient mice. For bone marrow transplant assays, hematopoietic stem/progenitor cells were harvested from in-house bred BL/6 mice using the EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit (Stem Cell Technologies). The cells were transduced with retrovirus for expression of the desired oncogenes, and subsequently injected through tail-vein injection into recipient BL/6 mice. The survival of the mice was recorded daily. All mice were kept in SPF or semi-SPF conditions in the KU Leuven animal facility. For ex vivo assays, cells were cultured in RPMI 1640 medium supplemented with 20% calf serum in 5% carbon dioxide at 37°C. Mouse experiments were approved and supervised by the KU Leuven ethical committee and conducted according to EU legislation (Directive 2010/63/EU).
Patient-Derived Xenograft (PDX) Samples
PDX sample X12 was a kind gift from prof. Jules Meijerinck (Princess Máxima Center for Pediatric Oncology, Utrecht). PDX sample XD82 was a kind gift of prof. Jean-Pierre Bourquin (Department of Oncology and Children's Research Center, University Children's Hospital Zurich, Zurich). Informed consent was obtained from all subjects. Patient PDX characteristics are shown in Figure S7. PDX samples X12 and XD82 were injected in NSG mice through tail vein injection: 1-3.106 cells (resuspended in 300 μL PBS) were injected per mouse. Expansion of the human leukemic cells was monitored by biweekly peripheral blood withdrawal and stained using human CD45 staining (BD Biosciences, 560363). Cells were analysed using MACSQuant Vyb (Miltenyi biotech). All data analysis was carried out using FlowJo software (Treestar). Moribund mice were sacrificed and human leukemic cells were harvested from the spleen. For ex vivo assays, cells were cultured in RPMI 1640 medium supplemented with 20% calf serum in 5% carbon dioxide at 37°C. For in vivo treatment studies, mice were injected with 2.106 X12 patient cells. When engraftment was established, mice were randomized into the different treatment groups (4-5 mice per group) according to the percentage human CD45 detected in peripheral blood. Mice were treated for 10 consecutive days. Imatinib was orally delivered (100 mg/kg/day). JQ1 (50 mg/kg/day) and ABT-199 (20 mg/kg/day) were delivered through intraperitoneal injection. Experiments on human samples were approved and supervised by the UZ Leuven ethical committee.
Ex Vivo Primary T Cells
Ex vivo primary T cells (pro T cells) were generated from freshly harvested mouse (BL/6) hematopoietic stem/progenitor cells as previously described (Bornschein et al., 2018, Gehre et al., 2015). The pro T cells were transduced with retrovirus to express EML1-ABL1-IRES-GFP, TLX1-IRES-mCherry or a combination of both. Then, the cells were grown for 4 days in the presence of immobilized DLL4, but without interleukin 7 (IL7) or stem cell factor (SCF). Cell number, viability and GFP/mCherry percentage was measured daily on a MACSQuant Vyb (Miltenyi).
Cell Lines
ALL-SIL cells (source: male) and PEER cells (source: female) were obtained from DSMZ. The cells were cultured in RPMI 1640 medium supplemented with 20% fetal calf serum (Invitrogen, CA, USA) in 5% carbon dioxide at 37°C.
Method Details
Flow Cytometry Analyses
Single-cell suspensions were prepared from spleen, thymus, bone marrow and lymph nodes. Peripheral blood was incubated in red blood cell lysis buffer (150 mM NH4Cl, 0.1 mM EDTA, 10 mM KHCO3) for 5-10 minutes prior to staining. Cells were then washed with PBS and stained for 20 minutes at 4°C. Stained cells were washed with PBS and then analysed on either a FACSCanto flow cytometer (BD Biosciences), a FACSverse flow cytometer (BD biosciences) or a MACSQuant Vyb (Miltenyi). Data was analysed with the FlowJo software (Tree Star). Antibodies are listed in the Key Resources Table.
Virus Production and Viral Transduction
Viral particles were produced by HEK293T cells transfected (using GeneJuice transfection reagent (Millipore)) with an ecopac packaging plasmid and the retroviral MSCV-EML1-ABL1-IRES-GFP, MSCV-STAT5 N642H-IRES-GFP or MSCV-TLX1-IRES-mCherry expression plasmids. The supernatant carrying the viral particles was harvested after 48 hr.
1.106 primary T cells were seeded in 2 mL RPMI 1640 supplemented with 20% fetal calf serum (Invitrogen), IL7 (20 ng/mL), SCF (20 ng/mL) and polybrene (8 μg/mL). 1 mL viral supernatant was added, and the cells were spinfected at 2500 rpm for 99 minutes at 30°C. 4 hours after spinfection, the cells were transferred to a mDLL4-coated plate, and the medium was changed into RPMI 1640 supplemented with 20% fetal calf serum (Invitrogen), IL7 (20 ng/mL) and SCF (20 ng/mL), to remove polybrene and remaining viral particles.
1.106 Hematopoietic stem/progenitor cells were seeded in 2 mL RPMI 1640 supplemented with 20% fetal calf serum (Invitrogen), IL3 (10 ng/mL), IL6 (10 ng/mL), SCF (50 ng/mL) and polybrene (8 μg/mL). 1 mL viral supernatant was added, and the cells were spinfected at 2500 rpm for 99 minutes at 30°C. 4 hours after spinfection, the medium was changed into RPMI 1640 supplemented with 20% fetal calf serum (Invitrogen), IL3 (10 ng/mL), IL6 (10 ng/mL) and SCF (50 ng/mL), to remove polybrene and remaining viral particles.
1.106 NA+TLX1 spleen cells were seeded in 2 mL RPMI 1640 supplemented with 20% fetal calf serum (Invitrogen) and polybrene (8 μg/mL). 1 mL STAT5 N642H-IRES-GFP viral supernatant was added, and the cells were spinfected at 2500 rpm for 99 minutes at 30°C. 4 hours after spinfection, the medium was changed into RPMI 1640 supplemented with 20% fetal calf serum (Invitrogen), to remove polybrene and remaining viral particles.
RNA Extraction, qRT-PCR and RNA-seq
RNA was extracted from tissue and cells using the Maxwell 16 LEV Simply RNA purification kit (Promega) according to the instructions of the manufacturer. RNA quality was measured using the 2100 BioAnalyzer (Agilent). cDNA synthesis was carried out using GoScript (Promega) and qRT-PCR was performed using the GoTaq qRT-PCR master mix (Promega) with the ViiA7 Real Time PCR system (Applied Biosystem). Primers used for qRT-PCR are listed in Table S3.
Libraries were sequenced on a HiSeq 2500 with 125bp single-end reads (Illumina). This RNA-sequencing data was first cleaned (i.e. removal of adapters and low quality reads) with fastq-MCF (ea-utils) and a quality control was perfomed with fastQC. The reads were then aligned with TopHat2 (Kim et al., 2013, Li et al., 2009) to the respective genomes, either Homo Sapiens (GRCh37/hg19) or Mus Musculus (mm10). To identify the gene expression HTSeq-count (Anders et al., 2015) was used to count the number of reads per gene. These read count numbers were then normalized to the sample size. Differential gene expression analysis was performed with the R-package DESeq2 (Love et al., 2014).
ChIPmentation ChIP-seq
ChIPmentation ChIP-seq was performed as described with minor modifications (Rendeiro et al., 2016, Schmidl et al., 2015). 20-40 million cells were washed in PBS and cross-linked with 1% formaldehyde for 10 min at room temperature and then quenched by addition of glycine (125 mM final concentration). For Nuclei isolation, cells were resuspended in 1X RSB buffer (10 mM Tris pH7.4, 10 mM NaCl, 3 mM MgCl2) and left on ice for 10 min to swell. Cells were collected by centrifugation and resuspended in RSBG40 buffer (10 mM Tris pH7.4, 10 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.5% NP40) with 1/10 v/v of 10% detergent (3.3% w/v sodium deoxycholate, 6.6% v/v Tween-40). Nuclei were collected by centrifugation and resuspended in L3B+ buffer (10 mM Tris-Cl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-Lauroylsarcosine, 0.2% SDS). Chromatin was fragmented to 200-400 bp using 20-25 cycles (30 s on, 30 s off, High) using the Bioruptor (Diagenode). The chromatin was supplemented with 1% Triton-X100 after fragmentation. Antibodies (listed in the Key Resources Table) were preconjugated to either magnetic protein A/G beads (Millipore) or magnetic protein-G beads (Celll Signaling Technology). Chromatin immunoprecipitation was carried out overnight. Tagmentation and library preparation was performed using the Nextera DNA library prep kit (Illumina). DNA was purified using triple sided SPRI bead clean-up (1.2X, 0.6X, 0.9X). (Agencourt AMPure Beads, Beckman Coulter). Sequencing was carried out by Illumina Hiseq 2000 (Illumina, San Diego, CA, USA). The raw ChIPmentation sequencing data was first cleaned with fastq-MCF (ea-utils) after which a quality control was performed with fastQC. Subsequently it was mapped to the human genome (GRCh37/hg19) or to the mouse genome (mm10) with Bowtie2 (Langmead and Salzberg, 2012). The peaks were called with the MACS2 software (Zhang et al., 2008) using the input signal as background. For visualization purposes the ChIP-signals were normalized with deeptools (Ramírez et al., 2014). To discover which motifs are enriched in the ChIP peaks the online tool RSAT peak-motifs was used (Thomas-Chollier et al., 2012).
Fast-ATAC-seq
Fast-ATAC-seq was carried out as described with minor modifications (Corces et al., 2016), Briefly, 100,000 cells were resuspended in transposase mix containing the tagmentation enzyme and 0,1% digitonin. Cells were incubated for 30 min at 37°C. Tagmented DNA was purified using MinElute columns (Qiagen). Library preparation was performed using the Nextera DNA library prep kit, Illumina. DNA was purified using triple-sided SPRI bead clean-up. (Agencourt AMPure Beads, Beckman Coulter) using the following ratio: 1.2X, 0.55X, 1.5X. Sequencing was carried out by Illumina Hiseq 2000 (Illumina, San Diego, CA, USA). The ATAC-sequencing reads were processed similarly as the ChIPmentation data. The peak calling with MACS2 was performed without any background signal. To identify differential peaks, all peak files were merged together with the bedtools software and transformed to a GFF file which could then be used by HTSeq-count to count the number of reads per peak. A differential peak analysis was then performed with the R package DESeq2.
Gene Set Enrichment Analysis
A ranked gene set enrichment analysis was performed with the BROAD GSEA software. For the RNA sequencing data a ranked gene list was constructed from the differential gene expression results, in which the ranking value was calculated as –sign(log2FC)∗log(padj). Different gene sets were used, such as the KEGG pathway database, and custom gene sets like the genes with an appearing ATAC-peak, or genes with certain ChIP-signals.
Western Blotting
Cell lysates were prepared using 1X Cell Lysis Buffer (Cell Signaling) containing protease inhibitor (Complete – EDTA-free, Roche) and 1 mM Na3VO4. Proteins were separated by SDS-PAGE (NuPAGE 3-8% Tris-Acetate or NuPAGE 4-12% Bis Tris, Invitrogen) and transferred to a nitrocellulose membrane using an iBlot Gel Transfer device (Thermo Fischer). Subsequent labelling was carried out using unlabelled primary antibodies. Western blot detection was performed with secondary antibodies conjugated with horseradish peroxidase (GE Healthcare). Images were acquired using a cooled charge-coupled device camera system (ImageQuant LAS-4000, GE Health Care).
RNA Interference
For siRNA studies, 1.106 ALL-SIL cells were electroporated in 400 μL serum-free RPMI 1640 medium supplemented with 4 μL of a 50 μM siRNA solution (final concentration of 200 nM) using the Gene Pulser Xcell™ system (Biorad) in 0,4 cm cuvettes. Immediately after electroporation, cells were transferred to 2 mL pre-warmed RPMI 1640 medium supplemented with 20% fetal calf serum. For antisense oligo knock-down experiments, 1.106 ALL-SIL cells were incubated with 5 μM or 10 μM antisense oligo in RPMI 1640 medium supplemented with 20% fetal calf serum. The medium was changed every 48 hours, and fresh antisense oligo was added each time. 1-2 days after siRNA electroporation or 2-6 days after antisense-oligo treatment, RNA was isolated using the Maxwell 16 LEV Simply RNA purification kit (Promega) according to the instructions of the manufacturer.
Immunohistochemistry
Spleen tissue was fixed overnight in 10% neutral buffered formalin (Sigma) and then transferred to 50% ethanol. The samples were processed for paraffin embedding (Thermo Scientific Excelsior AS Tissue Processor and HistoStar Embedding Workstation). Sections of 4 μm were mounted on Superfrost Plus Adhesion slides (Thermo Fisher Scientific) and stained with hematoxylin and eosin (Diapath). For CD3 and Cre stainings, heat-induced antigen retrieval was performed, and the sections were subsequently blocked with normal goat (Cre) or horse (CD3) serum. The sections were incubated overnight with the primary antibody at 4°C and incubated with the secondary antibody (conjugated with biotin-free HRP) for 30-45 min at room temperature. Visualisation was performed using a peroxidase substrate, and hematoxylin counterstaining was carried out.
In Vitro/Ex Vivo Inhibitor Treatments
For single-drug concentration studies, ALL-SIL, mouse leukemic cells, mouse leukemic cells transduced with an STAT5 N642H expression construct, or patient-derived xenograft cells were incubated overnight with either the small molecule inhibitors (imatinib, JQ1, ABT-199) or DMSO. To determine synergy between the different compounds, ALL-SIL, mouse leukemic cells or patient-derived xenograft cells were seeded into 96-well plates and the compounds were added in a randomized fashion using a D300e digital dispenser (Tecan). The DMSO concentration was normalized. Cell proliferation was measured after 48 hours using the ATPlite luminescence system (PerkinElmer) using a Victor multilabel plate reader. Data were analysed using the SynergyFinder software (Ianevski et al., 2017).
Quantification and Statistical Analysis
All statistical analyses were performed using Prism software (Graphpad software, CA, USA). For analysis of the mouse data, survival was calculated using the Kaplan-Meier method and two-sided p values were determined by the log-rank (Mantel Cox) test.
For qRT-PCR analyses, data are expressed as the mean ± standard deviation (SD). Comparisons between two groups were performed by unpaired Student t tests.
To identify statistical significance of co-occurring ChIP peaks, the regulatory regions of the genome were defined by the H3K27ac signal, and only the peaks within these regulatory regions were taken into account. To determine whether the overlap is greater than expected by chance, a hypergeometric distribution was used to calculate the expected overlap and the p value.
Data and Software Availability
The RNA-seq, ChIP-seq and ATAC-seq data have been deposited in the Gene Expression Omnibus (GEO) database via accession number GSE102209.
Acknowledgments
This work was supported by grants from KU Leuven (PF/10/016 SymBioSys), the Swiss Bridge Award, FWO-Vlaanderen, Foundation against Cancer (2014-120), European Research Counsel (ERC-consolidator 617340), and Interuniversity Attraction Poles (IAP) granted by the Federal Office for Scientific, Technical and Cultural Affairs, Belgium. M.V.B. holds a PhD fellowship strategic basic research of the Research Foundation – Flanders (FWO). M.B., S.B., and J.D.B. hold a PhD fellowship of the Research Foundation – Flanders (FWO).
Author Contributions
Conceptualization, M.V.B., S.D., C.E.d.B., and J.C.; Software, S.D.; Formal analysis, M.V.B., S.D., E.R., C.E.d.B., and J.C.; Investigation, M.V.B., S.D., M.B., S.B., J.D.B., N.M., E.G., R.V., and E.R.; Resources, J.P.M., J.-P.B., B.C.B., and A.E.K.; Writing – Original Draft, M.V.B., S.D., C.E.d.B., and J.C.; Writing – Review and Editing, M.V.B., S.D., M.B., S.B., J.D.B., J.-P.M., C.E.d.B., and J.C.; Funding Acquisition, M.V.B. and J.C.; Supervision, C.E.d.B. and J.C.
Declaration of Interests
The authors declare no competing interests.
Published: August 13, 2018
Footnotes
Supplemental Information includes seven figures and three tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.07.007.
Contributor Information
Charles E. de Bock, Email: charles.debock@kuleuven.vib.be.
Jan Cools, Email: jan.cools@kuleuven.vib.be.
Supplemental Information
References
- Adhikary S., Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Amorim S., 5his A., Gleeson M., Iyengar S., Magarotto V., Leleu X., Morschhauser F., Karlin L., Broussais F., Rezai K. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196–e204. doi: 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyoshi K., Nosaka T., Yamada K., Onishi M., Oka Y., Miyajima A., Kitamura T. Constitutive activation of STAT5 by a point mutation in the SH2 domain. J. Biol. Chem. 2000;275:24407–24413. doi: 10.1074/jbc.M909771199. [DOI] [PubMed] [Google Scholar]
- Bahr C., von Paleske L., Uslu V.V., Remeseiro S., Takayama N., Ng S.W., Murison A., Langenfeld K., Petretich M., Scognamiglio R. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553:515–520. doi: 10.1038/nature25193. [DOI] [PubMed] [Google Scholar]
- Bandapalli O.R., Schuessele S., Kunz J.B., Rausch T., Stutz A.M., Tal N., Geron I., Gershman N., Izraeli S., Eilers J. The activating STAT5B N642H mutation is a common abnormality in pediatric T cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014;99:e188–e192. doi: 10.3324/haematol.2014.104992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassan R., Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J. Clin. Oncol. 2011;29:532–543. doi: 10.1200/JCO.2010.30.1382. [DOI] [PubMed] [Google Scholar]
- Belver L., Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer. 2016;16:494–507. doi: 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- Berthon C., Raffoux E., Thomas X., Vey N., Gomez-Roca C., Yee K., Taussig D.C., Rezai K., Roumier C., Herait P. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–e195. doi: 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- de Bock C.E., Demeyer S., Degryse S., Verbeke D., Sweron B., Gielen O., Vandepoel R., Vicente C., Vanden Bempt M., Dagklis A. HOXA9 cooperates with activated JAK/STAT signaling to drive leukemia development. Cancer Discov. 2018;8:616–631. doi: 10.1158/2159-8290.CD-17-0583. [DOI] [PubMed] [Google Scholar]
- Bornschein S., Demeyer S., Stirparo R., Gielen O., Vicente C., Geerdens E., Ghesquière B., Aerts S., Cools J., de Bock C.E. Defining the molecular basis of oncogenic cooperation between TAL1 expression and Pten deletion in T-ALL using a novel pro-T cell model system. Leukemia. 2018;32:941–951. doi: 10.1038/leu.2017.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister T., Gökbuget N., Reinhardt R., Rieder H., Hoelzer D., Schwartz S. NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood. 2006;108:3556–3559. doi: 10.1182/blood-2006-04-014514. [DOI] [PubMed] [Google Scholar]
- Canté-Barrett K., Spijkers-Hagelstein J.A.P., Buijs-Gladdines J.G.C.A.M., Uitdehaag J.C.M., Smits W.K., van der Zwet J., Buijsman R.C., Zaman G.J.R., Pieters R., Meijerink J.P.P. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T cell acute lymphoblastic leukemia. Leukemia. 2016;30:1832–1843. doi: 10.1038/leu.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S., O’Reilly J., Romeo G., Cooney J. NUP214-ABL1 positive T cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk. Res. 2011;35:e131–e133. doi: 10.1016/j.leukres.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombet O., Lastrapes K., Zieske A., Morales-Arias J. Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with T cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr. Blood Cancer. 2012;59:333–334. doi: 10.1002/pbc.23327. [DOI] [PubMed] [Google Scholar]
- Dadi S., Le Noir S., Payet-Bornet D., Lhermitte L., Zacarias-Cabeza J., Bergeron J., Villarèse P., Vachez E., Dik W.A., Millien C. TLX homeodomain oncogenes mediate T cell maturation arrest in T-ALL via interaction with ETS1 and suppression of TCRα gene expression. Cancer Cell. 2012;21:563–576. doi: 10.1016/j.ccr.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Deenik W., Beverloo H.B., van der Poel-van de Luytgaarde S.C., Wattel M.M., van Esser J.W.J., Valk P.J.M., Cornelissen J.J. Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T cell acute lymphoblastic leukemia. Leukemia. 2009;23:627–629. doi: 10.1038/leu.2008.318. [DOI] [PubMed] [Google Scholar]
- Degryse S., de Bock C.E., Cox L., Demeyer S., Gielen O., Mentens N., Jacobs K., Geerdens E., Gianfelici V., Hulselmans G. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T cell acute lymphoblastic leukemia in a mouse model. Blood. 2014;124:3092–3100. doi: 10.1182/blood-2014-04-566687. [DOI] [PubMed] [Google Scholar]
- Durinck K., Van Loocke W., Van der Meulen J., Van de Walle I., Ongenaert M., Rondou P., Wallaert A., de Bock C.E., Van Roy N., Poppe B. Characterization of the genome-wide TLX1 binding profile in T cell acute lymphoblastic leukemia. Leukemia. 2015;29:2317–2327. doi: 10.1038/leu.2015.162. [DOI] [PubMed] [Google Scholar]
- Della Gatta G., Palomero T., Perez-Garcia A., Ambesi-Impiombato A., Bansal M., Carpenter Z.W., De Keersmaecker K., Sole X., Xu L., Paietta E. Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as tumor suppressor in T-ALL. Nat. Med. 2012;18:436–440. doi: 10.1038/nm.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehre N., Nusser A., von Muenchow L., Tussiwand R., Engdahl C., Capoferri G., Bosco N., Ceredig R., Rolink A.G. A stromal cell free culture system generates mouse pro-T cells that can reconstitute T cell compartments in vivo. Eur. J. Immunol. 2015;45:932–942. doi: 10.1002/eji.201444681. [DOI] [PubMed] [Google Scholar]
- Girardi T., Vicente C., Cools J., De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129:1113–1123. doi: 10.1182/blood-2016-10-706465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graux C., Cools J., Melotte C., Quentmeier H., Ferrando A., Levine R., Vermeesch J.R., Stul M., Dutta B., Boeckx N. Fusion of NUP214 to ABL1 on amplified episomes in T cell acute lymphoblastic leukemia. Nat. Genet. 2004;36:1084–1089. doi: 10.1038/ng1425. [DOI] [PubMed] [Google Scholar]
- Graux C., Stevens-Kroef M., Lafage M., Dastugue N., Harrison C.J., Mugneret F., Bahloula K., Struski S., Grégoire M.J., Nadal N. Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T cell acute lymphoblastic leukemia. Leukemia. 2009;23:125–133. doi: 10.1038/leu.2008.278. [DOI] [PubMed] [Google Scholar]
- Haddy T.B., Mosher R.B., Reaman G.H. Late effects in long-term survivors after treatment for childhood acute leukemia. Clin. Pediatr. (Phila) 2009;48:601–608. doi: 10.1177/0009922809332680. [DOI] [PubMed] [Google Scholar]
- Herranz D., Ambesi-Impiombato A., Palomero T., Schnell S.A., Belver L., Wendorff A.A., Xu L., Castillo-Martin M., Llobet-Navás D., Cordon-Cardo C. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014;20:1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C., Van de Sande B., Potier D., Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40:e114. doi: 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homminga I., Pieters R., Langerak A.W., de Rooi J.J., Stubbs A., Verstegen M., Vuerhard M., Buijs-Gladdines J., Kooi C., Klous P. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19:484–497. doi: 10.1016/j.ccr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Ianevski A., He L., Aittokallio T., Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–2415. doi: 10.1093/bioinformatics/btx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K., Graux C., Odero M.D., Mentens N., Somers R., Maertens J., Wlodarska I., Vandenberghe P., Hagemeijer A., Marynen P. Fusion of EML1 to ABL1 in T cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32) Blood. 2005;105:4849–4852. doi: 10.1182/blood-2004-12-4897. [DOI] [PubMed] [Google Scholar]
- De Keersmaecker K., Rocnik J.L., Bernad R., Lee B.H., Leeman D., Gielen O., Verachtert H., Folens C., Munck S., Marynen P. Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Mol. Cell. 2008;31:134–142. doi: 10.1016/j.molcel.2008.05.005. [DOI] [PubMed] [Google Scholar]
- De Keersmaecker K., Versele M., Cools J., Superti-Furga G., Hantschel O. Intrinsic differences between the catalytic properties of the oncogenic NUP214-ABL1 and BCR-ABL1 fusion protein kinases. Leukemia. 2008;22:2208–2216. doi: 10.1038/leu.2008.242. [DOI] [PubMed] [Google Scholar]
- De Keersmaecker K., Real P.J., Gatta G.D., Palomero T., Sulis M.L., Tosello V., Van Vlierberghe P., Barnes K., Castillo M., Sole X. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat. Med. 2010;16:1321–1327. doi: 10.1038/nm.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K., Atak Z.K., Li N., Vicente C., Patchett S., Girardi T., Gianfelici V., Geerdens E., Clappier E., Porcu M. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T cell acute lymphoblastic leukemia. Nat. Genet. 2013;45:186–190. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleppe M., Lahortiga I., El Chaar T., De Keersmaecker K., Mentens N., Graux C., Van Roosbroeck K., Ferrando A.A., Langerak A.W., Meijerink J.P.P. Deletion of the protein tyrosine phosphatase gene PTPN2 in T cell acute lymphoblastic leukemia. Nat. Genet. 2010;42:530–535. doi: 10.1038/ng.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmieder S., Burmeister T., Brüggemann M., Berkemeier A., Volpert S., Wieacker P., Silling G., Gökbuget N., Müller-Tidow C., Berdel W.E. Molecular monitoring in NUP214-ABL-positive T-acute lymphoblastic leukemia reveals clonal diversity and helps to guide targeted therapy. Leukemia. 2014;28:419–422. doi: 10.1038/leu.2013.272. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P.P., Fitzpatrick D.R., Beard C., Jessup H.K., Lehar S., Makar K.W., Pérez-Melgosa M., Sweetser M.T., Schlissel M.S., Nguyen S. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Buijs-Gladdines J.G.C.A.M., Canté-Barrett K., Stubbs A.P., Vroegindeweij E.M., Smits W.K., van Marion R., Dinjens W.N.M., Horstmann M., Kuiper R.P. IL-7 receptor mutations and steroid resistance in pediatric T cell acute lymphoblastic leukemia: a genome sequencing study. PLoS Med. 2016;13:e1002200. doi: 10.1371/journal.pmed.1002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Easton J., Shao Y., Maciaszek J., Wang Z., Wilkinson M.R., McCastlain K., Edmonson M., Pounds S.B., Shi L. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017;49:1211–1218. doi: 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan C.G., Zhang J., Harvey R.C., Collins-Underwood J.R., Schulman B.A., Phillips L.A., Tasian S.K., Loh M.L., Su X., Liu W. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA. 2009;106:9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinz S., Unser S., Rascle A. Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer. BMC Mol. Biol. 2016;17:10. doi: 10.1186/s12867-016-0063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui C.-H., Yang J.J., Hunger S.P., Pieters R., Schrappe M., Biondi A., Vora A., Baruchel A., Silverman L.B., Schmiegelow K. Childhood acute lymphoblastic leukemia: progress through collaboration. J. Clin. Oncol. 2015;33:2938–2948. doi: 10.1200/JCO.2014.59.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkw257. W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendeiro A.F., Schmidl C., Strefford J.C., Walewska R., Davis Z., Farlik M., Oscier D., Bock C. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat. Commun. 2016;7:11938. doi: 10.1038/ncomms11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert R.C., Roes J., Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada S., Scarborough J.D., Killeen N., Littman D.R., Baniahmad A., Muller M., Steiner C., Renkawitz R., Bienz M., Biggin M.D. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell. 1994;77:917–929. doi: 10.1016/0092-8674(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Schmidl C., Rendeiro A.F., Sheffield N.C., Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods. 2015;12:963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M., Kimura S., Isobe T., Yoshida K., Ueno H., Nakajima-Takagi Y., Wang C., Lin L., Kon A., Suzuki H. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017;49:1274–1281. doi: 10.1038/ng.3900. [DOI] [PubMed] [Google Scholar]
- Shi J., Whyte W.A., Zepeda-Mendoza C.J., Milazzo J.P., Shen C., Roe J.-S., Minder J.L., Mercan F., Wang E., Eckersley-Maslin M.A. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., Dayton B.D., Ding H., Enschede S.H., Fairbrother W.J. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Soulier J., Clappier E., Cayuela J.-M., Regnault A., García-Peydró M., Dombret H., Baruchel A., Toribio M.-L., Sigaux F. HOXA genes are included in genetic and biologic networks defining human acute T cell leukemia (T-ALL) Blood. 2005;106:274–286. doi: 10.1182/blood-2004-10-3900. [DOI] [PubMed] [Google Scholar]
- Stergianou K., Fox C., Russell N.H. Fusion of NUP214 to ABL1 on amplified episomes in T-ALL—implications for treatment. Leukemia. 2005;19:1680–1681. doi: 10.1038/sj.leu.2403877. [DOI] [PubMed] [Google Scholar]
- Thomas-Chollier M., Darbo E., Herrmann C., Defrance M., Thieffry D., van Helden J. A complete workflow for the analysis of full-size ChIP-seq (and similar) data sets using peak-motifs. Nat. Protoc. 2012;7:1551–1568. doi: 10.1038/nprot.2012.088. [DOI] [PubMed] [Google Scholar]
- Vacchio M.S., Wang L., Bouladoux N., Carpenter A.C., Xiong Y., Williams L.C., Wohlfert E., Song K.-D., Belkaid Y., Love P.E. A ThPOK-LRF transcriptional node maintains the integrity and effector potential of post-thymic CD4+ T cells. Nat. Immunol. 2014;15:947–956. doi: 10.1038/ni.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Bempt M., Mentens N., Vandenberghe P., Cools J., De Keersmaecker K. EML1–ABL1 is activated by coiled-coil-mediated oligomerization and induces t cell acute lymphoblastic leukemia or myeloproliferative disease in a mouse bone marrow transplant model. HemaSphere. 2018;2:1. doi: 10.1097/HS9.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie A., Imrichova H., Janky R., Aerts S. iRegulon and i-cisTarget: reconstructing regulatory networks using motif and track enrichment. Curr. Protoc. Bioinformatics. 2015;52 doi: 10.1002/0471250953.bi0216s52. [DOI] [PubMed] [Google Scholar]
- Vicente C., Schwab C., Broux M., Geerdens E., Degryse S., Demeyer S., Lahortiga I., Elliott A., Chilton L., La Starza R. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T cell acute lymphoblastic leukemia. Haematologica. 2015;100:1301–1310. doi: 10.3324/haematol.2015.130179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P., Ferrando A., Reinhardt R., Rieder H., Hoelzer D., Schwartz S. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng A.P., Ferrando A.A., Lee W., Morris J.P., Silverman L.B., Sanchez-Irizarry C., Blacklow S.C., Look A.T., Aster J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Whitfield J.R., Beaulieu M.-E., Soucek L. Strategies to inhibit Myc and their clinical applicability. Front. Cell Dev. Biol. 2017;5:10. doi: 10.3389/fcell.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro-Ohtani Y., Wang H., Zang C., Arnett K.L., Bailis W., Ho Y., Knoechel B., Lanauze C., Louis L., Forsyth K.S. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA. 2014;111:E4946–E4953. doi: 10.1073/pnas.1407079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenatti P.P., Ribeiro D., Li W., Zuurbier L., Silva M.C., Paganin M., Tritapoe J., Hixon J.A., Silveira A.B., Cardoso B.A. Oncogenic IL7R gain-of-function mutations in childhood T cell acute lymphoblastic leukemia. Nat. Genet. 2011;43:932–939. doi: 10.1038/ng.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X., Willi M., Shin H.Y., Hennighausen L., Wang C. Lineage-specific and non-specific cytokine-sensing genes respond differentially to the master regulator STAT5. Cell Rep. 2016;17:3333–3346. doi: 10.1016/j.celrep.2016.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.