

Abstract
Both the activation of the renin angiotensin aldosterone system (RAAS) and elevations of circulating Fibroblast Growth Factor-23 (FGF-23) have been implicated in the pathogenesis of left ventricular hypertrophy (LVH) in chronic kidney disease. To investigate potential cross-talk between RAAS and FGF-23, we administered angiotensin II (Ang II) to wild-type rodents and the Hyp mouse model of excess FGF-23. Ang II administration for four weeks to wild-type rodents resulted in significant increases in systolic blood pressure and LVH. Unexpectedly, FGF-23 circulating levels were increased by 1.5–1.7 fold in Ang II treated animals. In addition, Ang II treatment increased expression of FGF-23 message levels in bone, the predominant tissue for FGF-23 production, and induced expression of FGF-23 and its co-receptor α-Klotho in the heart, which normally does not express FGF-23 or α-Klotho in physiologically relevant levels. Hyp mice with elevated FGF-23 exhibited increased blood pressure and LVH at baseline. Ang II administration to Hyp mice resulted further increments in blood pressure and left ventricular hypertrophy, consistent with additive cardiovascular effects. These findings suggest that FGF-23 may participate in unexpected systemic and paracrine networks regulating hemodynamic and myocardial responses.
Introduction
Patients with chronic kidney disease (CKD) and end stage renal disease (ESRD) have high cardiovascular mortality associated with non-traditional risks factors1–3. Fibroblast growth factor-23 (FGF-23) has emerged as one of the most powerful predictors of adverse outcomes in these patients4–7. FGF-23 is a bone-derived hormone that regulates phosphate and 1,25(OH)2D metabolism through activation of the canonical FGF-23 receptor binary complex created by FGFR 1, 3 and 4 binding with α-Klotho (α-Kl), a type I membrane, β-glycosidase-like protein8–12. Progressive increases in circulating FGF-23 concentrations occur during the course of CKD, achieving levels that are several hundred times the normal range in advanced CKD and ESRD7,13. Elevations of FGF-23 are associated with adverse cardiovascular events and death. These adverse outcomes are attributed to effects of FGF-23 to stimulate left ventricular hypertrophy (LVH)14–16, and occur with small increments in circulating FGF-23 concentrations17,18.
The mechanism (s) whereby FGF-23 causes LVH uncertain, and multiple mechanisms have been proposed19. The leading hypothesis is that FGF-23 directly effects the heart to cause LVH through activation of FGFR4/PLCγ-dependent signaling in the myocardium16,20,21. This non-canonical, α-Kl-independent signaling pathway is controversial, because the tissue selectivity of FGF-23 is imparted by co-expression of α-Kl with FGFRs8, which is not expressed at physiological levels in the normal heart22. Alternatively, there are many kidney effects of FGF-23 mediated by activation of FGFRs/α-Kl in renal tubules that could lead to LVH and adverse cardiovascular outcomes. For example, FGF-23 may activate the renin-angiotensin-aldosterone system (RAAS), which is linked to a multitude of pathologic processes, including left ventricular hypertrophy, through suppression of 1,25(OH)2D, which would increase renin expression23. FGF-23 also reduces angiotensin converting enzyme 2 (ACE2) expression24, an enzyme which cleaves angiotensin II (Ang II) to generate vasodilatory angiotensin 1–7 peptides. RAAS activation and ACE2 insufficiency have been linked to cardiac hypertrophy and myocardial fibrosis25 and oxidative stress and inflammation26. In addition, FGF-23 administration to mice induces hypertension and LVH through stimulation of renal distal tubule sodium transport27. Finally, both FGF-23 suppresses kidney expression of α-Klotho24. α-Kl deficiency is linked to uremic cardiomyopathy through FGF-23 independent mechanisms28. Conversely, soluble Klotho (s-Kl) released into the circulation from ectodomain shedding is reported to exert cardioprotective effects29.
In the current study, we examined the cardiovascular interactions between Ang II and FGF-23. We found that Ang II administration to rodents causes LVH, increased circulating FGF-23 levels and ectopic expression of Fgf-23 and α-Klotho in the heart, whereas Ang II administration to Hyp mice with preexisting elevation of FGF-23 levels resulted in additive effects on blood pressure and LVH. These observations provide a new conceptual framework for understanding the role of FGF-23 in adverse cardiovascular outcomes.
Results
Angiotensin II-induces hypertension, cardiac hypertrophy and increased FGF-23 expression in rodents
Ang II infusion is an established method for inducing hypertension and cardiac hypertrophy in rodents30,31. Consistent with prior reports, Ang II administered by osmotic minipump for 4 weeks (n = 5 per group) resulted in significant increases in systolic blood pressure, from 115 ± 5.5 to 182 9.5 in rats (Fig. 1A). The heart-weight-to-body-weight ratio (HW/BW, mg/g) was 3.35 ± 0.08 in Ang II treated rats compared to 2.93 ± 0.03 in vehicle treated controls (Fig. 1B). In addition, Ang II increased the expression of the genes related to hypertrophy14,15,32, including Anp, Bnp and Trpc6, but not β-Mhc in rat hearts, as well as Foxo1 and Pdk4, factors regulating glucose oxidation in the heart33,34. (Fig. 1C). We found that serum creatinine was significantly lower after 3 and 4 weeks of Ang II treatment compared to controls (Fig. 1D), possibly related to pressure-mediated hyperfiltration. Ang II treated rats also exhibited a significant increase serum ACE2 levels after 4 weeks (Fig. 1D), suggesting that counter regulatory pathways were activated. Unexpectedly, intact serum FGF-23 levels were significantly increased in Ang II treated rats, reaching levels of 564 pg/ml by the 4th week of treatment (Fig. 1F).
Figure 1.

Ang II-induced hypertension and LVH in rats. (A) Blood pressure in Ang II treated and vehicle treated rats. (B) Cardiac hypertrophy as measured by heart weight/ body weight ratio in Ang II treated and vehicle treated rats. (C) Ang II increased the expression of the genes related to hypertrophy in the rat heart. (D) Serum concentrations of creatinine (D), ACE2 (E) and FGF-23 (F) in Ang II and vehicle treated rats. Animals treated with Ang II (35 μg/kg/h by minipump) or vehicle (saline) infusion for four weeks (n = 5/group). *p < 0.05 vs vehicle treated controls. All values are shown as mean ± SEM.
Ang II treatment, which is known to induce hypertension, significantly increased systolic blood pressure, from 98 ± 5 to 145 ± 7 mmHg in mice (Fig. 2A). The HW/BW ratio was 6.4 ± 0.4 in Ang II treated mice compared to 5.2 ± 0.2 in vehicle treated controls (Fig. 2B). The circulating FGF-23 level (pg/ml) increased from 185.5 ± 32.2 in vehicle treated mice to 321.8 ± 14.2 in Ang II treated mice (Fig. 2C). Anp, Bnp, β-Mhc, and Timp-1 were significantly increased in hearts from mice treated with Ang II compared to vehicle treated mice (Fig. 2D). Histological sections of the heart demonstrated hypertrophy of cardiomyocytes after Ang II administration (Fig. 2E,F). Using collagen-specific picrosirius red (PSR) staining for fibrosis, we observed minimal amount of collagen in the normal mouse heart (Fig. 2G), but cardiac interstitial fibrosis in the mice receiving Ang II infusion (Fig. 2H). Macrophages, staining positive for an ED-1 monoclonal antibody, were increased in myocardium in Ang II treated compared to vehicle treated mice (Fig. 2I,J). We found that Ang II infusion also resulted in glomerular sclerosis (Fig. 2K,L) in the kidney. In Fig. 2M, the myocyte size and cardiac collagen volume fraction were significantly increased 82% and 272% fold, respectively, in 4 weeks Ang II infusion mice compared to vehicle treated mice. We also found that T1Col, αSma, Timp-1 and Timp-2 were significantly increased in kidney of the mice receiving Ang II infusion compared to vehicle treated mice (Fig. 2N), consistent with the presence of renal fibrosis. Overall our findings are consistent with known effects of Ang II to cause LVH, cardiomyocyte hypertrophy and cardiac and kidney fibrosis35–38.
Figure 2.

Ang II induced hypertension and LVH in mice. (A) Blood pressure in Ang II treated and vehicle treated mice. (B) Cardiac hypertrophy as measured by heart weight/ body weight ratio in Ang II treated and vehicle treated mice. (C) Ang II increased serum FGF-23 levels in Mice. (D) Ang II increased the expression of the genes related to hypertrophy in the mouse heart. (E and F) H&E staining of heart hypertrophy in Ang II treated mice (F) compared to control mice (E). (F and H) Mouse fibrotic responses in response to 4 weeks of Ang II infusion. Increased interstitial fibrosis in Ang II treated mice (H) compared to vehicle treated mice (G) by picrosirius red (PSR) staining. (I,J) Mouse cardiac inflammatory in response to 4 weeks of Ang II infusion. ED-1 staining of the normal myocardium of mouse (I). ED-1 (is the most widely used monoclonal antibody clone directed against the CD68 protein) marker of positive macrophages are accumulated in the damaged myocardium (J). (K and L) Ang II-induced kidney injure in mice. PSR straining in normal mouse kidney (K) and the kidney from Ang II treated mice showing glomerular sclerosis (L). Magnification X200. (M) Comparison of myocyte size and cardiac collagen volume fraction in Ang II treated and vehicle treated mice. Myocyte size was assessed on 5 μm cross-sectional hematoxylin-eosin stained slices. The outer border of transverse sectioned myocytes was drawn and myocyte area was calculated using NIH Image J software. (N) Comparison of fibrosis related gene expression in kidney from Ang II treated and vehicle treated mice. Animals treated with Ang II (35 μg/kg/h by minipump) or vehicle (saline) infusion for four weeks.n = 4/group, *p < 0.05 vs vehicle treated controls. All values are shown as mean ± SEM.
Consistent with bone as a possible source for the increased circulating FGF-23, we found that Fgf-23 message expression was present in bone devoid of marrow of control mice, and was significantly increased in mouse bone after Ang II treatment (Fig. 3A). Efforts to test the effect of Ang II to directly stimulate FGF-23 production produced variable results (Supplemental Fig. 1); thus we are not able to determine if Ang II is regulating FGF-23 transcription in osteoblasts directly or through indirect effects mediated by Ang II effects on the kidney or other tissues, such as stimulating aldosterone-mediated increase in FGF-2339. Consistent with previous reports40, Fgf-23 was predominately expressed in bone, but was detected at very low levels in the heart, but and undetectable in the kidney of control mice. We detected significant increases in Fgf-23 message expression in the bone, but not kidney, of Ang II treated mice (Fig. 3A). We also observed increased expression of FGF-23 in the heart, but the increase was much lower than in bone, where FGF-23 is normally produced. We found that Ang II treated rats also had increments in Cyp24a1 and decrements in Npt2c and α-Kl in the kidney (Fig. 3B), consistent with increased FGF-23 effects on the kidney. Ace2 message expression was increased in the kidneys from Ang II treated rats, possibly due to effects of Ang II to increase this compensatory pathway and overriding effects of FGF-23 to suppress ACE2 (Fig. 3B). Interestingly, Ang II increased α-Kl message expression in the heart of mice, although the magnitude of α-Kl expression was low compared to the kidney, the predominate tissue that expresses α-Kl (Fig. 3C). The ectopic expression of FGF-23 along with α-Kl and the ubiquitous presence of FGFRs, suggests that Ang II may create a tissue environment for paracrine FGF-23 effects.
Figure 3.
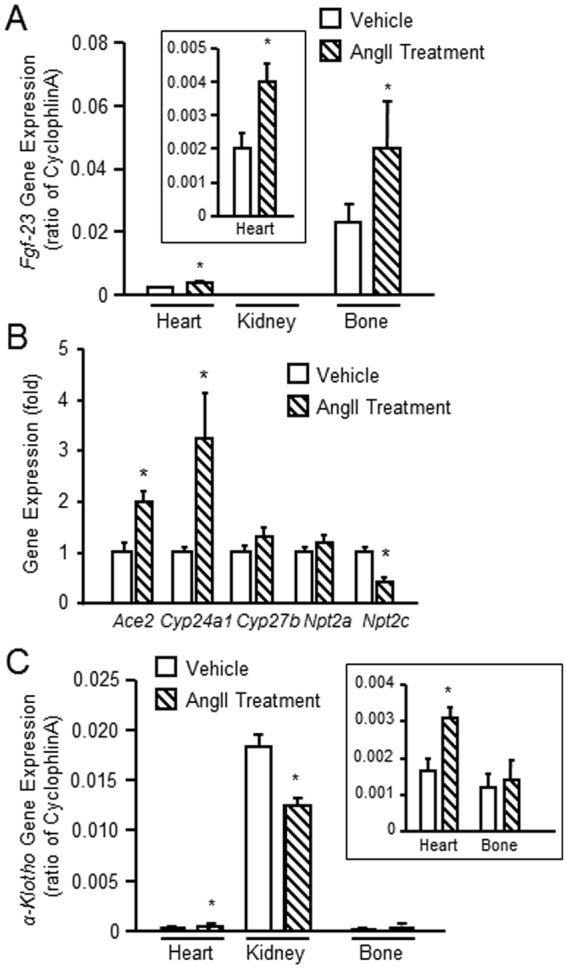
Ang II induced gene expression in heart, bone and kidney. (A) Comparison of Fgf-23 message expression in heart, kidney and bone (without marrow) from Ang II or vehicle treated mice. (B) Comparison of FGF-23 message expression in the kidney from rats with/without Ang II treatment. (C) Comparison of α-Kl expression in heart, kidney and bone (without marrow) from Ang II treated with vehicle treated mice. n = 4/group, *p < 0.05 vs vehicle treated controls. All values are shown as mean ± SEM.
Elevated FGF-23 is associated hypertension and cardiac hypertrophy in Hyp mice
Primary elevations of FGF-23 are associated with LVH in the X-linked hypophosphatemic rickets (Hyp) mouse model of X-linked hypophosphatemic rickets27. Hyp mice had markedly elevated circulating FGF-23 levels (Fig. 4A), that were 6-fold greater than the elevations observed in Ang II treated animals (Fig. 1F). Similar to prior reports, the heart weight in 20 and 40 week-old Hyp mice was increased by 15% and 22% compared to controls, and serum aldosterone significantly suppressed due to FGF-23 effects to increase renal sodium reabsorption (Fig. 4B,C). FGF-23 is predominately expressed in osteoblasts and osteocytes in bone41. We also found that Hyp mice, which have elevated FGF-23 production due to inactivating Phex mutations in bone, had increased Fgf-23 message expression in bone (Fig. 4D). Interestingly, however, Hyp mice also exhibited measurable and significantly increased FGF-23 message levels in heart compared to wild-type controls (Fig. 4D). Gene expression markers of cardiac hypertrophy, including Anp, Foxo1, and β-Mhc, but not Bnp and Ddk4, were also increased in the heart of Hyp mice (Fig. 4E). The renal gene expression profile in the kidney of Hyp mice was consistent with known actions of FGF-23 to suppress Ace2, Cyp27b1, Npt2a, Np2tc and increase in Cyp24a1 (Fig. 4F). In addition, α-Kl expression was decreased in the Hyp kidney, but α-Kl expression was slightly but significantly increased in the heart of Hyp mice (Fig. 4G).
Figure 4.

Increased FGF-23 is associated with LVH in Hyp mice. Comparison of serum FGF-23 levels (A), cardiac hypertrophy (B), and serum aldosterone levels (C) in Hyp and wild type mice at age of 20 or 40 weeks old. (D) Comparison of Fgf-23 expression in heart, kidney and bone (without marrow) from Hyp and wild type mice at age of 20 weeks old. (E) Comparison of gene expression markers of cardiac hypertrophy in heart from Hyp and wild type mice at age of 20 weeks old. (F) Comparison of FGF-23 regulated genes expression in the kidney from Hyp and wild type mice at age of 20 weeks old. (G) Comparison of α-Kl expression in heart, kidney and bone (without marrow) from Hyp and wild type mice at age of 20 weeks old. n = 4/group, *p < 0.05 vs vehicle treated controls. All values are shown as mean ± SEM.
Ang II and FGF-23 exhibit additive effects on hypertension and cardiomegaly in mice
To gain insights into the potential contribution of elevated FGF-23 to LVH observed in Ang II treated animals, we compared FGF-23 levels and the severity of LVH between Hyp and Ang II treated wild-type mice. We administered Ang II to Hyp mice to determine if the cardiovascular effects of FGF-23 and Ang II are additive. Eight week-old wild-type and Hyp mice were treated with 35 μg/kg/h Ang II by implanted minipump for 4 weeks. At baseline, systolic blood pressure (BP) were significantly higher in Hyp compared to in wild-type mice (Fig. 5A). Ang II administration further increased blood pressure in Hyp mice by ~17%, from a mean systolic blood pressure of 126.8 ± 3.4 to 148 ± 2.1 mmHg (Fig. 5A). In contrast, Ang II treatment of wild-type mice increased systolic blood pressure by ~ 63%, from 89.5 ± 1.9 to 145.8 ± 3.7 mmHg. The maximum blood pressure level induced by Ang II was not different between Hyp and wild-type mice. LVH was also present in Hyp mice at baseline compared to wild-type mice by HW/BW and echocardiographic parameters. Ang II increased HW/BW ratio in both Hyp and wild-type mice (~32% and ~48%, respectively) (Fig. 5B). Echocardiographs showed that Hyp mice had a greater left ventricular wall thickness at baseline compared to wild-type mice. Ang II elevated left ventricular wall thickness in wild-type mice, and resulted in additive effects to further increase LVH in Hyp mice. (Fig. 5C–F).
Figure 5.

Effect of Ang II on blood pressure and LVH in WT and Hyp mice. Four-week-old wild-type and Hyp mice were treated with Ang II for 4 weeks. Blood pressure, echocardiography and heart to body weight ratio were measured at the end of Ang II treatment. Hyp mice showed elevated blood pressure in the absence of Ang II treatment and blood pressure in Hyp mice was significantly increased by treatment of Ang II compared to control Hyp mice. Ang II treatment in WT mice increased blood pressure (A) and induced LVH (B). Untreated Hyp mice exhibited LVH and Ang II administration resulted in worsening of LVH in Hyp mice, as assessed by ratio of heart weight to body weight (B) and echocardiography (C). (D and E) Hyp mice showed higher LV mass and LV wall thickness, which were further increased after Ang II administration. (F) Ang II increased ejection fraction in WT mice and decreased EF in Hyp mice compared to wild type mice. Animals treated with Ang II (35 μg /kg/h by minipump) or vehicle (saline) infusion for four weeks. n = 4/group, *p < 0.05 and **p < 0.01 vs vehicle treated controls. All values are shown as mean ± SEM.
Discussion
Why FGF-23, a hormone that regulates mineral homeostasis, has hemodynamic effects is a physiological enigma. These studies provide a potential explanation by showing for the first time that Ang II administration increases circulating FGF-23 levels in animal models in vivo. Treatment of rodents with Ang II stimulated the expression of FGF-23 message in bone, the physiological site FGF-23 production40,42, as well as the ectopic expression of FGF-23 in the heart, which does not normally express FGF-23 in physiological amounts40. Similar to RAAS activation, the sympathetic nervous system (SNS) has recently been shown to stimulate FGF-23 production in bone through β-adrenergic signaling pathways43,44. Thus, activation of RAAS and SNS, which are key hemodynamic regulators associated with adverse outcomes in CKD45–47, are important regulators of FGF-23.
Elevations of FGF-23 are purported to contribute to adverse cardiovascular effects as evidence by the finding of hypertension and LVH in the Hyp mouse model of FGF-23 excess24,27. We also have found that β-Mhc expression was significantly increased in heart from Hyp mice (Fig. 4E), which is similar to findings from Dmp1 knockout mice, another hereditary model of excess FGF-2348. Moreover, the observation that elevations of FGF-23 exacerbate the severity of Ang II-induced hypertension and LVH in the Hyp mouse model, indicates that FGF-23 and RAAS may work through complementary pathways to enhance cardiovascular responses. Finally, small increments in FGF-23, as observed in Ang II treated animals, are associated with adverse cardiovascular outcomes in clinical observation studies49. Thus, the regulation of FGF-23 by RAAS and the additive cardiovascular effects of FGF-23 and Ang II potentially identifies a novel feed-forward endocrine network to enhance hemodynamic responses and leads to the ectopic production of FGF-23 and α-Klotho to locally reconstitute canonical FGF-23/FGFR/α-Kl signal in the myocardium7,15,19.
The mechanism whereby Ang II stimulates FGF-23 expression is not defined by our studies. Increased FGF-23 might result from direct effects to activate AT1 receptors in osteoblasts50. Efforts to test effects of Ang II to stimulate FGF-23 production in osteoblasts, however, produced variable results (Supplemental Fig. 1). Alternatively, increased FGF-23 might occur secondary to Ang II stimulation of aldosterone and TNF-α production, both which can increase FGF-2339,51, or suppression of Klotho expression in the kidney, leading to end organ resistance and secondary elevations in FGF-2352. Finally, Ang II stimulation of FGF-23 may play a role in skeletal homeostasis. Ang II excess has been implicated in the development of osteoporosis; and Ang II inhibits bone mineralization through activation of AT1 receptors in osteoblasts50,53–55. In this context, Ang II regulation of FGF-23 and bone mineralization might represent another component of the bone-kidney endocrine axis whereby FGF-23 coordinates bone mineralization and renal phosphate handling. Future studies will be needed to understand the mechanisms whereby Ang II increases the systemic an local tissue production of FGF-23, and define its contribution to the cardiotoxic actions of excess SNS and RAAS stimulation45.
We also do not define the exact mechanisms whereby FGF-23 exacerbates the cardiovascular effects of Ang II. Ang II activation of AT1 receptors causes hypertension and cardiac hypertrophy in the heart30 and stimulates renal sodium reabsorption and suppresses α-Klotho expression in the kidney56,57. There are several possibilities. First, kidney-specific deletion of α-Klotho, an FGF-23 regulated gene, also causes salt-sensitive hypertension in mice58. Consequently, Ang II and FGF-23 additive effects on renal tubular functions regulating blood pressure and cardiomegaly may account for enhanced hemodynamic responses. In support of this possibility, FGF-23 activation of FGFR/α-Klotho complexes in renal tubules to stimulate distal tubular sodium reabsorption27, and suppress Ace2 and α-Klotho expression19,24 may enhance Ang II hypertensive actions.
Alternatively, suppression of Ace2 by FGF-23 might prolong the actions of Ang II and prevent its conversion to the vasodilatory Ang 1–759. Interestingly, Ang II is reported to down-regulate ACE2 in the kidney and the heart60. However, we observed, possibly for the first time that circulating levels of ACE2 are increased in the circulation in response to Ang II treatment. This suggests that Ang II induces the ectodomain shedding of ACE2, and may account for the observed association between elevated circulating ACE2 and hypertension61.
Second, FGF-23 also suppresses 1,25(OH)2D, which is known to have hemodynamic effect through suppression of renin23. Finally, reduced Klotho may contribute to the cardiovascular effects of FGF-23, since sKlotho (sKL) released by ectodomain shedding of α-Klotho by the kidney has cardioprotective effects by downregulating TRPC6 channels in cardiomyocytes28,62–65, and administration of sKl inhibits RAS and normalizes blood pressure in mouse models of kidney diseases66.
Third, increased FGF-23, either systemically or locally, might act through the non-canonical direct activation of FGFRs in the heart by FGF-23 (i.e., α-Klotho independent effects)16 or through Ang II induction of ectopic expression of FGF-23 and α-Klotho in the heart and local reconstitute FGFR/α-Klotho signaling in the stressed heart. With regards to the latter, we found that Ang II administration increased FGF-23 and α-Klotho message levels in heart, a tissue that does not normally express FGF-23 or α-Klotho in physiological amounts. These findings are consistent with other studies showing ectopic expression of FGF-23 and α-Klotho in the heart under disease conditions16,67. Ang II administration to α-Kl transgenic mice, which overexpress α-Kl in the heart and other tissues, exacerbates LVH and cardiac fibrosis68, supports the notion that activation of ectopically expressed FGFR/α-Klotho complexes in the myocardium can lead to cardiac hypertrophy. Finally, FGF-23 could have direct effects on vascular smooth muscle and vascular calcification through Klotho-mediated nitric oxide synthesis and oxidative stress69–72.
FGF-23 is reported to be expressed in macrophages that do not normally express FGF-23 in response to stress and inflammation73. Ectopic expression of FGF-23 in macrophages and effects on cells in the myeloid lineage has been purported to regulate innate immune responses outcomes67,73,74. The AT1 receptor is expressed in immune cells and Ang II alters their inflammatory functions75. FGF-23 in activated macrophages infiltrating hypertrophic hearts may contribute to cardiac fibrosis and abnormal expression of FGF-23 in cardiomyocytes67,16. FGF-23 may also interact with innate immune responses through suppression of 1,25D production by the kidney56,76,77. Interestingly, in spite of seeing both cardiac and renal inflammation and fibrosis, we did not see ectopic expression of FGF-23 in the kidney in response to Ang II treatment. This contrasts to the expression of FGF-23 in the renal epithelium that is observed in polycystic kidneys that is thought to be induced by inflammation52.
Pharmacological blockade of FGF-23 or Ang II will be need to tease out the indirect effects of FGF-23 from those mediated by Ang II activation of angiotensin receptors78.The clinical importance of cross talk between RAAS and FGF-23, however, is suggested by the finding that FGF-23 levels are higher in patients with heart failure not treated with angiotensin converting enzyme inhibitors (ACEi) and patients in the top tertile of elevated serum FGF-23 exhibit a lower risk of adverse events after treatment with ACEi79. The Ang II and FGF-23 endocrine network may help explain why elevated FGF-23 is associated with enhanced responses to angiotensin-converting enzyme inhibitor (ACEi) therapy in patients with heart failure but without CKD79,80.
The presence of hypertension and LVH in patients with XLH and the Hyp mouse homologue of this disease is variably reported. Similar to our findings, Erben’s group found that Hyp mice have LVH due to FGF-23 dependent effects on sodium reabsorption in the kidney81, and patients with XLH are reported to have exercise induced increases in BP82. In contrast, several studies have failed to identify associations between elevated FGF-23 and HTN or LVH in Hyp and the Dmp1 null mouse model of autosomal recessive hypophosphatemia48,83–85. The reasons for these differences are unknown, but likely reflect age, dietary, genetic and/or environmental modifiers.
In summary, we propose a new schema for understanding cardiovascular homeostasis whereby Ang II stimulates the release of FGF-23 into the circulation and the ectopic expression of FGF-23 and its co-receptor α-Klotho in the heart; in turn, the systemic and local elevations of FGF-23 augment the cardiovascular effects of Ang II through multiple potential molecular mechanisms that include renal sodium absorption, suppression of 1,25(OH)2D3 and enhanced renin production, Ace2 expression and systemic effects of α-Klotho (Fig. 6). If so, FGF-23 may participate in previously unrecognized systemic and local regulatory networks whereby the sympathetic nervous system and renin angiotensin system control hemodynamics44 and FGF-23 may link SNS and RAAS to inflammation and oxidative stress67,86, thus contributing to adverse effects in CKD and other conditions, such as congestive heart failure. Additional investigations are needed to understand the afferent pathways whereby Ang II stimulates FGF-23, the relative importance of the multiple efferent pathways potentially mediating FGF-23 associated cardiac toxicity, and the feedback pathway that shuts off this Ang II and FGF-23 feed forward loop.
Figure 6.

Crosstalk between RAAS and FGF-23 pathways. A new schema of hemodynamic regulation suggests actions of Ang II to stimulate FGF-23 in bone (afferent limb), leading to additive renal effects with Ang II (efferent limb) to increase blood pressure and cardiomegaly. In this model, FGF-23 effects are mediated by activation of FGFR/α-Klotho complexes, whereas Ang II activates AT1 receptors in renal tubules to enhance sodium transport, and to suppress α-Klotho. The resulting positive sodium balance leads to increased blood pressure and the reduced levels of soluble Klotho (s-Kl) may enhance cardiotoxicity through s-Kl-dependent TRPC6 cardiac effects. In contrast, FGF-23 suppresses, whereas Ang II stimulates Ace2, making degradation of Ang II and formation of Ang 1–7 a possible point of differential control. FGF-23 also suppresses 1,25(OH)2D synthesis, an effect predicted to increase renin production by the kidney. This novel feed-forward endocrine pathway may contribute to the association between elevated FGF-23 and adverse cardiovascular outcomes. In addition, Ang II upregulates FGF-23 and α-Klotho expression the heart and macrophages to possible reconstitute FGFR/α-Klotho signaling leading to paracrine effects of FGF-23 to induce cardiac hypertrophy and stimulate inflammation.
Methods
Animals
Eight week old male SD rats and 4 month old male C57BL/6 J and male Hyp mice (C57BL/6-PhexHyp-2J/J) were treated with Ang II at the dose of 35 μg/kg/h respectively given subcutaneously by implanted minipump87,88. Vehicle treated littermate rats and mice served as controls. The animals were fed with ad lib regular chow, Harlan Teklad 2018 (including 1% calcium, 0.7% phosphorus, 1.5 IU/g Vitamin D3; Harlan Teklad, Madison, WI, USA). Heart, kidney, and bone were collected after 4 weeks of Ang II infusion, the tissues were used for RNA purification, real time RT-PCR, histology and immunohistochemistry. Cardiac collagen volume was detected by picrosirius red (PSR) staining in cardiac sections and quantitated using a computer image analysis system (NIH image 1.6) as we previously reported87. Cardiac hypertrophy was assessed by heart-weight-to-body-weight ratio89.
This study was approved by the University of Tennessee Health Science Center Animal Care and Use Committee. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Blood pressure measurements
Blood pressure was measured by the tail-cuff plethysmography method in unanesthetized mice using a Hatteras Instruments SC1000 Blood Pressure Analysis System as previously described61. Measurements were made on the day at end of study 4 weeks after mini implantation.
Histology and Immunohistochemistry
Mouse heart and kidney tissues were embedded into OCT compound (Tissue-Tek, Sakura Finetek USA; Torrance, CA, USA). Cryostat sections (6 μm) were air-dried, fixed in 10% buffered formalin for 5 min, and washed in phosphate-buffered saline (PBS) for 10 min. For H&E staining, the cryostat sections were rinsed in H2O, dipped into Mayer’s hematoxylin and agitated for 30 sec. Then the slide was rinsed in H2O for 1 min, and stained with 1% eosin Y solution for 10–30 sec, and dehydrated and mounted. Cardiac sections (6 m) were prepared to determine the fibrillar collagen accumulation by collagen-specific picrosirius red staining and observed by light microscopy as previously reported38,90. Collagen volume fraction of each section was determined using a computer image analyzing system (NIH image, 1.60), as previously reported38,90. Cardiac expression of ED-1 (a marker of macrophages) was detected by immunohistochemistry. Cryostat cardiac sections (6 μm) were incubated with a primary antibody against ED1 (Sigma, St. Louis, MO, USA) for 1 hour at room temperature. The sections were then incubated with the immunoglobulin G peroxidase–conjugated secondary antibody (Sigma) for 1 hour at room temperature and incubated with 0.5 mg/ml diaminobenzidine tetrahydrochloride 2-hydrate +0.05% hydrogen peroxide for 5 minutes. Negative control sections were incubated with the secondary antibody alone. All sections were counterstained with hematoxylin, dehydrated, mounted, and viewed by light microscopy91. We used Nikon-2 Optiphot-2 microscopy and at 20X objective lens. We analyzed 5 difference locations in per slide and total 3 slides per sample were analyzed.
Real time RT-PCR
For quantitative real-time RT-PCR assessment of the markers of hypertrophy, fibrosis and Fgf-23 expression (Supplemental Table 1), we isolated and reverse transcribed 2.0 µg of total RNA from the long bone, kidney and heart of mice with/without Ang II treatment as described previously92. PCR reactions contained 100 ng of template (cDNA or RNA), 300 nM each of forward and reverse primer, and 1X iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) in 50 µL. Samples were amplified for 40 cycles in an iCycler iQ Real-Time PCR Detection System (Bio-Rad) with an initial melt at 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute. PCR product accumulation was monitored at multiple points during each cycle by measuring the increase in fluorescence caused by the binding of SybrGreen I to dsDNA. The threshold cycle93 of tested-gene product from the indicated genotype was normalized to the Ct for cyclophilin A. Dissociation analysis was used to confirm the presence of a single transcript and lack of primer-dimer amplification in all PCR reactions.
Serum Biochemical Measurements
Blood was collected using a retroorbital bleeding technique. Serum was separated by using Serum Separator Tubes (BD Life Sciences, Franklin Lakes, NJ, USA). Serum FGF-23 levels were measured using an FGF-23 ELISA kit (Kainos Laboratories, Inc., Tokyo, Japan). This kit is measurement for intact FGF-23. Serum creatinine was measured, using Creatinine Liquicolor Test (Stanbio Laboratory, Boerne, TX) as described previously94. Serum ACE2 and aldosterone were measured by using Angiotensin II Converting Enzyme (ACE2) WLIA Kit (San Diego, CA, USA) and Aldosterone EIA kit from Cayman chemical (Ann Arbor, MI, USA), respectively.
Statistics
We evaluated differences between groups by one-way analysis of variance, followed by a post-hoc Tukey’s test. Significance was set at p < 0.05. All values are expressed as means ± SEM. All computations were performed using the Statgraphic statistical graphics system (STSC Inc.).
Electronic supplementary material
Acknowledgements
This work was supported by NIH NIDDK (1RO-1A045955, Darryl L Quarles) and Heart, Blood, and Lung Institute (1RO1-HL096503, Yao Sun).
Author Contributions
M.P., Y.S. and L.D.Q. conceived the experiments and analyzed the results and prepared the manuscript; M.P., R.Y., X.H., B.A., X.L. and Y.C. conducted the experiments. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-30098-1.
References
- 1.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. The New England journal of medicine. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 2.Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Archives of internal medicine. 2004;164:659–663. doi: 10.1001/archinte.164.6.659. [DOI] [PubMed] [Google Scholar]
- 3.Block GA, Kilpatrick RD, Lowe KA, Wang W, Danese MD. CKD-mineral and bone disorder and risk of death and cardiovascular hospitalization in patients on hemodialysis. Clinical journal of the American Society of Nephrology: CJASN. 2013;8:2132–2140. doi: 10.2215/CJN.04260413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fliser D, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. Journal of the American Society of Nephrology: JASN. 2007;18:2600–2608. doi: 10.1681/ASN.2006080936. [DOI] [PubMed] [Google Scholar]
- 5.Isakova T, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. Jama. 2011;305:2432–2439. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kendrick J, et al. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. Journal of the American Society of Nephrology: JASN. 2011;22:1913–1922. doi: 10.1681/ASN.2010121224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gutierrez OM, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. The New England journal of medicine. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, et al. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology. 2005;146:4647–4656. doi: 10.1210/en.2005-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li SA, et al. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell structure and function. 2004;29:91–99. doi: 10.1247/csf.29.91. [DOI] [PubMed] [Google Scholar]
- 11.Yamazaki, Y. et al. Anti-FGF23 Neutralizing Antibodies Demonstrate the Physiological Role and Structural Features of FGF23. J Bone Miner Res (2008). [DOI] [PubMed]
- 12.Tomiyama K, et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1666–1671. doi: 10.1073/pnas.0913986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isakova T, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international. 2011;79:1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez OM, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119:2545–2552. doi: 10.1161/CIRCULATIONAHA.108.844506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faul C, et al. FGF23 induces left ventricular hypertrophy. The Journal of clinical investigation. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabner A, et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell metabolism. 2015;22:1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirza MA, et al. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24:3125–3131. doi: 10.1093/ndt/gfp205. [DOI] [PubMed] [Google Scholar]
- 18.Mirza MA, Larsson A, Lind L, Larsson TE. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis. 2009;205:385–390. doi: 10.1016/j.atherosclerosis.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Kovesdy CP, Quarles LD. FGF23 from Bench to Bedside. American journal of physiology. Renal physiology, ajprenal. 2016;00606:02015. doi: 10.1152/ajprenal.00606.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vainikka S, Joukov V, Klint P, Alitalo K. Association of a 85-kDa serine kinase with activated fibroblast growth factor receptor-4. J Biol Chem. 1996;271:1270–1273. doi: 10.1074/jbc.271.3.1270. [DOI] [PubMed] [Google Scholar]
- 21.Drafahl KA, McAndrew CW, Meyer AN, Haas M, Donoghue DJ. The receptor tyrosine kinase FGFR4 negatively regulates NF-kappaB signaling. PloS one. 2010;5:e14412. doi: 10.1371/journal.pone.0014412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends in endocrinology and metabolism: TEM. 2015;26:22–29. doi: 10.1016/j.tem.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaidya A, Williams JS. The relationship between vitamin D and the renin-angiotensin system in the pathophysiology of hypertension, kidney disease, and diabetes. Metabolism: clinical and experimental. 2012;61:450–458. doi: 10.1016/j.metabol.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai B, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PloS one. 2012;7:e44161. doi: 10.1371/journal.pone.0044161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong, J. et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation122, 717–728, 718 p following 728, 10.1161/CIRCULATIONAHA.110.955369 (2010). [DOI] [PubMed]
- 26.Zhong J, et al. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension. 2011;57:314–322. doi: 10.1161/HYPERTENSIONAHA.110.164244. [DOI] [PubMed] [Google Scholar]
- 27.Andrukhova O, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO molecular medicine. 2014;6:744–759. doi: 10.1002/emmm.201303716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. Journal of the American Society of Nephrology: JASN. 2015;26:1150–1160. doi: 10.1681/ASN.2014040325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song S, Gao P, Xiao H, Xu Y, Si LY. Klotho suppresses cardiomyocyte apoptosis in mice with stress-induced cardiac injury via downregulation of endoplasmic reticulum stress. PloS one. 2013;8:e82968. doi: 10.1371/journal.pone.0082968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowley SD, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu YC, et al. Role of angiotensin AT1 and AT2 receptors in cardiac hypertrophy and cardiac remodelling. Clinical and experimental pharmacology & physiology. 2003;30:911–918. doi: 10.1111/j.1440-1681.2003.03942.x. [DOI] [PubMed] [Google Scholar]
- 32.Gruson D, et al. C-terminal FGF23 is a strong predictor of survival in systolic heart failure. Peptides. 2012;37:258–262. doi: 10.1016/j.peptides.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Gopal K, et al. FoxO1 regulates myocardial glucose oxidation rates via transcriptional control of pyruvate dehydrogenase kinase 4 expression. American journal of physiology. Heart and circulatory physiology. 2017;313:H479–H490. doi: 10.1152/ajpheart.00191.2017. [DOI] [PubMed] [Google Scholar]
- 34.Mori J, et al. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. American journal of physiology. Heart and circulatory physiology. 2013;304:H1103–1113. doi: 10.1152/ajpheart.00636.2012. [DOI] [PubMed] [Google Scholar]
- 35.Johnson RJ, et al. Renal injury from angiotensin II-mediated hypertension. Hypertension. 1992;19:464–474. doi: 10.1161/01.HYP.19.5.464. [DOI] [PubMed] [Google Scholar]
- 36.Ma J, et al. Accelerated fibrosis and collagen deposition develop in the renal interstitium of angiotensin type 2 receptor null mutant mice during ureteral obstruction. Kidney international. 1998;53:937–944. doi: 10.1111/j.1523-1755.1998.00893.x. [DOI] [PubMed] [Google Scholar]
- 37.Tsukamoto Y, et al. A novel heart failure mice model of hypertensive heart disease by angiotensin II infusion, nephrectomy, and salt loading. American journal of physiology. Heart and circulatory physiology. 2013;305:H1658–1667. doi: 10.1152/ajpheart.00349.2013. [DOI] [PubMed] [Google Scholar]
- 38.Zhao W, Ahokas RA, Weber KT, Sun Y. ANG II-induced cardiac molecular and cellular events: role of aldosterone. American journal of physiology. Heart and circulatory physiology. 2006;291:H336–343. doi: 10.1152/ajpheart.01307.2005. [DOI] [PubMed] [Google Scholar]
- 39.Zhang B, et al. Up-regulation of FGF23 release by aldosterone. Biochemical and biophysical research communications. 2016;470:384–390. doi: 10.1016/j.bbrc.2016.01.034. [DOI] [PubMed] [Google Scholar]
- 40.Liu S, et al. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Molecular endocrinology. 2009;23:1505–1518. doi: 10.1210/me.2009-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshiko Y, et al. Mineralized tissue cells are a principal source of FGF23. Bone. 2007;40:1565–1573. doi: 10.1016/j.bone.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, et al. Pathogenic role of Fgf23 in Hyp mice. American journal of physiology. Endocrinology and metabolism. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 43.Kawai M, Kinoshita S, Shimba S, Ozono K, Michigami T. Sympathetic activation induces skeletal Fgf23 expression in a circadian rhythm-dependent manner. J Biol Chem. 2014;289:1457–1466. doi: 10.1074/jbc.M113.500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fajol, A. et al. Enhanced FGF23 production in mice expressing PI3K-insensitive GSK3 is normalized by beta-blocker treatment. FASEB journal: official publication of the Federation of American Societies for Experimental Biology, 10.1096/fj.15-279943 (2015). [DOI] [PMC free article] [PubMed]
- 45.Ruster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. Journal of the American Society of Nephrology: JASN. 2006;17:2985–2991. doi: 10.1681/ASN.2006040356. [DOI] [PubMed] [Google Scholar]
- 46.Cachofeiro, V. et al. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney international. Supplement, S4-9, 10.1038/ki.2008.516 (2008). [DOI] [PubMed]
- 47.Ewen S, et al. The sympathetic nervous system in chronic kidney disease. Current hypertension reports. 2013;15:370–376. doi: 10.1007/s11906-013-0365-0. [DOI] [PubMed] [Google Scholar]
- 48.Wacker MJ, et al. Skeletal Muscle, but not Cardiovascular Function, Is Altered in a Mouse Model of Autosomal Recessive Hypophosphatemic Rickets. Frontiers in physiology. 2016;7:173. doi: 10.3389/fphys.2016.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leaf DE, et al. Fibroblast Growth Factor 23 Levels Associate with AKI and Death in Critical Illness. Journal of the American Society of Nephrology: JASN. 2017;28:1877–1885. doi: 10.1681/ASN.2016080836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakai K, et al. Angiotensin II suppresses osteoblastic differentiation and mineralized nodule formation via AT1 receptor in ROS17/2.8 cells. Archives of medical science: AMS. 2015;11:628–637. doi: 10.5114/aoms.2015.52369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito N, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Molecular and cellular endocrinology. 2015;399:208–218. doi: 10.1016/j.mce.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Spichtig D, et al. Renal expression of FGF23 and peripheral resistance to elevated FGF23 in rodent models of polycystic kidney disease. Kidney international. 2014;85:1340–1350. doi: 10.1038/ki.2013.526. [DOI] [PubMed] [Google Scholar]
- 53.Guan XX, Zhou Y, Li JY. Reciprocal roles of angiotensin II and Angiotensin II Receptors Blockade (ARB) in regulating Cbfa1/RANKL via cAMP signaling pathway: possible mechanism for hypertension-related osteoporosis and antagonistic effect of ARB on hypertension-related osteoporosis. International journal of molecular sciences. 2011;12:4206–4213. doi: 10.3390/ijms12074206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaneko K, et al. Physiological function of the angiotensin AT1a receptor in bone remodeling. J Bone Miner Res. 2011;26:2959–2966. doi: 10.1002/jbmr.501. [DOI] [PubMed] [Google Scholar]
- 55.Shimizu H, et al. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:2465–2475. doi: 10.1096/fj.07-098954. [DOI] [PubMed] [Google Scholar]
- 56.de Borst MH, Vervloet MG, ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. Journal of the American Society of Nephrology: JASN. 2011;22:1603–1609. doi: 10.1681/ASN.2010121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoon HE, et al. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol Dial Transplant. 2011;26:800–813. doi: 10.1093/ndt/gfq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou X, Chen K, Lei H, Sun Z. Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. Journal of the American Society of Nephrology: JASN. 2015;26:121–132. doi: 10.1681/ASN.2013101033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertension research: official journal of the Japanese Society of Hypertension. 2009;32:533–536. doi: 10.1038/hr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koka V, et al. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol. 2008;172:1174–1183. doi: 10.2353/ajpath.2008.070762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circulation research. 2006;98:463–471. doi: 10.1161/01.RES.0000205761.22353.5f. [DOI] [PubMed] [Google Scholar]
- 62.Lindberg K, et al. The kidney is the principal organ mediating klotho effects. Journal of the American Society of Nephrology: JASN. 2014;25:2169–2175. doi: 10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xie J, et al. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun. 2012;3:1238. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu, M. C. et al. Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney international, 10.1016/j.kint.2016.10.034 (2017). [DOI] [PMC free article] [PubMed]
- 65.Kuwahara K, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. The Journal of clinical investigation. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou L, et al. Klotho Ameliorates Kidney Injury and Fibrosis and Normalizes Blood Pressure by Targeting the Renin-Angiotensin System. Am J Pathol. 2015;185:3211–3223. doi: 10.1016/j.ajpath.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han X, et al. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS letters. 2016;590:53–67. doi: 10.1002/1873-3468.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu X, et al. Differential Regulatory Role of Soluble Klothos on Cardiac Fibrogenesis in Hypertension. American journal of hypertension. 2016;29:1140–1147. doi: 10.1093/ajh/hpw062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lindberg K, et al. Arterial klotho expression and FGF23 effects on vascular calcification and function. PloS one. 2013;8:e60658. doi: 10.1371/journal.pone.0060658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Six I, et al. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PloS one. 2014;9:e93423. doi: 10.1371/journal.pone.0093423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silswal N, et al. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. American journal of physiology. Endocrinology and metabolism. 2014;307:E426–436. doi: 10.1152/ajpendo.00264.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richter B, Haller J, Haffner D, Leifheit-Nestler M. Klotho modulates FGF23-mediated NO synthesis and oxidative stress in human coronary artery endothelial cells. Pflugers Archiv: European journal of physiology. 2016;468:1621–1635. doi: 10.1007/s00424-016-1858-x. [DOI] [PubMed] [Google Scholar]
- 73.Han X, Quarles LD. Multiple faces of fibroblast growth factor-23. Current opinion in nephrology and hypertension. 2016;25:333–342. doi: 10.1097/MNH.0000000000000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rossaint J, et al. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J Clin Invest. 2016;126:962–974. doi: 10.1172/JCI83470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rudemiller NP, Crowley SD. Interactions Between the Immune and the Renin-Angiotensin Systems in Hypertension. Hypertension. 2016;68:289–296. doi: 10.1161/HYPERTENSIONAHA.116.06591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bacchetta J, Salusky IB, Hewison M. Beyond mineral metabolism, is there an interplay between FGF23 and vitamin D in innate immunity? Pediatric nephrology. 2013;28:577–582. doi: 10.1007/s00467-012-2336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bacchetta J, et al. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2013;28:46–55. doi: 10.1002/jbmr.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zanchi C, et al. Renal expression of FGF23 in progressive renal disease of diabetes and the effect of ACE inhibitor. PloS one. 2013;8:e70775. doi: 10.1371/journal.pone.0070775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wohlfahrt P, et al. Association of Fibroblast Growth Factor-23 Levels and Angiotensin-Converting Enzyme Inhibition in Chronic Systolic Heart Failure. JACC. Heart failure. 2015;3:829–839. doi: 10.1016/j.jchf.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 80.Udell JA, et al. Fibroblast growth factor-23, cardiovascular prognosis, and benefit of angiotensin-converting enzyme inhibition in stable ischemic heart disease. Journal of the American College of Cardiology. 2014;63:2421–2428. doi: 10.1016/j.jacc.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Erben RG. Update on FGF23 and Klotho signaling. Molecular and cellular endocrinology. 2016;432:56–65. doi: 10.1016/j.mce.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 82.Nehgme R, Fahey JT, Smith C, Carpenter TO. Cardiovascular abnormalities in patients with X-linked hypophosphatemia. The Journal of clinical endocrinology and metabolism. 1997;82:2450–2454. doi: 10.1210/jcem.82.8.4181. [DOI] [PubMed] [Google Scholar]
- 83.Liu, E. S. et al. Increased circulating FGF23 does not lead to cardiac hypertrophy in the male Hyp mouse model of XLH. Endocrinology, 10.1210/en.2018-00174 (2018). [DOI] [PMC free article] [PubMed]
- 84.Slavic S, et al. Genetic Ablation of Fgf23 or Klotho Does not Modulate Experimental Heart Hypertrophy Induced by Pressure Overload. Scientific reports. 2017;7:11298. doi: 10.1038/s41598-017-10140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takashi Y, et al. Patients with FGF23-related hypophosphatemic rickets/osteomalacia do not present with left ventricular hypertrophy. Endocrine research. 2017;42:132–137. doi: 10.1080/07435800.2016.1242604. [DOI] [PubMed] [Google Scholar]
- 86.Yuki M, et al. Expression of Fgf23 in activated dendritic cells and macrophages in response to immunological stimuli in mice. Biological & pharmaceutical bulletin. 2015 doi: 10.1248/bpb.b14-00276. [DOI] [PubMed] [Google Scholar]
- 87.Meng W, et al. Autocrine and paracrine function of Angiotensin 1-7 in tissue repair during hypertension. American journal of hypertension. 2014;27:775–782. doi: 10.1093/ajh/hpt270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trachet, B. et al. Ascending Aortic Aneurysm in Angiotensin II-Infused Mice: Formation, Progression, and the Role of Focal Dissections. Arterioscler Thromb Vasc Biol, 10.1161/ATVBAHA.116.307211 (2016). [DOI] [PubMed]
- 89.Rocha R, et al. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart. Endocrinology. 2002;143:4828–4836. doi: 10.1210/en.2002-220120. [DOI] [PubMed] [Google Scholar]
- 90.Sun Y, Ratajska A, Weber KT. Inhibition of angiotensin-converting enzyme and attenuation of myocardial fibrosis by lisinopril in rats receiving angiotensin II. The Journal of laboratory and clinical medicine. 1995;126:95–101. [PubMed] [Google Scholar]
- 91.Zhao W, et al. Platelet-derived growth factor involvement in myocardial remodeling following infarction. Journal of molecular and cellular cardiology. 2011;51:830–838. doi: 10.1016/j.yjmcc.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pi M, et al. Impaired osteoblast function in GPRC6A null mice. J Bone Miner Res. 2010;25:1092–1102. doi: 10.1359/jbmr.091037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dehghan, A. et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation123, 731–738, CIRCULATIONAHA.110.948570 [pii] 10.1161/CIRCULATIONAHA.110.948570 (2011). [DOI] [PMC free article] [PubMed]
- 94.Liu S, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. Journal of the American Society of Nephrology: JASN. 2006;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.