

Summary
The discovery of mutant or fusion kinases that drive oncogenesis, and the subsequent approval of specific inhibitors for these enzymes, has been instrumental in the management of some cancers. However, acquired resistance remains a significant problem in the clinic, limiting the long-term effectiveness of most of these drugs. Here we demonstrate a general strategy to overcome this resistance through drug-induced MEK cleavage (via direct procaspase-3 activation) combined with targeted kinase inhibition. This combination effect is shown to be general across diverse tumor histologies (melanoma, lung cancer, and leukemia) and driver mutations (mutant BRAF or EGFR, fusion kinases EML4-ALK and BCR-ABL). Caspase-3-mediated degradation of MEK kinases results in sustained pathway inhibition and substantially delayed or eliminated resistance in cancer cells in a manner far superior to combinations with MEK inhibitors. These data suggest the generality of drug-mediated MEK kinase cleavage as a therapeutic strategy to prevent resistance to targeted anticancer therapies.
Keywords: Caspase activation, cancer, targeted therapy, kinases, resistance, apoptosis
TOC image
Rapid onset of resistance to targeted kinase inhibitors limits their use in treating advanced cancers. Peh et al. show that combination of diverse kinase inhibitors with a procaspase-3 activating compound (PAC-1), leads to degradation of MEK1/2, dramatically delaying acquired resistance.
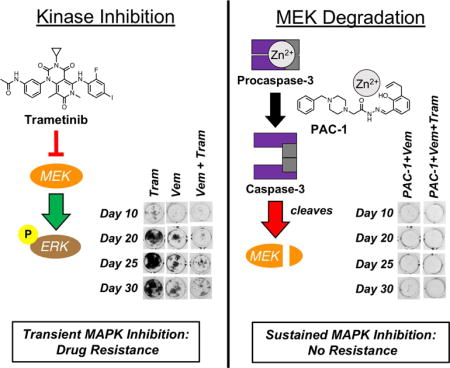
Introduction
Overexpression (Leicht et al., 2007; Paul and Mukhopadhyay, 2004), mutation (Vogelstein et al., 2013), or fusion (Mertens et al., 2015; Stransky et al., 2014) of kinases that affect cell proliferation and survival pathways drive tumorigenesis in numerous cancers. Specific targeting of these oncogenic kinases with inhibitors has led to dramatic responses in large fractions of patients with advanced disease (Gharwan and Groninger, 2016; Gross et al., 2015). However, response to kinase inhibitors is often short-lived due to the rapid onset of resistance to these drugs (Chong and Janne, 2013; Daub et al., 2004; Groenendijk and Bernards, 2014; Holohan et al., 2013). Various resistance mechanisms exist to reactivate the cell proliferation and survival pathways. In particular, reactivation of the mitogen-activated protein kinase (MAPK) pathway is responsible for acquired resistance to a large number of clinically approved inhibitors, including those targeting mutant BRAF (Lito et al., 2013; Wagle et al., 2011), mutant EGFR (Gazdar, 2009), EML4-ALK (Lin et al., 2017), or BCR-ABL (Hare et al., 2007) kinases.
Recognizing that reactivation of the MAPK pathway diminishes the clinical efficacy of kinase inhibitors, and that MEK1/2 kinases are the ultimate gatekeeper kinases of the MAPK pathway (Caunt et al., 2015), upfront combination therapy with a MEK1/2 inhibitor (e.g. trametinib or cobimetinib) has been investigated with several classes of kinase inhibitors in an effort to delay resistance (Eberlein et al., 2015; Hrustanovic et al., 2015; Ma et al., 2014; Tanizaki et al., 2012; Tricker et al., 2015). Clinically, the combination of MEK1/2 and mutant BRAF inhibitors extends progression-free and overall survival in the treatment of metastatic BRAFV600E melanomas (Ascierto et al., 2016; Long et al., 2015). However, resistance to this dual therapy invariably occurs after a year of therapy initiation, in part due to secondary mutations on MEK1 and MEK2 kinases that abolish anticancer efficacy (Long et al., 2014; Moriceau et al., 2015; Shi et al., 2014; Wagle et al., 2011).
Given the transient and differential inhibition of MEK1/2 activity with the clinically used inhibitors (Gilmartin et al., 2011; Woodfield et al., 2016), we hypothesized that combination therapy with a small molecule capable of inducing enzymatic degradation of MEK1/2 kinases would have an advantage over direct inhibition, resulting in low-or-no resistance when used with a wide range of clinically approved kinase inhibitors. Detailed proteomics experiments have shown that MEK1/2 kinases are cleaved by caspase-3 during apoptosis (Dix et al., 2008; Mahrus et al., 2008), and it has been widely reported that procaspase-3 is overexpressed in a variety of cancers relative to healthy tissues (Fink, 2001; Nakopoulou et al., 2001; Persad et al., 2004; Putt et al., 2006; Roth and Hergenrother, 2016; Sadowska et al., 2014). While evasion of apoptosis, through a variety of mechanisms, is regarded as a hallmark of cancer (Hanahan and Weinberg, 2011), studies suggest that overexpression of procaspase-3 can drive oncogenesis (Cartwright et al., 2017; Ichim et al., 2015; Liu et al., 2015). These observations imply that activation of procaspase-3 to caspase-3 and subsequent caspase-3 mediated degradation of MEK can occur selectively in cancer cells relative to healthy cells. An additional advantage of direct procaspase-3 activation is the ability to bypass defects in the apoptotic circuitry commonly found upstream of procaspase-3 in cancer cells (Johnstone et al., 2002; Pommier et al., 2004).
PAC-1 is a selective procaspase-3 activating compound that synergizes with vemurafenib, a BRAFV600E inhibitor, in numerous melanoma cell lines harboring the V600E mutation in BRAF to delay onset of acquired resistance (Peh et al., 2016), suggesting the feasibility of this strategy. Here we assess PAC-1 in combination with four different clinically approved inhibitors targeting four different kinases that signal through the MAPK pathway. These combinations dramatically enhance caspase-3 activity and induce degradation of MEK1/2 kinases. We report that adding PAC-1 to kinase inhibitors targeting BRAFV600E (vemurafenib), EGFRT790M (osimertinib), EML4-ALK (ceritinib), or BCR-ABL (imatinib) enhances MEK1 and MEK2 degradation, leading to durable inhibition of MEK1/2 and ERK1/2 phosphorylation, enhanced apoptotic cell death, and markedly delayed or eliminated acquired resistance. As PAC-1 is currently being evaluated in clinical trials in human cancer patients (NCT02355535, NCT03332355), the results presented herein can be rapidly translated to combination clinical trials with the various targeted kinase inhibitors, studies that could provide significant benefit to cancer patients.
Results
Caspase-3 activity is significantly enhanced in cells treated with PAC-1 and diverse kinase inhibitors
As shown in Figs. 1A and B, PAC-1 significantly increases the caspase-3 activity in osimertinib-treated non-small cell lung cancer (NSCLC) cell lines H1975 (EGFRL858R+T790M) and PC-9 GR (EGFRex19del+T790M). Increased PARP-1 cleavage and disappearance of the procaspase-3 band were also observed in both cell lines when treated with the combination (Figs. 1A and 1B). A similar effect is also observed in H3122 NSCLC cells (harboring the EML4-ALK fusion) co-treated with PAC-1 and ceritinib (Fig. 1C) and K-562 chronic myelogenous leukemia (CML) cells (harboring the BCR-ABL fusion) treated with PAC-1 and imatinib (Fig. 1D). The enhancement of caspase-3 activity observed at longer timepoints (Fig. 1) was determined to be synergistic using a two-way ANOVA test. As a result of increased caspase-3 activity, a significantly larger population of cells treated with the combination of PAC-1 and osimertinib/ceritinib/imatinib die via apoptosis (Figs. S1A–C). Collectively these results demonstrate that, in addition to general cytotoxins and inhibitors against BRAFV600E (Botham et al., 2016; Huang et al., 2016; Peh et al., 2016; Razi et al., 2015), PAC-1 is able to broadly enhance the caspase-3 activity of kinase inhibitors targeted to EGFRT790M, EML4-ALK, and BCR-ABL.
Figure 1.
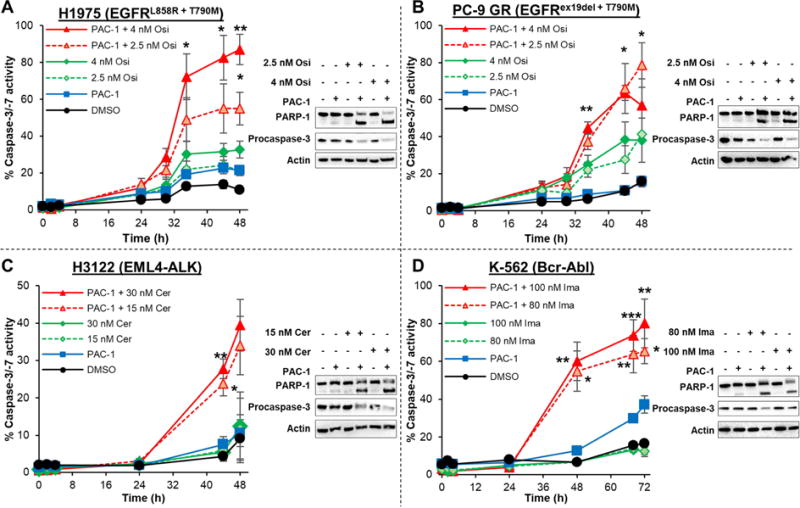
Enhancement of caspase-3 activity following co-treatment of cancer cells with PAC-1 and diverse targeted kinase inhibitors. Negligible increases in caspase-3 activity or PARP-1 cleavage was observed in (A) H1975 and (B) PC-9 GR NSCLC cells treated with DMSO, single-agent PAC-1 (5 μM) or osimertinib. In cells treated with PAC-1 + osimertinib, dramatic increases in caspase-3 activity was observed as early as 36 h post-treatment. Significant PARP-1 cleavage and reduction in procaspase-3 levels were observed after 48 h, consistent with results obtained from the caspase-3 activity assay. (C) H3122 NSCLC cells were treated with PAC-1 (5 μM) + ceritinib for varying periods of time and a significant increase in caspase-3 activity is observed. Increased PARP-1 cleavage and reduction in procaspase-3 levels were also observed after a 48 h treatment. (D) Significant enhancement of caspase-3 activity was also observed in K-562 cells treated with PAC-1 (7.5 μM) + imatinib with negligible single-agent activity. Following 48 h of PAC-1 + imatinib treatment, increased PARP-1 cleavage and procaspase-3 activation were also observed. Values shown are averages of at least 3 experiments, error bars are s.e.m., p values shown for two-way ANOVA analysis to determine if the combination is different from an additive effect of individual agents are statistically different (* p<0.05, ** p<0.01, *** p<0.001). See also Figure S1.
Caspase-3 activity leads to degradation of MEK kinases
Activation of an executioner caspase such as caspase-3 leads to the cleavage of hundreds of proteins in the cell. Intriguingly, the protein substrates for caspase-3 tend to be found in protein complexes or signaling pathways that govern cell fate and survival (Mahrus et al., 2008). Proteome-wide identification of caspase-3 substrates by the Wells (Mahrus et al., 2008) and Cravatt (Dix et al., 2008) laboratories have independently shown that both MEK1 and MEK2 kinases are cleaved during caspase-3-mediated apoptosis. Moreover, it has also been shown that MEK1 and MEK2 are the only kinases that phosphorylate ERK1/2 (Roskoski Jr, 2012; Shaul and Seger, 2007), serving as the critical gatekeepers of ERK1/2 activity (Caunt et al., 2015). Given the observation that addition of PAC-1 to diverse kinase inhibitors leads to enhanced apoptosis, we hypothesized that the dramatic increase in caspase-3 activity leads to MEK1 and MEK2 degradation, inhibiting downstream pro-survival signaling.
To investigate this hypothesis, levels of MEK1 and MEK2 kinases in BRAFV600E cell lines were probed following treatment with PAC-1 + vemurafenib or trametinib + vemurafenib. In both A375 and SK-MEL-5 cells treated with PAC-1 + vemurafenib for 48 h, dramatic reduction in procaspase-3, MEK1, and MEK2 levels were observed, suggesting that procaspase-3 activation led to MEK1 and MEK2 degradation (Fig. 2A). In contrast, when these two cell lines are treated with trametinib and vemurafenib, no observable change in the levels of procaspase-3, MEK1, and MEK2 was detected (Fig. 2A). It is worth noting that MEK1/2 cleavage products are transiently stable (Dix et al., 2008), making their detection after a 48 hour treatment challenging.
Figure 2.
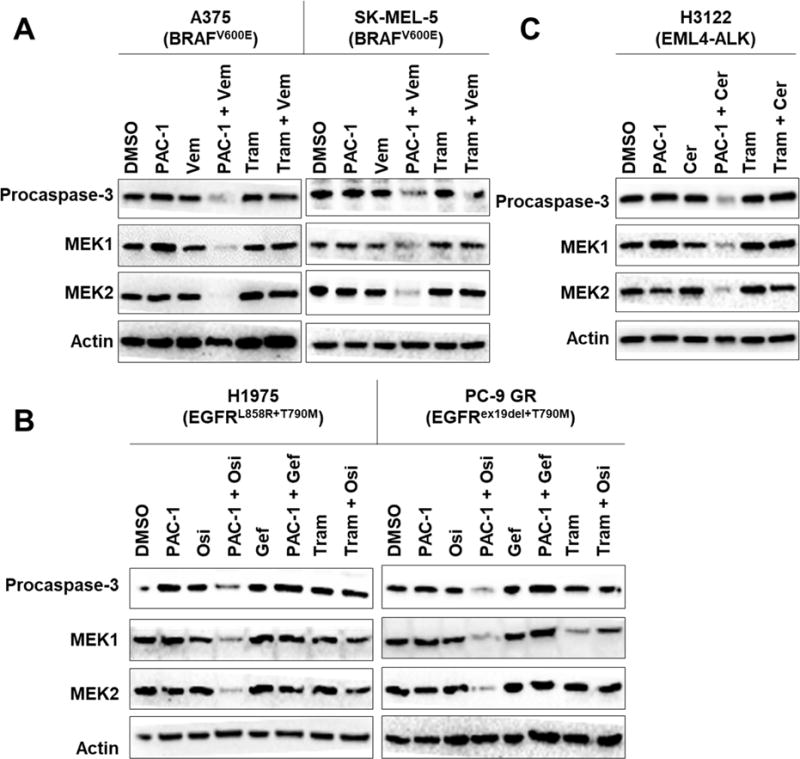
PAC-1 combination therapies lead to caspase-3 activation and degradation of MEK1 and MEK2 kinases. (A) Procaspase-3 activation leads to dramatic reduction in MEK1 and MEK2 levels in A375 and SK-MEL-5 cells treated with PAC-1 (5 μM) + vemurafenib (10 μM) for 48 hours. This reduction was not observed in cells treated with trametinib (30 nM) + vemurafenib. (B) H1975 and PC-9 GR cells treated with PAC-1 (5 μM) + osimertinib (4 nM) for 48 hours led to procaspase-3 activation and corresponding degradation of MEK1 and MEK2 kinases. This was not observed in cells treated with PAC-1 + gefitinib (4 nM) or trametinib (30 nM) + osimertinib. (C) MEK1 and MEK2 degradation were similarly observed in H3122 cells treated with PAC-1 (5 μM) + ceritinib (30 nM) for 48 hours but not in cells treated with trametinib (30 nM) + ceritinib. See also Figure S2 and S3.
To investigate the generality of these results, changes in procaspase-3, MEK1, and MEK2 levels in EGFRT790M, EML4-ALK, and BCR-ABL cells were assessed following combination treatment with PAC-1 and kinase inhibitors specific for those alterations. In both H1975 and PC-9 GR cells, treatment with PAC-1 and osimertinib (4 nM) led to dramatic reduction in procaspase-3, MEK1, and MEK2 levels, but not when the inactive inhibitor gefitinib (Gazdar, 2009) (Fig. S2A) was used (Fig. 2B). Using a lower concentration of osimertinib (2.5 nM) also led to degradation of MEK1 and MEK2 kinases (Fig. S2B). Importantly, co-treatment with trametinib and osimertinib did not lead to reduction in MEK1 and MEK2 levels in either cell lines (Fig. 2B), similar to that observed in BRAFV600E cell lines. Degradation of MEK1 and MEK2 was also seen in H3122 cells when they were co-treated with PAC-1 + ceritinib (Fig. 2C), even when at reduced concentrations of ceritinib (Fig. S2C). Co-treatment with trametinib and ceritinib did not lead to reduction in MEK1 and MEK2 levels in H3122 cells (Fig. 2C), consistent with data obtained in BRAFV600E and EGFRT790M cells. Extensive procaspase-3 activation was observed in K-562 cells treated with either PAC-1 + imatinib or trametinib + imatinib, and degradation of MEK1 and MEK2 kinases was also observed in both samples (Fig. S2D). Varying the concentration of imatinib used also led reduction in MEK1 and MEK2 levels (Fig. S2E).
To provide further mechanistic understanding on the importance of caspase-3 activity in mediating the synergistic cell death observed in the PAC-1/drug combination, Q-VD-OPh, a general caspase inhibitor, was used. In these experiments protection of apoptotic cell death (Fig. S3A–D) as well as attenuated MEK1/2 cleavage (Fig. S3E–G) were both observed when Q-VD-OPh was added concurrently to cells treated with PAC-1/drug combinations. These results suggest that inhibition of caspase-3 activity, in particular, its activity in cleaving MEK1 and MEK2 kinases, was sufficient to abolish the dramatic synergy observed in cells treated with the PAC-1/drug combinations. Taken together, our results indicate the importance of caspase-3-induced MEK1/2 cleavage in mediating the synergistic apoptotic cell death, and the generality of this observation in diverse cancer types.
Degradation of MEK1 and MEK2 kinases leads to sustained inhibition of MEK1/2 and ERK1/2 phosphorylation
Significant inhibition (>80%) of ERK1/2 phosphorylation is necessary for the clinical efficacy of targeted kinase inhibitors like vemurafenib (Bollag et al., 2010). Since reactivation of ERK1/2 phosphorylation is commonly observed in resistant tumors (Lito et al., 2013; Wagle et al., 2011), a MEK1/2 inhibitor has been added to the treatment regimen to achieve sustained ERK1/2 inhibition. While clinically approved MEK1/2 inhibitors are effective in preventing ERK1/2 phosphorylation, the inhibition of ERK1/2 activity disrupts the negative feedback on RAF, resulting in RAF hyper-activation and hyper-phosphorylation of MEK1/2 (Caunt et al., 2015). The rebound in MEK1/2 phosphorylation subsequently leads to pathway reactivation. Development of “feedback buster” MEK1/2 inhibitors such as trametinib (Hatzivassiliou et al., 2013; Lito et al., 2014) is intended to mitigate the rebound but that effect is transient (Gilmartin et al., 2011). Therefore, sustained inhibition of both MEK1/2 and ERK1/2 remains challenging despite the availability of numerous MEK1/2 inhibitors.
Given that enhanced caspase-3 activity led to degradation of MEK1/2 kinases, we hypothesized that PAC-1 combination therapies would lead to sustained inhibition of MEK1/2 and ERK1/2 phosphorylation. Addition of PAC-1 to vemurafenib led to inhibition of ERK1/2 phosphorylation in both A375 (Figs. 3A and 3B) and SK-MEL-5 cells (Fig. 3A), consistent with our previous work (Peh et al., 2016). While sustained inhibition of MEK1/2 phosphorylation was observed when cells were treated with vemurafenib + PAC-1 due to the degradation of MEK1 and MEK2 kinases, a dramatic rebound in MEK1/2 phosphorylation was rapidly seen in trametinib + vemurafenib treated cells (Fig. 3B). This observation is consistent with a previous report detailing the transient (6 h) effect of trametinib in inhibiting MEK1/2 phosphorylation in mutant BRAF melanoma cells (Gilmartin et al., 2011). These results suggest the distinct advantage of drug-induced degradation of MEK1 and MEK2 kinases as an effective strategy to inhibit both MEK1/2 and ERK1/2 activity.
Figure 3.
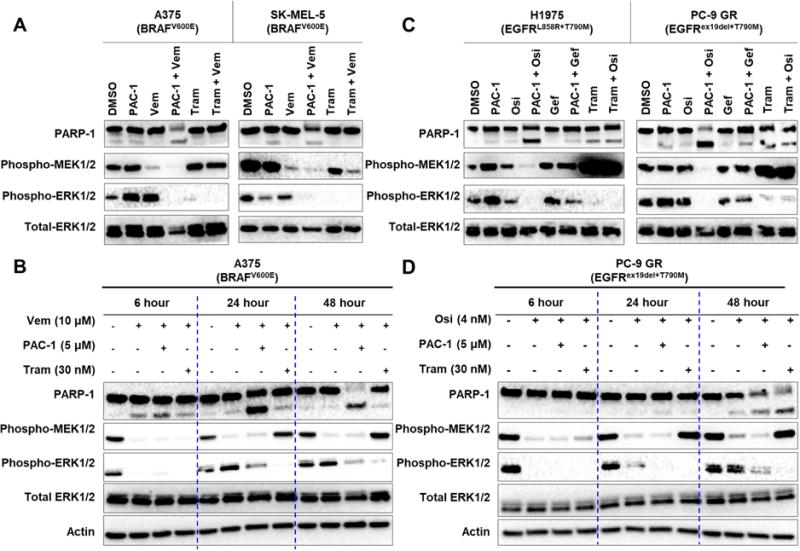
Cells treated with PAC-1 and vemurafenib or osimertinib have sustained inhibition of MAPK signaling. (A) A375 and SK-MEL-5 melanoma cells were treated with PAC-1 (5 μM), vemurafenib (10 μM), trametinib (30 nM), or the indicated combinations for 48 h. Inhibition of both ERK1/2 and MEK1/2 phosphorylation was only observed in cells treated with PAC-1 + vemurafenib. (B) Timecourse of phospho-MEK1/2 and phospho-ERK1/2 inhibition in A375 cells treated with DMSO, vemurafenib, vemurafenib + PAC-1 or vemurafenib + trametinib for 6, 24, or 48 hours. (C) After 48 hours of treatment with PAC-1 (5 μM) + osimertinib (4 nM), sustained inhibition of both MEK1/2 and ERK1/2 phosphorylation were observed in H1975 and PC-9 GR cells. Treatment with PAC-1 + gefitinib (4 nM) for a similar time period did not lead to similar observations. Sustained inhibition of MEK1/2 phosphorylation was also not observed in cells treated with trametinib (30 nM) + osimertinib. (D) Timecourse of phospho-MEK1/2 and phospho-ERK1/2 inhibition in PC-9 GR cells treated with DMSO, osimertinib, osimertinib + PAC-1 or osimertinib + trametinib for 6, 24, or 48 hours. See also Figure S4.
To explore the generality of this effect, H1975 and PC-9 GR cells were treated with PAC-1 and osimertinib and probed for changes ERK1/2 and MEK1/2 phosphorylation. As seen in Figs. 3C and 3D, EGFRT790M cells treated with PAC-1 and osimertinib led to sustained loss of ERK1/2 and MEK1/2 phosphorylation, as a result of MEK1 and MEK2 degradation. Using PAC-1 and gefitinib, no corresponding reduction in ERK1/2 or MEK1/2 phosphorylation is observed (Fig. 3C), indicating that the effect with osimertinib is specific for the EGFRT790M target. At a lower concentration of osimertinib, sustained inhibition of MEK1/2 and ERK1/2 phosphorylation (Fig. S4A) corresponding to degradation of MEK1 and MEK2 kinases was also observed (Fig. S2B). Rapid rebound of MEK1/2 phosphorylation similarly occurs within 24 hour of treatment with osimertinib and trametinib but not in cells treated with osimertinib and PAC-1 (Fig. 3D), mirroring the effect seen in BRAFV600E cells.
In a similar fashion, H3122 cells (EML4-ALK) were treated with PAC-1 + ceritinib or trametinib + ceritinib and probed for changes in ERK1/2 and MEK1/2 phosphorylation. In this case, H3122 cells co-treated with PAC-1 + ceritinib for 48 hours also led to sustained reduction in ERK1/2 and MEK1/2 phosphorylation (Figs. S4B and S4D), which can be attributed to the caspase-3-mediated degradation of MEK1 and MEK2 kinases. When used in combination with PAC-1, even lower concentration of ceritinib cause sustained inhibition of MEK1/2 and ERK1/2 phosphorylation (Fig. S4C), due to degradation of MEK1 and MEK2 kinases (Fig. S2C). Similarly, transient inhibition of MEK1/2 phosphorylation was also observed in H3122 cells treated with trametinib and ceritinib (Fig. S4D), consistent with results seen in BRAFV600E and EGFRT790M cells.
Finally, in K-562 cells expressing BCR-ABL, co-treatment with PAC-1 and imatinib also lead to sustained inhibition of ERK1/2 and MEK1/2 phosphorylation (Fig. S4E) due to extensive degradation of MEK1 and MEK2 kinases (Fig. S2D). It should be noted that rebound of MEK1/2 phosphorylation was not observed in cells treated with trametinib + imatinib for 48 hours (Fig. S4E), since degradation of MEK1 and MEK2 kinases was also observed in this cell line (Fig. S2D). Similarly, varying the concentration of imatinib used also led reduction in phospho-MEK1/2 and phospho-ERK1/2 levels (Fig. S4F) due to degradation of MEK1 and MEK2 (Fig. S2E). Collectively, our results demonstrate the ability of PAC-1, in combination with diverse kinase inhibitors, to provide sustained inhibition of ERK1/2 and MEK1/2 phosphorylation, a result generally not observed in the combinations of these targeted kinase inhibitors with the MEK1/2 inhibitor trametinib.
PAC-1 + vemurafenib is more efficacious than trametinib + vemurafenib in eliminating acquired resistance
Knowing that secondary activating mutations on MEK kinases are commonly found in melanomas resistant to BRAFi + MEKi (Long et al., 2014; Moriceau et al., 2015; Shi et al., 2014; Wagle et al., 2011), we hypothesized that PAC-1 + vemurafenib would be more efficacious than trametinib + vemurafenib in delaying resistance in A375 cells. The rationale behind the hypothesis is that PAC-1 + vemurafenib significantly enhances apoptotic cell death leading to degradation of MEK kinases to further inhibit ERK1/2 phosphorylation. To test this hypothesis, A375 cells were treated with PAC-1, vemurafenib, trametinib, and their respective combinations for up to 30 days. Consistent with previous work, resistant colonies were visibly present in A375 cells treated with single-agent vemurafenib as early as 20 days post treatment (Figs. 4A and 4B). In cells treated with trametinib + vemurafenib, resistant colonies were first noted after 25 days of continuous treatment. Following 30 days of treatment, more resistant colonies were visible in A375 cells treated with trametinib and vemurafenib, indicating the presence of BRAFi + MEKi resistance (Figs. 4A and 4B). However, emergence of resistant colonies were not observed in A375 cells treated with PAC-1 and vemurafenib in the presence or absence of trametinib following 30 days of treatment (Figs. 4A and 4B), indicating that the double or triple combination of PAC-1 is significantly more effective in eliminating the onset of vemurafenib resistance as compared to the clinically used BRAFi + MEKi combination.
Figure 4.

PAC-1 combination therapies substantially delay or eliminate acquired resistance. (A) A375 cells were treated with indicated concentrations of PAC-1, vemurafenib, trametinib, or the respective combinations for up to 30 days. Cells were fixed and stained with SRB dye before imaging. (B) Quantification of (A), data reported is the mean and standard error of three independent experiments. (C) PC-9 GR cells were treated with indicated concentrations of PAC-1, osimertinib, trametinib, or the respective combinations for up to 35 days. Cells were imaged as described in (B). See also Figure S5. (D) Quantification of (C), data reported is the mean and standard error of two independent experiments. (E) H3122 cells were treated with indicated concentrations of PAC-1, ceritinib, trametinib, or the respective combinations for up to 32 days. Image shown is representative of two independent experiments. (F) Zoom in view of H3122 cells treated PAC-1 + ceritinib, or trametinib + ceritinib for 32 days. Visibly more resistant colonies were seen in cells treated to trametinib + ceritinib compared to PAC-1 + ceritinib.
The combination of PAC-1 and osimertinib/ceritinib is effective in eliminating resistance in EGFRT790M and EML4-ALK cells
The ability of the combination of PAC-1 and osimertinib to delay acquired resistance in EGFRT790M cells was investigated. In this case, H1975 and PC-9 GR cells were treated with indicated concentrations of PAC-1 and/or osimertinib for up to 28 days. In both cell lines, 8 days of single-agent PAC-1 (2 μM) treatment had minimal cytotoxic effect compared to DMSO-treated samples. On the other hand, both single-agent osimertinib (30 nM) and the combination of PAC-1 and osimertinib were very effective inhibiting cell proliferation (Fig. S5). Following 28 days of drug treatment, resistant clones were clearly visible in PC-9 GR and H1975 cells treated only with osimertinib, in contrast to cells treated with both PAC-1 and osimertinib (Fig. S5). These results suggest that the combination of PAC-1 and osimertinib is effective in dramatically delaying or eliminating the onset of osimertinib resistance in EGFRT790M cell lines. Experiments were then conducted to compare the PAC-1 + osimertinib combination versus trametinib + osimertinib in delaying resistance in PC-9 GR cells. In this case, PAC-1 or trametinib (5 nM) as single agents had minimal cytotoxic effect as compared to DMSO-treated cells following 8 days of treatment. As expected, treatment with osimertinib, PAC-1 + osimertinib, or trametinib + osimertinib for 8 days was effective in inhibiting cell proliferation. Consistent with Fig. S5A, resistant clones were visible in PC-9 GR cells after 28 days of treatment with single-agent osimertinib but not in cells treated with PAC-1 and osimertinib (Figs. 4C and D). No resistant clones were also visible in cells treated with trametinib and osimertinib after 28 days. While there was a dramatic increase in the number of resistant clones present in cells treated with single-agent osimertinib after 35 days of treatment, no resistant clones were observable in cells treated with either PAC-1 + osimertinib or trametinib + osimertinib (Figs. 4C and D). This observation suggests that the combination of PAC-1 + osimertinib is equipotent, but not more efficacious in delaying resistance as trametinib + osimertinib.
Finally, the ability of PAC-1 + ceritinib to delay acquired resistance in EML4-ALK cells was investigated. Here, H3122 cells were treated with indicated concentrations of PAC-1, trametinib, ceritinib, or the respective combinations for up to 32 days. Single-agent PAC-1 (2 μM) or trametinib (5 nM) treatment had minimal cytotoxic effect compared to DMSO-treated samples after 8 days of treatment. Treatment with ceritinib, PAC-1 + ceritinib, or trametinib + ceritinib for 8 days was effective in inhibiting cell proliferation. (Fig. 4E). Resistant clones were visible in H3122 cells after 20 days of treatment with single-agent ceritinib but not in cells treated with either PAC-1 and osimertinib or trametinib and ceritinib (Fig. 4E). After 32 days of treatment, there was a dramatic increase in the number of resistant clones present in cells treated with single-agent ceritinib but few resistant clones were observable in cells treated with PAC-1 + ceritinib (Fig. 4E). In cells treated with trametinib + ceritinib, a number of resistant clones were clearly visible, indicating the presence of ALKi + MEKi resistant H3122 cells (Figs. 4E and 4F).
In summary, the combination of kinase inhibitors targeting BRAFV600E, EGFRT790M, EML4-ALK, and BCR-ABL with PAC-1, leads to enhanced procaspase-3 activation and degradation of MEK1 and MEK2 kinases (Fig. 5). The degradation of MEK kinases then leads to sustained inhibition of MEK1/2 and ERK1/2 signaling. The combined effect of increased apoptotic cell death and sustained inhibition of the MAPK pathway that is observed in the PAC-1 combination therapies work in tandem to dramatically delay or eliminate resistance.
Figure 5.
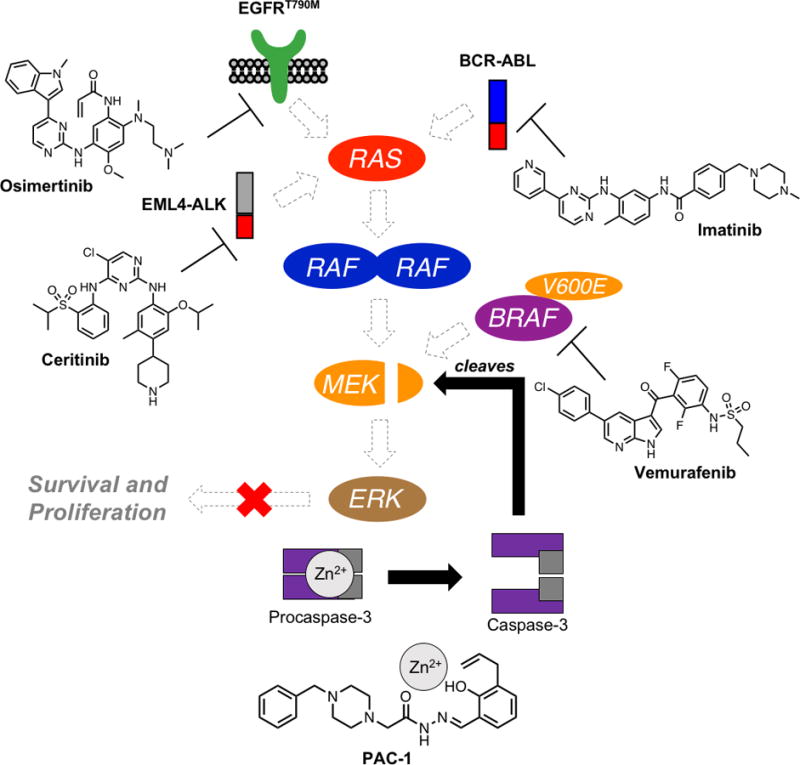
Proposed mechanism of action of PAC-1 combination therapy with clinically approved kinase inhibitors examined in this work. Shown kinase inhibitors target key oncogenic driver kinases, leading to transient inhibition of signaling through MEK. Of critical importance is the ability of PAC-1 treatment to induce cleavage of MEK kinases. This MEK cleavage, in conjunction with upstream pathway inhibition, potently abolishes ERK phosphorylation and hinders pro-survival and proliferation signaling.
Discussion
Significant progress has been made in understanding the mechanisms of acquired resistance to targeted kinase inhibitors. This understanding has translated into combination therapies for BRAFV600E melanomas and next-generation inhibitors for mutant EGFR and fusion EML4-ALK and BCR-ABL kinases. Unfortunately, cancer cells rapidly circumvent inhibition by these next-generation inhibitors via alternative resistance mechanisms, necessitating the development of newer drugs to combat resistant tumors. Moreover, a large proportion of drug-induced resistance remains unexplained, meaning that newer drugs only benefit a small population of patients with molecularly defined resistance mechanisms.
Our results show that co-treatment of a procaspase-3 activator, PAC-1, with diverse targeted kinase inhibitors at clinically relevant concentrations (Bollag et al., 2010; Cross et al., 2014; Kitagawa et al., 2013; Marsilje et al., 2013) (see Supplementary Info Table 1 for predicted effects of each kinase inhibitor on MEK1/2 phosphorylation and caspase activity) is broadly effective in enhancing caspase-3 activity and apoptotic cell death across diverse tumor histologies and driver mutations. The resultant caspase-3 activity leads to enzymatic degradation of both MEK1 and MEK2 kinases and sustained inhibition of both MEK1/2 and ERK1/2 phosphorylation. While sustained ERK1/2 inhibition can be achieved with MEK1/2 inhibitors, this disrupts the negative feedback on RAF kinases, leading to the paradoxical hyper-phosphorylation of MEK1/2. Trametinib was developed as a “feedback buster” to minimize MEK1/2 hyper-phosphorylation (Hatzivassiliou et al., 2013; Lito et al., 2014) but the inhibitory effect is relatively transient as shown in the literature (Gilmartin et al., 2011) and consistent with our results (Fig. 3). In contrast, we now show that caspase-3 mediated degradation of MEK1 and MEK2 kinases is an excellent strategy to inactivate ERK1/2, without the corresponding rebound in MEK1/2 phosphorylation. Our results are supported by the abolishment of ERK1/2 phosphorylation observed upon genomic knockdown of both MEK1 and MEK2 (Lee et al., 2011). Due to the critical role of MEK1/2 kinases in regulating the MAPK pathway (Caunt et al., 2015), its sustained inhibition can be advantageous in significantly delaying the onset of acquired resistance.
In contrast to direct procaspase-3 activation using PAC-1, non-specific induction of apoptosis using general cytotoxins such as doxorubicin can lead to hyper-activation of ERK1/2 due to the cellular stress induced by these agents (McCubrey et al., 2006; McCubrey et al., 2007). This observation underscores the importance of using a direct procaspase-3 activator instead of general cytotoxins, in combination with targeted kinase inhibitors, to avoid the paradoxical reactivation of ERK1/2 phosphorylation.
Our results also show that addition of 1–2 μM of PAC-1 (a concentration easily achieved in human patients (Danciu et al., 2016)) is effective in delaying acquired resistance to vemurafenib, osimertinib, and ceritinib in BRAFV600E melanoma, EGFRT790M, and EML4-ALK NSCLCs respectively. Moreover, there is a marked benefit of combining PAC-1 with targeted kinase inhibitors as compared to MEK1/2 inhibition (with trametinib) in combination with targeted kinase inhibitors, in dramatically delaying or eliminating resistance. Two mechanisms are likely in play to account for this observation. First, enhanced apoptosis observed in cells treated with PAC-1 combination therapies likely impedes the emergence of resistant clones, as the vast majority of cancer cells are killed. Second, sustained MEK1/2 and ERK1/2 inhibition severely compromise the cells ability to proliferate and form resistant colonies.
Targeted kinase inhibitors have had a dramatic impact on cancer treatment, but resistance has seriously limited the durability of this effect. Instead of developing new drugs for each resistance mechanism, in this work we have identified a potentially generalizable strategy to eliminate or substantially delay the resistance to targeted anticancer therapies, and have successfully demonstrated its efficacy in cancers driven by BRAFV600E, EGFRT790M, and EML4-ALK kinases. Given that PAC-1 is currently being evaluated in clinical trials (NCT02355535, NCT03332355), and the kinase inhibitors used in this study are already approved by the FDA, the preclinical data results presented herein can inform the design of future trials to investigate PAC-1 combination therapies that may result in delayed or eliminated resistance.
STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Beta-Actin HRP | Cell Signaling Technology | Product # 5125 |
| Caspase-3 (Full Length) (Rabbit) | Cell Signaling Technology | Product # 9662 |
| PARP-1 (Rabbit) | Cell Signaling Technology | Product # 9532 |
| MEK1 (Rabbit) | Cell Signaling Technology | Product # 9146 |
| MEK2 (Rabbit) | Cell Signaling Technology | Product # 9147 |
| p-MEK1/2 (Ser217/Ser221) (Rabbit) | Cell Signaling Technology | Product # 9121 |
| p-ERK1/2 (Thr202/Tyr204) (Rabbit) | Cell Signaling Technology | Product # 4370 |
| Total ERK1/2 (Rabbit) | Cell Signaling Technology | Product # 4695 |
| Annexin V-FITC | Southern Biotechnology | Cat # 10040-02 |
| IgG (Rabbit) HRP | Cell Signaling Technology | Product # 7074 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PAC-1 | Putt, et al., 2006 | N/A |
| Raptinal | Palchaudhuri, et al., 2015 | N/A |
| Vemurafenib | LC Laboratories | Cat # V-2800 |
| Gefitinib | Cayman Chemicals | Cat # 13166 |
| Osimertinib | MedChem Express | Cat # HY-15772 |
| Ceritinib | MedChem Express | Cat # HY-15656 |
| Imatinib | MedChem Express | Cat # HY-15463 |
| Trametinib | MedChem Express | Cat # HY-10999 |
| BSA | Research Products International Corp. | Cat # A30075-100.0 |
| Sulforhodamine B (SRB) | Sigma Aldrich | Cat # 230162 |
| Acetic Acid | Fisher Scientific | Cat # A38-212 |
| Propidium Iodide | Sigma Aldrich | Cat # 81845 |
| Ac-DEVD-AFC | Cayman Chemicals | Cat # 14459 |
| HEPES | Fisher Scientific | Cat # BP310-1 |
| Sodium Chloride | Fisher Scientific | Cat # S271-500 |
| DTT | Sigma Aldrich | DTT-RO ROCHE |
| EDTA | Fisher Scientific | Cat # S311-100 |
| Triton-X-100 | Fisher Scientific | Cat # BP151-100 |
| Q-VD-OPh | Cayman Chemical | Cat # 15260 |
| RIPA Buffer | Cold Spring Harbor Protocols | doi:10.1101/pbd.rec10617 |
| Protease Inhibitor Cocktail III EDTA-Free | Calbiochem | Cat # 539134 |
| Phosphatase Inhibitor Cocktail IV | BioVision | Cat # K282-1 |
| SDS (Tris/Glycine) Buffer (10×) | BioRad | Cat # 161-0732 |
| Sucrose | EMD Millipore | Cat # SX1075 |
| Tris Base | Fisher Scientific | Cat # BP152-500 |
| Calcium Chloride | Fisher Scientific | Cat # AC349610 |
| Critical Commercial Assays | ||
| Pierce BCA Reagents A/B | Thermo Fisher Scientific | Cat # 23225 |
| Experimental Models: Cell Lines | ||
| A375 | ATCC | Cat # CRL-1619 |
| K-562 | ATCC | Cat # CCL-243 |
| PC-9 GR | Prof. Eric Haura, Moffitt Cancer Center | N/A |
| H3122 | Prof. Eric Haura, Moffitt Cancer Center | N/A |
| H1975 | Prof. Eric Haura, Moffitt Cancer Center | N/A |
| SK-MEL-5 | ATCC | Cat # HTB-70 |
| Software and Algorithms | ||
| ANOVA 2 Way T Test | OriginPro V10 | https://www.originlab.com/ |
| FCS Express V5 | De Novo software | https://www.denovosoftware.com/ |
| Two Way T-Test | Microsoft Excel 16.12 | https://products.office.com/en-us/home |
| Other | ||
| Mini-PROTEAN TGX Gels (4–20%) | BioRad | Cat # 456-1096 |
| PDVF Membrane | Millipore | Cat # IPVH00010 |
| Stripping Buffer | Thermo Fisher | Cat # 21059 |
| SuperSignal West Pico | Thermo Fisher | Cat # 34577 |
| BD LSR II Flow Cytometer | BD Biosciences | n/a |
| GelDoc XR | BioRad | Cat # 1708195 |
| ChemiDoc Touch | BioRad | Cat # 17001401 |
| SpectraMax M3 | Molecular Devices | Cat # M3 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paul Hergenrother (hergenro@illinois.edu).
Experimental model and subject details
A375, K-562, and SK-MEL-5 were obtained from ATCC. PC-9 GR, H1975, and H3122 were provided by Prof. Eric Haura (Moffitt Cancer Center). PC-9 GR, H1975, and H3122 were cultured in RPMI 1640 supplemented with 10% FBS (Gemini). A375 and SK-MEL-5 were cultured in DMEM + 10% FBS. K-562 was cultured in IMDM + 10% FBS. All cells were cultured at 37 °C with 5% CO2. Sex of human cell lines: A375 (Female, 54 years old), K-562 (Female, 53 years old), SK-MEL-5 (Female, 24 years old), H3122 (Female, age unknown), H1975 (Female, age unknown), PC-9 GR (Female, age unknown).
Methods Details
Cell line authentication
All human cell lines used in this study (PC-9 GR, H1975, SK-MEL-5, A375, K-562, and H3122) have been authenticated using the PowerPlex16HS Assay (Promega) as described previously (Peh et al., 2016): 15 Autosomal Loci, X/Y at the University of Arizona Genetics Core (UAGC). >1 million cells were harvested and lysed using the cell lysis buffer (50 mM Tris, 50 mM EDTA, 25 mM sucrose, 100 mM NaCl, 1% SDS, pH 8). DNA extraction and short tandem repeats (STRs) profiling for each cell line were carried out at the UAGC. The resulting autosomal STR profiles were compared to reference databases such as ATCC, DSMZ, and JCRB.
Cell viability assay
1000 cells were seeded per well in a 96-well plate and allowed to adhere before DMSO solutions of osimertinib, gefitinib, or PAC-1 were added to each well. Final concentration of DMSO in each well is 0.5%. At the end of 5 days, viability was assessed by the sulforhodamine B (SRB) assay sulforhodamine B (SRB) assay (Vichai and Kirtikara, 2006). Briefly, 100 μL of 10% trichloroacetic acid (TCA) was added in each well and the plate was incubated for at least 1 hour at 4°C. After the 4°C incubation, the plate was washed with water and allowed to dry for at least 1 hour at room temperature. 100 μL of SRB dye (1% w/v) was added to each well and incubated for 30 minutes at room temperature. At the end of the 30 minutes incubation, the plate was washed with 1% acetic acid solution and allowed to dry at room temperature. Finally, 200 μL of Tris solution (pH >10) was added to each well to dissolve the SRB dye. The absorbance of each well was read with a SpectraMax M3 plate reader (Molecular Devices) at 510 nm.
Caspase-3/-7 activity assay
For the EGFRT790M cell lines, 4,000 cells were seeded in each well of 96-well plates and allowed to adhere overnight. The next day, indicated concentrations of PAC-1 or osimertinib, were added and treated for 0, 2, 4, 24, 30, 35, 44 and 48 hour. K-562 cells were seeded at 3,000 cells per well and treated with indicated concentrations of PAC-1 or imatinib for 0, 2, 4, 24, 48, 68, and 72 hour. H3122 cells were seeded at 4,000 cells per well and allowed to adhere overnight. The cells were then treated with indicated concentrations of PAC-1 or ceritinib for 0, 2, 4, 24, 44, and 48 hour. 10 μM raptinal (Palchaudhuri et al., 2015) was used as the positive control throughout the experiment. After indicated incubation times, the cells were lysed and caspase-3/-7 activity was assessed via addition of bifunctional lysis and activity buffer (200 mM HEPES, 400 mM NaCl, 40 mM DTT, 0.4 mM EDTA, 1% Triton-X, pH 7.4) with 50 μM of fluorogenic Ac-DEVD-AFC substrate (λex=405 nm, λem=505 nm). Plates were pre-incubated at 37°C for 30 minutes in the SpectraMax M3 (Molecular Devices) plate reader and then read for 30 minutes at 3-minute intervals. Slopes for each well were calculated and averaged over six technical replicates. Activity is normalized to the maximal and minimal activity observed within the assay.
Immunoblotting
Cells were lysed using RIPA buffer containing phosphatase (BioVision) and protease inhibitor cocktail (Calbiochem). Protein concentration was determined using the BCA assay (Pierce). Cell lysates containing 8–20 μg of protein were loaded into each lane of 4–20% gradient gels (BioRad) and ran for SDS-PAGE. Proteins were transferred onto PDVF membrane (Millipore) for Western blot analysis. Blots were blocked with BSA for one hour followed by incubation with primary antibody overnight (manufacturer’s recommended dilutions). Secondary antibody was incubated for one hour. Blots were then imaged with a ChemiDoc Touch after incubation with SuperSignal West Pico Solution following manufacturer’s protocols.
Assessment of apoptosis by flow cytometry
For the EGFRT790M cell lines, 40,000 cells were seeded in 12-well plates and allowed to adhere overnight. The next day, indicated concentrations of PAC-1 or osimertinib were added and allowed to incubate at 37°C for 48 hours. K-562 cells were seeded at 30,000 and incubated with PAC-1 or imatinib for 72 hours. In 12-well plates, 40,000 H3122 cells were seeded and allowed to adhere overnight. The next day, they were incubated with PAC-1 or ceritinib for 48 hours. After the indicated incubation period, cells were harvested and resuspended in 450 μL of cold buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) premixed with Annexin V-FITC and PI dyes. Samples were analyzed on a BD Biosciences LSRII flow cytometer, and data analysis was performed using FSC Express Version5.
Q-VD-OPh Protection by flow cytometry
A375 (BRAFV600E) cells were seeded with 70,000 cells/well in 12-well plates and allowed to adhere overnight. The next day, indicated concentrations of PAC-1, vemurafenib, trametinib, and/or Q-VD-OPh were added and cells were incubated at 37°C for 48 hours. For the EGFRT790M cell line, PC-9 GR, 40,000 cells were seeded in 12-well plates and allowed to adhere overnight. The next day, indicated concentrations of PAC-1, osimertinib, and Q-VD-OPh were added and allowed to incubate at 37°C for 48 hours. In 12-well plates, 40,000 H3122 cells were seeded and allowed to adhere overnight. The next day, they were incubated with PAC-1, ceritinib, and/or Q-VD-OPh for 48 hours. K-562 cells were seeded with 35,000 cells/well in 12-well plates. The next day, indicated concentrations of PAC-1, imatinib, and/or Q-VD-OPh were added and cells were incubated at 37°C for 48 hours. After the indicated incubation period, cells were harvested and resuspended in 450 μL of cold buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4) premixed with Annexin V-FITC and PI dyes. Samples were analyzed on a BD Biosciences LSRII flow cytometer, and data analysis was performed using FSC Express Version5.
Long term experiments in BRAFV600E cell lines
This assay was performed as described previously (Peh et al., 2016). Briefly, 100–250 cells were seeded and allowed to adhere overnight. The next day, cells were treated with indicated concentrations of PAC-1, vemurafenib or trametinib for 10, 20, 25 or 30 days. Media was refreshed with new compounds added every 3–4 days. At the end of the incubation period, cells were fixed 10% trichloroacetic acid, stained with SRB, imaged using GelDoc XR (BioRad), and absorbance at 510 nm read using SpectraMax M3 (Molecular Devices) plate reader.
Long term experiments with EGFRT790M cell lines
In 12 well plates, PC-9 GR or H1975 cells were seeded at 2,000 cells per well and allowed to adhere overnight. The next day, cells were treated with indicated concentrations of PAC-1 or osimertinib for 8 or 28 days. Media was refreshed every 3–4 days with new compounds. For experiments comparing the effect of PAC-1 combination versus trametinib combination, PC-9 GR cells were seeded at 10,000 cells per well in 6 well plates and allowed to adhere overnight. The next day, cells were treated with indicated concentrations of PAC-1, osimertinib, or 5 nM trametinib for 8, 28, or 35 days. Media was refreshed every 3–4 days with new compounds. At the end of the incubation period, cells were fixed 10% trichloroacetic acid, stained with SRB, imaged using GelDoc XR, and absorbance at 510 nm read using SpectraMax M3 (Molecular Devices) plate reader.
Long term experiments with EML4-ALK cell line
H3122 cells were seeded at 10,000 cells per well in 6 well plates and allowed to adhere overnight. The next day, cells were treated with indicated concentrations of PAC-1, osimertinib, or 5 nM trametinib for 8, 20, or 32 days. Media was refreshed every 3–4 days with new compounds. At the end of the incubation period, cells were fixed 10% trichloroacetic acid, stained with SRB, imaged using GelDoc XR, and absorbance at 510 nm read using SpectraMax M3 (Molecular Devices) plate reader.
Quantification and statistical analysis
Data are presented as mean values ± standard error of the mean (s.e.m.). Levels of significance were determined by two-way t-test for control versus experimental groups using Microsoft Excel. To determine if the increase in caspase-3 activity was synergistic, two-way ANOVA analysis was performed for DMSO-, single-agent-versus combination-treated samples using OriginPro (Version 10, Origin Lab). No analyses were performed to determine whether the data met the assumptions of this statistical approach. Statistical values, including the number of independent replicates and statistical significance, are reported in the Figure Legends.
Data and software availability
All data generated in this study will be made available upon request to the corresponding author, Prof. Paul Hergenrother (hergenro@illinois.edu).
Supplementary Material
Significance.
The clinical utility of targeted anticancer therapies is limited by the rapid onset of resistance. In drug-resistant clones reactivation of downstream signaling via MEK1/2 kinases is often observed, thus inhibition of MEK1/2 has become an attractive strategy to delay resistance; however, drug-mediated MEK1/2 inhibition provides only temporary shutdown of downstream signaling and modest survival benefit. As a promising anticancer strategy we show that drug-induced degradation of MEK1 and MEK2 broadly enhances the cell death mediated by a diverse set of approved kinase inhibitors, including for melanoma, lung cancer, and leukemia. This loss of MEK1 and MEK2 leads to sustained inhibition of downstream signaling, dramatically delaying or eliminating the onset of resistance in cancer cells.
Highlights.
PAC-1 enhances caspase-3 activity and apoptosis induced by diverse kinase inhibitors
Caspase-3 mediated MEK degradation sustains inhibition of MEK phosphorylation
PAC-1 combination therapies dramatically delay or eliminate acquired resistance
Acknowledgments
This work is supported by the NIH (R01-CA120439), the Melanoma Research Alliance (Award ID 412305), and the University of Illinois. M.W.B. is a member of the NIH Chemistry-Biology Interface Training Program (T32-GM070421). We thank Prof. Eric Haura (Moffitt Cancer Center) for providing the PC-9 GR, H1975, and H3122 cell lines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions
J.P. and P.J.H. designed the experiments and analyzed the data; J.P., M.W.B., and H.M.S. performed the experiments. J.P., M.W.B., and P.J.H. wrote the manuscript.
Declaration of Interests
The University of Illinois has filed patents on some of this work.
References
- Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248–1260. doi: 10.1016/S1470-2045(16)30122-X. [DOI] [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botham RC, Roth HS, Book AP, Roady PJ, Fan TM, Hergenrother PJ. Small-Molecule Procaspase-3 Activation Sensitizes Cancer to Treatment with Diverse Chemotherapeutics. ACS Cent Sci. 2016;2:545–559. doi: 10.1021/acscentsci.6b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright IM, Liu X, Zhou M, Li F, Li CY. Essential roles of Caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. eLife. 2017;6:e26371. doi: 10.7554/eLife.26371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MRV, Ward RA, Mellor MJ, et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danciu OC, Nicholas MK, Holdhoff M, Venepalli NK, Hergenrother PJ, Tarasow TM, Dudek AZ. Phase I study of procaspase activating compound-1 (PAC-1) in the treatment of advanced malignancies. J Clin Oncol. 2016;34(Suppl) doi: 10.1038/s41416-022-02089-7. abstr TPS2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov. 2004;3:1001–1010. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Cravatt BF. Global Mapping of the Topography and Magnitude of Proteolytic Events in Biological Systems. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, Meador CB, Spitzler P, Ichihara E, Ross SJ, et al. Acquired resistance to mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75:2489–2500. doi: 10.1158/0008-5472.CAN-14-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink DSWH, Selzer E, Lucas T, Wolff K, Pehamberger H, Eichler HG, Jansen B. Elevated procaspase levels in human melanoma. Melanoma Res. 2001;11:385–393. doi: 10.1097/00008390-200108000-00009. [DOI] [PubMed] [Google Scholar]
- Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28:S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol. 2016;13:209–227. doi: 10.1038/nrclinonc.2015.213. [DOI] [PubMed] [Google Scholar]
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- Groenendijk FH, Bernards R. Drug resistance to targeted therapies: Déjà vu all over again. Mol Oncol. 2014;8:1067–1083. doi: 10.1016/j.molonc.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–1789. doi: 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hare T, Eide CA, Deininger MWN. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–2249. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JFM, Zak M, Peck A, Orr C, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis A, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK–positive lung cancer. Nat Med. 2015;21:1038–1047. doi: 10.1038/nm.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JQ, Liang HL, Zhang XC, Xie Z, Jin TE. Synergistic antitumor activity of pro-apoptotic agent PAC-1 with cisplatinum by the activation of CASP3 in pulmonary adenocarcinoma cell line H1299. Asia Pac J Clin Oncol. 2016;12:41–51. doi: 10.1111/ajco.12419. [DOI] [PubMed] [Google Scholar]
- Ichim G, Lopez J, Ahmed Shafiq U, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley Joel S, Mason Susan M, Athineos D, et al. Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol Cell. 2015;57:860–872. doi: 10.1016/j.molcel.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: A Link between Cancer Genetics and Chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, Akita K, Kirii Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. 2013;18:110–122. doi: 10.1111/gtc.12022. [DOI] [PubMed] [Google Scholar]
- Lee CS, Dykema KJ, Hawkins DM, Cherba DM, Webb CP, Furge KA, Duesbery NS. MEK2 Is Sufficient but Not Necessary for Proliferation and Anchorage-Independent Growth of SK-MEL-28 Melanoma Cells. Plos One. 2011;6:e17165. doi: 10.1371/journal.pone.0017165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G. Raf kinases: Function, regulation and role in human cancer. BBA - Mol Cell Res. 2007;1773:1196–1212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, Riely GJ, Shaw AT. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017;7:137–155. doi: 10.1158/2159-8290.CD-16-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, Saborowski M, Kastenhuber E, Fellmann C, Ohara K, et al. Disruption of CRAF-Mediated MEK Activation Is Required for Effective MEK Inhibition in KRAS Mutant Tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, He Y, Li F, Huang Q, Kato Takamitsu A, Hall Russell P, Li CY. Caspase-3 Promotes Genetic Instability and Carcinogenesis. Mol Cell. 2015;58:284–296. doi: 10.1016/j.molcel.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–451. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, Zhu LJ, Hutchinson L, Cerny J, Khoury HJ, et al. A therapeutically targetable mechanism of BCR-ABL–independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6:252ra121. doi: 10.1126/scitranslmed.3009073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA. Global Sequencing of Proteolytic Cleavage Sites in Apoptosis by Specific Labeling of Protein N Termini. Cell. 2008;134:866–876. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J Med Chem. 2013;56:5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, LaHair MM, Franklin RA. Reactive Oxygen Species-Induced Activation of the MAP Kinase Signaling Pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EWT, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the RAF/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15:371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu Clarissa C, Koya Richard C, Samatar Ahmed A, Khanlou N, Braun J, et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting but Result in Melanoma Drug Addiction. Cancer Cell. 2015;27:240–256. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakopoulou L, Alexandrou P, Stefanaki K, Panayotopoulou E, Lazaris AC, Davaris PS. Immunohistochemical expression of caspase-3 as an adverse indicator of the clinical outcome in human breast cancer. Pathobiology. 2001;69:266–273. doi: 10.1159/000064337. [DOI] [PubMed] [Google Scholar]
- Palchaudhuri R, Lambrecht MJ, Botham RC, Partlow KC, van Ham TJ, Putt KS, Nguyen LT, Kim SH, Peterson RT, Fan TM, et al. A Small Molecule that Induces Intrinsic Pathway Apoptosis with Unparalleled Speed. Cell Rep. 2015;13:2027–2036. doi: 10.1016/j.celrep.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul MK, Mukhopadhyay AK. Tyrosine kinase – Role and significance in Cancer. Int J Med Sci. 2004;1:101–115. doi: 10.7150/ijms.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh J, Fan TM, Wycislo KL, Roth HS, Hergenrother PJ. The combination of vemurafenib and procaspase-3 activation is synergistic in mutant BRAF melanomas. Mol Cancer Ther. 2016;15:1859–1869. doi: 10.1158/1535-7163.MCT-16-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad R, Liu C, Wu TT, Houlihan PS, Hamilton SR, Diehl AM, Rashid A. Overexpression of caspase-3 in hepatocellular carcinomas. Mod Pathol. 2004;17:861–867. doi: 10.1038/modpathol.3800146. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004;23:2934–2949. doi: 10.1038/sj.onc.1207515. [DOI] [PubMed] [Google Scholar]
- Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- Razi SS, Rehmani S, Li X, Park K, Schwartz GS, Latif MJ, Bhora FY. Antitumor activity of paclitaxel is significantly enhanced by a novel proapoptotic agent in non– small cell lung cancer. J Surg Res. 2015;194:622–630. doi: 10.1016/j.jss.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr MEK1/2 dual-specificity protein kinases: Structure and regulation. Biochem Biophys Res Commun. 2012;417:5–10. doi: 10.1016/j.bbrc.2011.11.145. [DOI] [PubMed] [Google Scholar]
- Roth HS, Hergenrother PJ. Derivatives of Procaspase-Activating Compound 1 (PAC-1) and their Anticancer Activities. Curr Med Chem. 2016;23:201–241. doi: 10.2174/0929867323666151127201829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowska A, Car H, Pryczynicz A, Guzinska-Ustymowicz K, Kowal KW, Cepowicz D, Kedra B. Expression of apoptotic proteins in human colorectal cancer and metastatic lymph nodes. Pathol Res Pract. 2014;210:576–581. doi: 10.1016/j.prp.2014.04.023. [DOI] [PubMed] [Google Scholar]
- Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. BBA - Mol Cell Res. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizaki J, Okamoto I, Takezawa K, Sakai K, Azuma K, Kuwata K, Yamaguchi H, Hatashita E, Nishio K, Janne PA, et al. Combined effect of ALK and MEK inhibitors in EML4–ALK-positive non-small-cell lung cancer cells. Br J Cancer. 2012;106:763–767. doi: 10.1038/bjc.2011.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR mutant Lung Cancer. Cancer Discov. 2015;5:960–971. doi: 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, MacConaill LE, Hahn WC, et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield SE, Zhang L, Scorsone KA, Liu Y, Zage PE. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer. 2016;16:172. doi: 10.1186/s12885-016-2199-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.