

Abstract
Cold-water corals provide critical habitats for a multitude of marine species, but are understudied relative to tropical corals. Primnoa pacifica is a cold-water coral prevalent throughout Alaskan waters, while another species in the genus, Primnoa resedaeformis, is widely distributed in the Atlantic Ocean. This study examined the V4-V5 region of the 16S rRNA gene after amplifying and pyrosequencing bacterial DNA from samples of these species. Key differences between the two species’ microbiomes included a robust presence of bacteria belonging to the Chlamydiales order in most of the P. pacifica samples, whereas no more than 2% of any microbial community from P. resedaeformis comprised these bacteria. Microbiomes of P. resedaeformis exhibited higher diversity than those of P. pacifica, and the two species largely clustered separately in a principal coordinate analysis. Comparison of P. resedaeformis microbiomes from samples collected in two submarine canyons revealed a significant difference between locations. This finding mirrored significant genetic differences among the P. resedaeformis from the two canyons based upon population genetic analysis of microsatellite loci. This study presents the first report of microbiomes associated with these two coral species.
Introduction
Tropical corals provide critical habitat for a vast number of marine species1, and recent expansion of deep-water marine research has revealed that cold-water coral ecosystems are abundant and equally critical to marine biodiversity2. Deep-sea and other cold-water corals are habitats for a wide variety of animals, including dozens of fish species3–6. Cold-water corals are also critical habitats for thousands of invertebrate species7. The biodiversity associated with cold-water corals continues to increase when microscopic associates are taken into account. All corals host rich microbial communities, and the microbiomes of cold-water corals are increasingly the focus of investigation into the complex interactions and symbioses among the members of the coral holobiont. Much of the research into the microbiomes of cold-water corals has centered on Lophelia pertusa and other stony corals8–17. However, deep-sea gorgonian coral microbiomes are beginning to be investigated18–20.
Only a few cold-water gorgonian coral genera have been the subject of microbiome comparisons between species. Microbiomes of two temperate Muricea species (M. californica and M. fruticosa) from the kelp forests of southern California were recently compared, revealing that the two species harbored distinct microbial communities21. A survey of temperate Mediterranean Eunicella species (E. singularis, E. cavolini, and E. verrucosa) found each species had a unique core microbiome. However, there was overlap of shared sequences, particularly Endozoicomonas, between the species22. In contrast, two deep-sea Anthothela species from the western Atlantic Ocean had essentially indistinguishable bacterial communities20. Further, samples of the two Anthothela species were collected in two different submarine canyons, but their geographic origin had no impact on the microbiome composition.
In this study, two species of Primnoa corals were sampled for the purpose of characterizing and comparing their microbial communities. The corals were sampled in two different ocean basins: P. pacifica samples were collected from one location in the Gulf of Alaska in the Pacific Ocean, and P. resedaeformis samples were collected from two submarine canyons in the North Atlantic Ocean, approximately 140 km apart. Collection of two different species enabled us to compare their microbiomes and determine which microbes were core to the genus and which were core to each species. In light of reports showing that cold-water corals have conserved bacterial communities13,20, we hypothesized that the microbiomes of Primnoa corals contain core bacterial species common to all members of the genus. We further hypothesized that P. resedaeformis and P. pacifica contain bacteria common to each coral species but not part of the core genus microbiome.
Collection of P. resedaeformis from separate submarine canyons allowed us to examine whether corals of the same species in different locations contained distinctive microbial communities. We expected the P. resedaeformis populations to have similar microbiomes based on a study of Anthothela coral microbiomes from the same locations20. When significant microbiome variation between canyons was detected, we analyzed microsatellite loci of the P. resedaeformis samples to examine whether host population genetics could be an underlying factor.
Results
Six samples of Primnoa pacifica were collected from Tracy Arm Fjord in the Gulf of Alaska in 2011 and 2012, at depths ranging from approximately 10 to 16 m (Table 1). Primnoa resedaeformis samples were collected from Baltimore Canyon in 2012 (nine samples) and from Norfolk Canyon in 2013 (five samples) in the Atlantic Ocean at depths ranging from approximately 410 to 580 m (Norfolk) and 380 to 500 m (Baltimore) (Table 1). DNA was extracted from each sample as described in the Materials and Methods section. After amplification and sequencing of the 16S rRNA gene, sequence analysis was conducted in order to examine the microbial communities of each sample.
Table 1.
Primnoa sample locations and environmental parameters.
| Sample | Year | Collection Location | Ocean Basin | Temp (°C) | Depth (m) | Salinity (psu) |
|---|---|---|---|---|---|---|
| PR_BC_01 | 2012 | Baltimore Canyon | Atlantic | 6.2 | 450 | 35.1 |
| PR_BC_02 | 2012 | Baltimore Canyon | Atlantic | 9.0 | 383 | 35.2 |
| PR_BC_03 | 2012 | Baltimore Canyon | Atlantic | 7.4 | 443 | 35.1 |
| PR_BC_04 | 2012 | Baltimore Canyon | Atlantic | 7.4 | 443 | 35.1 |
| PR_BC_05 | 2012 | Baltimore Canyon | Atlantic | 7.5 | 430 | 35.0 |
| PR_BC_06 | 2012 | Baltimore Canyon | Atlantic | 7.5 | 431 | 34.9 |
| PR_BC_07 | 2012 | Baltimore Canyon | Atlantic | 7.3 | 506 | 35.1 |
| PR_BC_08 | 2012 | Baltimore Canyon | Atlantic | 7.3 | 494 | 35.1 |
| PR_BC_09 | 2012 | Baltimore Canyon | Atlantic | 7.6 | 500 | 35.1 |
| PR_BC_10 | 2012 | Baltimore Canyon | Atlantic | 7.6 | 508 | 35.1 |
| PR_NC_01 | 2012 | Norfolk Canyon | Atlantic | 6.2 | 535 | 35.0 |
| PR_NC_02 | 2012 | Norfolk Canyon | Atlantic | 6.6 | 523 | 35.1 |
| PR_NC_03 | 2012 | Norfolk Canyon | Atlantic | 6.3 | 434 | 35.0 |
| PR_NC_04 | 2013 | Norfolk Canyon | Atlantic | 10.8 | 411 | 35.5 |
| PR_NC_05 | 2013 | Norfolk Canyon | Atlantic | 9.0 | 441 | 35.2 |
| PR_NC_06 | 2013 | Norfolk Canyon | Atlantic | 9.0 | 441 | 35.3 |
| PR_NC_07 | 2013 | Norfolk Canyon | Atlantic | 6.3 | 498 | 35.0 |
| PR_NC_08 | 2013 | Norfolk Canyon | Atlantic | 6.3 | 498 | 35.0 |
| PR_NC_09 | 2013 | Norfolk Canyon | Atlantic | 6.6 | 479 | 35.1 |
| PR_NC_10 | 2013 | Norfolk Canyon | Atlantic | 5.5 | 576 | 35.0 |
| PP_GA_01 | 2012 | Gulf of Alaska | Pacific | 5.0 | 9.8 | 30.1 |
| PP_GA_02 | 2012 | Gulf of Alaska | Pacific | 5.0 | 13.1 | 30.1 |
| PP_GA_03 | 2012 | Gulf of Alaska | Pacific | 5.0 | 11.6 | 30.1 |
| PP_GA_04 | 2012 | Gulf of Alaska | Pacific | 5.0 | 16.2 | 30.1 |
| PP_GA_05 | 2011 | Gulf of Alaska | Pacific | 4.6 | 13.4 | 26.9 |
| PP_GA_06 | 2011 | Gulf of Alaska | Pacific | 4.6 | 12.8 | 26.9 |
| PP_GA_07 | 2011 | Gulf of Alaska | Pacific | 4.6 | 12.5 | 26.9 |
Samples beginning with “PR” are P. resedaeformis. Samples beginning with “PP” are P. pacifica. Date and time of collection and latitude and longitude for each sample are provided in Supplementary Table S2. Samples in bold were analyzed in this study.
Bacterial diversity
Bacterial diversity of each sample was assessed through alpha diversity measurements including ACE richness (abundance-based coverage estimator), Chao1 richness, Shannon index, reciprocal Simpson index, and Simpson evenness index23–26 (Table 2). By every diversity measurement, the Atlantic P. resedaeformis samples exhibited higher average richness and evenness than the Pacific P. pacifica samples. The P. resedaeformis samples also exhibited a wider range of diversity measurements than the P. pacifica samples. Not only did the P. resedaeformis samples collectively have higher mean diversity (mean ACE richness 260.51, mean Chao1 richness 253.18, mean Shannon index 4.62, mean reciprocal Simpson index 9.70, mean Simpson evenness 0.05) than P. pacifica (mean ACE richness 125.31, mean Chao1 richness 117.76, mean Shannon index 1.87, mean reciprocal Simpson index 2.91, mean Simpson evenness 0.03), but they also included the samples with the highest individual diversity measurements. The samples with the highest values for ACE richness and Chao1 richness (sample PR_BC_02, with ACE richness of 448.65 and Chao1 richness of 414.56) as well as reciprocal Simpson index and Simpson evenness (sample PR_BC_09, with reciprocal Simpson index of 19.55 and Simpson evenness of 0.116) both came from Baltimore Canyon, while the sample with the highest Shannon index (which, like the Simpson index, incorporates richness and evenness26,27) came from Norfolk Canyon (sample PR_NC_06, with a Shannon index of 6.12). The least diverse samples came from the Gulf of Alaska.
Table 2.
Alpha diversity associated with each Primnoa sample analyzed in this study. Samples from Baltimore Canyon and Norfolk Canyon are P. resedaeformis; samples from the Gulf of Alaska are P. pacifica.
| Sample | Collection Location | Sequence Reads* | OTUs | ACE Richness | Chao1 Richness | Shannon Index | Reciprocal Simpson Index | Simpson Evenness |
|---|---|---|---|---|---|---|---|---|
| PR_BC_01 | Baltimore Canyon | 22,448 | 84 | 132.83 | 115.50 | 1.92 | 1.88 | 0.022 |
| PR_BC_02 | Baltimore Canyon | 17,031 | 314 | 448.65 | 414.56 | 5.68 | 12.63 | 0.040 |
| PR_BC_03 | Baltimore Canyon | 23,285 | 66 | 91.51 | 91.00 | 1.36 | 1.44 | 0.022 |
| PR_BC_04 | Baltimore Canyon | 11,425 | 207 | 291.87 | 273.50 | 4.58 | 7.26 | 0.035 |
| PR_BC_05 | Baltimore Canyon | 7,546 | 219 | 270.61 | 265.50 | 5.21 | 11.40 | 0.052 |
| PR_BC_06 | Baltimore Canyon | 14,653 | 297 | 427.07 | 402.22 | 5.22 | 11.02 | 0.037 |
| PR_BC_07 | Baltimore Canyon | 6,773 | 235 | 304.82 | 300.28 | 5.25 | 10.09 | 0.043 |
| PR_BC_08 | Baltimore Canyon | 5,024 | 115 | 128.05 | 128.13 | 4.37 | 7.39 | 0.064 |
| PR_BC_09 | Baltimore Canyon | 4,261 | 168 | 234.50 | 233.81 | 5.28 | 19.55 | 0.116 |
| PR_NC_04 | Norfolk Canyon | 2,561 | 178 | 242.39 | 233.62 | 4.75 | 10.84 | 0.061 |
| PR_NC_06 | Norfolk Canyon | 3,176 | 273 | 313.93 | 318.02 | 6.12 | 14.66 | 0.054 |
| PR_NC_07 | Norfolk Canyon | 4,753 | 183 | 238.74 | 232.88 | 5.01 | 9.89 | 0.054 |
| PR_NC_08 | Norfolk Canyon | 5,820 | 241 | 294.42 | 289.37 | 5.21 | 6.32 | 0.026 |
| PR_NC_10 | Norfolk Canyon | 2,557 | 159 | 227.78 | 246.14 | 4.78 | 11.39 | 0.072 |
| Mean (standard deviation) for P. resedaeformis | 260.51 (102.0) | 253.18 (95.5) | 4.62 (1.3) | 9.70 (4.7) | 0.05 (0.025) | |||
| PP_GA_01 | Gulf of Alaska | 43,163 | 32 | 78.36 | 84.50 | 0.31 | 1.06 | 0.033 |
| PP_GA_02 | Gulf of Alaska | 44,282 | 53 | 101.29 | 86.83 | 0.71 | 1.17 | 0.022 |
| PP_GA_04 | Gulf of Alaska | 22,140 | 100 | 159.72 | 145.15 | 2.81 | 3.38 | 0.034 |
| PP_GA_05 | Gulf of Alaska | 23,068 | 98 | 160.37 | 152.47 | 1.95 | 1.82 | 0.019 |
| PP_GA_06 | Gulf of Alaska | 22,242 | 141 | 179.60 | 176.36 | 4.24 | 8.60 | 0.061 |
| PP_GA_07 | Gulf of Alaska | 35,370 | 47 | 72.49 | 61.25 | 1.22 | 1.45 | 0.031 |
| Mean (standard deviation) for P. pacifica | 125.31 (46.8) | 117.76 (46.1) | 1.87 (1.5) | 2.91 (2.9) | 0.03 (0.02) | |||
*Samples were rarefied to 2,557 sequences before calculation of diversity metrics.
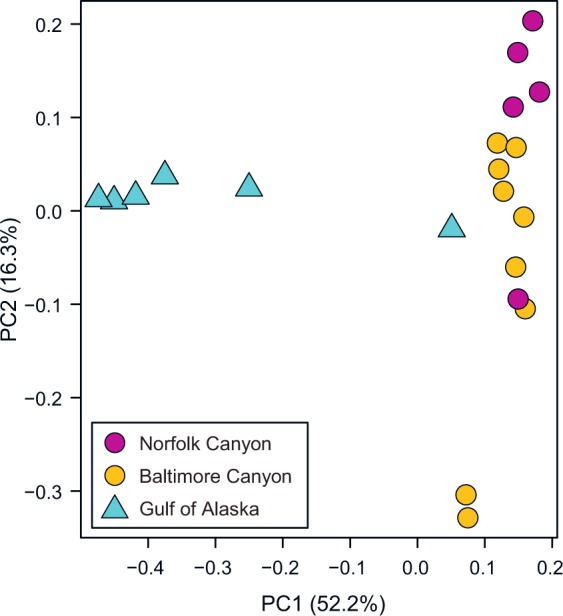
Beta diversity measurements were visualized using principal coordinate analysis (PCoA). Similarity matrices were computed using four metrics: weighted and unweighted UniFrac (which incorporate phylogenetic distance), Bray-Curtis, and Sorensen-Dice. Analysis using the weighted UniFrac similarity matrix (Fig. 1) explained the greatest amount of variation among the samples. The metrics based on presence/absence (unweighted UniFrac and Sorensen-Dice) displayed greater separation between the two Primnoa species than the abundance-weighted metrics (weighted UniFrac and Bray-Curtis), and more overlap between the Norfolk Canyon and Baltimore Canyon P. resedaeformis samples. Analysis using the Bray-Curtis metric explained less of the variation among the samples, but revealed greater separation between P. resedaeformis samples from the two canyons.
Figure 1.
Principal coordinate analysis (PCoA) plot of weighted UniFrac distance. PCoA was used to plot beta diversity of coral-associated bacterial communities using the weighted UniFrac distance matrix.
Bacterial community composition
Analysis of the taxonomic makeup of their bacterial communities revealed differences between the two species of Primnoa. Bacteria from the Rhabdochlamydiaceae family (in the Chlamydiales order) dominated five of the six P. pacifica samples (Fig. 2). In those samples, Rhabdochlamydiaceae bacteria constituted from 49% to 97% of the bacterial communities, with three communities comprising more than 80% from that family. In contrast, no P. resedaeformis bacterial community consisted of more than 0.5% Rhabdochlamydiaceae. The P. resedaeformis samples instead had greater abundances of several families in the Proteobacteria phylum, including Xanthomonadaceae, Pseudomonadaceae, Pseudoalteromonadaceae, Moraxellaceae, and an unspecified Kiloniellales family (Fig. 2).
Figure 2.

Relative abundance of bacterial families (or lowest identifiable phylogenetic level) in Primnoa samples. Bacterial groups shown present at ≥5% relative abundance in at least one sample. All remaining taxa are summarized under “Other”.
Of the families comprising at least 5% of any sample’s bacterial community, three families were absent from the P. pacifica samples, appearing only in P. resedaeformis samples (Lachnospiraceae, Mycoplasmataceae, and Pseudoaltermonadaceae), while three families appeared in every sample (Moraxellaceae, Pseudomonadaceae, and Rhodobacteraceae) (Fig. 2). However, the Moraxellaceae family played a larger role in the P. resedaeformis samples than in the P. pacifica samples. That family represented more than 5% of the bacterial community in all but three of the Canyon samples, but comprised less than 1% of every Alaska sample but one (in which it represented 1.7%) (Supplementary Table S1).
ANOSIM and SIMPER analyses
Analysis of similarity (ANOSIM) between the P. resedaeformis and P. pacifica samples was conducted using the weighted UniFrac distance between each pair of samples (because that metric explained the greatest amount of variation among the samples). The ANOSIM R statistic was 0.839, showing a significantly high level of dissimilarity between the two species (p = 0.0001). Average similarity of each pair of samples within P. resedaeformis (SIMPER) was 31.65%. (Note that SIMPER analysis is based on Bray-Curtis dissimilarity, rather than weighted UniFrac distance, which means that the SIMPER analyses incorporate differences in abundance of OTUs but not phylogenetic distance between OTUs.) The largest contributors to that similarity were members of Kiloniellales (5.87%) and two Acinetobacter OTUs that totaled 5.64%. Within P. pacifica, average similarity of each pair of samples was 39.28%. The largest contributor to that similarity was the Chlamydiales OTU, at 37.54%. The next nearest contributor, an unassigned OTU, contributed 4.28%. The average dissimilarity between the two species was 83.32%. Again, the Chlamydiales OTU, the greatest contributor, was responsible for 7% of the dissimilarity, while Kiloniellales contributed 3.25%.
Core microbiomes
The set of OTUs shared among all samples represents the Primnoa genus core microbiome (Fig. 3, Supplementary Table S1). Six OTUs appear in all Primnoa samples, from five microbial genera: two OTUs in the Pseudomonas genus, and one OTU each from the Lysobacter, Bacillus, Acinetobacter, and Propionibacterium genera. These Primnoa genus core OTUs comprise from 0.35% of the relative abundance in a P. pacifica sample to 54.2% in a P. resedaeformis sample from Baltimore Canyon (Fig. 3).
Figure 3.

Relative abundance of genus core microbiome (OTUs found in all Primnoa samples), species core microbiome (OTUs found in all samples of P. pacifica or all samples of P. resedaeformis), and individual microbiome (remaining variable OTUs present in each coral colony).
The species core microbiome for P. resedaeformis consists of four OTUs (Supplementary Table S1). Three OTUs were classified to the family level (Vibrionaceae, Pirellulaceae, and Rhodobacteraceae) and one to the genus level (Sphingobium). These OTUs make up 0.3% to 11.6% relative abundance of the P. resedaeformis samples. The species core microbiome for P. pacifica consisted of 19 OTUs (Supplementary Table S1), and constituted 18.8% to 97.3% of the bacterial community for the Pacific samples (Fig. 3). The largest contributor to the species core microbiome for P. pacifica was the Chlamydiales OTU. In four of the six samples, more than 74% of the entire microbiome comprised this OTU; in the other two samples, this OTU contributed 49.1% (PP_GA_04) and 0.013% (PP_GA_06). With one exception (PP_GA_06, the P. pacifica sample that is low in Rhabdochlamydiaceae), the individual microbiomes of the Pacific samples make up a much smaller portion of the entire microbiome than they do in the Atlantic samples.
Inter-canyon comparison of P. resedaeformis
Microsatellite profiling of the host populations in this study revealed a clear genetic difference between the two canyons. First, a Bayesian clustering analysis (STRUCTURE) recovered two distinct groupings based upon canyon of origin (Fig. 4). The genetic assignment of Norfolk Canyon P. resedaeformis samples suggest a few instances of mixed ancestry from Baltimore Canyon, which is concordant with larval transport following the slow southwestward flow of shelf and slope waters in the Mid-Atlantic Bight28. Second, a pairwise estimate of FST allele frequency-based measure of population differentiation29 between P. resedaeformis canyon populations was high and significant (FST = 0.117, p < 0.001). None of the loci appeared to be under selection based on the FST-outlier test performed using LOSITAN30. Third, genetic assignment methods correctly assigned individuals to their canyon of origin at 97% and 93% to Baltimore and Norfolk Canyons, respectively. This high assignment success is only slightly less than the 100% correct assignment to species using a portion of the same microsatellite markers for P. resedaeformis and P. pacifica31.
Figure 4.
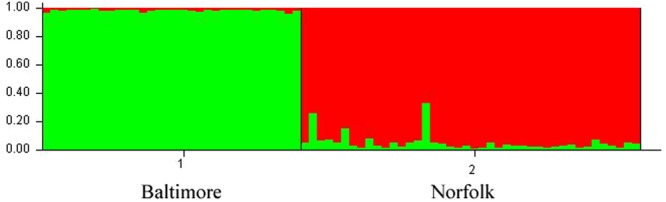
Bayesian clustering of Primnoa resedaeformis individuals from the Mid-Atlantic Bight canyons (Baltimore Canyon N = 32; Norfolk Canyon N = 42) based upon multilocus genotypes at eight microsatellite loci. Within bar plots, each P. resedaeformis individual is represented by a vertical bar partitioned into sections with lengths proportional to estimated probability of membership into K clusters, with the optimal number of clusters at K = 2.
ANOSIM/SIMPER
Additional ANOSIM and SIMPER analyses were conducted to analyze similarity between samples from Baltimore Canyon and Norfolk Canyon. The difference between bacterial communities from the two canyons was weaker (R = 0.254) than the difference between the two species, and the difference was barely significant (p = 0.047). However, the detection of a significant (albeit weak) difference between bacterial communities of the two P. resedaeformis populations is consistent with the microsatellite analysis that identified two distinct clusters of canyon samples (Fig. 4).
Considering each set of Canyon samples separately, the Norfolk Canyon samples had an average within-group similarity of 38.84%, while the Baltimore Canyon samples had an average within-group similarity of 34.65%. For Norfolk Canyon, the primary contributor to the similarity of the samples was Acinetobacter, which contributed (among two OTUs) nearly 12% of the average similarity. In Baltimore Canyon, the highest-contributing OTU was Kiloniellales (7.38%). Between the two canyons, the bacterial communities in P. resedaeformis were on average 72.35% dissimilar. Together, two Acinetobacter OTUs contributed just over 4% to this dissimilarity. Beyond that, no individual OTU contributed more than 3% to this dissimilarity. It is noteworthy that in contrast to the findings here, the microbiomes of the two Anthothela species studied in Lawler et al.20 exhibited no difference in bacterial community composition despite originating from the same two canyons sampled here.
Bacterial community composition
Differences emerged in the bacterial communities between the two submarine canyons. Two Norfolk Canyon samples contained moderate amounts of Mycoplasmataceae (12–15%), while Mycoplasmataceae did not exceed 3.6% of the bacterial community in any Baltimore Canyon sample (and were undetected in four samples) (Fig. 2). The Moraxellaceae family stood out because it was present in every Norfolk Canyon sample, ranging from 11% to 40% of those samples (Fig. 2). In contrast, only one Baltimore Canyon sample had a bacterial community that consisted of more than 10% Moraxellaceae (18% of sample PR_BC_08).
Discussion
Diversity in microbiomes of gorgonian corals
Temperate gorgonians in the Mediterranean have been shown to have very low diversity bacterial microbiomes, up to 90% composed of one or a few species, mainly of the genus Endozoicomonas22,32. It does not appear that deep-sea sibling species share this trait; for example, temperate Paramuricea clavata was dominated by Endozoicomonas32, but deep-sea Paramuricea placomus had no detectible Endozoicomonas, and no bacterial family represented more than 44% of the microbiome19. In this study, all but one of the P. pacifica samples (obtained from ca. 16 m) were dominated by Chlamydiae, much as Mediterranean gorgonian microbiomes are dominated by Endozoicomonas, whereas the P. resedaeformis samples obtained from the deep sea showed much more diversity (Fig. 2, Table 2). Because we were unable to sample at equivalent depths, we cannot separate differences based on species versus those due to depth or ocean basin, but mention this as a potential trend.
Unlike the comparison in this study, previous species comparisons within the same cold-water gorgonian genera have all been made using co-occurring species: Muricea spp. in California21, Eunicella spp. in the Mediterranean22, and Anthothela spp. in Atlantic submarine canyons20. The Anthothela microbiomes were not significantly different from each other. However, this may be due to the extremely low sequence divergence between the host species20. The three Eunicella species had significant overlap in their microbiomes, but this is largely due to the dominance of a single bacterial genus (Endozoicomonas), which kept the total diversity low. Even though E. singularis has zooxanthellae while the other two species (E. cavolini and E. verrucosa) do not, no obvious microbiome structuring was observed22. In contrast, the two Muricea species had very different microbiomes, and it was speculated that the presence of a photosymbiont in one species might be driving that difference21. Here, though both Primnoa species are azooxanthellate, they come from two different oceans, and it is interesting that the dissimilarity between the species is on par with that seen in co-occurring Muricea species.
Comparing microbiomes of P. resedaeformis and P. pacifica
While the weighted UniFrac metric explains the greatest amount of variation among the samples (Fig. 1), the other beta diversity matrices provide additional insights. Both the Sorensen-Dice and unweighted UniFrac indices reveal a clear separation between P. pacifica and P. resedaeformis, indicating that the differences between the species’ microbiomes are being driven by the presence/absence of specific taxa (e.g., the Rhabdochlamydiaceae family in P. pacifica). The dominance of just one taxon in all but one of the P. pacifica samples’ microbiomes highlights the lower diversity of those samples reflected in the alpha diversity metrics. These two indices (Sorensen-Dice and unweighted UniFrac) also show overlap of the P. resedaeformis samples from different canyons. Further, the Bray-Curtis metric, which is based on OTUs, has no overlap between the canyon samples at all whereas weighted UniFrac, which factors in phylogenetic relatedness, shows one Norfolk Canyon sample clustering with the Baltimore Canyon samples (Fig. 1). In contrast, diversity matrices that factor in abundance of taxa as well as presence/absence (weighted UniFrac and Bray-Curtis) show much cleaner separation between the two canyon populations, indicating that it is not unique taxa that characterize the two canyon populations, but rather abundance ratios (e.g., higher relative abundance of Moraxellaceae in Norfolk samples; Fig. 2).
Though it is unclear why one P. pacifica sample (PP_GA_06) clustered with the P. resedaeformis samples (Fig. 1; Fig. 2), environmental conditions are unlikely to be the cause. The Alaskan coral was sampled at a temperature of 4.6 °C (versus 7.4 to 9 °C for the closest Atlantic samples), depth of 12.8 m (versus 383 to 500 m), and salinity of 26.9 psu (versus 35 psu). Given that the environmental conditions of the outlying P. pacifica sample were similar to those of the other Alaska samples, and dissimilar from those of the canyon samples, those conditions cannot account for its clustering with the canyon samples. However, it is not necessarily surprising that it clustered near the more diverse canyon samples, because its bacterial community was the most diverse of all the Alaska samples by every measure (Table 2).
Core genus microbiome
The core genus microbiome consists of six OTUs, including Propionibacterium. The Propionibacterium OTU is noteworthy because the same OTU appears in the core microbiomes of two other cold-water corals: Paramuricea placomus19 and Anthothela sp.20. While the Propionibacterium OTU detected in Primnoa is two bases longer than the same OTU in Paramuricea and Anthothela, the three sequences are identical over the 331 bases they share. The similarity of the sequence of this persistent microbiome member among the three cold-water corals may be due to the close phylogenetic relationship among the three coral species; all fall within the Alcyonacea order. Not only is Propionibacterium a persistent member of the core microbiome of cold-water gorgonians, but it has also been identified as a rare but conserved member of the core microbiome of many stony tropical coral species33–38. However, its function in these microbiomes remains unknown39.
The other five core Primnoa OTUs (Supplementary Table S1) are also members of the Paramuricea placomus microbiome. The Bacillus and Lysobacter OTUs are identical to their counterparts found in P. placomus. The Acinetobacter OTU is two nucleotides longer in Primnoa than in P. placomus, but the OTUs are 99% identical over the 329 nucleotides they share. Comparing one set of core Pseudomonas OTUs (NCUR_OTU2 in Primnoa and OTU 4474944 in P. placomus) reveals that they are 99% identical over their shared 329 nucleotides. The other set of core Pseudomonas OTUs (OTU 4406538 in Primnoa and OTU 4478861 in P. placomus), both 331 nucleotides long, are 96% identical. The complete identity or high similarity of these core microbial OTUs in two different cold-water octocoral families (Plexauridae for Paramuricea, Primnoidae for Primnoa) allows us to speculate that these bacterial species may play a critical role in the functioning of the coral holobiont, at least for members of the Alcyonacea order.
The bacteria that are core to the Primnoa genus also appear in the microbiomes of many other corals, including tropical scleractinians. All five families that make up the Primnoa genus core have been documented in the microbiome of Porites lutea36. Three families (Pseudomonadaceae, Bacillaceae, and Moraxellaceae) are core members of the mucus microbiome of Mussimilia hispida40. In addition, Pseudomonadaceae are core members of the Coelastrea aspera microbiome34 and associate with numerous other tropical stony corals, including Stylophora pistillata41, Pachyseris speciosa37, Fungia echinata42, Orbicella faveolata43, O. annularis43, and Astrangia poculata44, whose range extends from tropical to temperate regions. This family of bacteria also populates the microbiomes of tropical soft corals45,46, as well as cold-water gorgonians19. Pseudomonads may assist their coral hosts in a broad range of metabolic functions47, which could account for their wide distribution in coral species. In particular, sulfur cycling genes have been identified in Pseudomonas species48, as well as genes implicated in the degradation of hydrocarbons, including crude oil49,50. Degradation of polycyclic aromatic hydrocarbons may also be performed by Xanthomonadaceae bacteria51, found as part of the core Primnoa microbiome as well as in the microbiome of Porites lutea36.
Moraxellaceae bacteria appear in the microbiomes of many of the same coral species as Pseudomonadaceae37,41–43,52. Like Pseudomonads, some members of the Moraxellaceae family can break down oil17,53, and may use that ability to recycle carbon for corals. Bacillaceae bacteria also populate the microbiomes of soft19,54 and stony54 corals, including tropical scleractinians36,40,54, and may benefit the corals by producing antimicrobial compounds that prevent host infection54,55.
Core species microbiome: Primnoa pacifica.
Significance of Chlamydiae sequences
Despite the dominance of Rhabdochlamydiaceae sequences in the P. pacifica samples, the sequences of DNA amplified from one of those samples in this study using Chlamydiales 23S primers did not match that family when queried against the GenBank nucleotide database. Rather, the top hits for those sequences were to Simkania negevensis, a member of the Simkaniaceae family in the Chlamydiales order. This suggests that Sanger sequencing did not capture the dominant Chlamydiales OTU present in the Alaska samples. However, there is some ambiguity about the lower-level classification of the OTU dominating the P. pacifica samples. Though Greengenes (within QIIME) identifies that OTU as a member of the Rhabdochlamydia genus (family Rhabdochlamydiaceae), the Ribosomal Database Project (RDP) Classifier56 identifies it as a member of the Parachlamydia genus in the Parachlamydiaceae family. It is not uncommon for databases to provide conflicting assignments at lower classification levels for a variety of reasons, including mislabeled sequences, errors in sequencing, and disproportionate population of databases by human-health-related bacteria rather than environmental samples57. Thus while we can confidently assign the OTU to the Chlamydiales order, database discrepancies prevent us from definitively classifying it to a family or genus.
We could find no reports of bacteria from the Rhabdochlamydiaceae family associated with corals or marine invertebrates. Chlamydiales sequences were recently found to be abundant in the microbiomes of cold-water sponges from the North Sea58. Relatively high percentages (>14%) of the bacterial communities associated with the sponges consisted of members of the Chlamydiales order. However, the sequences in that study that could be classified further were in the Parachlamydiaceae family, and are not closely related to the Chlamydiales sequences detected here. The only reports linking corals to Chlamydiales occur at the order, class, or phylum level, or through a histological study rather than a sequence-based study. Because none of the studies targeted the same region of the 16S gene as our study, we could not directly compare their sequences to ours. However, a Chlamydia-like bacteria was found in the microbiome of a shallow-water stony coral, Isopora palifera, comprising up to 33% of the bacterial communities in some samples off the coast of Taiwan59. At the phylum level, representatives of the Chlamydia phylum were reported in 2012 as 2% of the microbiome of a sample of Pocillopora verrucosa, a stony coral sampled in the Red Sea60. Members of the Chlamydiae class were also recently found in Red Sea P. verrucosa at abundances of nearly 25% of several samples’ microbiomes41. In the histological study, Work and Aeby61 examined an archive of coral tissue samples to search for cell-associated microbial aggregates. In their examination of 131 species and 36 genera of corals, they found that one species, Acropora acuminata, stained positive with a Gimenez stain, indicating the presence of chlamydia- or rickettsia-like bacteria.
Chlamydia in general are obligate intracellular bacteria, and have long been studied as animal/human pathogens. Now that their diverse presence in the environment as symbionts has been discovered62, we can speculate on what role they might play in a symbiosis. Chlamydiales bacteria may fulfill a metabolic function that Primnoa cannot themselves perform. Wagner and Horn62 suggest that the nucleotide transport proteins encoded by Chlamydia genomes may assist in exchanging bacterial ADP with host ATP. Alternately, these Chlamydiales sequences may reflect intracellular symbionts of amoebae that may be unrecognized members of these coral microbiomes63. In any case, given the relatively recent discovery of these bacteria in coral microbiomes, and the limited number of such reports, their role in coral-associated microbial communities is unknown.
Remaining members of core P. pacifica microbiome
Most remaining members of the core species microbiome for P. pacifica (Supplementary Table S1) have also been documented in other coral hosts’ microbiomes. Two families are also part of the Primnoa core genus microbiome (Pseudomonadaceae and Moraxellaceae, discussed above). Rhodobacteraceae are particularly widespread. Most recently, Rhodobacteraceae have been reported as core members of Mussimilia hispida mucus64, and appear in numerous other stony corals11,35–37,43,44,63. Many gorgonian corals also host Rhodobacteraceae19,21,46,65,66. This family’s presence in such a wide variety of coral hosts suggests that the bacteria serve a general function for the coral animal, but it has been hypothesized that their ability to oxidize thiosulfate as well as break down chitinous exoskeletons may provide those benefits to their coral hosts14.
Another member of the P. pacifica species core, Bradyrhizobiaceae, is commonly found in other coral microbiomes19,35,36,46,63, and may assist its hosts by fixing nitrogen67–69. Rhodocyclaceae bacteria, found in Porites lutea36, may degrade aromatic hydrocarbons51,70, while both Rhodocyclaceae and Comamonadaceae (found in soft, stony, temperate, and tropical corals19,36,37,71) have been linked to denitrification72,73. The SAR324 clade, a group of Deltaproteobacteria, are capable of a variety of metabolic pathways74 and thus could provide flexibility to their coral host. Streptococcus bacteria have been reported in other corals’ microbiomes34,36,75, but their function as part of the coral holobiont is not yet known. Both Staphylococcus36,37,41,54,75 and Enterobacteriaceae11,19,36,37,46,76 may act as opportunistic pathogens14,77,78 in the corals whose microbiomes they inhabit.
Core species microbiome: P. resedaeformis.
The core species microbiome for P. resedaeformis is much smaller than that of P. pacifica, containing only four members (families Rhodobacteraceae, Pirellulaceae, Vibrionaceae, and Sphingobium in the Sphingomonadaceae family). These bacteria commonly associate with corals. As noted above in discussion of the P. pacifica core species microbiome, members of the Rhodobacteraceae family are widespread in coral microbiomes, and may serve either general or particularized functions for the host. Pirellulaceae bacteria have been reported both in scleractinian11,36,44 and gorgonian19,66 corals, and have been hypothesized to play a role in the nitrogen cycle within the coral holobiont19,20,79,80.
In addition to appearing as core members of the P. resedaeformis microbiome, Vibrionaceae bacteria were recently identified as core members of the microbiome of stony coral Cladocora caespitosa81, and appear in the microbiomes of corals worldwide19,21,22,35–37,41,42,45,54,63–65,71,76,82–85. Though Vibrios are often associated with coral disease, their widespread association with healthy corals suggests an important function in the normal functioning of the coral holobiont. Vibrionaceae bacteria may fix nitrogen for the host68,86, and may also produce biosurfactant to degrade oil87. Sphingobium species also degrade polycyclic aromatic hydrocarbons88, and members of the Sphingomonadaceae family have recently been documented in other coral hosts11,36,37,44.
Inter-canyon comparison of P. resedaeformis populations
Microbiome diversity and host influence
A study of the soft coral Lobophytum pauciflorum’s microbiome found a higher abundance of Spirochaetes- and Rhodobacteraceae-related sequences in male versus female corals89. We did not see evidence of this pattern. However, we were only able to determine sex for 11 out of 20 samples, and only two of those were male (Supplementary Table S2). It has been suggested that microbiome composition and specificity may be influenced by the reproductive mode of the host coral, linking selection of specific bacterial groups to the host’s life history41. In a study of two tropical corals, the species that spawned (and therefore acquired its microbiome from the environment every time) always had the same species of Endozoicomonas, whereas the brooding species had a strong regional signal in its Endozoicomonas species, attributed to vertical transmission41. Primnoa resedaeformis is a broadcast spawner with external fertilization90, and thus any regional signal we observe in the microbiomes between the canyons cannot be attributed to host reproductive strategy.
Given the unique genetic signatures of P. resedaeformis in Baltimore and Norfolk Canyons, a possible explanation for the differences in microbiome composition detected between canyons may be the influence of host genotype on the microbiome. The possibility of coral host genotype affecting its bacterial associates has been proposed11,91 given that several studies have shown that coral microbiome or bacterial symbiont phylogenies correlated to host phylogeny92,93. Studies focused on the endangered tropical stony coral Acropora cervicornis found coral genotype to be linked to differences in thermal tolerance and bleaching94,95, growth rate94, and disease susceptibility96,97. Recent studies have also linked coral microbiome shifts to thermal tolerance98 and altered growth rates99, and there is an obvious link between the microbiome and disease susceptibility. Another recent soft coral study found that during stress experiments, the observed microbial profiles could be linked back to coral colonies, suggesting host genotype was driving their results89. Therefore, we suggest that the microbiome patterns in concert with the host population genetics observed between these two canyons may be another piece of evidence connecting coral microbiome selection to coral host genotype.
We cannot exclude the possibility that differences in the microbiomes between the two populations of canyon samples (P. resedaeformis) could stem from differences in environmental conditions under which the populations were sampled. However, the differences in the environmental conditions we measured were relatively minor (Table 1) and thus unlikely to be a factor in the differences in microbiomes between the two populations. Seasonality may play a role, given that the Baltimore Canyon samples were collected in August 2012, while the Norfolk Canyon samples were collected in May 2013 (Table S2). Differential abundance of several bacterial OTUs varied seasonally in the microbiomes of Mediterranean gorgonian corals (three Eunicella species and one Leptogorgia species), though no pattern to the variation could be detected76. The microbiome of temperate gorgonian Paramuricea clavata also varied with the season100. Seasonal shifts in relative bacterial abundance have also been observed in microbiomes associated with numerous non-gorgonian corals44,59,71,101–103. Note, however, that Anthothela species collected from the same two canyons at the same time points as the P. resedaeformis collections did not display any difference in their microbiomes20,104.
Moreover, it is possible that diet differs between the two populations of P. resedaeformis, given that there are differences in turbidity between the two canyons, which may affect the corals’ nutrition. While Baltimore Canyon has a persistent thick nepheloid layer105, turbidity layers in Norfolk Canyon are thinner and more uniform106. In addition, seasonal changes in the composition of particulate matter have been documented in Baltimore Canyon105, indicating that coral diet could change seasonally. However, two populations of Anthothela species sampled in the same canyons contain nearly identical microbiomes20, and we can find no information to suggest that Anthothela and Primnoa corals have different diets. Nonetheless, the observed differences in the microbiomes of P. resedaeformis between the two canyons may result from differences in diet, seasonality, host genotype, or a combination of those factors.
Conclusion
This study provides the first report of the microbial community associated with two Primnoa species of cold-water coral: P. pacifica from shallow water in Gulf of Alaska fjords, and P. resedaeformis from two submarine canyons in the western North Atlantic Ocean. One of the prominent differences between the two species’ microbiomes is the abundance of Chlamydiales bacteria in P. pacifica. The P. resedaeformis samples showed higher average mean diversity than those of P. pacifica. While five taxa (six OTUs) were core microbiome members conserved within the genus, principal coordinate analysis and ANOSIM statistics demonstrated that the two species’ microbiomes were relatively distinct. Within P. resedaeformis, a subtle but significant difference in bacterial communities was detectable between two submarine canyons. This pattern mirrors population genetic isolation demonstrated between the host coral population using microsatellite markers, raising the possibility that host genotype may play a role in differences in microbiomes between corals of the same species.
Materials and Methods
Sample sites and collection
Primnoa pacifica samples from Tracy Arm Fjord, Gulf of Alaska (Pacific Ocean) were collected at depths of 9.8 m to 16.2 m using SCUBA during research cruises in September 2011 and January 2012. Divers wearing clean nitrile gloves donned at the surface sampled the first P. pacifica colony located on the dive (i.e., no other corals were touched before collecting each of these samples). Small pieces of coral were broken from the colony by hand and placed into sterile 50-mL tubes (Fisher Scientific, Pittsburgh, PA). Environmental parameters were recorded for all samples (Table 1, Supplementary Table S2). Upon return to the surface, the ambient seawater in the tubes was decanted and replaced with RNAlater solution (Ambion, Waltham, MA). The tubes were kept at 4 °C overnight and then moved to −20 °C for long-term storage.
Primnoa resedaeformis samples were collected from Baltimore Canyon in the western North Atlantic Ocean (Table 1; Supplementary Table S2) on the NOAA ship Nancy Foster at depths of 383 m to 508 m using the remotely operated vehicle (ROV) Kraken II (National Undersea Research Technology and Education Center, University of Connecticut). Small pieces of colonies were collected using the ROV’s manipulator arm and placed into PVC tubes that had been washed, sterilized with ethanol, and filled with freshwater while the ROV was on deck. Care was taken to isolate samples from different coral colonies by placing only one sample per tube. Upon recovery of the ROV, the samples were removed from the tubes using ethanol-sterilized forceps, trimmed if necessary with ethanol-sterilized shears, and placed into 50-mL tubes. The tubes were filled with RNAlater and placed at 4 °C overnight, then transferred to −20 °C for long term storage. Georeferenced P. resedaeformis genetics samples were also stored in PVC tubes on the ROV. DNA from these samples was stabilized in 95% ethanol and Whatman FTA Technology Classic cards (GE Healthcare Bio-Sciences, Pittsburgh, PA). Additional P. resedaeformis samples were collected from Norfolk Canyon in the western North Atlantic Ocean on the NOAA ship Ronald H. Brown at depths of 411 m to 576 m using the ROV Jason II (Deep Submergence Laboratory, Woods Hole Oceanographic Institution) as described for the Baltimore Canyon samples.
Sex determination
Coral samples collected for sex determination were placed into 10% fully buffered formalin during the cruise and then transferred into 70% ethanol in the laboratory. Sex of the coral samples was determined by histological examination of oocytes and spermatocysts as described in Mercier and Hamel90. Briefly, the calcified skeleton of each sample was dissolved in 10% hydrochloric acid, then rinsed in distilled water and dehydrated through a series of ethanol baths. The tissues were then cleared, embedded into paraffin, and sliced into 8-µm sections. After mounting and staining, images of the sections were taken using a digital camera attached to a compound microscope.
Nucleic acid extraction and 16S rRNA gene sequencing
DNA was extracted from the corals using the MoBio PowerPlant DNA Isolation Kit (Carlsbad, CA) according to the manufacturer’s instructions as modified by Sunagawa et al.92,107. Extractions were performed in triplicate and then pooled for each sample. Extracted DNA was quantified using the PicoGreen DNA quantification kit (Invitrogen, Grand Island, NY). For microsatellite genotyping, total DNA was isolated from preserved coral tissue and/or FTA card hole-punches using the Gentra PureGene Tissue kit (QIAGEN, Germantown, MD). The DNA was eluted in 30 µL of molecular grade water. DNA extracted from the coral samples was amplified using primers 563F and 926 R, which target the V4-V5 region of the 16S rRNA gene: forward primer 5′-AYTGGGYDTAAAGNG, reverse primer 5′-CCGTCAATTYYTTTRAGTTT108. The DNA was then sequenced by 454 pyrosequencing using GS FLX Titanium chemistry following Roche 454′s standard protocol for amplicons. Twenty samples were sequenced on each plate, so barcodes were attached to the primers before amplification (Integrated DNA Technologies, Inc., Coralville, IA). Amplification and 454 pyrosequencing were conducted by Selah Genomics (Greenville, SC).
Bioinformatics and statistical analyses
Bioinformatic analysis of the sequences was performed using QIIME version 1.9.1109. A total of 1,517,466 raw reads were generated from 27 individual coral samples. Sequences were screened based on the following quality parameters: sequence length between 200 and 700 bp, minimum average quality score of 25, minimum quality score window of 50, maximum of one primer mismatch, maximum of six ambiguous bases, and a maximum six-homopolymer run110. Next, the sequences were denoised110,111. After screening and denoising, 409,886 sequences remained. Next, samples with a low yield of sequences were removed from the analysis. One sample (PR_BC_10) did not produce any reads, while three other samples (PR_NC_01, PR_NC_05, and PR_NC_09) each produced fewer than 2,500 reads. After removal of these samples, 23 samples and 404,999 sequences remained.
An open-reference method with a 97% similarity threshold112 was used to select OTUs in order to avoid discarding sequences that were not a perfect match to the Greengenes reference database release 13_8113,114. Chimeras were removed and the OTUs picked using usearch61115. Alignment was done with PyNAST (version 1.2.2)116. Representative sequences from each OTU were selected, assigned a taxonomic classification using uclust115, and used to generate a phylogenetic tree117. Non-bacterial sequences (i.e., Eukarya, Archaea, chloroplast, and mitochondria) and absolute singletons (defined as OTUs present only once in the analysis) were removed. After these steps, three samples had less than 2,500 sequences and were removed from the analysis (PR_NC_02, PR_NC_03, and PP_GA_03). Twenty samples and 321,578 sequences remained, with each individual sample library containing over 2,500 sequences. Samples were randomly rarefied to the size of the smallest remaining library (2,557 sequences) before diversity metrics were calculated118. A comprehensive list of scripts and parameters used in this analysis is presented as part of a USGS data release119 (https://doi.org/10.5066/F7P55KMJ).
Alpha and beta diversity measurements and OTU relative abundances were calculated using QIIME v. 1.9.1109. Beta diversity metrics were visualized via principal coordinate analysis (PCoA) using the vegan package120 in R121. PRIMER-E122 was used to calculate analysis of similarities (ANOSIM) from the weighted UniFrac distance matrix using 9999 permutations. PRIMER-E was also used to conduct SIMPER analysis, which quantifies within-group similarities and between-group dissimilarities, and identifies the OTUs that are responsible for each. Because SIMPER analysis is based on Bray-Curtis dissimilarity, the analysis incorporates differences in abundance of OTUs but not phylogenetic distance between OTUs. Relative abundance column graphs were prepared in R121. The core microbiome (set of shared OTUs) was derived from the OTU table prepared by QIIME before the rarefaction step. To be considered a member of the core genus microbiome, an OTU had to be present in 100% of samples. OTUs were designated as members of core species microbiomes when they were present in 100% of that species’ samples. The core microbiomes were analyzed in Excel and visualized with R.
23S Chlamydiales gene from P. pacifica
Because the QIIME analysis assigned some sequences from the P. pacifica samples to the Rhabdochlamydia genus (family Rhabdochlamydiaceae, order Chlamydiales), we attempted to amplify the 23S rRNA gene specific to the Chlamydiales order from bacterial DNA extracted from those samples. Primers U23F (5′-GATGCCTTGGCATTGATAGGCGATGAAGGA) and 23SIGR (5′-TGGCTCATCATGCAAAAGGCA)123 were used to amplify a product of approximately 600 bp. The 50-µL reaction mixture contained 25 µL AmpliTaq Gold 360 Master Mix (Applied Biosystems, Foster City, CA), 0.2 µM concentration of each primer, and 1 µL of template. Reaction conditions of Everett et al.123 were followed after 15 min of initial denaturation at 95 °C. One Alaska sample, PP_GA_01, yielded a visible PCR product. The product was cloned into the pDrive vector using the PCR Cloning Plus kit (QIAGEN, Germantown, MD) and used to transform competent cells. Inserts in positive transformants were sequenced by Eurofins Genomics (Louisville, KY). Vector sequences were identified with VecScreen124 and trimmed using EnzymeX v.3.3.3 (Nucleobytes, Amsterdam, The Netherlands). Sequences were aligned with Clustal Omega as implemented by EMBL-EBI125,126, revealing two variations of the sequences. The sequences were analyzed using BLASTN against the nt database127.
Microsatellite genotyping and analyses
Next-generation sequencing was used to develop microsatellite loci for both Primnoa species sampled in this study31. Eight of these loci amplified successfully and were polymorphic in the Atlantic Canyons samples: Prim014, Prim026, Prim060, Prim068, Prim069, Prim074, Prim094, and Prim096. Loci were amplified singly via PCR following conditions in Morrison et al.31, with a final reaction volume of 20 µL.
In order to describe genetic relationships between populations, an allele frequency model-based Bayesian clustering approach128 was implemented in STRUCTURE v. 2.3.4129. This method infers the number of genetic clusters (K) from multi-locus genotype data by minimizing Hardy-Weinberg equilibrium and linkage disequilibrium among loci within groups and assigning individuals (probabilistically) to each cluster. Models utilizing collection location information to inform prior probability that an individual sample comes from a particular population have proven useful when detecting genetic structure in small datasets or when structuring is weak129. Therefore, we included sample location information to weight the model in favor of outcomes that are correlated with sampling information. Settings for all runs also included an admixture model (i.e., individuals may have mixed ancestry), correlated allele frequencies130, and 200,000 Markov chain Monte Carlo iterations after a burn-in of 50,000 iterations. Ten independent chains were run to test each value of K. The optimum number of clusters was determined by evaluating the values of K as the highest mean likelihood of the probability of the number of clusters given the data observed, ln Pr(X|K)128, and ΔK131. This information was compiled and graphed using STRUCTURE Harvester v.0.56.1132. Additionally, population genetic differentiation among canyons was examined through a pairwise FST allele frequency-based estimate29. Significance of the FST value was tested using 999 pairwise population permutations in GenAlEx v. 6.5b4133,134. Assignment tests were performed in GenAlEx using the leave-one-out procedure. Microsatellite loci were searched to identify those potentially under selection using LOSITAN (Antao et al.30), which implements FST-outlier tests. Simulations were run under infinite alleles and stepwise mutation models using default settings (50,000 replications).
Electronic supplementary material
Acknowledgements
Thanks are extended to the captains and the crews of the NOAA ships Nancy Foster and Ronald H. Brown, as well as to the Kraken II ROV team and Jason II ROV team. Thanks also to Captain Dan Foley and the crew of the MV Steller for their assistance. Special thanks to G. Boland and R. Green (BOEM) for their coordination efforts, as well S. Viada (CSA) for support during the overall project. Betsy Boynton (USGS) prepared final versions of all the figures. Funding for this project was provided by the U.S. Geological Survey’s Ecosystems Mission Area, Environments Program through the Outer Continental Shelf study on Mid-Atlantic Canyons. The Gulf of Alaska field research was supported by funding from the Alaska Fisheries Science Center. Additional funding was sponsored by the National Oceanographic Partnership Program and supplied by the Bureau of Ocean Energy Management (BOEM) contract number M10PC00100 (contracted to CSA Ocean Sciences, Inc.). Ship time and ROV assets were funded by the NOAA Office of Ocean Exploration. The findings and conclusions in this paper are those of the authors and the U.S. Geological Survey but do not necessarily represent the views of the National Marine Fisheries Service, NOAA. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the US government.
Author Contributions
D.B.G. analyzed data, interpreted results, prepared figures and tables, and took the lead in writing the manuscript. C.A.K. conceived and planned the experiments, collected samples, analyzed data, interpreted results, and wrote the manuscript. C.L.M. analyzed data, interpreted results, and wrote the population genetics methods and results sections of the manuscript. M.A.G. collected samples, extracted DNA, prepared tables, and wrote the molecular methods section of the manuscript. R.P.S. collected samples and provided gender data for P. pacifica. R.G.W. collected samples and provided gender data for P. pacifica. S.D.B. collected samples and provided gender data for P. resedaeformis. S.W.R. collected samples. All authors reviewed drafts of the manuscript.
Data availability
The raw data files associated with the 16 S sequences in this study have been submitted to the NCBI Sequence Read Archive under Bioproject number PRJNA348705 and are also available from the USGS data release119 (https://doi.org/10.5066/F7P55KMJ). The bar codes used in sequencing can be found in mapping files, which are part of the data release. The USGS data release also contains the bioinformatics workflow, including scripts and parameters for each step. The 23 S sequences have been submitted to GenBank and assigned accession numbers KY010287 and KY010288; they are also part of the USGS data release. A separate USGS data release135 presents the microsatellite genotypes for Primnoa resedaeformis (https://doi.org/10.5066/F7B27T74).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-30901-z.
References
- 1.Reaka-Kudla, M. L. The global biodiversity of coral reefs: a comparison with rain forests in Biodiversity II: Understanding and Protecting Our Biological Resources (eds Reaka-Kudla, M. L., Wilson, D. E. & Wilson, E. O.) Ch. 7, 83–108 (Joseph Henry Press, 1997).
- 2.Roberts JM, Wheeler AJ, Freiwald A. Reefs of the deep: The biology and geology of cold-water coral ecosystems. Science. 2006;312:543–547. doi: 10.1126/science.1119861. [DOI] [PubMed] [Google Scholar]
- 3.Ross SW, Quattrini AM. The fish fauna associated with deep coral banks off the southeastern United States. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 2007;54:975–1007. doi: 10.1016/j.dsr.2007.03.010. [DOI] [Google Scholar]
- 4.Stone RP. Coral habitat in the Aleutian Islands of Alaska: depth distribution, fine-scale species associations, and fisheries interactions. Coral Reefs. 2006;25:229–238. doi: 10.1007/s00338-006-0091-z. [DOI] [Google Scholar]
- 5.Stone RP, Masuda MM, Karinen JF. Assessing the ecological importance of red tree coral thickets in the eastern Gulf of Alaska. ICES J. Mar. Sci. 2015;72:900–915. doi: 10.1093/icesjms/fsu190. [DOI] [Google Scholar]
- 6.Ross SW, Rhode M, Quattrini AM. Demersal fish distribution and habitat use within and near Baltimore and Norfolk Canyons, US middle Atlantic slope. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 2015;103:137–154. doi: 10.1016/j.dsr.2015.06.004. [DOI] [Google Scholar]
- 7.Buhl-Mortensen L, Mortensen PB. Symbiosis in deep-water corals. Symbiosis. 2004;37:33–61. [Google Scholar]
- 8.Meistertzheim AL, et al. Patterns of bacteria-host associations suggest different ecological strategies between two reef building cold-water coral species. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 2016;114:12–22. doi: 10.1016/j.dsr.2016.04.013. [DOI] [Google Scholar]
- 9.Schöttner S, et al. Inter- and intra-habitat bacterial diversity associated with cold-water corals. ISME J. 2009;3:756–759. doi: 10.1038/ismej.2009.15. [DOI] [PubMed] [Google Scholar]
- 10.Schöttner S, Wild C, Hoffmann F, Boetius A, Ramette A. Spatial scales of bacterial diversity in cold-water coral reef ecosystems. PLoS One. 2012;7:e32093. doi: 10.1371/journal.pone.0032093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kellogg CA, Goldsmith DB, Gray MA. Biogeographic comparison of Lophelia-associated bacterial communities in the western Atlantic reveals conserved core microbiome. Front. Microbiol. 2017;8:796. doi: 10.3389/fmicb.2017.00796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galkiewicz JP, Pratte ZA, Gray MA, Kellogg CA. Characterization of culturable bacteria isolated from the cold-water coral Lophelia pertusa. FEMS Microbiol. Ecol. 2011;77:333–346. doi: 10.1111/j.1574-6941.2011.01115.x. [DOI] [PubMed] [Google Scholar]
- 13.Kellogg CA, Lisle JT, Galkiewicz JP. Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 2009;75:2294–2303. doi: 10.1128/AEM.02357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neulinger SC, Järnegren J, Ludvigsen M, Lochte K, Dullo W-C. Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (Scleractinia) and their implications for the coral’s nutrition, health, and distribution. Appl. Environ. Microbiol. 2008;74:7272–7285. doi: 10.1128/AEM.01777-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Bleijswijk J, et al. Microbial assemblages on a cold-water coral mound at the SE Rockall Bank (NE Atlantic): interactions with hydrography and topography. Biogeosciences. 2015;12:4483–4496. doi: 10.5194/bg-12-4483-2015. [DOI] [Google Scholar]
- 16.Hansson L, Agis M, Maier C, Weinbauer MG. Community composition of bacteria associated with cold-water coral Madrepora oculata: within and between colony variability. Mar. Ecol. Prog. Ser. 2009;397:89–102. doi: 10.3354/meps08429. [DOI] [Google Scholar]
- 17.Röthig T, Yum LK, Kremb SG, Roik A, Voolstra CR. Microbial community composition of deep-sea corals from the Red Sea provides insight into functional adaption to a unique environment. Sci. Rep. 2017;7:44714. doi: 10.1038/srep44714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray MA, Stone RP, McLaughlin MR, Kellogg CA. Microbial consortia of gorgonian corals from the Aleutian Islands. FEMS Microbiol. Ecol. 2011;76:109–120. doi: 10.1111/j.1574-6941.2010.01033.x. [DOI] [PubMed] [Google Scholar]
- 19.Kellogg CA, Ross SW, Brooke SD. Bacterial community diversity of the deep-sea octocoral Paramuricea placomus. PeerJ. 2016;4:e2529. doi: 10.7717/peerj.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawler SN, et al. Coral-associated bacterial diversity is conserved across two deep-sea Anthothela species. Front. Microbiol. 2016;7:458. doi: 10.3389/fmicb.2016.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holm, J. B. & Heidelberg, K. B. Microbiomes of Muricea californica and M. fruticosa: comparative analyses of two co-occurring eastern Pacific octocorals. Front. Microbiol. 7 (2016). [DOI] [PMC free article] [PubMed]
- 22.van de Water JAJM, et al. Comparative assessment of Mediterranean gorgonian-associated microbial communities reveals conserved core and locally variant bacteria. Microb. Ecol. 2016;73:446–478. doi: 10.1007/s00248-016-0858-x. [DOI] [PubMed] [Google Scholar]
- 23.Chao A. Nonparametric estimation of the number of classes in a population. Scand. Stat. Theory Appl. 1984;11:265–270. [Google Scholar]
- 24.Chao A, Lee S-M. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 1992;87:210–217. doi: 10.1080/01621459.1992.10475194. [DOI] [Google Scholar]
- 25.Simpson E. Measurement of diversity. Nature. 1949;163:688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 26.Shannon, C. A mathematical theory of communication. Bell System Technical Journal27, 379–423, 623–565 (1948).
- 27.Morris EK, et al. Choosing and using diversity indices: insights for ecological applications from the German Biodiversity Exploratories. Ecol. Evol. 2014;4:3514–3524. doi: 10.1002/ece3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Packer, D. B., Boelke, D., Guida, V. & McGee, L.-A. State of deep coral ecosystems in the northeastern US region: Maine to Cape Hatteras in The State of Deep Coral Ecosystems of the United States. (eds Lumsden, S. E., Hourigan, T. F., Bruckner, A. W. & Dorr, G.) 195–232 (NOAA Technical Memorandum CRCP-3, 2007).
- 29.Weir BS, Cockerham CC. Estimating F‐statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 30.Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G. LOSITAN: a workbench to detect molecular adaptation based on a F ST-outlier method. BMC Bioinformatics. 2008;9:323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison C, Springmann MJ, Shroades K, Stone RP. Development of twelve microsatellite loci in the red tree corals Primnoa resedaeformis and Primnoa pacifica. Conserv. Genet. Resour. 2015;7:763–765. doi: 10.1007/s12686-015-0455-1. [DOI] [Google Scholar]
- 32.Vezzulli L, Pezzati E, Huete-Stauffer C, Pruzzo C, Cerrano C. 16 SrDNA pyrosequencing of the Mediterranean gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PLoS One. 2013;8:e67745. doi: 10.1371/journal.pone.0067745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ainsworth TD, et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015;9:2261–2274. doi: 10.1038/ismej.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sweet MJ, Brown BE, Dunne RP, Singleton I, Bulling M. Evidence for rapid, tide-related shifts in the microbiome of the coral Coelastrea aspera. Coral Reefs. 2017;36:815–828. doi: 10.1007/s00338-017-1572-y. [DOI] [Google Scholar]
- 35.Glasl B, et al. Microbiome variation in corals with distinct depth distribution ranges across a shallow–mesophotic gradient (15–85 m) Coral Reefs. 2017;36:447–452. doi: 10.1007/s00338-016-1517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pootakham W, et al. High resolution profiling of coral-associated bacterial communities using full-length 16S rRNA sequence data from PacBio SMRT sequencing system. Sci. Rep. 2017;7:2774. doi: 10.1038/s41598-017-03139-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez-Agreda A, Leggat W, Bongaerts P, Ainsworth TD. The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio. 2016;7:e00560–00516. doi: 10.1128/mBio.00560-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hester ER, Barott KL, Nulton J, Vermeij MJ, Rohwer FL. Stable and sporadic symbiotic communities of coral and algal holobionts. ISME J. 2015;10:1157. doi: 10.1038/ismej.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Water JA, Allemand D, Ferrier-Pagès C. Host-microbe interactions in octocoral holobionts-recent advances and perspectives. Microbiome. 2018;6:64. doi: 10.1186/s40168-018-0431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leite DC, et al. Broadcast spawning coral Mussismilia hispida can vertically transfer its associated bacterial core. Front. Microbiol. 2017;8:176. doi: 10.3389/fmicb.2017.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neave MJ, et al. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 2017;11:186–200. doi: 10.1038/ismej.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badhai J, Ghosh TS, Das SK. Composition and functional characterization of microbiome associated with mucus of the coral Fungia echinata collected from Andaman Sea. Front. Microbiol. 2016;7:936. doi: 10.3389/fmicb.2016.00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer JL, Rodgers JM, Dillard BA, Paul VJ, Teplitski M. Epimicrobiota associated with the decay and recovery of Orbicella corals exhibiting dark spot syndrome. Front. Microbiol. 2016;7:893. doi: 10.3389/fmicb.2016.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharp KH, Pratte ZA, Kerwin AH, Rotjan RD, Stewart FJ. Season, but not symbiont state, drives microbiome structure in the temperate coral Astrangia poculata. Microbiome. 2017;5:120. doi: 10.1186/s40168-017-0329-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Correa H, Haltli B, Duque C, Kerr R. Bacterial communities of the gorgonian octocoral Pseudopterogorgia elisabethae. Microb. Ecol. 2013;66:972–985. doi: 10.1007/s00248-013-0267-3. [DOI] [PubMed] [Google Scholar]
- 46.Shirur KP, Jackson CR, Goulet TL. Lesion recovery and the bacterial microbiome in two Caribbean gorgonian corals. Mar. Biol. 2016;163:238. doi: 10.1007/s00227-016-3008-6. [DOI] [Google Scholar]
- 47.Röthig T, Ochsenkühn MA, Roik A, Merwe R, Voolstra CR. Long‐term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol. Ecol. 2016;25:1308–1323. doi: 10.1111/mec.13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng Y-X, Qiao Z-Y, Yu Y, Li H-R, Luo W. Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of Arctic Kongsfjorden. Sci. Rep. 2016;6:33031. doi: 10.1038/srep33031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Izmalkova TY, et al. Diversity of Oil-Degrading Microorganisms in the Gulf of Finland (Baltic Sea) in Spring and in Summer. Microbiology. 2018;87:261–271. doi: 10.1134/S0026261718020054. [DOI] [Google Scholar]
- 50.Liu W, Duan X, Wu L, Masakorala K. Biosurfactant Production by Pseudomonas aeruginosa SNP0614 and its Effect on Biodegradation of Petroleum. Appl. Biochem. Microbiol. 2018;54:155–162. doi: 10.1134/S0003683818020060. [DOI] [Google Scholar]
- 51.Jayamani I, Cupples AM. Stable isotope probing and high-throughput sequencing implicate Xanthomonadaceae and Rhodocyclaceae in ethylbenzene degradation. Environ. Eng. Sci. 2015;32:240–249. doi: 10.1089/ees.2014.0456. [DOI] [Google Scholar]
- 52.Cai L, et al. Exploring coral microbiome assemblages in the South China Sea. Sci. Rep. 2018;8:2428. doi: 10.1038/s41598-018-20515-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al-Dahash LM, Mahmoud HM. Harboring oil-degrading bacteria: A potential mechanism of adaptation and survival in corals inhabiting oil-contaminated reefs. Mar. Pollut. Bull. 2013;72:364–374. doi: 10.1016/j.marpolbul.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 54.Pereira, L. B., Palermo, B. R., Carlos, C. & Ottoboni, L. M. Diversity and antimicrobial activity of bacteria isolated from different Brazilian coral species. FEMS Microbiol. Lett. 364 (2017). [DOI] [PubMed]
- 55.Ahila N, et al. Bio-prospecting of coral (Porites lutea) mucus associated bacteria, Palk Bay reefs, Southeast coast of India. Microb. Pathog. 2017;113:113–123. doi: 10.1016/j.micpath.2017.09.056. [DOI] [PubMed] [Google Scholar]
- 56.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollock J, Glendinning L, Wisedchanwet T, Watson M. The madness of microbiome: Attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 2018;84:e02627–02617. doi: 10.1128/AEM.02627-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naim MA, et al. Host-specific microbial communities in three sympatric North Sea sponges. FEMS Microbiol. Ecol. 2014;90:390–403. doi: 10.1111/1574-6941.12400. [DOI] [PubMed] [Google Scholar]
- 59.Chen C-P, Tseng C-H, Chen CA, Tang S-L. The dynamics of microbial partnerships in the coral Isopora palifera. ISME J. 2011;5:728. doi: 10.1038/ismej.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee OO, et al. Spatial and species variations in bacterial communities associated with corals from the Red Sea as revealed by pyrosequencing. Appl. Environ. Microbiol. 2012;78:7173–7184. doi: 10.1128/AEM.01111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Work TM, Aeby GS. Microbial aggregates within tissues infect a diversity of corals throughout the Indo-Pacific. Mar. Ecol. Prog. Ser. 2014;500:1–9. doi: 10.3354/meps10698. [DOI] [Google Scholar]
- 62.Wagner M, Horn M. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr. Opin. Biotechnol. 2006;17:241–249. doi: 10.1016/j.copbio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 63.Apprill A, Weber LG, Santoro AE. Distinguishing between microbial habitats unravels ecological complexity in coral microbiomes. mSystems. 2016;1:e00143–00116. doi: 10.1128/mSystems.00143-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leite, D. C., Salles, J. F., Calderon, E. N., Elsas, J. D. & Peixoto, R. S. Specific plasmid patterns and high rates of bacterial co‐occurrence within the coral holobiont. Ecol. Evol. (2018). [DOI] [PMC free article] [PubMed]
- 65.Keller-Costa T, et al. The gorgonian coral Eunicella labiata hosts a distinct prokaryotic consortium amenable to cultivation. FEMS Microbiol. Ecol. 2017;93:fix143. doi: 10.1093/femsec/fix143. [DOI] [PubMed] [Google Scholar]
- 66.McCauley, E. P., Haltli, B., Correa, H. & Kerr, R. G. Spatial and temporal investigation of the microbiome of the Caribbean octocoral Erythropodium caribaeorum. FEMS Microbiol. Ecol. 92 (2016). [DOI] [PubMed]
- 67.Godoy-Vitorino F, Ruiz-Diaz CP, Rivera-Seda A, Ramírez-Lugo JS, Toledo-Hernández C. The microbial biosphere of the coral Acropora cervicornis in Northeastern Puerto Rico. PeerJ. 2017;5:e3717. doi: 10.7717/peerj.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krediet CJ, Ritchie KB, Paul VJ, Teplitski M. Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proceedings of the Royal Society of London B: Biological Sciences. 2013;280:20122328. doi: 10.1098/rspb.2012.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lema KA, Willis BL, Bourne DG. Corals form characteristic associations with symbiotic nitrogen-fixing bacteria. Appl. Environ. Microbiol. 2012;78:3136–3144. doi: 10.1128/AEM.07800-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vila J, Tauler M, Grifoll M. Bacterial PAH degradation in marine and terrestrial habitats. Curr. Opin. Biotechnol. 2015;33:95–102. doi: 10.1016/j.copbio.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 71.Rubio‐Portillo E, et al. Structure and temporal dynamics of the bacterial communities associated to microhabitats of the coral Oculina patagonica. Environ. Microbiol. 2016;18:4564–4578. doi: 10.1111/1462-2920.13548. [DOI] [PubMed] [Google Scholar]
- 72.Ginige MP, Keller J, Blackall LL. Investigation of an acetate-fed denitrifying microbial community by stable isotope probing, full-cycle rRNA analysis, and fluorescent in situ hybridization-microautoradiography. Appl. Environ. Microbiol. 2005;71:8683–8691. doi: 10.1128/AEM.71.12.8683-8691.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan ST, Horiba Y, Yamamoto M, Hiraishi A. Members of the family Comamonadaceae as primary poly (3-hydroxybutyrate-co-3-hydroxyvalerate)-degrading denitrifiers in activated sludge as revealed by a polyphasic approach. Appl. Environ. Microbiol. 2002;68:3206–3214. doi: 10.1128/AEM.68.7.3206-3214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheik CS, Jain S, Dick GJ. Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environ. Microbiol. 2014;16:304–317. doi: 10.1111/1462-2920.12165. [DOI] [PubMed] [Google Scholar]
- 75.Brück TB, Brück WM, Santiago-Vázquez LZ, McCarthy PJ, Kerr RG. Diversity of the bacterial communities associated with the azooxanthellate deep water octocorals Leptogorgia minimata, Iciligorgia schrammi, and Swiftia exertia. Marine Biotechnology. 2007;9:561–576. doi: 10.1007/s10126-007-9009-1. [DOI] [PubMed] [Google Scholar]
- 76.van de Water JA, et al. Seasonal stability in the microbiomes of temperate gorgonians and the red coral Corallium rubrum across the Mediterranean Sea. Microb. Ecol. 2018;75:274–288. doi: 10.1007/s00248-017-1006-y. [DOI] [PubMed] [Google Scholar]
- 77.Blackall LL, Wilson B, Oppen MJ. Coral—the world’s most diverse symbiotic ecosystem. Mol. Ecol. 2015;24:5330–5347. doi: 10.1111/mec.13400. [DOI] [PubMed] [Google Scholar]
- 78.Daniels C, et al. Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Frontiers in Marine Science. 2015;2:62. doi: 10.3389/fmars.2015.00062. [DOI] [Google Scholar]
- 79.Mohamed NM, Saito K, Tal Y, Hill RT. Diversity of aerobic and anaerobic ammonia-oxidizing bacteria in marine sponges. ISME J. 2010;4:38. doi: 10.1038/ismej.2009.84. [DOI] [PubMed] [Google Scholar]
- 80.Zehr JP, Kudela RM. Nitrogen cycle of the open ocean: from genes to ecosystems. Ann. Rev. Mar. Sci. 2011;3:197–225. doi: 10.1146/annurev-marine-120709-142819. [DOI] [PubMed] [Google Scholar]
- 81.Rubio-Portillo E, Kersting DK, Linares C, Ramos-Esplá AÁ, Antón J. Biogeographic differences in the microbiome and pathobiome of the coral Cladocora caespitosa in the Western Mediterranean Sea. Front. Microbiol. 2018;9:22. doi: 10.3389/fmicb.2018.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beatty DS, Clements CS, Stewart FJ, Hay ME. Intergenerational effects of macroalgae on a reef coral: major declines in larval survival but subtle changes in microbiomes. Mar. Ecol. Prog. Ser. 2018;589:97–114. doi: 10.3354/meps12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franco T, Califano G, Gonçalves AC, Cúcio C, Costa R. Draft genome sequence of Vibrio sp. strain Evh12, a bacterium retrieved from the gorgonian coral Eunicella verrucosa. Genome announcements. 2016;4:e01729–01715. doi: 10.1128/genomeA.01729-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harder T, Lau SC, Dobretsov S, Fang TK, Qian P-Y. A distinctive epibiotic bacterial community on the soft coral Dendronephthya sp. and antibacterial activity of coral tissue extracts suggest a chemical mechanism against bacterial epibiosis. FEMS Microbiol. Ecol. 2003;43:337–347. doi: 10.1111/j.1574-6941.2003.tb01074.x. [DOI] [PubMed] [Google Scholar]
- 85.Shiu J-H, et al. Dynamics of coral-associated bacterial communities acclimated to temperature stress based on recent thermal history. Sci. Rep. 2017;7:14933. doi: 10.1038/s41598-017-14927-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chimetto LA, et al. Vibrios dominate as culturable nitrogen-fixing bacteria of the Brazilian coral Mussismilia hispida. Syst. Appl. Microbiol. 2008;31:312–319. doi: 10.1016/j.syapm.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 87.Hu X, Wang C, Wang P. Optimization and characterization of biosurfactant production from marine Vibrio sp. strain 3B-2. Front. Microbiol. 2015;6:976. doi: 10.3389/fmicb.2015.00976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fu B, et al. Sphingobium sp. FB3 degrades a mixture of polycyclic aromatic hydrocarbons. Int. Biodeterior. Biodegradation. 2014;87:44–51. doi: 10.1016/j.ibiod.2013.10.024. [DOI] [Google Scholar]
- 89.Wessels W, Sprungala S, Watson SA, Miller DJ, Bourne DG. The microbiome of the octocoral Lobophytum pauciflorum: minor differences between sexes and resilience to short-term stress. FEMS Microbiol. Ecol. 2017;93:fix013. doi: 10.1093/femsec/fix013. [DOI] [PubMed] [Google Scholar]
- 90.Mercier A, Hamel J-F. Contrasting reproductive strategies in three deep-sea octocorals from eastern Canada: Primnoa resedaeformis, Keratoisis ornata, and Anthomastus grandiflorus. Coral Reefs. 2011;30:337–350. doi: 10.1007/s00338-011-0724-8. [DOI] [Google Scholar]
- 91.Hernandez-Agreda A, Gates RD, Ainsworth TD. Defining the core microbiome in corals’ microbial soup. Trends Microbiol. 2017;25:125–140. doi: 10.1016/j.tim.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Sunagawa S, Woodley CM, Medina M. Threatened corals provide underexplored microbial habitats. PLoS One. 2010;5:e9554. doi: 10.1371/journal.pone.0009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.La Rivière M, Garrabou J, Bally M. Evidence for host specificity among dominant bacterial symbionts in temperate gorgonian corals. Coral Reefs. 2015;34:1087–1098. doi: 10.1007/s00338-015-1334-7. [DOI] [Google Scholar]
- 94.Ladd MC, Shantz AA, Bartels E, Burkepile DE. Thermal stress reveals a genotype-specific tradeoff between growth and tissue loss in restored Acropora cervicornis. Mar. Ecol. Prog. Ser. 2017;572:129–139. doi: 10.3354/meps12169. [DOI] [Google Scholar]
- 95.Drury C, Manzello D, Lirman D. Genotype and local environment dynamically influence growth, disturbance response and survivorship in the threatened coral, Acropora cervicornis. PLoS One. 2017;12:e0174000. doi: 10.1371/journal.pone.0174000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Muller EM, van Woesik R. Genetic susceptibility, colony size, and water temperature drive white-pox disease on the coral Acropora palmata. PLoS One. 2014;9:e110759. doi: 10.1371/journal.pone.0110759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vollmer SV, Kline DI. Natural disease resistance in threatened staghorn corals. PLoS One. 2008;3:e3718. doi: 10.1371/journal.pone.0003718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ziegler M, Seneca FO, Yum LK, Palumbi SR, Voolstra CR. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat. Commun. 2017;8:14213. doi: 10.1038/ncomms14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thurber RV, et al. Macroalgae decrease growth and alter microbial community structure of the reef-building coral. Porites astreoides. PLoS One. 2012;7:e44246. doi: 10.1371/journal.pone.0044246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.La Rivière M, Roumagnac M, Garrabou J, Bally M. Transient shifts in bacterial communities associated with the temperate gorgonian Paramuricea clavata in the Northwestern Mediterranean Sea. PLoS One. 2013;8:e57385. doi: 10.1371/journal.pone.0057385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J, et al. Bacterial dynamics within the mucus, tissue and skeleton of the coral Porites lutea during different seasons. Sci. Rep. 2014;4:7320. doi: 10.1038/srep07320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ceh J, Van Keulen M, Bourne DG. Coral-associated bacterial communities on Ningaloo Reef, Western Australia. FEMS Microbiol. Ecol. 2010;75:134–144. doi: 10.1111/j.1574-6941.2010.00986.x. [DOI] [PubMed] [Google Scholar]
- 103.Kimes NE, et al. The Montastraea faveolata microbiome: ecological and temporal influences on a Caribbean reef‐building coral in decline. Environ. Microbiol. 2013;15:2082–2094. doi: 10.1111/1462-2920.12130. [DOI] [PubMed] [Google Scholar]
- 104.Fernandez-Arcaya U, et al. Ecological role of submarine canyons and need for canyon conservation. Frontiers in Marine Science. 2017;4:00005. doi: 10.3389/fmars.2017.00005. [DOI] [Google Scholar]
- 105.Prouty NG, et al. Seasonal variability in the source and composition of particulate matter in the depositional zone of Baltimore Canyon, US Mid-Atlantic Bight. Deep Sea Research Part I: Oceanographic Research Papers. 2017;127:77–89. doi: 10.1016/j.dsr.2017.08.004. [DOI] [Google Scholar]
- 106.Brooke S, et al. Distributions and habitat associations of deep-water corals in Norfolk and Baltimore Canyons, Mid-Atlantic Bight, USA. Deep Sea Research Part II: Topical Studies in Oceanography. 2017;137:131–147. doi: 10.1016/j.dsr2.2016.05.008. [DOI] [Google Scholar]
- 107.Baker, E. J. & Kellogg, C. A. Comparison of three DNA extraction kits to establish maximum yield and quality of coral-associated microbialDNA. US Geological Survey Open-File Report 2014–1066, http://pubs.usgs.gov/of/2014/1066 (2014).
- 108.Claesson MJ, et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010;38:e200. doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 111.Quince C, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 112.Rideout JR, et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2:e545. doi: 10.7717/peerj.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 116.Caporaso JG, et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Price MN, Dehal PS, Arkin AP. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gihring TM, Green SJ, Schadt CW. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ. Microbiol. 2012;14:285–290. doi: 10.1111/j.1462-2920.2011.02550.x. [DOI] [PubMed] [Google Scholar]
- 119.Kellogg, C. A. & Goldsmith, D. B. Cold-water coral microbiomes (Primnoa spp.) from Gulf of Alaska, Baltimore Canyon, and Norfolk Canyon: raw data, https://doi.org/10.5066/F7P55KMJ (2017).
- 120.Oksanen, J. et al. Vegan: Community Ecology Package. R package version 2.4-0, https://CRAN.R-project.org/package=vegan (2016).
- 121.R: A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria, 2015).
- 122.Clarke KR. Non-parametric multivariate analyses of changes in community structure. Austral. Ecol. 1993;18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 123.Everett KD, Hornung LJ, Andersen AA. Rapid detection of the Chlamydiaceae and other families in the order Chlamydiales: three PCR tests. J. Clin. Microbiol. 1999;37:575–580. doi: 10.1128/jcm.37.3.575-580.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.National Center for Biotechnology Information. VecScreen. https://www.ncbi.nlm.nih.gov/tools/vecscreen/ (2016).
- 125.Sievers F, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li W, et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43:W580–W584. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 128.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009;9:1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 132.Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 133.Peakall R, Smouse P. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28:2537–2539. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Peakall R, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Resour. 2006;6:288–295. doi: 10.1111/j.1471-8286.2005.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Morrison, C. L., Coykendall, D. K. & Sanders, L. Microsatellite genotypes for four cold-water coral species (Lophelia pertusa, Desmophyllum dianthus, Paragorgia arborea, Primnoa resedaeformis) from Baltimore and Norfolk Canyons used to assess patterns of connectivity, https://doi.org/10.5066/F7B27T74 (2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data files associated with the 16 S sequences in this study have been submitted to the NCBI Sequence Read Archive under Bioproject number PRJNA348705 and are also available from the USGS data release119 (https://doi.org/10.5066/F7P55KMJ). The bar codes used in sequencing can be found in mapping files, which are part of the data release. The USGS data release also contains the bioinformatics workflow, including scripts and parameters for each step. The 23 S sequences have been submitted to GenBank and assigned accession numbers KY010287 and KY010288; they are also part of the USGS data release. A separate USGS data release135 presents the microsatellite genotypes for Primnoa resedaeformis (https://doi.org/10.5066/F7B27T74).