

Abstract
A systematic study of the addition of C‐based nucleophiles to fluorinated lactones based on 2‐deoxy‐2‐fluoro‐d‐pyranoses is disclosed. This high yielding, α‐selective process was found to be independent on the nature or configuration [(R)‐C(sp3)–F, (S)‐C(sp3)–F] of the substituent at C2. Representative, fluorinated analogues of Trehalose, Carminic acid, and the spirocyclic cores of Tofogliflozin and Papulacandin D are also reported. These glycomimics constitute a valuable series of 19F NMR active probes for application in structural biology.
Keywords: Fluorinated compounds, Carbohydrates, Fluorine, Glycosides, Bioisostere, Natural products
Introduction
The diversity of mammalian and bacterial “glycospace” provides a rich platform from which to design tailored glycomimetics for applications in medicine1 Through relatively modest structural modifications, the natural role of complex carbohydrates in molecular recognition events can be harnessed for therapeutic purposes.2 Subtle, often single site, alterations are sufficient to markedly enhance a biological response3 or even to engage pathways that elude the native sugars.4 Of the plethora of structural modifications employed in glycomimic design, formation of C‐glycosides is arguably one of the most common (Figure 1). These scaffolds mitigate the intrinsic risk of enzymatic degradation by replacing the C(sp3)–O bond of the glycosidic link with a more robust C(sp3)‐C(sp3/sp2/sp) bond. A number of successful pharmaceuticals contain this structural feature, including a series of SGLT‐2 inhibitors such as Dapagliflozin (1).5 It is also important to note that the aryl C‐glycoside motif is found in numerous natural products,6 including Carminic acid (2) and Papulacandin D (3). Whilst this approach is logical, the intrinsic challenges associated with the generation of crowded, often quaternary,7 stereocentres must be considered.
Figure 1.
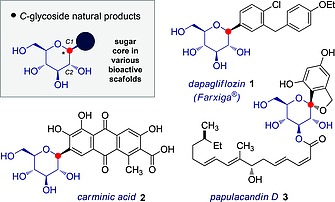
Selected examples of C‐glycosides.
A second strategy in devising metabolically robust glycomimics is the strategic OH → F substitution at C2 of pyranoside glycosyl donors.8 This seemingly subtle modification introduces several notable features that include enhanced hydrolytic stability,9 retention of the electronegativity intrinsic to the native structure, and the ability to direct glycosylation selectivity.3, 4, 8, 10 A classic bioisostere of the hydroxyl group,11 fluorine also functions as a valuable 19F NMR probe12 (Figure 2, upper) and allows various physicochemical parameters to be modulated such as lipophilicity, metabolic stability, and the pK a values of neighbouring groups.13 Despite the rich body of literature describing fluoro‐C‐glycosides and fluoro‐carbasugars,14 strategies that simultaneously exploit C‐glycoside formation and fluorine installation at C2 appear conspicuously absent from the glycosylation repertoire. To address this, a systematic study of the addition of various C‐based nucleophiles to fluorinated lactones based on 2‐deoxy‐2‐fluoro‐d‐pyranoses is disclosed.
Figure 2.
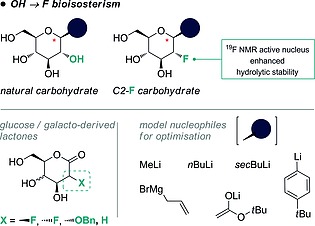
An overview of the scope of this study.
Results and Discussion
Specifically, benzylated lactones based on d‐glucose and d‐galactose were investigated in which the substituent at C2 was varied (X = F, OBn and H). In the case of the fluorinated electrophiles, both the gluco‐ and manno‐configured systems were studied to assess any possible influence of the configuration on reaction efficiency. The nucleophile series was composed of simple organometallic species that were either commercially available or could be easily prepared by simple halogen–metal exchange (Figure 2, lower).
Preliminary studies to assess the comparative efficiency and selectivity of additions were performed with lactones L1 15 and L2 (X = F and OBn, respectively. Table 1). For this purpose, commercially available organo‐lithium reagents were employed (Nu‐1 = sBuLi, Nu‐2 = nBuLi and Nu‐3 = MeLi).
Table 1.
Exploring the effect of C2‐substituent and nucleophile on C‐glycosylationa
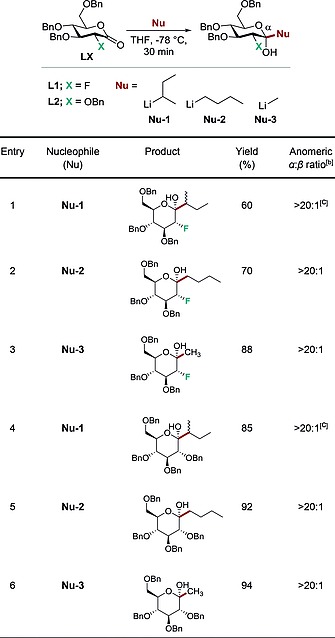
Reactions were performed with the specified lactone (LX) (0.1 mmol, 1.0 equiv.) and the specified nucleophile (0.11 mmol, 1.1 equiv.) in 0.58 mL of THF.
Selectivity determined by 19F NMR analysis of the crude reaction mixture.
1:1 mixture of diastereomers with respect to the sec‐butyl moiety.
Reactions were performed in THF at –78 °C for 0.5 h prior to being quenched, and the α/β ratio of the product was determined by 19F NMR spectroscopy and NOE experiments of the crude mixture4 Importantly, care must be exercised during the work‐up to prevent epimerisation at C2. This was observed when utilising MeOH and furnished an inseparable mixture of gluco‐ and manno‐configured analogues. This issue could be easily circumvented by performing an aqueous work up (full details in the Supporting Information)
As is immediately evident from Table 1, the addition of nucleophiles Nu‐1, Nu‐2 and Nu‐3 to the fluorinated lactone L1 proceeded smoothly to deliver the corresponding products exclusively as the α‐anomer (> 20:1, up to 88 % yield, entries 1, 2 and 3) irrespective of the substituent at C2. This clear manifestation of the anomeric effect16 would prove useful in subsequent transformations (vide infra). Comparable behaviour was also observed with the 2‐OBn derivative L2 (entries 4, 5 and 6) where the α‐anomer predominated and synthetically useful yields were observed (up to 94 %).
Encouraged by these preliminary results, the nucleophile scope was extended to include aryllithium (Nu‐4), lithium enolate (Nu‐5) and allylmagnesium species (Nu‐6) (Table 2). Moreover, since L1 and L2 displayed closely similar behaviour in the initial investigation, 2‐deoxy lactones (L3) were evaluated. Since hydrogen is often substituted by fluorine in molecular editing processes, this transformation may provide facile access to another class of bioisosteres.11 For completeness, the manno‐configured lactone L4 was included in this comparison to assess the effect of the configuration at C2 on the reaction outcome. Initially, the independent additions of Nu‐4, Nu‐5 and Nu‐6 to the 2‐deoxy lactone L3 were performed (entries 1, 2 and 3).
Table 2.
Exploring the effect of the lactone on C‐glycosylationa
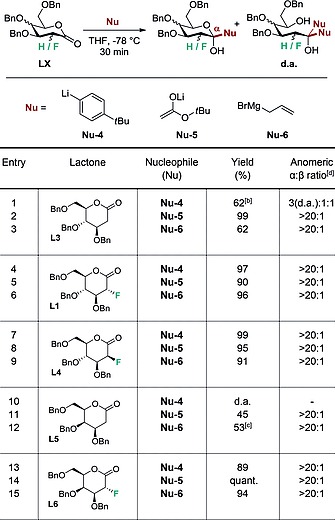
Reactions were performed with the specified lactone (LX) (0.1 mmol, 1.0 equiv.) and the specified nucleophile (0.11 mmol, 1.1 equiv.) in toluene/THF (2:1) or THF.
Mixture of product and double addition (d.a.).
Mixture of product and opened form of the product.
Selectivity determined by 19F NMR analysis of the crude reaction mixture.
Whilst employing Nu‐5 and Nu‐6 delivered the expected products (α/β > 20:1, entries 2 and 3, respectively), double addition was observed with the aryllithium Nu‐4 (entry 1). In contrast, the gluco‐configured lactone L‐1 was smoothly converted into the expected α‐product, irrespective of the nucleophile employed (entries 4, 5 and 6; 97, 96 and 90 % yield, respectively). Inverting the configuration of the C2 fluorine had no effect on selectivity, and again furnished the product glycosides in good yields (99, 95, 91 % yield; entries 7, 8 and 9, respectively). The 2‐deoxy‐galactose derived lactone L‐5 proved troublesome under the general condition of the study. Double addition was observed with Nu‐4 (entry 10), where with Nu‐5 and Nu‐6, a significant reduction in yield was noted (45 and 53 %, entries 11 and 12, respectively). Reintroducing the fluorine substituent at C2 was found to significantly enhanced the reaction efficiency (89 %, quant., 94 % yield, entries 13, 14 and 15, respectively.)
Having validated this class of fluorinated lactones as competent electrophiles en route to C‐glycosides, a series of natural and non‐natural products were prepared employing L1 as a common foundation (Scheme 1). In the preparation of a fluorinated analogue of trehalose (6), L1 was smoothly converted to 4 by the addition of MeLi at –78 °C in THF. The expected, α‐configured product was isolated in quantitative yield, before being processed to the disaccharide core. This was achieved by standard glycosylation conditions employing the TCA donor 5 (TMSOTf, CH2Cl2) to furnish the fluorinated glycomimetic 6 (30 %) as a mixture of diastereoisomers (α/β = 1.8:1). Next, a masked analogue of Carminic acid (9) was prepared by exposure to the appropriate aryllithium reagent with concomitant reduction with Et3SiH and BF3 ·Et2O. This first fluorinated analogue of Carminic acid 9 was isolated in 80 % yield, again with exclusive β selectivity. By extension, an addition/deprotection/cyclisation sequence provided an efficient and α‐selective route to compound 11, which is a structural analogue of the cores of Tofogliflozin (12) and Papulacandin D (3). Finally, the addition of allylmagnesium bromide, followed by hydroboration/oxidation generated the alcohol 14. When exposed to CSA, this material readily cyclised to furnish the fluorinated spiro‐systems 15 further underscoring the versatility of L1 as a starting material.
Scheme 1.
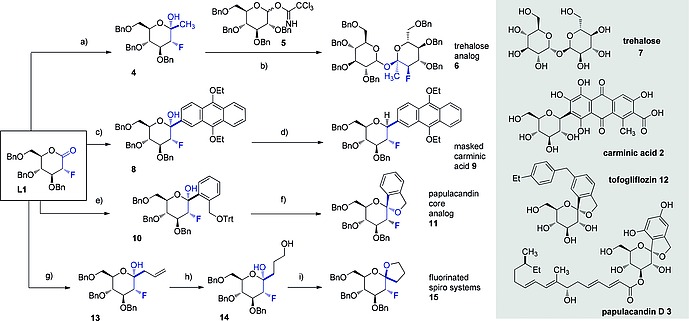
a) MeLi, THF, –78 °C, 30 min. quant. α/β = 1:0; b) 4, 5, TMSOTf, CH2Cl2, –78 °C to r.t. overnight, 30 %, αα/αβ = 1.8:1; c) nBuLi, ArBr (see SI), THF, –78 °C, 1 h, 73 %, α/β = 1:0; d) Et3SiH, BF3 ·Et2O, CH2Cl2/MeCN, –10 °C to r.t., 3 h, 80 %, α/β = 0:1; e) nBuLi, ArBr (see SI), THF/toluene (1:2), –78 °C, 1 h, 73 %, α/β = 1:0; f) Et3SiH, BF3 ·Et2O, MeCN, –40 °C to 0 °C, 2 h, 69 %, α/β = 1:0; g) Allylmagnesium bromide, THF, –78 °C, 30 min., 96 %, α/β = 1:0; h) 9‐BBN, H2O2 (30 % in H2O), NaOH (3 m aq.), overnight, r.t., 50 %; i) CSA, dioxane, 80 °C, 8 h, 53 %, α/β = 1:1.
Conclusion
Motivated by rapid advances in glycobiology, a series of glycomimetics have been prepared that exploit the stability advantages of C‐glycosides and 2‐fluorosugars over native systems. This study demonstrates that whilst the OBn → F substitution at C2 is extremely well tolerated, the fluorinated lactones are more robust towards the addition of organometallic reagents than the corresponding 2‐deoxy lactones. The α‐selectivity observed for the 2‐OBn and 2‐F sugars can be exploited in subsequent transformations. In this study, a series of natural and non‐natural product analogues have been prepared in a stereoselective manner. Exploiting these systems in the context of chemical biology will be the focus of future efforts from this laboratory.
Experimental Section
Supporting Information (see footnote on the first page of this article): Full experimental details are provided in the supporting information.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the WWU Münster, the Deutsche Forschungsgemeinschaft (DFG): “Cells in Motion, Cluster of Excellence”; and the European Research Council (ERC‐2013‐StG Starter Grant to RG ‐ Project number 336376‐ChMiFluorS); Initial Training Network, FLUOR21 (AS), funded by the FP7 Marie Curie Actions of the European Commission (FP7‐PEOPLE‐2013‐ITN‐607787).
References
- 1.a) Werz D. B., Ranzinger R., Herget S., Adibekian A., Lieth C.‐W. and Seeberger P. H., ACS Chem. Biol., 2007, 2, 685–691; [DOI] [PubMed] [Google Scholar]; b) Bertozzi C. R. and Kiessling L. L., Science, 2001, 291, 2357–2364; [DOI] [PubMed] [Google Scholar]; c) Dube D. H. and Bertozzi C. R., Nat. Rev., 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- 2. Ernst B. and Magnani J. L., Nat. Rev. Drug Discovery, 2009, 8, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kieser T. J., Santschi N., Nowack L., Kehr G., Kuhlmann T., Albrecht S. and Gilmour R., ACS Chemical Neurosci., 2018, 9, 1159–1165. [DOI] [PubMed] [Google Scholar]
- 4. Sadurni A., Kehr G., Ahlqvist M., Peilot Sjögren H., Kankkonen C., Knerr L. and Gilmour R., Chem. Eur. J., 2018, 24, 2832–2836. [DOI] [PubMed] [Google Scholar]
- 5. Aguillón A. R., Mascarello A., Segretti N. D., de Azevedo H. F. Z., Guimaraes C. R. W., Miranda L. S. M. and de Souza R. O. M. A., Org. Process Res. Dev., 2018, 22, 467–488. [Google Scholar]
- 6. Kitamura K., Ando Y., Matsumoto T. and Suzuki K., Chem. Rev., 2018, 118, 1495–1598. [DOI] [PubMed] [Google Scholar]
- 7. Bera S., Chatterjee B. and Mondal D., RSC Adv., 2016, 6, 77212–77242. [Google Scholar]
- 8. Bucher C. and Gilmour R., Angew. Chem. Int. Ed., 2010, 49, 8724–8728; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2010, 122, 8906. [Google Scholar]
- 9. Street I. P., Kempton J. B. and Withers S. G., Biochemistry, 1992, 31, 9970–9978. [DOI] [PubMed] [Google Scholar]
- 10.a) Bucher C. and Gilmour R., Synlett, 2011, 1043–1046; [Google Scholar]; b) Durantie E., Bucher C. and Gilmour R., Chem. Eur. J., 2012, 18, 8208–8215; [DOI] [PubMed] [Google Scholar]; c) Santschi N. and Gilmour R., Eur. J. Org. Chem., 2015, 6983–6987; [Google Scholar]; d) Aiguabella N., Holland M. C. and Gilmour R., Org. Biomol. Chem., 2016, 14, 5534–5538. [DOI] [PubMed] [Google Scholar]
- 11.a) O'Hagan D., J. Fluorine Chem., 2010, 131, 1071–1081; [Google Scholar]; b) Ojima I., J. Org. Chem., 2013, 78, 6358–6383; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J. and Meanwell N. A., J. Med. Chem., 2015, 58, 8315–8359; [DOI] [PubMed] [Google Scholar]; d) Meanwell N. A., J. Med. Chem. 2018, https://doi.org/:10.1021/acs.jmedchem.7b01788 [DOI] [PubMed] [Google Scholar]
- 12. Chenm H., Viel S., Ziarelli F. and Peng L., Chem. Soc. Rev., 2013, 42, 7971–7982. [DOI] [PubMed] [Google Scholar]
- 13.a) Müller K., Faeh C. and Diederich F., Science, 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]; b) Purser S., Moore P. R., Swallow S. and Gouverneur V., Chem. Soc. Rev., 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- 14. Leclerc E., Pannecoucke X., Ethève‐Quelquejeu M. and Sollogoub M., Chem. Soc. Rev., 2013, 42, 4270–4283. [DOI] [PubMed] [Google Scholar]
- 15.The fluorinated lactone was prepared according to Waschke D., Leshch Y., Thimm J., Himmelreich U. and Thiem J., Eur. J. Org. Chem., 2012, 948–959. [Google Scholar]
- 16.a) Kirby A. J., in: The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, Springer Verlag, New York, 1982; [Google Scholar]; b) Thiehoff C., Rey Y. P. and Gilmour R., Isr. J. Chem., 2017, 57, 92–100. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information