

Abstract
Oxidative stress is a major contributor to secondary injury signaling cascades following traumatic brain injury (TBI). The role of lipid peroxidation in the pathophysiology of a traumatic insult to neural tissue is increasingly recognized. As the methods to quantify lipid peroxidation have gradually improved, so has the understanding of mechanistic details of lipid peroxidation and related signaling events in the injury pathogenesis. While free-radical mediated, non-enzymatic lipid peroxidation has long been studied, recent advances in redox lipidomics have demonstrated the significant contribution of enzymatic lipid peroxidation to TBI pathogenesis. Complex interactions between inflammation, phospholipid peroxidation, and hydrolysis define the engagement of different cell death programs and the severity of injury and outcome. This review focuses on enzymatic phospholipid peroxidation after TBI, including the mechanism of production, signaling roles in secondary injury pathology, and temporal course of production with respect to inflammatory response. In light of the newly identified phospholipid oxidation mechanisms, we also discuss possible therapeutic targets to improve neurocognitive outcome after TBI. Finally, we discuss current limitations in identifying oxidized phospholipids and possible methodologic improvements that can offer a deeper insight into the region-specific distribution and subcellular localization of phospholipid oxidation after TBI.
Keywords: redox lipidomics, inflammation, apoptosis, ferroptosis, efferocytosis, lipid mediator
Graphical Abstract
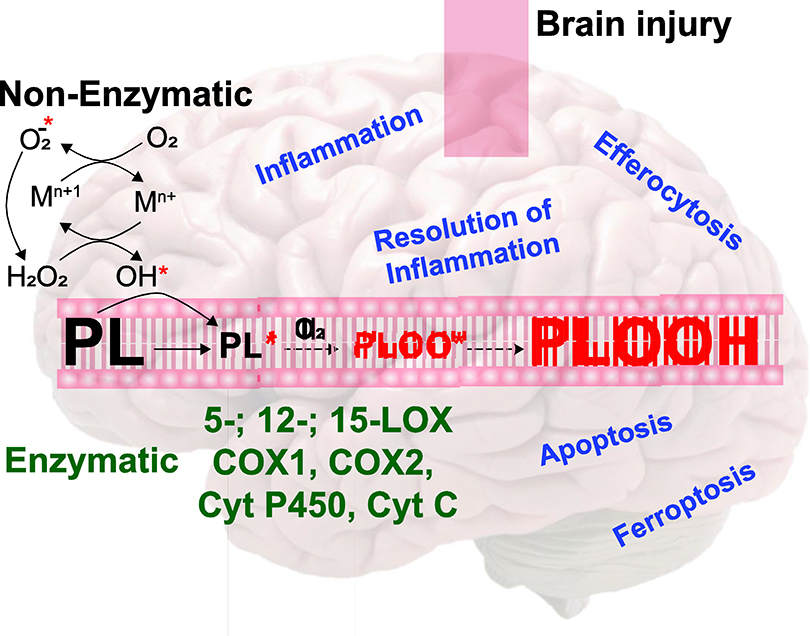
INTRODUCTION
Traumatic brain injury (TBI) affects over 1.7 million individuals each year in the United States alone [1]. Fortunately, survival following TBI has modestly improved over the past several years. However, it remains that fewer than half of patients with severe TBI maintain moderate or good functional status in long-term follow-up [2–4]. This positive but limited success can be attributed to the implementation of early aggressive, standardized treatments at all stages of the acute care continuum [5]. Yet, the longitudinal burden of TBI remains multifaceted, negatively impacting societal participation, mood, quality of life, and financial stability for both the patient and caregivers [6, 7]. Therefore, therapies that ameliorate the chronic burden of TBI by reducing neuronal injury and improving functional outcomes are sorely needed. Neuronal cell death is a major contributor to functional outcome after TBI [8, 9]. Accordingly, the interdependent and multimodal nature of TBI pathogenesis necessitates comprehensive understanding of the factors that contribute to both neuronal survival and death in the pursuit of effective pharmacotherapy [10].
The complexity of the brain requires equally multifaceted and diversified signaling networks to maintain homeostasis and respond to external stresses. Indeed, the pathophysiology of TBI is a complex and dynamic process including both primary and secondary injury with multitude of small molecule, enzymatic, and lipid-based signaling mediators involved. The primary injury of TBI refers to damage incurred from the mechanical forces of trauma. Consequently, secondary injury is initiated at the time of the insult and develops in the post-injury period through signaling of multiple interconnected molecular pathways, including but not limited to: inflammation, excitotoxicity, ion/fluid imbalance, oxidative stress, and mitochondrial dysfunction [11, 12]. The pro-inflammatory and pro-oxidative states observed following TBI stand apart; innumerable studies in pre-clinical and clinical settings recognize their critical and interdependent contributions to secondary injury evolution and their role as prime targets for therapeutic intervention [13, 14]. Whilst the importance of generalized oxidative stress after TBI is well recognized, the role of the oxidized signaling mediators, particularly those enzymatically regulated, is not completely understood. Phospholipids (PLs) are the most abundant and diverse of all the various oxidizable lipids found in the brain and are subsequently a major source of oxidized lipid derivatives. Enzymatically regulated oxidation of PLs serve a critical and under acknowledged role in both the pathogenesis and resolution of injury after brain trauma. In this focused review, we discuss the identification, sources and temporal course of formation, and signaling roles of oxidized PL and their derivatives in cell survival and death following TBI while identifying past and prospective targets of lipid oxidation for neuroprotective intervention. In this focused review, we discuss the identification, sources and temporal course of formation, and signaling roles of oxidized PL and their derivatives in cell survival and death following TBI while identifying past and prospective targets of lipid oxidation for neuroprotective intervention.
TBI pathogenesis and the role of oxidative stress
TBI is a physical injury to the brain tissue due to a mechanical force caused by a jolt, blast, compression or penetrating external object. The primary damage caused by the mechanical force expands in time as a result of a number of secondary injury cascades that include disruption of ionic channels, release of excitatory neurotransmitters, oxidative stress, mitochondrial dysfunction, and inflammation [15]. The time course of these secondary injury mechanisms differ with oxidative stress and mitochondrial dysfunction lasting weeks after injury, thus presenting themselves as potential therapeutic targets. TBI can cause temporary or permanent damage to the brain function owing to a complex disease process. Despite improvement in the overall mortality owing to advances in emergency and critical care of severe TBI patients, a significant portion of them (≥50%) endure neurological deficits affecting multiple domains of function [4]. Due to the heterogeneity of TBI, its classification depends on the data availability and purpose of the classification. The mode of classification includes: 1) etiological, 2) symptomatic 3) prognostic and 4) pathoanatomic. These classifications can serve as waymarks towards treatment strategies and have been reviewed thoroughly in the literature [16]. To understand the pathophysiology of TBI, numerous animal models have been developed. These models used extensively in preclinical setup and generally mimic focal cortical contusion (such as controlled cortical impact) or diffuse axonal injury (such as weight drop model). A discussion of different TBI animal models is beyond the scope of this review and the reader is referred to excellent reviews on this subject [17, 18].
While the contribution of oxidative stress to TBI pathogenesis is well-established, initial studies focused on the realization of oxidative stress through three major non-enzymatic mechanisms [19]. 1) Release of iron and other transition metals [20]. Free iron, commonly released through heme oxygenase-mediated breakdown of heme, and other transition metals have potential for biological toxicity, primarily through the production of highly potent hydroxyl radicals via Haber-Weiss, Fenton-like, and Fenton reactions. [20, 21]. TBI leads to mishandling of transition metals. TBI-induced intracranial hemorrhage leading to erythrocyte lysis is a major source of free heme and Fe2+. Though the vast majority of transition metals are sequestrated by storage proteins, such as ferritin and metallothioneins, to minimize the redox-related toxicity of unbound transition metals [22–25]. Highly reactive hydroxyl radicals, formed by the interaction of transition metals and soluble hydroperoxides such as hydrogen peroxide (H2O2), initiate a chain reaction of lipid peroxidation [21]. 2) Generation of superoxide and peroxynitrite. Dysregulation of the electron transport chain following TBI results in the partial reduction of oxygen and serves as the major source of superoxide generation [26]. Superoxide by itself is relatively nonreactive, however it can interact with nitric oxide (NO) to produce peroxynitrite at a rate an order of magnitude higher than its dismutation to hydrogen peroxide (H2O2) [27, 28]. Furthermore, inducible NO synthase is activated after TBI and generates NO in a calcium-independent fashion fueling formation of peroxynitrite [29]. 3) Decrease in antioxidant defenses. Cellular protection against superoxide involves superoxide dismutases (SOD) and antioxidants such as vitamin C and E [30]. H2O2 formed by enzymatic or non-enzymatic dismutation of superoxide is efficiently removed by catalase or glutathione peroxidase (GPx) [30]. Despite the inherent protective capacities of healthy cells, TBI-induced pro-oxidative processes can overrun innate antioxidant reserves and other defense systems against transition metals [31]. Resultant oxidative stress leads to injurious oxidative and nitrative modifications of DNA, protein, and lipids that disrupt normal function [32, 33]. In addition to alteration of membrane fluidity and integrity, oxidation of lipids produces a diverse range of signaling molecules.
While non-enzymatic oxidation mechanisms contribute to the overall oxidative burden immediately following TBI, the role of these non-enzymatic mechanisms beyond the acute post-traumatic period is unclear. More recently, the contribution of specific, enzymatic oxidation to oxidative stress following TBI has been increasingly appreciated. Adding to the burden of lipid oxidation in this setting is the overexpression of lipid peroxidation enzymes after brain trauma - namely members of the cyclooxygenase (COX) [34] family, lipoxygenase (LOX) [35] family, cytochrome (cyt) p450 [36], and cyt c [37]. Unlike non-enzymatic lipid oxidation which occurs immediately following injury, enzymatic oxidation of lipids results in delayed and persistent signaling. In the next section, we will briefly review the commonly studied markers of lipid peroxidation in clinical and experimental TBI and the significance of lipid peroxidation for clinical outcome.
Detection of lipid peroxidation after TBI
Various markers of oxidative stress have been studied in TBI (Table 1). In this review, we will focus on lipid peroxidation markers. A number of lipid oxidation markers have been shown to increase in brain tissue, serum, and cerebrospinal fluid (CSF) after experimental and clinical TBI. The primary product of lipid oxidation is hydroperoxides, which have a short half-life. Thus, the study of lipid peroxidation in TBI has largely relied on the detection of more stable reaction end products including isoprostanes, malondialdehyde (MDA), and hydroxynonenal (HNE) [38]. Increased 4-HNE or HNE-modified proteins have been detected in brain tissue using various experimental models of TBI. Numerous clinical studies have observed an increase in F2-isoprostanes and MDA in plasma or CSF following severe TBI [31, 39–43]. In addition to providing information on the extent of oxidation in the brain, elevations in F2-isoprostanes [43] and thiobarbituric acid (TBA)-reactive substances (TBARS) [42, 44–46] - a surrogate marker of MDA - have been correlated with neurological outcome and mortality following TBI in humans.
Table 1.
Oxidative stress markers commonly used in the studies of TBI
| oxidative stress markers | Analysis Method | Reference |
|---|---|---|
| Lipid peroxidation markers | ||
| Isoprostanes | Immuno assay/LC-MS | [19, 31, 39, 43] |
| Oxidized phospholipids | LC-MS | [33, 47, 48] |
| Oxidized free fatty acids | LC-MS | [49–51] |
| 4-Hydroxynanoenal | Immuno assay/LC-MS | [52] |
| Malondialdehyde | Immuno assay/LC-MS | [41, 42] |
| Protein oxidation Markers | ||
| Protein carbonyls | Immuno assay/LC-MS | [53, 54] |
| Protein adducts | Immuno histochemistry | [53, 55] |
| DNA oxidation products | ||
| 8-hydroxy-2’-deoxyguanosine (8-OHdG) | LC-MS | [56, 57] |
| Enzymes related to oxidative stress | ||
| NADPH oxidase | Spectroscopy | [58] |
| Xanthine oxidase | Spectroscopy | [59] |
| Nitric oxide synthase | Immuno assay | |
| Catalase | Spectroscopy | [60] |
| Glutathione peroxidase | Spectroscopy | [60, 61] |
| Antioxidants | ||
| Ascorbate | Spectroscopy | [31] |
| Glutathione | Spectroscopy | [62] |
| Vitamin E | HPLC | [46] |
While isoprostanes, MDA, and HNE are the most commonly evaluated markers of lipid oxidation in clinical samples and can provide generalized information on the extent of oxidation and neurological outcome, these methodologies are not without flaws. First, there are limitations to the methods used to detect lipid peroxidation end products. For example, detection of MDA continues to rely upon reaction with TBA for derivatization; however, TBA can react non-specifically with other aldehydes including carbohydrates, thus confounding the measurement of MDA [63, 64]. This is particularly problematic in diseases like TBI, where hyperglycemia is frequently seen after injury and is associated with neurological outcome and mortality [65]. Although separation of MDA-TBA from other TBARs using high-performance liquid chromatography (HPLC) can improve assay specificity [66], methods which detect end products of lipid peroxidation have an additional inherent flaw. Though measurement of end products of lipid peroxidation provides useful information about general lipid oxidation after TBI, important information about the species’ origin and time course of their generation are lost. Through advances in HPLC and mass spectrometry-based oxidative lipidomics, hydroperoxy lipid species can now be detected and quantified [38, 48, 51, 67]. The advent of oxidative lipidomics [68] has begun to reveal the diversity of lipid oxidation after TBI and elucidate enzymatic mechanisms of lipid oxidation that could potentially be targeted. Understanding the source and signaling associated with lipid oxidation is important in therapeutic targeting and may explain why general antioxidative treatment strategies have so far failed in clinical translation. In the next section, we will briefly review these strategies.
Targeting oxidative stress and lipid oxidation via antioxidants
With strong evidence of increased lipid peroxidation following TBI and a correlation with neurological outcome, antioxidant therapy targeting oxidation of lipids was an obvious therapeutic strategy following brain trauma. These therapies include nonspecific antioxidants such as vitamin C and vitamin E that function through free radical scavenging mechanisms, as well as antioxidant enzymes including superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPX). Despite the success of antioxidant therapy targeting lipid oxidation in experimental models of TBI, protective effects of antioxidant therapy largely have not translated in the large multicenter randomized controlled trials in TBI patients [69]. Three major reasons for this failure include: 1) Relatively slow reaction rate of antioxidants with oxidants thus requiring non-physiological or unachievable in vivo concentrations for their antioxidant activity [70]. In this regard, vitamin E is an exception that is a potent antioxidant at relatively low concentrations. However, the reaction rates for effective scavenging by Vitamin E are limited to hydroperoxyl radicals [71, 72]. 2) Insufficient understanding of the role of oxidative stress in the pathophysiology of disease processes due to lack of proper oxidative stress markers [73]. 3) Insufficient attention to the enzymatic reactions in which free radical intermediates can be formed. In this case, random chemical reactions and respective rate constants although important do not define the overall protective potency. Therefore, unless the mechanisms and sources of lipid peroxidation are better understood, blanket use of free radical scavengers and antioxidants may not be fully successful. Better understanding of mechanisms of lipid peroxidation, identification, and quantification of oxidized lipid species are needed. With the advent of oxidative lipidomics that includes a combination of various separation techniques and mass spectrometry over the last 10 years, our understanding of targets of lipid peroxidation and of the essential role oxidized lipids play in signaling during brain injury and repair have improved. In the next section, we will focus on the different signaling roles PLs play in neuronal health and disease.
Brain phospholipids and their signaling in TBI
Lipids are of critical importance to brain structure and function, comprising approximately 60% of brain weight. The brain lipidome is rich and complex consisting of 5,713 putative species of which 2,330 have been identified [74, 75]. PLs contribute substantially to the diversity and abundance of brain lipids, comprising 35–50% of total lipid weight [76, 77] and 60% of the number of lipids [75]. Polyunsaturated fatty acids (PUFAs) contain one or more methylene-interrupted dienes and are enriched at the sn2 position of PLs. Similar to di-acylated PLs, the tetra-acylated PL cardiolipin (CL) in the brain is also enriched with PUFAs. PUFAs are prone to non-enzymatic free radical and enzymatic oxidation due to the ease of abstraction of hydrogens from the methylene bridges. Therefore, PUFA-containing PLs form one the most important groups of oxidizable molecules in the brain’s cellular milieu. Despite the diversity of the PL head group and acyl chain composition, enzymatic mechanisms enable selective oxidation in a time-dependent manner. In the following section, we will review the mechanisms of PL oxidation and the associated cellular signaling that have been described after TBI.
Signaling by oxidized free fatty acids
Release of free fatty acids (FFA) from PLs is one of the earliest events following mechanical insult. Elevations of FFA in the injured brain are reported as early as 5 minutes after experimental TBI and remain up to 24 hr after injury [78]. In humans with severe TBI, polyunsaturated FFA have been shown to remain elevated up to a week after injury in CSF [79, 80]. This increase in FFA is due to calcium-dependent activation of phospholipase A2 (PLA2) resulting in the hydrolysis of esterified PUFAs from the sn2 position of PLs [81]. Concurrent with the increase in FFA, expression of lipid peroxidation enzymes including LOX [48] and COX [82] also increase after injury. This results in massive enzymatic FFA oxidation, which peaks at 1 hr after injury [51]. Various oxidized FFA products such as eicosanoids, docosanoids, and octadecanoids are elevated after TBI. More specifically, well-characterized lipid mediators [82] including hydroxyoctadecadienoic acids (HODEs), hydroxyeicosatetraenoic acids (HETEs), DiHETEs, prostaglandins, lipoxins, neuroprotectins, and resolvins [83] were among the 244 oxidized FFA observed following TBI [51]. Oxidized FFA are dynamic lipid mediators that regulate both the promotion and resolution of inflammation [84]. Production of pro-inflammatory oxidized FFA occurs by 1 hr after impact, whereas production of anti-inflammatory oxidized FFA predominates by 24 hr following injury. The early increase in oxidized FFA correlates with other early pro-inflammatory events such as microglia activation [85], blood brain barrier (BBB) damage [86], and neutrophil infiltration [87]. The later increase in the anti-inflammatory and resolving signals correlates with other events in the resolution of inflammation (Fig 1) [88]. The PLA2-mediated hydrolysis of fatty acids from PLs generates an additional lipid product, lysophospholipids (lyso-PL), with roles in various signaling pathways. Among the various lyso-PLs, lysophosphatidylcholine has known involvement in disruption of the BBB and induction of T-cell and neutrophil response [89]. Additionally, lyso-phosphatidic acid is involved in the initiation of neuropathic pain [90] via G protein-coupled receptor (GPCR)-mediated signaling [91]. As an alternative mechanism of the production of PUFA lipid mediators, oxidation of different PLs, particularly CLs, and their subsequent hydrolysis by Calcium independent phospholipases A2 leading to the release of oxygenated PUFA has been documented [47].
Fig 1. Production and signaling of oxidized free fatty acids (FFA-ox) after traumatic brain injury (TBI).
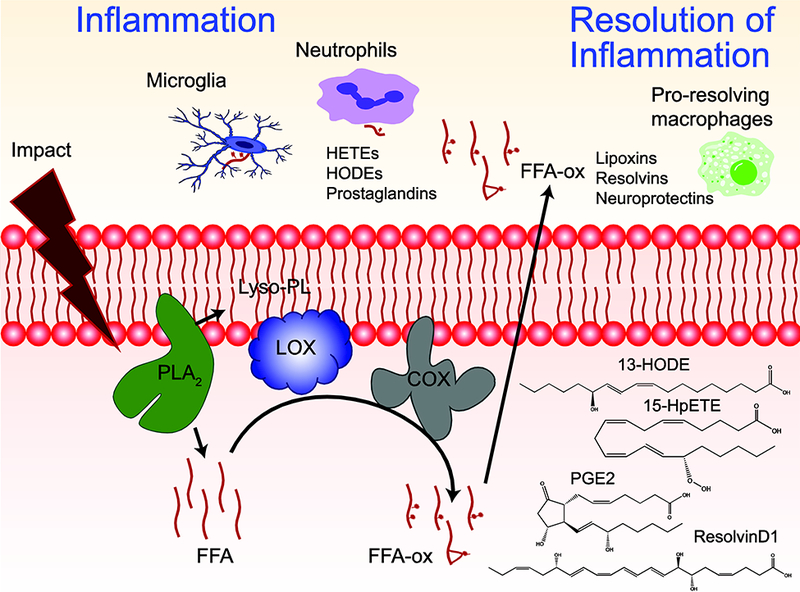
Schema showing the mechanism of production of FFA-ox signals after TBI. Early after injury, phospholipase A2 (PLA2) hydrolyzes phospholipid (PL) to generate lyso-PL and free fatty acid (FFA) which can subsequently be oxidized by lipoxygenases (LOX) and cyclooxygenases (COX). At early stages after injury, FFA-ox such as hydroxyeicosatetraenoic acids (HETEs), hydroxyoctadecadienoic acids (HODEs), and prostaglandins act as important inflammatory mediators initiating microglial activation and neutrophil infiltration. At later stages after injury, FFA-ox such as lipoxins, resolvins, and neuroprotectins contribute to the resolution of inflammation. Structures of representative oxidized fatty acids are depicted
Signaling by oxidized phosphatidylethanolamine
Direct enzymatic oxidation of PLs, resulting in the initiation of cell death pathways in the injured brain, is also triggered by TBI [33]. As an early response, oxidation of CLs by cyt c takes place in mitochondria and leads to the execution of apoptosis [67]. Another death pathway realized in TBI is ferroptosis - the recently described, iron-dependent form of cell death [92]. Ferroptosis is triggered by doubly- and triply-oxygenated phosphatidylethanolamine (PE), especially those containing omega-6 fatty acids such as arachidonic acid (AA) and adrenic acid (AdA) [65]. Production of these oxidized PE is accelerated by the formation of a transient protein complex through the binding of phosphatidylethanolamine-binding protein 1 (PEBP1) and 15-lipoxygenase (15LO) [48]. This complex allosterically alters the substrate specificity of 15LO from FFA to AA-PE. Under normal conditions, hydroperoxy-PLs are reduced to less active hydroxy-PLs by the action of glutathione peroxidase 4 (GPX4) in the presence of glutathione (GSH) [93]. The accumulation of ferroptotic signal molecules can occur as a result of overproduction by the 15LO/PEBP1 complex or by inefficient clearance by the GPX4/GSH system. Unlike apoptosis, there are no specific morphological features of ferroptotically dying cells revealed by conventional (fluorescence) or electron microscopy. However, a recent study identified the complex of 15-LO with PEBP1 as a required stage of ferroptosis leading to the production of pro-ferroptotic hydroperoxy-arachidonoyl-phosphatidylethanolamines [48]. These complexes can be visualized by high-resolution fluorescence microscopy using dual object recognition protocol[94] [48]. Application of this technique showed remarkably increased contents of these complexes in the ferroptotically dying neurons after TBI. This is illustrated in the new Fig 2 where the 15-LO-2/PEBP1 complexes are comparatively shown for the normal vs injured brain. These studies suggest that ferroptotic cell death is initiated early (1 hr) after the impact progressing up to 24 hr post-injury (Fig 3) [48]. Though the exact role and mechanisms of ferroptotic cell death in disease pathology is yet to be explored, the number of neurological diseases where ferroptosis has been shown to play an important pathogenic role is growing [95]. Therapies targeting ferroptosis, such as Ferrostatin-1 or inhibitors of LOX, have been shown to reduce neuronal death and infarct size in animal models of intracerebral hemorrhage [96]. Similarly another ferroptosis inhibitor, Liproxstatin-1 given after ischemia reperfusion injury was shown to improve motor function in rats [97].
Fig 2. Ferroptosis in injured Brain.
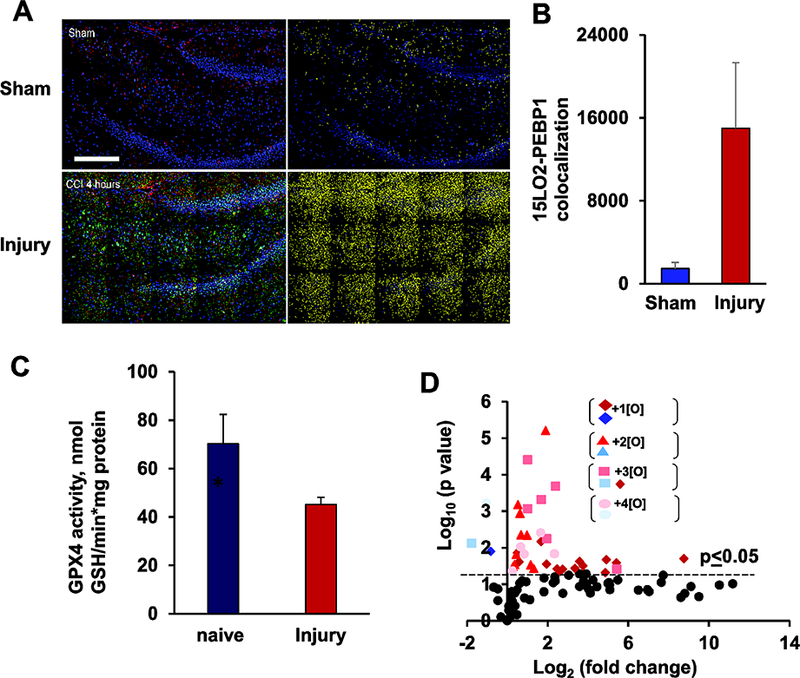
Ferroptosis in injured (Control cortical impact) rat brain is shown using various indicators of ferroptosis. (A) Co-localization of PEBP1 and 15LO2 in brain tissue. Stitched image showing high resolution large area confocal scanning of 3 × 5 image fields. Left: the overlaid emissions for the immunolocalization of PEBP1 (red), 15LO2 (green), and nuclei (blue). Right: co-localization analysis for 15LO2 and PEBP1, with the number of spots having both proteins appearing yellow. Scale bar, 200 μm. (B) Number of co-localized 15LO2 and PEBP1 in brain tissue 4 hr after injury. (C) Changes in GPX4 activity in rat brain cortex at 4 hr after injury. (D) Volcano plot demonstrates changes in the content of PEox at 1 hr post injury. The figure has been reproduced from Wenzel et. al., [48] with permission from Elsevier.
Fig 3. Production and signaling of phosphatidylethanolamine (PE) oxidation after traumatic brain injury (TBI).
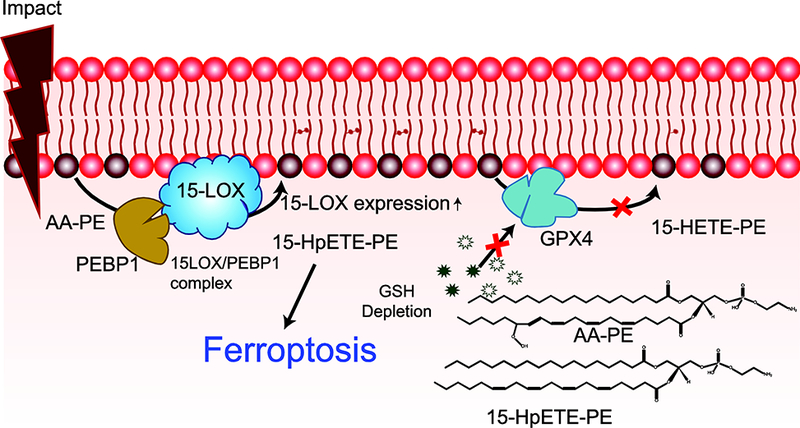
After TBI, the expression of 15-LOX and its formation of a complex with PE binding protein 1 (PEBP1) increase. This complex oxidizes arachidonyl (AA)-PE to 15-hydroperoxyeicosatetraenoic acid-PE (15-HpETE-PE). The glutathione (GSH) and glutathione peroxidase 4 (GPX4) system responsible for the reduction of 15-HpETE-PE to 15-hydroxyeicosatetraenoic acid-PE (15-HETE-PE) is ineffective post-TBI, resulting in 15-HpETE-PE accumulation and ferroptosis. Structure of AA-PE and 15-HpETE-PE are shown in inset.
Signaling by oxidized cardiolipin
As indicated above, oxidation of CL and apoptosis occurs early during TBI. CL is typically confined to the inner membrane of mitochondria but can be externalized to the mitochondrial outer membrane upon the changes in mitochondrial physiology. As this process requires at least three translocations of CL across the inner and outer mitochondrial membranes, several mechanisms may be implicated in the transmembrane migration of CLs including: Ca2+-dependent phase transition [98], Ca2+-dependent migration by phospholipid scramblase 3 [99], and nicotinamide diphosphate kinase D (NDPKD)-catalyzed translocation [100]. At this new location, CL can form a complex with cyt c, the electron shuttle between complex III and IV of the electron transport chain. CL binding destabilizes the structure of cyt c [101] resulting in its conversion to a peroxidase [37]. CL is oxidized by this CL/cyt c peroxidase using H2O2, generating more than 150 unique oxidized CL products in the injured brain [67]. Oxidation of CL results in cytosolic release of pro-apoptotic cyt c to initiate apoptosis after TBI (Fig 4) [37]. The temporal profile of CL oxidation in the injured brain indicates the initiation of apoptosis 3 hr following TBI [33]. Various cell types in brain exhibit varying levels of sensitivity towards apoptosis. For example, neurons are sensitive to apoptotic cell death while microglia are resistant [21]. In TBI, the apoptosis regulation is also mediated by various other factors such as, B-cell lymphoma-2 (Bcl-2) family of proteins [102] and mitogen-activated protein kinase (MAPK) signal-transduction pathways [103]. Studies suggest that TBI sets into motion both apoptotic as well regulated necrotic cell death pathways such ferroptosis [8, 104]. The exact regulating factors determining initiation of apoptosis versus ferroptosis after TBI is not known. It is possible that mitochondrial injury beyond the repair capacity might predispose the neurons to trigger apoptotic cell death pathway via oxidation of CLs.
Fig 4. Production and signaling of oxidized cardiolipin (CL-ox) after traumatic brain injury (TBI).
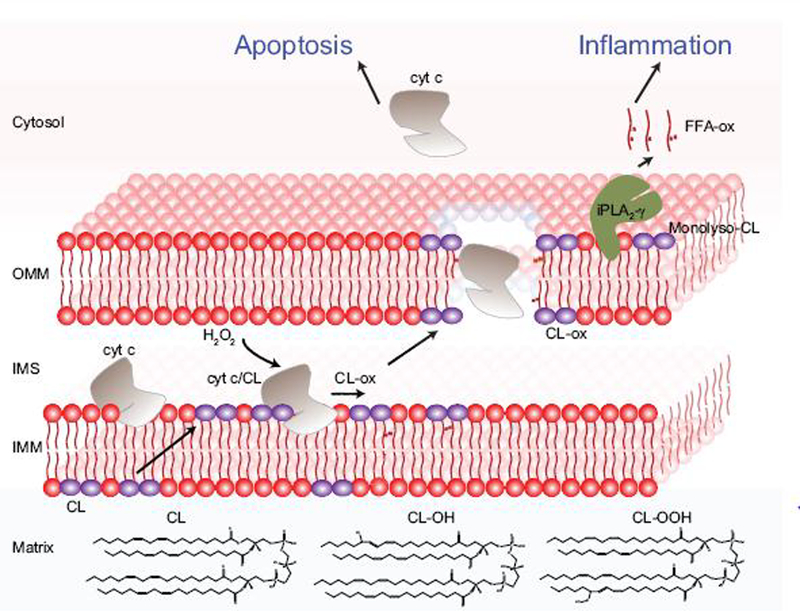
After TBI, cardiolipin (CL) translocate from the inner leaflet to the outer leaflet of the inner mitochondrial membrane (IMM). Once in the outer leaflet, CL binds cytochrome (cyt) c, an electron shuttle of the mitochondrial electron transport chain, to form a complex with peroxidase activity. Using hydrogen peroxide (H2O2) as an oxidizing equivalent, this enzyme oxidizes CL. This CL-specific oxidation leads to the release of cyt c from the intermembrane space (IMS) into the cytosol triggering apoptosis. Oxidized CL can be hydrolyzed by calcium-independent phospholipase A2 (iPLA2γ) to generate monolyso-CL and oxidized free fatty acids (FFA-ox) involved in inflammatory signaling. Inset showing the structure of cardiolipin and oxidized cardiolipin.
In addition to the role of CL oxidation in apoptosis, oxidized CL species can be further hydrolyzed by the calcium-independent phospholipase A2 (iPLA2-γ) producing oxidized FFA and monolyso-CL (MLCL). Like oxidized FFA produced via the classical lipid mediator pathway described above, mitochondrial-based lipid mediator production contributes to inflammatory signaling by oxidized FFA [47]. MLCL is also implicated in the activation of apoptosis. Increased MLCL aids in outer mitochondrial membrane (OMM) localization of truncated BH3 interacting domain protein (tBID), leading to OMM permeabilization and release of apoptogenic factors [105, 106]. Interestingly, cells devoid of iPLA2-γ were resistant to in vitro TBI using a mechanical stretch model of injury, supporting a role for MLCL in the activation of apoptosis (unpublished results). Furthermore, increased MLCL content due to mutation in CL remodeling enzyme tafazzin results in cognitive defects, such as lower visual spatial skills and mathematical performance in patients with Barth syndrome [107–109]. Barth syndrome is an X-linked inherited disease that is caused solely by the impairment of taffazin to reacylate CL leading to dilated cardiomyopathy, skeletal myopathy and neutropenia [110].
While the oxidation of CL in the OMM induces apoptosis and inflammation, externalized CL is involved in several other key signaling events. Externalized CL can interact with the key autophagy protein, microtubule-associated-protein-1-light chain-3 (LC3), to initiate autophagosome formation and thus mitophagy [99]. As mitochondrial dysfunction plays a crucial role in secondary injury following TBI, removal of damaged mitochondria via mitophagy is important in preventing mitochondrial-derived oxidative and inflammatory signaling [111]. In TBI, inhibition of mitophagy has been shown to increase inflammation, while activation of mitophagy reduces inflammation [112]. Mitochondria with externalized CL which fail to undergo mitophagy can be released to the systemic circulation. Once outside the cell, externalized CL is recognized by CD36 scavenger receptors on phagocytes and can modulate inflammatory response by TLR4-mediated pathways [113].
Signaling by oxidized phosphatidylserine
As previously discussed, CL oxidation by the CL/cyt c peroxidase results in the release of cyt c into the cytoplasm where it is involved in the initiation of apoptosis. cyt c displays high affinity towards anionic phospholipids, and thus once in the cytoplasm it can bind to anionic phosphatidylserine (PS) in the plasma membrane. This cyt c-PS complex again enables the peroxidase activity of cyt c with resulting oxidation of PS [114]. Along with caspase-mediated changes in phospholipid flippase and scramblase activity, oxidized PS can function as a non-enzymatic scramblase leading to PS externalization [115–118] which serves as an “eat me” signal in efferocytosis. Oxidized PS is an even more potent “eat me” signal than non-oxidized PS for the recruitment of phagocytes to the vicinity of apoptotic cells (Fig 5). Studies indicate that oxidized PS is a ligand for the scavenger receptor CD36 and MFG-E8 in mediating macrophage response [119, 120]. In line with its role in the clearance of apoptotic cells, PS oxidation occurs only by 24 hr. Efferocytosis serves a critical role in modulating the inflammatory response and promoting injury resolution [67, 68]. Importantly another lipid mediator implicated in resolution of inflammation, resolvins, are also pronounced during this time, indicating synergetic effects of oxidized lipid mediators in the resolution of inflammation. Aside from its role in efferocytosis and inflammation resolution, oxidized PS may also directly bind to protein kinase C inhibitor therefore preventing the inactivation of blood coagulation factor [121]. To date, no studies have directly connected PS oxidation and coagulation status in TBI, but it is possible that oxidized PS may prevent injury propagation through an unexplored role in promotion of coagulation.
Fig 5. Production and signaling of oxidized phosphatidylserine (PS-οx) after traumatic brain injury (TBI).
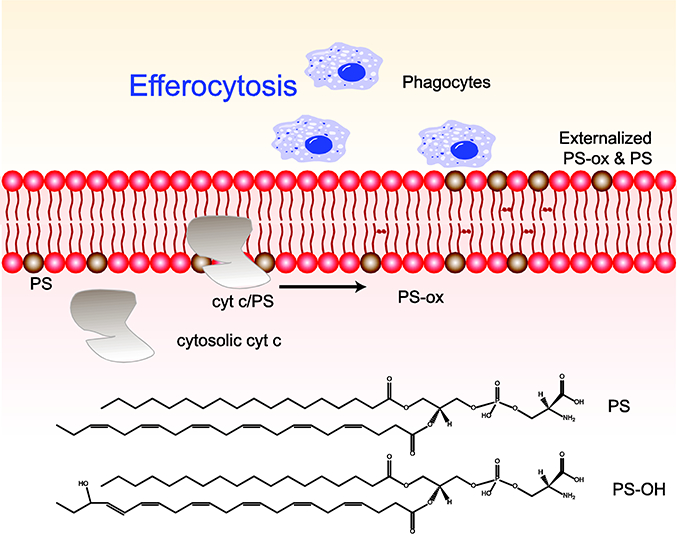
The cytochrome (cyt) c released from mitochondria into the cytosol during apoptosis can bind to phosphatidylserine (PS) in the inner leaflet of the plasma membrane to form a peroxidase. This PS-specific oxidation leads to oxidation of PS. Externalization of PS and PS-οχ to the outer leaflet of the plasma membrane results in efferocytosis by serving as “eat me” signals for phagocytic cells. Structures of PS and PS-ox also shown in the figure.
Targeting phospholipid oxidation signaling in TBI
The oxidized PL signaling pathways present both potential targets and challenges when considering the design of effective neuroprotective therapies. However, opportunity does exist for the design of small molecule inhibitors to target common pathways involved in the synthesis and signaling of oxidized PL in TBI. Common and non-specific drug targets in preventing lipid peroxidation have been reviewed previously [30]. With new insight into targets and time course of lipid peroxidation gained through mass spectrometry-based lipidomics, here we will focus on new possibilities of drug targeting aimed at signaling by oxidized PL (Table 2.).
Table 2.
List of potential Drug/small molecule to inhibit phospholipid oxidation pathways.
| Drug/Small molecule | Pathway | Model | Reference |
|---|---|---|---|
| Imidazole-substituted derivatives of stearic acid and oleic acid | cyt C/CL peroxidase activity | Mouse whole body irradiation | [125] |
| nitroxide-based electron scavengers (i.e. XJB-5–131, JP4– 039 | cyt C/CL peroxidase activity | Rat TBI, global cerebral ischemia/reperfusion | [9, 67] |
| (R)-bromoenol lactone [(R)-BEL] | Hydrolysis of Clox | Mouse whole body irradiation | [47] |
| N-acetylcysteine | GSH precursor, Ferroptosis inhibitor | Human TBI | [126] |
| ebselen | GPx mimetic, ferroptosis inhibitor | Human stroke | [127] |
| Baicalein | 15LOX, ferroptosis inhibitor | Rat TBI, Cerebral Ischemia | [128, 129] |
Inflammation mediated by FFA oxidation is a major driving force in TBI pathogenesis and thus a viable option for secondary injury prevention [122]. Targeting this process can include both PLA2 and lipid oxidation enzymes. Despite the success of enzyme inhibition or genetic knockdown in animal models, nearly all clinical trials targeting FFA oxidation or inflammation demonstrated limited or no success [123]. It is now recognized neuro-inflammation is necessary for tissue regeneration and repair. Early inflammation is required for removal of dead cells generated during the impact but chronic inflammation is deleterious. Identification of a switc between beneficial and harmful inflammation will be critical in designing therapies targeting FFA oxidation or inflammation [124].
Interfering with CL oxidation and thereby preventing apoptosis is another avenue for development of neuroprotective therapies in TBI. One option is to prevent the peroxidase activity of cyt C/CL complex by irreversibly blocking the accessibility to the oxidizing heme in cyt c. Proof-of-concept studies have demonstrated that mitochondrial-targeted imidazole-substituted derivatives of stearic acid and oleic acid (TPP-ISA and TPP-IOA) inhibit cyt C/CL peroxidase activity while reducing caspase activation and cell death following γ-irradiation injury [130]. Alternatively, ongoing post-injury generation of H2O2 necessary for cyt C/CL activity can be inhibited through treatment with mitochondria-targeted nitroxide-based electron scavengers (i.e. XJB-5–131, JP4–039). XJB-5–131 has been shown to reduce lesion volume, neuronal cell death, and improve functional outcome following experimental TBI [67]. Reducing the abundance of oxidizable CL species using mitochondrial-targeted TPP-conjugated oleic acid is also effective in prevention of apoptotic cell death [67]. Finally, iPLA2γ, which hydrolyzes oxidized acyl chains including those esterified to CL, can be specifically and potently inhibited by (R)-bromoenol lactone [(R)-BEL], leading to significant reduction in oxidized FFA following γ-irradiation [47]. The potential neuroprotective role of (R)-BEL is worth exploring in TBI. Collectively, TPP-ISA, TPP-IOA, XJB-5–131, (R)-BEL, and other mechanistically similar approaches may provide protection after TBI by limiting generation and subsequent release of CL-generated lipid mediators to their distal sites of action.
Due to the highly regulated nature of ferroptosis, this cell death pathway may offer a better option with multiple avenues for therapeutic targeting. The targets for prevention of ferroptosis include: 1) augmenting the defense system which remove oxidized PE products, 2) preventing the formation of 15LOX/PEBP1 complex, thereby reducing the rate of PE oxidation, and 3) directly preventing PE oxidation with 15LOX inhibition. The defense system which removes oxidized PE, namely GSH and GPX4, can be augmented with N-acetylcysteine (NAC) [126] and ebselen [127] supplementation. NAC is a precursor for GSH production and showed protective effects in adult TBI patients [131, 132]; however, the protective effects have not borne out in pediatric TBI [133]. Early administration of ebselen, a GPX mimetic that reduces hydroperoxy-PL, showed significant improvement in stroke patients [134, 135]. Furthermore, ebselen treatment ameliorated neurological injury in a rat model of TBI [136]. 15LOX inhibitors such as baicalein can improve functional outcome after TBI [128, 129], while various 15LOX inhibitors were shown to be protective in stroke models [137, 138]. Other small molecule inhibitors such as Liproxstatin and Ferrostatin-1 also reduce oxidized PE accumulation and decrease ferroptosis. As previously discussed, accumulation of oxidized AA- or AdA-containing PE results in ferroptosis, therefore inhibition of acyl-coA synthetase long-chain family member 4 (ACSL4) and thus the formation of AA- and AdA-esterified PE may also protect against TBI [139].
Future perspectives.
The current understanding of oxidized PL signaling has identified promising treatment targets for mitigation of TBI, however, several outstanding questions remain unanswered regarding PL oxidation in TBI. Brain regions perform specific neuronal functions [140] and display differential proteomes [141] and metabolomes [142]. A recent metabolomics analysis of different brain regions showed significant PL species variation across the brain [142]. In line with the functional and metabolomic differences, the susceptibility of different brain regions to injury significantly differs [143–145]. Together, these findings strongly suggest that different brain regions may exhibit differential pathogenesis and lipid signaling. Though information on the temporal course of lipid oxidation after TBI can be studied using liquid chromatography mass spectrometry (LC-MS) analysis of brain homogenates, spatial information about the lipid mediators is lost with this methodology. Unlike LC-MS methods in which each lipid species or classes are separated prior to mass analysis, imaging methods can acquire information on complex mixture of lipids with a wide range of concentrations; however, the identification of low-abundance, signaling PL is difficult with imaging mass spectrometry techniques. Using a combination of various tissue treatments to remove abundant lipids and enhance ionization, we have utilized imaging mass spectrometry to identify the specific loss of PUFA-containing CL species from the contusional cortex, hippocampi (CA1 and CA3), and thalamus after experimental TBI [146, 147]. Although the low abundance of oxidized lipids precluded their detection, the specific loss of PUFA-containing CL indicated that oxidation may explain the decrease in tissue CL after TBI. Recently, oxidatively truncated CL species were imaged using Desorption Electrospray Ionization Mass Spectrometry (DESI-MS) in thyroid oncocytic tumors [148]. The latest advances in imaging mass spectrometry methods including gas cluster ion beam-secondary ion mass spectrometry (GCIB-SIMS), sub-micrometer focused ionization beams, and enhanced and high-resolution mass detection techniques could enable cellular-level imaging of PL in the near future.
In addition to the importance of brain region-specific oxidized PL signaling, the implications of subcellular location of PLs cannot be ignored. The cellular distribution of PLs is not uniform, with the enrichment of several PL classes in specific subcellular compartments. For example, CL is restricted to the mitochondria [149], PE is enriched in mitochondria as well as endoplasmic reticulum, and PS is confined to the plasma membrane [150]. Identification of oxidized PL does not only provide information related to the mechanism and time course of injury, it can also provide insight to the cellular organelle involved in the injury mechanism. Understanding of oxidized PL signaling may aid in the development of organelle-targeting drugs to increase the treatment specificity and efficacy. While CL oxidation is clearly linked to the mitochondria, our understanding of the subcellular location of all other PL oxidation pathways is limited. Combining the strength of subcellular organelle lipidomics [151, 152] and oxidative lipidomics can offer a solution to identify the exact location of oxidized PL production.
Conclusions
The pathophysiology of TBI is a complex and dynamic process which involves signaling through multiple pathways including inflammation and cell death. While oxidative stress and lipid peroxidation have long been implicated in TBI pathogenesis, our understanding of the mechanisms of lipid peroxidation and their role in the regulation of disease pathogenesis remain limited. With the advent of oxidative lipidomics, we have begun to unravel the complexity and diversity of oxidized lipid mediators and the time course of their production after TBI. While free radical-based non-enzymatic reactions cannot be ignored, a substantial portion of lipid peroxidation following TBI is generated through tightly controlled, enzymatically regulated mechanisms that may be a leading cause of secondary injury. Such enzymatic lipid peroxidation produces distinct mediators during different stages of inflammation and activation of specific cell death pathways. This temporal differentiation of signaling events offer many advantages as certain phase of otherwise deleterious processes are essential in the prognosis of the TBI. In addition to providing new targets for therapeutic intervention, our current knowledge of PL oxidation in TBI provides a possible explanation as to why previous clinical trials targeting lipid oxidation were unsuccessful and what changes in treatment strategy are needed to improve outcomes after TBI. Despite the improved understanding of lipid signaling pathways in the context of TBI, further study is needed. Future research should aim to identify exact targets for neuro-therapeutic intervention, spatial and temporal signaling, and association of (oxidized) lipids with cell death and repair pathways.
Acknowledgements
The work is supported, in part, by NIH grants (P01HL114453, U19AI068021, NS076511, NS061817, NS084604, CA165065).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- [1].Faul M, Xu L, Wald MM, Coronado V, Dellinger AM, Traumatic brain injury in the United States: national estimates of prevalence and incidence, 2002–2006, Injury Prevention 16(Suppl 1) (2011) A268. [Google Scholar]
- [2].Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, Guzman BR, Hemphill JD, Surveillance for traumatic brain injury-related deaths-United States, 1997–2007, Morbidity and mortality weekly report. Surveillance summaries (Washington, D.C. : 2002) 60(5) (2011) 1–32. [PubMed] [Google Scholar]
- [3].Maas AIR, Stocchetti N, Bullock R, Moderate and severe traumatic brain injury in adults, The Lancet Neurology 7(8) (2008) 728–741. [DOI] [PubMed] [Google Scholar]
- [4].Andelic N, Hammergren N, Bautz-Holter E, Sveen U, Brunborg C, Roe C, Functional outcome and health-related quality of life 10 years after moderate-to-severe traumatic brain injury, Acta neurologica Scandinavica 120(1) (2009) 16–23. [DOI] [PubMed] [Google Scholar]
- [5].Andriessen TM, Horn J, Franschman G, van der Naalt J, Haitsma I, Jacobs B, Steyerberg EW, Vos PE, Epidemiology, severity classification, and outcome of moderate and severe traumatic brain injury: a prospective multicenter study, Journal of neurotrauma 28(10) (2011) 2019–31. [DOI] [PubMed] [Google Scholar]
- [6].Humphreys I, Wood RL, Phillips CJ, Macey S, The costs of traumatic brain injury: a literature review, ClinicoEconomics and Outcomes Research: CEOR 5 (2013) 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wachelder EM, Moulaert VRMP, van Heugten C, Verbunt JA, Bekkers SCAM, Wade DT, Life after survival: Long-term daily functioning and quality of life after an out-of-hospital cardiac arrest, Resuscitation 80(5) (2009) 517–522. [DOI] [PubMed] [Google Scholar]
- [8].Stoica BA, Faden AI, Cell death mechanisms and modulation in traumatic brain injury, Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 7(1) (2010) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ji J, Baart S, Vikulina AS, Clark RSB, Anthonymuthu TS, Tyurin VA, Du L, Croix C.M. St, Tyurina YY, Lewis J, Skoda EM, Kline AE, Kochanek PM, Wipf P, Kagan VE, Bayır H, Deciphering of Mitochondrial Cardiolipin Oxidative Signaling in Cerebral Ischemia-Reperfusion, Journal of Cerebral Blood Flow & Metabolism 35(2) (2014) 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Puyal J, Ginet V, Clarke PG, Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection, Progress in neurobiology 105 (2013) 24–48. [DOI] [PubMed] [Google Scholar]
- [11].Werner C, Engelhard K, Pathophysiology of traumatic brain injury, British Journal of Anaesthesia 99(1) (2007) 4–9. [DOI] [PubMed] [Google Scholar]
- [12].Prins M, Greco T, Alexander D, Giza CC, The pathophysiology of traumatic brain injury at a glance, Disease Models & Mechanisms 6(6) (2013) 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cornelius C, Crupi R, Calabrese V, Graziano A, Milone P, Pennisi G, Radak Z, Calabrese EJ, Cuzzocrea S, Traumatic brain injury: oxidative stress and neuroprotection, Antioxidants & redox signaling 19(8) (2013) 836–53. [DOI] [PubMed] [Google Scholar]
- [14].Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A, Oxidative stress in traumatic brain injury, Current medicinal chemistry 21(10) (2014) 1201–11. [DOI] [PubMed] [Google Scholar]
- [15].Werner C, Engelhard K, Pathophysiology of traumatic brain injury, BJA: British Journal of Anaesthesia 99(1) (2007) 4–9. [DOI] [PubMed] [Google Scholar]
- [16].Saatman KE, Duhaime A-C, Bullock R, Maas AIR, Valadka A, Manley GT, Classification of Traumatic Brain Injury for Targeted Therapies, Journal of neurotrauma 25(7) (2008) 719–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xiong Y, Mahmood A, Chopp M, Animal models of traumatic brain injury, Nature reviews. Neuroscience 14(2) (2013) 128–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Phipps HW, Systematic Review of Traumatic Brain Injury Animal Models, Methods in molecular biology (Clifton, N.J.) 1462 (2016) 61–88. [DOI] [PubMed] [Google Scholar]
- [19].Bayır H, Kochanek PM, Kagan VE, Oxidative stress in immature brain after traumatic brain injury, Developmental neuroscience 28(4–5) (2006) 420–31. [DOI] [PubMed] [Google Scholar]
- [20].Chang EF, Claus CP, Vreman HJ, Wong RJ, Noble-Haeusslein LJ, Heme regulation in traumatic brain injury: relevance to the adult and developing brain, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 25(11) (2005) 1401–17. [DOI] [PubMed] [Google Scholar]
- [21].Hall ED, Vaishnav RA, Mustafa AG, Antioxidant therapies for traumatic brain injury, Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 7(1) (2010) 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF, Heme and iron metabolism: role in cerebral hemorrhage, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 23(6) (2003) 629–52. [DOI] [PubMed] [Google Scholar]
- [23].Giralt M, Penkowa M, Lago N, Molinero A, Hidalgo J, Metallothionein-1+ 2 protect the CNS after a focal brain injury, Experimental neurology 173(1) (2002) 114–128. [DOI] [PubMed] [Google Scholar]
- [24].Hidalgo J, Metallothioneins and brain injury: What transgenic mice tell us, Environmental Health and Preventive Medicine 9(3) (2004) 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nisenbaum EJ, Novikov DS, Lui YW, The Presence and Role of Iron in Mild Traumatic Brain Injury: An Imaging Perspective, Journal of neurotrauma 31(4) (2014) 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kontos HA, Wei EP, Superoxide production in experimental brain injury, Journal of neurosurgery 64(5) (1986) 803–7. [DOI] [PubMed] [Google Scholar]
- [27].Gray B, Carmichael AJ, Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance, The Biochemical journal 281 ( Pt 3) (1992) 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kissner R, Nauser T, Kurz C, Koppenol W, Peroxynitrous Acid - Where is the Hydroxyl Radical?, IUBMB Life 55(10–11) (2003) 567–572. [DOI] [PubMed] [Google Scholar]
- [29].Bayır H, Kagan VE, Borisenko GG, Tyurina YY, Janesko KL, Vagni VA, Billiar TR, Williams DL, Kochanek PM, Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 25(6) (2005) 673–84. [DOI] [PubMed] [Google Scholar]
- [30].Anthonymuthu TS, Kenny EM, Bayır H, Therapies targeting lipid peroxidation in traumatic brain injury, Brain research 1640(Pt A) (2016) 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bayır H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM, Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children, Pediatric research 51(5) (2002) 571–8. [DOI] [PubMed] [Google Scholar]
- [32].Chen X, Guo C, Kong J, Oxidative stress in neurodegenerative diseases, Neural Regeneration Research 7(5) (2012) 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bayır H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis, Annals of neurology 62(2) (2007) 154–169. [DOI] [PubMed] [Google Scholar]
- [34].Hickey RW, Adelson PD, Johnnides MJ, Davis DS, Yu Z, Rose ME, Chang YF, Graham SH, Cyclooxygenase-2 activity following traumatic brain injury in the developing rat, Pediatric research 62(3) (2007) 271–6. [DOI] [PubMed] [Google Scholar]
- [35].Zhang L, Zhang WP, Hu H, Wang ML, Sheng WW, Yao HT, Ding W, Chen Z, Wei EQ, Expression patterns of 5-lipoxygenase in human brain with traumatic injury and astrocytoma, Neuropathology : official journal of the Japanese Society of Neuropathology 26(2) (2006) 99–106. [DOI] [PubMed] [Google Scholar]
- [36].Harris LK, Black RT, Golden KM, Reeves TM, Povlishock JT, Phillips LL, Traumatic brain injury-induced changes in gene expression and functional activity of mitochondrial cytochrome C oxidase, Journal of neurotrauma 18(10) (2001) 993–1009. [DOI] [PubMed] [Google Scholar]
- [37].Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors, Nature chemical biology 1(4) (2005) 223. [DOI] [PubMed] [Google Scholar]
- [38].Niki E, Biomarkers of lipid peroxidation in clinical material, Biochimica et biophysica acta 1840(2) (2014) 809–17. [DOI] [PubMed] [Google Scholar]
- [39].Bayır H, Adelson PD, Wisniewski SR, Shore P, Lai Y, Brown D, Janesko-Feldman KL, Kagan VE, Kochanek PM, Therapeutic hypothermia preserves antioxidant defenses after severe traumatic brain injury in infants and children, Critical care medicine 37(2) (2009) 689–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bayır H, Marion DW, Puccio AM, Wisniewski SR, Janesko KL, Clark RS, Kochanek PM, Marked gender effect on lipid peroxidation after severe traumatic brain injury in adult patients, Journal of neurotrauma 21(1) (2004) 1–8. [DOI] [PubMed] [Google Scholar]
- [41].Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Signoretti S, Amorini AM, Fazzina G, Lazzarino G, Biochemical analysis of the cerebrospinal fluid: evidence for catastrophic energy failure and oxidative damage preceding brain death in severe head injury: a case report, Clinical biochemistry 38(1) (2005) 97–100. [DOI] [PubMed] [Google Scholar]
- [42].Lorente L, Martin MM, Abreu-Gonzalez P, Ramos L, Argueso M, Caceres JJ, Sole-Violan J, Lorenzo JM, Molina I, Jimenez A, Association between Serum Malondialdehyde Levels and Mortality in Patients with Severe Brain Trauma Injury, Journal of neurotrauma 32(1) (2015) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yen HC, Chen TW, Yang TC, Wei HJ, Hsu JC, Lin CL, Levels of F2-isoprostanes, F4-neuroprostanes, and total nitrate/nitrite in plasma and cerebrospinal fluid of patients with traumatic brain injury, Free radical research 49(12) (2015) 1419–30. [DOI] [PubMed] [Google Scholar]
- [44].Kasprzak HA, Wozniak A, Drewa G, Wozniak B, Enhanced lipid peroxidation processes in patients after brain contusion, Journal of neurotrauma 18(8) (2001) 793–7. [DOI] [PubMed] [Google Scholar]
- [45].Nayak C, Nayak D, Bhat S, Raja A, Rao A, Relationship between neurological outcome and early oxidative changes in erythrocytes in head injury patients, Clinical chemistry and laboratory medicine 45(5) (2007) 629–33. [DOI] [PubMed] [Google Scholar]
- [46].Paolin A, Nardin L, Gaetani P, Rodriguez YBR, Pansarasa O, Marzatico F, Oxidative damage after severe head injury and its relationship to neurological outcome, Neurosurgery 51(4) (2002) 949–54; discussion 954–5. [DOI] [PubMed] [Google Scholar]
- [47].Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, Mohammadyani D, Klein-Seetharaman J, Jackson TC, Kochanek PM, Pitt BR, Greenberger JS, Vladimirov YA, Bayır H, Kagan VE, A mitochondrial pathway for biosynthesis of lipid mediators, Nature chemistry 6(6) (2014) 542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wenzel SE, Tyurina YY, Zhao J, Croix CMS, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals, Cell 171(3) (2017) 628–641. e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].ELLIS EF, POLICE RJ, RICE LY, GRABEEL M, HOLT S, Increased plasma PGE2, 6-keto-PGF1α, and 12-HETE levels following experimental concussive brain injury, Journal of neurotrauma 6(1) (1989) 31–37. [DOI] [PubMed] [Google Scholar]
- [50].Westcott J, Murphy R, Stenmark K, Eicosanoids in human ventricular cerebrospinal fluid following severe brain injury, Prostaglandins 34(6) (1987) 877–887. [DOI] [PubMed] [Google Scholar]
- [51].Anthonymuthu TS, Kenny EM, Amoscato AA, Lewis J, Kochanek PM, Kagan VE, Bayır H, Global assessment of oxidized free fatty acids in brain reveals an enzymatic predominance to oxidative signaling after trauma, Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1863(10) (2017) 2601–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA, Oxidative stress in head trauma in aging, Free Radical Biology and Medicine 41(1) (2006) 77–85. [DOI] [PubMed] [Google Scholar]
- [53].Ansari MA, Roberts KN, Scheff SW, Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury, Free radical biology & medicine 45(4) (2008) 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lazarus RC, Buonora JE, Jacobowitz DM, Mueller GP, Protein carbonylation after traumatic brain injury: cell specificity, regional susceptibility, and gender differences, Free Radical Biology and Medicine 78 (2015) 89–100. [DOI] [PubMed] [Google Scholar]
- [55].Rama Rao KV, Iring S, Younger D, Kuriakose M, Skotak M, Alay E, Gupta RK, Chandra N, A Single Primary Blast-Induced Traumatic Brain Injury in a Rodent Model Causes Cell-Type Dependent Increase in Nicotinamide Adenine Dinucleotide Phosphate Oxidase Isoforms in Vulnerable Brain Regions, Journal of neurotrauma (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Itoh T, Satou T, Nishida S, Tsubaki M, Imano M, Hashimoto S, Ito H, Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury in rats, Neurochemical Research 35(2) (2010) 348–355. [DOI] [PubMed] [Google Scholar]
- [57].Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP, Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death, J Neurochem 69(1) (1997) 232–45. [DOI] [PubMed] [Google Scholar]
- [58].Zhang G-G, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW, Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury, PloS one 7(4) (2012) e34504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Solaroglu I, Okutan O, Kaptanoglu E, Beskonakli E, Kilinc K, Increased xanthine oxidase activity after traumatic brain injury in rats, Journal of Clinical Neuroscience 12(3) (2005) 273–275. [DOI] [PubMed] [Google Scholar]
- [60].Goss JR, Taffe KM, Kochanek PM, DeKosky ST, The antioxidant enzymes glutathione peroxidase and catalase increase following traumatic brain injury in the rat, Experimental neurology 146(1) (1997) 291–294. [DOI] [PubMed] [Google Scholar]
- [61].Kerman M, Kanter M, Coşkun KK, Erboga M, Gurel A, Neuroprotective effects of Caffeic acid phenethyl ester on experimental traumatic brain injury in rats, Journal of Molecular Histology 43(1) (2012) 49–57. [DOI] [PubMed] [Google Scholar]
- [62].Xiong Y, Peterson P, Lee C, Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats, Journal of neurotrauma 16(11) (1999) 1067–1082. [DOI] [PubMed] [Google Scholar]
- [63].Moselhy HF, Reid RG, Yousef S, Boyle SP, A specific, accurate, and sensitive measure of total plasma malondialdehyde by HPLC, Journal of Lipid Research 54(3) (2013) 852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tsikas D, Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges, Analytical biochemistry 524 (2017) 13–30. [DOI] [PubMed] [Google Scholar]
- [65].Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H, Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis, Nature Chemical Biology 13 (2016) 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lykkesfeldt J, Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by HPLC with fluorescence detection: comparison with ultraviolet-visible spectrophotometry, Clinical chemistry 47(9) (2001) 1725–7. [PubMed] [Google Scholar]
- [67].Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury, Nature neuroscience 15(10) (2012) 1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Anthonymuthu TS, Kim-Campbell N, Bayır H, Oxidative lipidomics: applications in critical care, Current opinion in critical care 23(4) (2017) 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bains M, Hall ED, Antioxidant therapies in traumatic brain and spinal cord injury, Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1822(5) (2012) 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Warnholtz A, Munzel T, Why do antioxidants fail to provide clinical benefit?, Trials 1(1) (2000) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nimse SB, Pal D, Free radicals, natural antioxidants, and their reaction mechanisms, Rsc Advances 5(35) (2015) 27986–28006. [Google Scholar]
- [72].Traber MG, Stevens JF, Vitamins C and E: Beneficial effects from a mechanistic perspective, Free radical biology & medicine 51(5) (2011) 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Steinhubl SR, Why Have Antioxidants Failed in Clinical Trials?, The American Journal of Cardiology 101(10, Supplement) (2008) S14–S19. [DOI] [PubMed] [Google Scholar]
- [74].O’Brien JS, Sampson EL, Lipid composition of the normal human brain: gray matter, white matter, and myelin, Journal of Lipid Research 6(4) (1965) 537–544. [PubMed] [Google Scholar]
- [75].Bozek K, Wei Y, Yan Z, Liu X, Xiong J, Sugimoto M, Tomita M, Pääbo S, Sherwood Chet C., Hof Patrick R., Ely John J., Li Y, Steinhauser D, Willmitzer L, Giavalisco P, Khaitovich P, Organization and Evolution of Brain Lipidome Revealed by Large-Scale Analysis of Human, Chimpanzee, Macaque, and Mouse Tissues, Neuron 85(4) 695–702. [DOI] [PubMed] [Google Scholar]
- [76].Rouser G, Bauman A, Kritchevsky G, Heller D, O’Brien JS, Quantitative chromatographic fractionation of complex lipid mixtures: Brain lipids, Journal of the American Oil Chemists Society 38(10) (1961) 544. [Google Scholar]
- [77].Sastry PS, Lipids of nervous tissue: composition and metabolism, Progress in lipid research 24(2) (1985) 69–176. [DOI] [PubMed] [Google Scholar]
- [78].Dhillon H, Donaldson D, Dempsey R, Prasad MR, Regional levels of free fatty acids and Evans blue extravasation after experimental brain injury, Journal of neurotrauma 11(4) (1994) 405–415. [DOI] [PubMed] [Google Scholar]
- [79].Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, Michael DB, Phillis JW, Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury, Neuroscience letters 349(2) (2003) 136–8. [DOI] [PubMed] [Google Scholar]
- [80].Farooqui AA, Horrocks LA, Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly, The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 12(3) (2006) 245–60. [DOI] [PubMed] [Google Scholar]
- [81].Shohami E, Shapira Y, Yadid G, Reisfeld N, Yedgar S, Brain Phospholipase A2 Is Activated After Experimental Closed Head Injury in the Rat, Journal of Neurochemistry 53(5) (1989) 1541–1546. [DOI] [PubMed] [Google Scholar]
- [82].Kunz T, Marklund N, Hillered L, Oliw EH, Cyclooxygenase-2, prostaglandin synthases, and prostaglandin H2 metabolism in traumatic brain injury in the rat, Journal of neurotrauma 19(9) (2002) 1051–1064. [DOI] [PubMed] [Google Scholar]
- [83].Serhan CN, Chiang N, Van Dyke TE, Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators, Nature Reviews Immunology 8 (2008) 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tam VC, Quehenberger O, Oshansky CM, Suen R, Armando AM, Treuting PM, Thomas PG, Dennis EA, Aderem A, Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation, Cell 154(1) (2013) 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W-B, ATP mediates rapid microglial response to local brain injury in vivo, Nature Neuroscience 8 (2005) 752. [DOI] [PubMed] [Google Scholar]
- [86].Ersahin M, Toklu HZ, Erzik C, Cetinel S, Akakin D, Velioglu-Ogunc A, Tetik S, Ozdemir ZN, Sener G, Yegen BC, The anti-inflammatory and neuroprotective effects of ghrelin in subarachnoid hemorrhage-induced oxidative brain damage in rats, Journal of neurotrauma 27(6) (2010) 1143–55. [DOI] [PubMed] [Google Scholar]
- [87].Clark RS, Schiding JK, Kaczorowski SL, Marion DW, Kochanek PM, Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models, Journal of neurotrauma 11(5) (1994) 499–506. [DOI] [PubMed] [Google Scholar]
- [88].David S, Kroner A, Repertoire of microglial and macrophage responses after spinal cord injury, Nature Reviews Neuroscience 12 (2011) 388. [DOI] [PubMed] [Google Scholar]
- [89].Ousman SS, David S, Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord, Glia 30(1) (2000) 92–104. [PubMed] [Google Scholar]
- [90].Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H, Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling, Nature medicine 10(7) (2004) 712. [DOI] [PubMed] [Google Scholar]
- [91].Yung YC, Stoddard NC, Mirendil H, Chun J, Lysophosphatidic acid signaling in the nervous system, Neuron 85(4) (2015) 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell 149(5) (2012) 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Angeli JPF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice, Nature cell biology 16(12) (2014) 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Rizk A, Paul G, Incardona P, Bugarski M, Mansouri M, Niemann A, Ziegler U, Berger P, Sbalzarini IF, Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh, Nature protocols 9(3) (2014) 586. [DOI] [PubMed] [Google Scholar]
- [95].Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC, Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC, Neurobiology of disease 94 (2016) 169–78. [DOI] [PubMed] [Google Scholar]
- [96].Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, Ying M, Koehler RC, Stockwell BR, Wang J, Inhibition of neuronal ferroptosis protects hemorrhagic brain, JCI Insight 2(7) (2017) e90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Tuo Q.z., Lei P, Jackman KA, Li X.l., Xiong H, Li X.l., Liuyang Z.y., Roisman L, Zhang S.t., Ayton S, Wang Q, Crouch PJ, Ganio K, Wang X.c., Pei L, Adlard PA, Lu Y.m., Cappai R, Wang J.z., Liu R, Bush AI, Tau-mediated iron export prevents ferroptotic damage after ischemic stroke, Molecular Psychiatry 22 (2017) 1520. [DOI] [PubMed] [Google Scholar]
- [98].Schug ZT, Gottlieb E, Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis, Biochimica et Biophysica Acta (BBA)-Biomembranes 1788(10) (2009) 2022–2031. [DOI] [PubMed] [Google Scholar]
- [99].Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells, Nature cell biology 15(10) (2013) 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kagan V, Jiang J, Huang Z, Tyurina Y, Desbourdes C, Cottet-Rousselle C, Dar H, Verma M, Tyurin V, Kapralov A, NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy, Cell death and differentiation 23(7) (2016) 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hanske J, Toffey JR, Morenz AM, Bonilla AJ, Schiavoni KH, Pletneva EV, Conformational properties of cardiolipin-bound cytochrome c, Proceedings of the National Academy of Sciences 109(1) (2012) 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Graham SH, Chen J, Clark RS, Bcl-2 family gene products in cerebral ischemia and traumatic brain injury, Journal of neurotrauma 17(10) (2000) 831–41. [DOI] [PubMed] [Google Scholar]
- [103].Mori T, Wang X, Jung JC, Sumii T, Singhal AB, Fini ME, Dixon CE, Alessandrini A, Lo EH, Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 22(4) (2002) 444–52. [DOI] [PubMed] [Google Scholar]
- [104].Raghupathi R, Cell death mechanisms following traumatic brain injury, Brain pathology (Zurich, Switzerland) 14(2) (2004) 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJA, Petit PX, Vaz FM, Gottlieb E, Cardiolipin provides an essential activating platform for caspase-8 on mitochondria, The Journal of Cell Biology 183(4) (2008) 681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Degli Esposti M, Cristea I, Gaskell S, Nakao Y, Dive C, Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death, Cell death and differentiation 10(12) (2003) 1300. [DOI] [PubMed] [Google Scholar]
- [107].Mazzocco MM, Kelley RI, Preliminary evidence for a cognitive phenotype in Barth syndrome, American Journal of Medical Genetics Part A 102(4) (2001) 372–378. [DOI] [PubMed] [Google Scholar]
- [108].Mazzocco MM, Henry AE, Kelly RI, Barth syndrome is associated with a cognitive phenotype, Journal of developmental and behavioral pediatrics: JDBP 28(1) (2007) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Raches D, Mazzocco MM, Emergence and nature of mathematical difficulties in young children with Barth syndrome, Journal of Developmental & Behavioral Pediatrics 33(4) (2012) 328–335. [DOI] [PubMed] [Google Scholar]
- [110].Jefferies JL, Barth syndrome, American journal of medical genetics. Part C, Seminars in medical genetics 163c(3) (2013) 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RS, Autophagy in acute brain injury: feast, famine, or folly?, Neurobiology of disease 43(1) (2011) 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wei L, Zhang Y, Yang C, Wang Q, Zhuang Z, Sun Z, Neuroprotective effects of ebselen in traumatic brain injury model: involvement of nitric oxide and p38 mitogen-activated protein kinase signalling pathway, Clinical and experimental pharmacology & physiology 41(2) (2014) 134–8. [DOI] [PubMed] [Google Scholar]
- [113].Balasubramanian K, Maeda A, Lee JS, Mohammadyani D, Dar HH, Jiang JF, Croix C.M. St., Watkins S, Tyurin VA, Tyurina YY, Klöditz K, Polimova A, Kapralova VI, Xiong Z, Ray P, Klein-Seetharaman J, Mallampalli RK, Bayır H, Fadeel B, Kagan VE, Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity, Science Signaling 8(395) (2015) ra95–ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jiang J, Serinkan BF, Tyurina YY, Borisenko GG, Mi Z, Robbins PD, Schroit AJ, Kagan VE, Peroxidation and externalization of phosphatidylserine associated with release of cytochrome c from mitochondria, Free Radic Biol Med 35(7) (2003) 814–25. [DOI] [PubMed] [Google Scholar]
- [115].Matsura T, Oxidized Phosphatidylserine: Production and Bioactivities, Yonago Acta Medica 57(4) (2014) 119–127. [PMC free article] [PubMed] [Google Scholar]
- [116].Segawa K, Nagata S, An apoptotic ‘eat me’signal: phosphatidylserine exposure, Trends in cell biology 25(11) (2015) 639–650. [DOI] [PubMed] [Google Scholar]
- [117].Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S, Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure, Science 344(6188) (2014) 1164–1168. [DOI] [PubMed] [Google Scholar]
- [118].Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S, Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members, Journal of Biological Chemistry 288(19) (2013) 13305–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL, Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells, The Journal of experimental medicine 203(12) (2006) 2613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B, Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells, Cell Death Differ 11(8) (2004) 943–5. [DOI] [PubMed] [Google Scholar]
- [121].Malleier JM, Oskolkova O, Bochkov V, Jerabek I, Sokolikova B, Perkmann T, Breuss J, Binder BR, Geiger M, Regulation of protein C inhibitor (PCI) activity by specific oxidized and negatively charged phospholipids, Blood 109(11) (2007) 4769–76. [DOI] [PubMed] [Google Scholar]
- [122].Corps KN, Roth TL, McGavern DB, Inflammation and neuroprotection in traumatic brain injury, JA MA neurology 72(3) (2015) 355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Bergold PJ, Treatment of traumatic brain injury with anti-inflammatory drugs, Experimental Neurology 275 (2016) 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Russo MV, McGavern DB, Inflammatory neuroprotection following traumatic brain injury, Science (New York, N.Y.) 353(6301) (2016) 783–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L, Peterson J, Huang Z, Jiang J, Samhan-Arias AK, Maeda A, Feng W, Wasserloos K, Belikova NA, Tyurin VA, Wang H, Fletcher J, Wang Y, Vlasova II, Klein-Seetharaman J, Stoyanovsky DA, Bayîr H, Pitt BR, Epperly MW, Greenberger JS, Kagan VE, A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death, Nature Communications 2 (2011) 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA, N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency, Current opinion in pharmacology 7(4) (2007) 355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Azad GK, Tomar RS, Ebselen, a promising antioxidant drug: mechanisms of action and targets of biological pathways, Molecular biology reports 41(8) (2014) 4865–79. [DOI] [PubMed] [Google Scholar]
- [128].Chen SF, Hsu CW, Huang WH, Wang JY, Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury, British journal of pharmacology 155(8) (2008) 1279–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Liang W, Huang X, Chen W, The Effects of Baicalin and Baicalein on Cerebral Ischemia: A Review, Aging and disease 8(6) (2017) 850–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Jiang J, Bakan A, Kapralov AA, Silva KI, Huang Z, Amoscato AA, Peterson J, Garapati VK, Saxena S, Bayır H, Designing inhibitors of cytochrome c/cardiolipin peroxidase complexes: mitochondria-targeted imidazole-substituted fatty acids, Free Radical Biology and Medicine 71 (2014) 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Amen DG, Wu JC, Taylor D, Willeumier K, Reversing brain damage in former NFL players: implications for traumatic brain injury and substance abuse rehabilitation, Journal of psychoactive drugs 43(1) (2011) 1–5. [DOI] [PubMed] [Google Scholar]
- [132].Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B, Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study, PLoS One 8(1) (2013) e54163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Clark RSB, Empey PE, Bayır H, Rosario BL, Poloyac SM, Kochanek PM, Nolin TD, Au AK, Horvat CM, Wisniewski SR, Bell MJ, Phase I randomized clinical trial of N-acetylcysteine in combination with an adjuvant probenecid for treatment of severe traumatic brain injury in children, PLoS One 12(7) (2017) e0180280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ogawa A, Yoshimoto T, Kikuchi H, Sano K, Saito I, Yamaguchi T, Yasuhara H, Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial, Cerebrovascular diseases (Basel, Switzerland) 9(2) (1999) 112–8. [DOI] [PubMed] [Google Scholar]
- [135].Yamaguchi T, Sano K, Takakura K, Saito I, Shinohara Y, Asano T, Yasuhara H, Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group, Stroke 29(1) (1998) 12–7. [DOI] [PubMed] [Google Scholar]
- [136].Kim EH, Tolhurst AT, Szeto HH, Cho SH, Targeting CD36-mediated inflammation reduces acute brain injury in transient, but not permanent, ischemic stroke, CNS neuroscience & therapeutics 21(4) (2015) 385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Yigitkanli K, Pekcec A, Karatas H, Pallast S, Mandeville E, Joshi N, Smirnova N, Gazaryan I, Ratan RR, Witztum JL, Montaner J, Holman TR, Lo EH, van Leyen K, Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke, Ann Neurol 73(1) (2013) 129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].van Leyen K, Holman TR, Maloney DJ, The potential of 12/15-lipoxygenase inhibitors in stroke therapy, Future medicinal chemistry 6(17) (2014) 1853–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H, Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis, Nat Chem Biol 13(1) (2017) 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL, A default mode of brain function, Proceedings of the National Academy of Sciences 98(2) (2001) 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch U-K, Philips M-A, Rossner MJ, Mann M, Simons M, Cell type-and brain region-resolved mouse brain proteome, Nat Neurosci 18(12) (2015) 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Ivanisevic J, Epstein Adrian A., Kurczy Michael E., Benton Paul H., Uritboonthai W, Fox Howard S., Boska Michael D., Gendelman Howard E., Siuzdak G, Brain Region Mapping Using Global Metabolomics, Chemistry & Biology 21(11) 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].McAllister TW, Neurobiological consequences of traumatic brain injury, Dialogues in clinical neuroscience 13(3) (2011) 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Anderson KJ, Miller KM, Fugaccia I, Scheff SW, Regional distribution of fluoro-jade B staining in the hippocampus following traumatic brain injury, Experimental neurology 193(1) (2005) 125–130. [DOI] [PubMed] [Google Scholar]
- [145].Pulsinelli WA, Brierley JB, Plum F, Temporal profile of neuronal damage in a model of transient forebrain ischemia, Annals of neurology 11(5) (1982) 491–498. [DOI] [PubMed] [Google Scholar]
- [146].Sparvero LJ, Amoscato AA, Fink AB, Anthonymuthu T, New LA, Kochanek PM, Watkins S, Kagan VE, Bayır H, Imaging mass spectrometry reveals loss of polyunsaturated cardiolipins in the cortical contusion, hippocampus, and thalamus after traumatic brain injury, J Neurochem 139(4) (2016) 659–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Tian H, Sparvero LJ, Amoscato AA, Bloom A, Bayır H.l., Kagan VE, Winograd N, Gas Cluster Ion Beam Time-of-Flight Secondary Ion Mass Spectrometry High-Resolution Imaging of Cardiolipin Speciation in the Brain: Identification of Molecular Losses after Traumatic Injury, Analytical chemistry 89(8) (2017) 4611–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Zhang J, Yu W, Ryu SW, Lin J, Buentello G, Tibshirani R, Suliburk J, Eberlin LS, Cardiolipins Are Biomarkers of Mitochondria-Rich Thyroid Oncocytic Tumors, Cancer Research 76(22) (2016) 6588–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Hatch GM, Cardiolipin: biosynthesis, remodeling and trafficking in the heart and mammalian cells, International journal of molecular medicine 1(1) (1998) 33–74. [DOI] [PubMed] [Google Scholar]
- [150].Vance JE, Steenbergen R, Metabolism and functions of phosphatidylserine, Progress in lipid research 44(4) (2005) 207–234. [DOI] [PubMed] [Google Scholar]
- [151].Andreyev AY, Fahy E, Guan Z, Kelly S, Li X, McDonald JG, Milne S, Myers D, Park H, Ryan A, Subcellular organelle lipidomics in TLR-4-activated macrophages, Journal of lipid research 51(9) (2010) 2785–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Aviram R, Manella G, Kopelman N, Neufeld-Cohen A, Zwighaft Z, Elimelech M, Adamovich Y, Golik M, Wang C, Han X, Asher G, Lipidomics Analyses Reveal Temporal and Spatial Lipid Organization and Uncover Daily Oscillations in Intracellular Organelles, Molecular Cell 62(4) 636–648. [DOI] [PubMed] [Google Scholar]