

Abstract
In connection with our research program on the development of new isatin-based anticancer candidates, herein we report the synthesis of two novel series of thiazolidinone-isatin conjugates (4a–n) and thiazolo[3,2-a]benzimidazolone-isatin conjugates (7a–d), and in vitro evaluation of their antiproliferative activity towards two breast cancer cell lines; triple negative MDA-MB-231, and MCF-7. Compounds 4m and 7b emerged as the most active congeners against MDA-MB-231 cells (IC50 = 7.6 ± 0.5 and 13.2 ± 1.1 µM, respectively). Compounds 4m and 7b were able to provoke apoptosis in MDA-MB-231 cells, evidenced by the up-regulation of Bax and down-regulation of Bcl-2, besides boosting caspase-3 levels. Hybrid 4m induced a fourfold increase in the percentage of cells at Sub-G1, with concurrent arrest in G2-M phase by 2.5-folds. Furthermore, hybrid 4m resulted in a sixfold increase in the percentage of annexin V-FITC positive apoptotic MDA-MB-231 cells as compared with the control. Moreover, the cytotoxic activities of the active conjugates were assessed towards two nontumorigenic cell lines (breast MCF-10A and lung WI-38) where both conjugates 4m and 7b displayed mean tumor selectivity index: 9.6 and 13.9, respectively. Finally, several ADME descriptors were predicted for the active conjugates via a theoretical kinetic study.
Keywords: triple-negative breast cancer, isatin-thiazolidinone hybrids, anticancer, apoptosis, QSAR
1. Introduction
Breast cancer has become the most frequently diagnosed malignancy among women, and the second leading cause of cancer-related deaths in women [1,2,3]. The deficiency in the maintenance of genomic integrity, excessive exposure to estrogens and advanced age are considered as the leading reasons for a high breast cancer risk [4,5]. Breast cancer is considered as a diverse group of diseases with several intrinsic tumor subtypes that have diverse treatment modalities and long-term survival probabilities. The immunohistochemical expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER-2) forms the platform of characterization of clinically defined breast cancer subtypes [4]. Particularly, triple-negative breast cancers (TNBCs) lacking the expression of the (ER), (PR) and (HER-2) represent the major cause of breast cancer mortality due to their metastatic potential, invasiveness and the lack of suitable molecular treatment targets [6,7,8].
Despite an early diagnosis and the diverse therapeutic regimens available for breast cancer treatment, the development of chemo-resistance, the disease relapse, and the mortality rate are still constantly on the rise [4]. This finding highlights the need for new drug leads as more effective and affordable approaches for breast cancer therapies.
Pertaining to its wide presence endogenously in human and other mammalian tissues, isatin 1, Figure 1, has emerged as a promising privileged scaffold that is endowed with diverse biological activity [9,10,11,12,13], primarily anticancer activity [14]. During the last two decades, there is a growing interest regarding development of several isatin derivatives as promising drug candidates for the treatment of different human malignancies. These research efforts have led to FDA approval of two drugs and the discovery of many isatin derivatives with diverse cellular and enzymatic targets.
Figure 1.

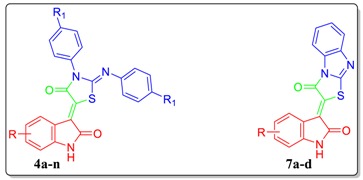
Structures of isatin-based approved anticancer drugs (II and III), some reported isatin-based hybrids with potent antiproliferative activity against breast cancer (VII–IX), and the target conjugates (4a–n and 7a–d).
Sunitinib (Sutent®) II (Figure 1), an isatin-based multitarget tyrosine kinase inhibitor, was granted its FDA approval in 2006 for the treatment of imatinib-resistant gastrointestinal stromal tumors (GIST) and advanced metastatic renal cell carcinoma (RCC) [15]. By 2014, Nintedanib (III, Ofev®, Figure 1), an orally available isatin-based triple-angiokinase inhibitor, was approved by the FDA for the management of idiopathic pulmonary fibrosis [16]. One year later, the European Medicines Agency approved Nintedanib, under a trade name Vargatef®, as a second-line treatment in combination with docetaxel for patients with advanced non-small cell lung cancer of adenocarcinoma who have progressed to first-line chemotherapy [17].
Over the last few years, isatin-based hybrids have attracted considerable attention as promising anticancer agents [18,19,20,21,22,23,24]. In this context, numerous research groups adopted a hybridization approach for the development of diverse isatin-thiazolidine/thiazolidinone [25,26,27,28,29] (such as compound IV, Figure 1) and isatin-benzimidazole (such as compound V, Figure 1) [30] hybrids as potent antiproliferative agents towards different breast cancer cell lines. Recently, our research group has focused on the design and synthesis of novel and potent isatin-based hybrids as promising anticancer agents [31,32,33,34,35,36,37,38,39,40]. In 2017, we developed two novel series of thiazolidinone-isatin hybrids as effective anticancer agents targeting tumor-associated carbonic anhydrase isoform IX. Among these hybrids, compound VI (Figure 1) displayed potent activity against MCF-7 breast cancer cell line (IC50 = 3.96 ± 0.21 μM) with induction of the intrinsic apoptotic mitochondrial pathway in MCF-7 cells [41].
Taking the above into account, herein we adopted the hybrid pharmacophore approach to design and synthesize two novel series of thiazolidinone-isatin (4a–n) and thiazolo[3,2-a]benzimidazolone-isatin conjugates (7a–d) (Figure 1), with the goal of developing potent antiproliferative agents toward TNBC MDA-MB-231, and MCF-7 breast cancer cell lines. Furthermore, the most potent antiproliferative congeners were evaluated for their apoptosis induction potential in MDA-MB-231 cells, to gain insight into the mechanism of the anticancer activity for the prepared hybrids. Finally, the cytotoxic activity of the active conjugates was tested towards nontumorigenic human normal lung fibroblast cell line (WI-38) to investigate the safety of the newly prepared conjugates.
2. Results
2.1. Chemistry
The target conjugates 4a–n and 7a–d were synthesized adopting the chemical pathways outlined in Scheme 1 and Scheme 2. In Scheme 1, preparation of conjugates 4a–n was accomplished via two synthetic routes. The first route comprised a one-pot three-component synthesis, involving a Knoevenagel condensation, followed by cyclization among isatins 1a–g, utilising an equimolar amount of N,N′-diphenyl/p-tolyl thiourea 2a,b and bromoacetic acid 3 in refluxing glacial acetic acid in the presence of sodium acetate to furnish conjugates 4a–n. This route proved successful to prepare conjugates 4a–n in high yields; 70–86% (Scheme 1). Alternatively, N,N′-diphenyl/p-tolyl thiourea 2a,b reacted with bromoacetic acid 3 to furnish compounds 5a,b, which subsequently condensed with isatins 1a–g in acetic acid in the presence of sodium acetate to afford the target conjugates 4a–n, with overall 55–68% yield over the two steps (Scheme 1).
Scheme 1.

Synthesis of target hybrids 4a–n; reagents and conditions: (i) glacial acetic acid/sodium acetate/reflux 3–4 h.
Scheme 2.

Synthesis of target hybrids 7a–d; Reagents and conditions: (i) glacial acetic acid/sodium acetate/reflux 2 h; (ii) ethanol/ KOH/reflux 1 h; (iii) acetic anhydride/pyridine 100 °C 1 h.
Similar to Scheme 1, target conjugates 7a–d were prepared in a one-pot three-component reaction of isatins 1c, e–g with equimolar amounts of 2-mercaptobenzimidazole 6 and bromoacetic acid 3 in refluxing glacial acetic acid in the presence of sodium acetate to produce conjugates 7a–d, in good yields; 80–88% (Scheme 2). The latter conjugates prepared through cyclization of 2-((benzimidazol-2-yl)thio)acetic acid 8 into compound 9, with subsequent condensation with isatins 1c, e–g, with low overall yield; 45–61%.
Postulated structures of the newly synthesized conjugates 4a–n and 7a–d were in full agreement with their spectral and elemental analyses data.
2.2. Biological Evaluation
2.2.1. In Vitro Antiproliferative Activity
The in vitro antiproliferative activity of the newly synthesized conjugates 4a–n and 7a–d was evaluated against breast cancer MDA-MB-231 and MCF-7 cell lines, following the Crystal Violet (CV) cell cytotoxicity assay [41]. Doxorubicin was included in this assay as a positive control, and Sunitinib was used as a reference drug. The results were expressed as median growth inhibitory concentration (IC50) values that represent the compounds concentrations required to afford a 50% inhibition of cell growth after 48 h of incubation, compared to untreated controls (Table 1).
Table 1.
In vitro antiproliferative activity of hybrids 4a–n and 7a–d against MDA-MB-231 and MCF-7 breast cancer cell lines.
| Compound | R | R1 | IC50 (µM) a | |
|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | |||
| 4a | H | H | NA b | NA b |
| 4b | 5-F | H | 108.3 ± 8.2 | 58.5 ± 3.2 |
| 4c | 5-Cl | H | 24.1 ± 2.1 | 29.2 ± 2.5 |
| 4d | 5-Br | H | 15.7 ± 0.9 | 17.1 ± 1.1 |
| 4e | 5-OCH3 | H | 42.6 ± 3.4 | 52.9 ± 5.0 |
| 4f | 5-CH3 | H | 60.9 ± 4.2 | 22.8 ± 2.0 |
| 4g | 5,7-(CH3)2 | H | 43.6 ± 3.1 | 47.4 ± 3.9 |
| 4h | H | CH3 | 51.7 ± 2.1 | 56.0 ± 3.7 |
| 4i | 5-F | CH3 | 152.1 ± 11.5 | 104.9 ± 7.5 |
| 4j | 5-Cl | CH3 | 50.8 ± 3.5 | NA b |
| 4k | 5-Br | CH3 | NA b | NA b |
| 4l | 5-OCH3 | CH3 | 47.1 ± 4.1 | 53.5 ± 3.6 |
| 4m | 5-CH3 | CH3 | 7.6 ± 0.5 | 8.4 ± 0.5 |
| 4n | 5,7-(CH3)2 | CH3 | 36.3 ± 2.9 | 23.7 ± 1.9 |
| 7a | 5-Cl | - | 47.2 ± 3.8 | 127.3 ± 9.9 |
| 7b | 5-OCH3 | - | 13.2 ± 1.7 | 21.7 ± 1.5 |
| 7c | 5-CH3 | - | 40.3 ± 2.5 | 48.2 ± 2.7 |
| 7d | 5,7-(CH3)2 | - | 23.5 ± 1.0 | 27.6 ± 2.4 |
| Dox. | 4.7 ± 0.4 | 3.8 ± 0.4 | ||
| Sunitinib | 5.5 ± 0.5 | 3.4 ± 0.3 | ||
a IC50 values are the mean ± S.D. of three separate experiments; b NA: Compounds having IC50 value > 200 µM.
Breast cancer is composed of multiple subtypes with distinct morphologies and clinical implications. In this study, the sensitivity of two different breast cancer subtypes towards the target conjugates was tested to examine if the target compounds possess selective growth inhibitory activity toward certain subtype. MCF-7 is a Luminal A breast cancer cells which is hormone-receptor positive (estrogen-receptor and/or progesterone-receptor positive), HER2 negative, and has low levels of the protein Ki-67, which helps control how fast cancer cells grow. Luminal A cancers are low-grade, tend to grow slowly and have the best prognosis. Whereas, MDA-MB-231 is a triple-negative/basal-like breast cancer cell line, which is hormone-receptor negative (estrogen-receptor and progesterone-receptor negative) and HER2 negative. This type of cancer is more common in women with BRCA1 gene mutations.
From the obtained results, it was obvious that many of the target conjugates 4a–n and 7a–d have excellent to modest growth inhibitory activity against the tested breast cancer cell lines; MDA-MB-231 and MCF-7. Also, it was noted that MDA-MB-231 cells is more sensitive to the influence of the target conjugates than MCF-7 cells, especially for the thiazolobenzimidazole conjugates 7a–d.
Concerning activity against MDA-MB-231 cells, conjugate 4m was the most active one and showed potent antiproliferative activity with IC50 7.6 ± 0.5 μM, in comparison to the standard drug doxorubicin (IC50 = 4.7 ± 0.4 μM). Furthermore, compounds 4c, 4d, 7b and 7d displayed good activity against MDA-MB-231 cells with IC50 values of 24.1 ± 2.1, 15.7 ± 0.9, 13.2 ± 1.1 and 23.5 ± 1.0 μM, respectively. Moreover, conjugates 4e, 4g, 4l, 4n, 7a and 7c possessed moderate activity with an IC50 range 36.3 ± 2.9–47.1 ± 4.1 µM.
On the other hand, examination of the antiproliferative activity in MCF-7 cells elucidated that conjugate 4m had the best growth inhibitory activity (IC50 = 8.4 ± 0.5), with 2.2-fold decreased activity than doxorubicin (IC50 = 3.80 ± 0.4 μM). Furthermore, hybrid 4g showed good antiproliferative activity against MCF-7 cancer cell line (IC50 = 17.1 ± 1.1). Also, hybrids 4c, 4f, 4n, 7b and 7d were moderately active towards MCF-7 cells with IC50 values of 29.2 ± 2.5, 22.8 ± 2.0, 23.7 ± 1.9, 21.7 ± 1.5 and 27.6 ± 2.4, respectively.
It is worth highlighting that both target thiazolidinone-isatin conjugates (4a–n) and thiazolo[3,2-a]benzimidazolone-isatin conjugates (7a–d), didn’t display significant selectivity in their growth inhibitory activities towards the tested cell line; TNBC MDA-MB-231 or MCF-7.
2.2.2. In Vitro Cytotoxic Activity towards Nontumorigenic Human WI-38 and MCF-10A Cells
The cytotoxic activity of the active conjugates towards MDA-MB-231 cells (4c–e, 4g, 4l–n and 7a–d) was examined against nontumorigenic human lung fibroblast cell line (WI-38) and human breast epithelial cell line (MCF-10A) to investigate the potential safety of the newly prepared conjugates towards the normal cells. Cultures derived from human fibrocystic mammary tissue (MCF-10A) are nontumorigenic and possess the features of primary cultures of breast tissue including dome formation [42]. The results were expressed as IC50 values, and selectivity index was calculated (Table 2). Most of the tested hybrids (4c–e, 4g, 4l–n and 7a–d) exhibited non-significant cytotoxic impact towards WI-38 and MCF-10A cells with IC50 range 49.1 ± 1.9–353.0 ± 17.1 and 73.1 ± 2.7–250.3 ± 9.8 μM, respectively. Compounds 4m and 7a–d displayed good selectivity index range 6.0–13.9, thereby providing a good safety profile as anticancer agents.
Table 2.
In vitro cytotoxic activity against nontumorigenic cell lines (WI-38 and MCF-10A), and selectivity index (MCF-10A/ MDA-MB-231) for the active compounds.
| Compound | IC50 (µM) a | Selectivity Index | ||
|---|---|---|---|---|
| Lung WI-38 | Breast MCF-10A | MDA-MB-231 | ||
| 4c | 87.2 ± 5.2 | 112.6 ± 5.9 | 24.1 ± 2.1 | 4.7 |
| 4d | 65.5 ± 4.2 | 80.7 ± 3.4 | 15.7 ± 0.9 | 5.1 |
| 4e | 193.1 ± 10.6 | 89.6 ± 5.1 | 42.6 ± 3.4 | 2.1 |
| 4g | 120.0 ± 7.8 | 141.7 ± 7.9 | 43.6 ± 3.1 | 3.25 |
| 4l | 176.2 ± 11.3 | 157.4 ± 6.8 | 47.2 ± 4.1 | 3.3 |
| 4m | 49.1 ± 1.9 | 73.1 ± 2.7 | 7.6 ± 0.5 | 9.6 |
| 4n | 218.3 ± 13.5 | 154.0 ± 5.2 | 36.3 ± 2.9 | 4.2 |
| 7a | 353.0 ± 17.1 | 233.8 ± 9.6 | 47.2 ± 3.8 | 5.0 |
| 7b | 121.1 ± 6.9 | 183.9 ± 8.0 | 13.2 ± 1.1 | 13.9 |
| 7c | 192.0 ± 10.8 | 242.2 ± 11.5 | 40.3 ± 2.5 | 6.0 |
| 7d | 234.0 ± 15.4 | 250.3 ± 9.8 | 23.5 ± 1.00 | 10.7 |
a IC50 values are the mean ± S.D. of three separate experiments.
2.2.3. Apoptosis Induction in TNBC MDA-MB-231 Cells
In the current medical era, induction of apoptosis in cancer cells has emerged as one of the most successful strategies for the development of cancer therapies [43,44,45]. As mentioned before, 4m and 7b emerged as the most active hybrids towards MDA-MB-231 cells. Consequently, we examined the ability of conjugates 4m and 7b to provoke apoptosis in MDA-MB-231 cells to determine the principle mechanism for their antiproliferative activity.
Effects on Mitochondrial Apoptosis Pathway Proteins Bcl-2 and Bax
Several members of the human Bcl-2 family of apoptosis-regulating proteins have been discovered, including antiapoptotic proteins such as Bcl-2 protein, and structurally similar pro-apoptotic proteins such as Bax protein, the first anti-death gene identified [44,45,46]. Bcl-2-family proteins regulate all major types of cell death, including apoptosis, necrosis and autophagy, therefore operating as nodal points at the convergence of diverse pathways with broad relevance to oncology.
In our study, we investigated the effect of conjugates 4m and 7b towards the level of the antiapoptotic Bcl2 and the level of the pro-apoptotic Bax, (Table 3, Figure 2). As displayed in Table 2, hybrid 4m induced the protein expression of Bax with 11.1 folds of the control while 7.3 folds were evaluated for hybrid 7b. On the other hand, treatment of MDA-MB-231 cancer cells with conjugates 4m and 7b significantly downregulated the expression levels of the antiapoptotic protein Bcl-2 by 66.6 and 55.1%, respectively, compared to the control.
Table 3.
Impact of hybrids 4m and 7b on the expression levels of Bcl-2 and Bax and on the active caspase-3 level in MDA-MB-231 cancer cells treated with each hybrid at its IC50 concentration.
| Compound | Bax (pg/mg of Total Protein) | Bcl-2 (ng/mg of Total Protein) | Bax/Bcl-2 | Caspase-3 (ng/mg of Total Protein) |
|---|---|---|---|---|
| 4m | 274.8 ± 13.5 (11.1) * | 2.1 ± 0.1 (0.33) * | 130.5 | 0.19 ± 0.01 (6.4) * |
| 7b | 180.0 ± 7.3 (7.3) * | 2.8 ± 0.1 (0.45) * | 64.1 | 0.15 ± 0.01 (4.9) * |
| Control | 24.7 ± 1.7 | 6.3 ± 0.2 | 3.9 | 0.03 ± 0.01 |
* Numbers given between parentheses are the numbers of folds of control.
Figure 2.

Effect of compounds 4m and 7b on the protein levels of (A) Bax; (B) Bcl-2; (C) Bax/Bcl2 ratio in MDA-MB-231 cells treated with the compounds at their IC50 concentrations against control (1% DMSO). Data are mean ±S.D. (n = 3). The experiment was done in triplicates. * Significantly different from control at p < 0.05.
The ratio between Bax (apoptosis inducer) and Bcl-2 (apoptosis suppressor) is an important parameter and gave more profound insight into the apoptotic activity of the compounds as a determining factor for cell fate regulation, and a key indicator of therapeutic response to chemotherapy [47]. Calculating the values for Bax/Bcl2 ratio indicated that conjugates 4m and 7b boosted the Bax/Bcl-2 ratio by 33- and 16-folds, respectively, compared to the control. Conclusively, the ability of conjugates 4m and 7b to up-regulate Bax level, down-regulate Bcl2 level while significantly boosting the Bax/Bcl2 ratio proves their effectiveness as apoptosis inducers.
Effects on the Levels of Active Caspase-3 (Key Executor of Apoptosis)
The down-regulation of the antiapoptotic Bcl-2 leads to an increase in the levels of free pro-apoptotic Bax which in turn accumulates at the inner mitochondrial membrane forming channels, altering membrane permeability. Accordingly, the apoptotic factors can leak into the cytoplasm resulting in the caspases cascade activation. As a key executioner protease, caspase-3 is activated by upstream initiator caspases as caspase-9 [48]. Thence, the elevated Bax/Bcl-2 ratios obtained herein triggered the examination of the protein expression levels of active caspases-3. Treatment of TNBC MDA-MB-231 cells with conjugates 4m and 7b led to a significant increase in the active caspase-3 levels by 6.4 and 4.9 folds, respectively, compared to control (Table 3, Figure 3).
Figure 3.

Effect of compounds 4m and 7b on the protein levels of caspase-3 in MDA-MB-231 cells treated with the compounds at their IC50 concentrations against control (1% DMSO). Data are mean ±S.D. (n = 3). The experiments were done in triplicate. * Significantly different from control at p < 0.05.
Cell Cycle Analysis
The effect of hybrid 4m on cell cycle progression was evaluated in TNBC MDA-MB-231 cells after 24 h of treatment (Figure 4). Such impact was illustrated by DNA flow cytometric assay where MDA-MB-231 cells were treated with conjugate 4m at its IC50 concentration.
Figure 4.

Effect of compound 4m on the phases of cell cycle of MDA-MB-231 cells. * Significantly different from control at p < 0.05. (Two-way ANOVA test).
As displayed in Figure 4, exposure of MDA-MB-231 cells to hybrid 4m resulted in a significant rise in the percentage of cells at Sub-G1 by 4-folds, with concurrent significant arrest in the G2-M phase by 2.5-folds compared to control. Arrest of G2-M phase and alteration of the Sub-G1 phase were significant remarks for hybrid 4m to prompt apoptosis in TNBC MDA-MB-231 cells.
Annexin V-FITC Apoptosis Assay
Annexin V-based flow cytometry assay elucidates either that cell death is achieved via programmed apoptosis or nonspecific necrosis [49]. The apoptotic impact of conjugate 4m was further proved by Annexin V-FITC/PI (AV/PI) dual staining analysis to examine the occurrence of phosphatidylserine externalization (Figure 5). Flow cytometric analysis revealed that treatment of TNBC MDA-MB-231 cells with conjugate 4m resulted in a significant elevation in the percent of annexin V-FITC-positive apoptotic cells, including both the early and late apoptotic phases (UR + LR), from 1.92% to 11.43% which represents about 6-folds increase as compared with the control.
Figure 5.

Effect of compound 4m on the percentage of annexin V-FITC-positive staining in MDA-MB-231 cells. The experiments were done in triplicates. The four quadrants identified as: LL, viable; LR, early apoptotic; UR, late apoptotic; UL, necrotic. * Significantly different from control at p < 0.05. (One-way ANOVA test).
In conclusion, enhanced expression of the pro-apoptotic protein Bax in addition to the reduced expression of the antiapoptotic protein Bcl-2, as well as the up-regulated active caspase-3 levels together with a harmonized increase in the Bax/Bcl-2 ratio, suggests that the antiproliferative effect of the target conjugates might be attributed, at least in part, to the induction of the intrinsic apoptotic mitochondrial pathway.
Collectively, these data have highlighted conjugate 4m as an ideal lead compound for further optimization and development as an effective anti-breast cancer therapy.
2.3. In Silico ADME Profiling
In order to examine drug-like physicochemical and pharmacokinetics properties of the target conjugates, certain ADME descriptors for the prepared conjugates (4a–n and 7a–d) were assessed through a computer-aided theoretical kinetic study using Discovery Studio software (Accelrys, San Diego, CA, USA), [50,51] Table 4.
Table 4.
Computer-aided ADME study for the synthesized conjugates 4a–m and 7a–d.
| Compound | AlogP98 a | PSA_2D b | Solubility c | Solubility Level d | Absorption Level e | CYP2D6 f |
|---|---|---|---|---|---|---|
| 4a | 4.476 | 62.087 | −6.305 | 1 | 0 | 0 |
| 4b | 4.682 | 62.087 | −6.605 | 1 | 0 | 0 |
| 4c | 5.141 | 62.087 | −7.005 | 1 | 0 | 0 |
| 4d | 5.225 | 62.087 | −7.079 | 1 | 0 | 0 |
| 4e | 4.46 | 71.017 | −6.292 | 1 | 0 | 0 |
| 4f | 4.962 | 62.087 | −6.765 | 1 | 0 | 0 |
| 4g | 5.449 | 62.087 | −7.226 | 1 | 0 | 0 |
| 4h | 5.449 | 62.087 | −7.197 | 1 | 0 | 0 |
| 4i | 5.654 | 62.087 | −7.484 | 1 | 1 | 0 |
| 4j | 6.113 | 62.087 | −7.885 | 1 | 1 | 0 |
| 4k | 6.197 | 62.087 | −7.958 | 1 | 1 | 0 |
| 4l | 5.432 | 71.017 | −7.159 | 1 | 0 | 0 |
| 4m | 5.935 | 62.087 | −7.645 | 1 | 1 | 0 |
| 4n | 6.421 | 62.087 | −8.093 | 0 | 1 | 0 |
| 7a | 3.581 | 64.021 | −6.005 | 1 | 0 | 0 |
| 7b | 2.9 | 72.951 | −5.327 | 2 | 0 | 0 |
| 7c | 3.403 | 64.021 | −5.771 | 2 | 0 | 0 |
| 7d | 3.889 | 64.021 | −6.263 | 1 | 0 | 0 |
a Lipophilicity descriptor. b Polar surface area. c Solubility parameter.(0:−2 = optimal, −2:−4 = good, −4:−6 = low, −6:−8 = very low). d Solubility level. (0 = extremely low, 1 = very low but possible, 2 = low, 3 = good, 4 = optimal). e Absorption level. (0 = good, 1 = moderate, 2 = low, 3 = very low). f CYP2D6 inhibition. (0 = noninhibitor, 1 = inhibitor).
The tested conjugates exhibited low aqueous solubility levels, except compounds 7b and 7c, which showed better solubility levels. The tested conjugates displayed good levels of human intestinal absorption, except compounds 4i–k, 4m and 4n, which showed moderate intestinal absorption levels. On the other hand, the investigated conjugates were predicted to be CYP2D6 non-inhibitors, Table 4.
3. Conclusions
In summary, herein we report the synthesis of two novel series of thiazolidinone-isatin conjugates (4a–n) and thiazolo[3,2-a]benzimidazolone-isatin conjugates (7a–d). All the prepared conjugates were evaluated for their antiproliferative activity towards breast cancer cell lines MDA-MB-231, and MCF-7. Conjugates 4m and 7b were the most active members towards MDA-MB-231 (IC50 = 7.6 ± 0.51 and 13.2 ± 1.07 µM, respectively). Conjugates 4d and 4m emerged as the most active derivatives against MCF-7 cells with IC50 values of 17.1 ± 1.14 and 8.4 ± 0.47 μM, respectively. Conjugates 4m and 7b induced apoptosis in MDA-MB-231 cells, confirmed by the up-regulation of the Bax, downregulation of the Bcl-2 and boosting caspase-3 levels. Compound 4m arrested the G2-M phase by 2.5-folds with concurrent significant increase in the percentage of cells at Sub-G1 by 4-folds, compared to control. Also, 4m showed significant increase in the percentage of annexin V-FITC positive apoptotic cells from 1.92% to 11.43%, approximately a 6-folds increase as compared with the control. Finally, the cytotoxic activity of the active conjugates (4c–e, 4g, 4l–n and 7a–d) were tested towards nontumorigenic human lung fibroblast WI-38 cell line and breast MCF-10A cell line, and compounds 4m and 7a–d displayed good selectivity index range 6.0–13.9, thereby providing a good safety profile as anticancer agents.
4. Experimental
4.1. Chemistry
4.1.1. General
Using a Stuart melting point apparatus, melting points were measured and uncorrected. Using Schimadzu FT-IR 8400S spectrophotometer (Shimadzu, Kyoto, Japan), Infrared (IR) Spectra were recorded as KBr disks. NMR Spectra were recorded on a Bruker spectrophotometer (Bruker, Karlsruhe, Germany). 1H spectrum was run at 400 MHz and 13C spectrum was run at 100 MHz in deuterated dimethylsulfoxide (DMSO-d6). All coupling constant (J) values are given in hertz. The abbreviations used are as follows: s, singlet; d, doublet; m, multiplet. Elemental analyses were done at the Regional Center for Microbiology and Biotechnology, Al-Azhar University, Egypt. Mass spectral data were given by a GCMS-QP1000 EX spectrometer (Shimadzu, Kyoto, Japan) at 70 e.V. High-resolution mass spectra were obtained using a Bruker MicroTOF spectrometer (Bruker Daltonics, Bremen, Germany). Compounds 5a,b [52], 8 [53], and 9 [54] were previously prepared.
4.1.2. General Procedure for the Preparation of Target Compounds 4a–n
Route A:
A mixture of isatins 1a–g (1 mmoL) with an equimolar amount of N,N′-diphenyl/(p-tolyl)thiourea 2a,b (1 mmoL) and bromoacetic acid 3 (0.14 g, 1 mmoL) in glacial acetic acid (10 mL) in the presence of sodium acetate (0.16 gm, 2 mmoL), was heated under reflux for 3 h. The formed solid was filtered off while hot, washed with hot ethanol, dried and recrystallized from DMF to furnish the target hybrids 4a–n.
Route B:
To a hot solution of N,N′-diphenyl/(p-tolyl)thiourea 2a,b (1 mmoL) and sodium acetate (0.16 gm, 2 mmoL) in glacial acetic acid (10 mL), bromoacetic acid 3 (0.14 gm, 1 mmoL) was added. The reaction mixture was refluxed for 4 h then allowed to cool to room temperature. The obtained solid was filtered off, washed with water and dried to give intermediates 3a,b. The later intermediates were added, without further purification, to a stirred solution of the appropriate isatin derivative 1a–g (1 mmoL) and sodium acetate (0.16 gm, 2 mmoL) in glacial acetic acid (5 mL) then the reaction mixture was refluxed for 3 h. The formed solid was filtered off, washed with ethanol and recrystallized from DMF to give compounds 4a–n.
5-(2-Oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4a)
Red powder (yield 83%), m.p. > 300 °C; IR (KBr, ν cm−1): 3251 (NH) and 1687 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 6.95 (d, J = 7.7 Hz, 1H, Ar-H), 6.99 (d, J = 7.3 Hz, 2H, Ar-H), 7.04 (t, J = 7.3 Hz, 1H, Ar-H), 7.17 (t, J = 7.4 Hz, 1H, Ar-H), 7.35–7.43 (m, 3H, Ar-H), 7.49–7.54 (m, 1H, Ar-H), 7.58 (d, J = 7.7 Hz, 4H, Ar-H), 8.77 (d, J = 7.7 Hz, 1H, Ar-H), 11.18 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 397.0 [M+, 100]; Anal. Calcd. for C23H15N3O2S: C, 69.51; H, 3.80; N, 10.57; found C, 69.34; H, 3.91; N, 10.83; HRMS m/z 398.09583 M+ + 1, calcd for C23H15N3O2S: 398.09577.
5-(5-Fluoro-2-oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4b)
Orange powder (yield 75%), m.p. > 300 °C; IR (KBr, ν cm−1): 3343 (NH) and 1683 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 6.81–6.87 (m, 3H, Ar-H), 7.22 (d, J = 7.8 Hz, 2H, Ar-H), 7.42–7.60 (m, 7H, Ar-H), 8.57 (d, J = 7.2 Hz, 1H, Ar-H), 11.25 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C23H14FN3O2S: C, 66.50; H, 3.40; N, 10.11; found C, 66.78; H, 3.26; N, 10.40.
5-(5-Chloro-2-oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4c)
Light brown powder (yield 80%), m.p. > 300 °C; IR (KBr, ν cm−1): 3257 (NH) and 1687 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 6.89 (d, J = 8.4 Hz, 1H, Ar-H), 6.97 (d, J = 7.6 Hz, 2H, Ar-H), 7.24–7.33 (m, 3H, Ar-H), 7.51–7.63 (m, 6H, Ar-H), 8.71 (d, J = 2.0 Hz, 1H, Ar-H), 11.29 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C23H14ClN3O2S: C, 63.96; H, 3.27; N, 9.73; found C, 64.23; H, 3.41; N, 10.01.
5-(5-Bromo-2-oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4d)
Red powder (yield 85%), m.p. > 300 °C; IR (KBr, ν cm−1): 3308 (NH) and 1689 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 6.93 (d, J = 8.3 Hz, 1H, Ar-H), 6.99 (d, J = 7.3 Hz, 2H, Ar-H), 7.20 (t, J = 7.4 Hz, 1H, Ar-H), 7.42 (t, J = 7.8 Hz, 2H, Ar-H), 7.65–7.49 (m, 6H, Ar-H), 8.96 (d, J = 2.0 Hz, 1H, Ar-H), 11.37 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C23H14BrN3O2S: C, 57.99; H, 2.96; N, 8.82; found C, C, 58.17; H, 3.08; N, 9.04; HRMS m/z 476.00635 M+ + 1, calcd for C23H14BrN3O2S: 476.00629.
5-(5-Methoxy-2-oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4e)
Red powder (yield 70%), m.p. > 300 °C; IR (KBr, ν cm−1): 3340 (NH) and 1692 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 3.72 (s, 3H, -OCH3), 6.86 (d, J = 8.5 Hz, 1H, Ar-H), 7.05–6.94 (m, 3H, Ar-H), 7.19 (t, J = 7.4 Hz, 1H, Ar-H), 7.41 (t, J = 7.8 Hz, 2H, Ar-H), 7.57–7.46 (m, 1H, Ar-H), 7.59 (d, J = 4.3 Hz, 4H, Ar-H), 8.48 (d, J = 2.6 Hz, 1H, Ar-H), 11.02 (s, 1H, NH isatin, D2O exchangeable); 13C-NMR (DMSO-d6, 100 MHz) δ ppm: 56.10, 111.14, 113.90, 118.24, 121.09, 121.31 (2C), 125.03, 125.50, 129.18 (2C), 129.56 (2C), 129.85 (2C), 132.14, 135.05, 137.54, 137.57, 147.76, 153.77, 154.83, 166.10, 169.05; MS, m/z [%]: 427 [M+, 100]; Anal. Calcd. for C24H17N3O3S: C, 67.43; H, 4.01; N, 9.83; found C, 67.70; H, 4.19; N, 10.02; HRMS m/z 428.10632 M+ + 1, calcd for C24H17N3O3S: 428.10634.
5-(5-Methyl-2-oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4f)
Red powder (yield 75%), m.p. > 300 °C; IR (KBr, ν cm−1): 3285 (NH) and 1684 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.28 (s, 3H, -CH3), 6.83 (d, J = 8.0 Hz, 1H, Ar-H), 6.98 (d, J = 7.2 Hz, 2H, Ar-H), 7.18 (d, J = 7.2 Hz, 2H, Ar-H), 7.39 (t, J = 7.6 Hz, 2H, Ar-H), 7.52–7.59 (m, 5H, Ar-H), 8.64 (s, 1H, Ar-H), 11.10 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 411 [M+, 100]; Anal. Calcd. for C24H17N3O2S: C, 70.06; H, 4.16; N, 10.21; found C, 69.84; H, 4.38; N, 10.38.
5-(5,7-Dimethyl-2-oxoindolin-3-ylidene)-3-phenyl-2-(phenylimino)thiazolidin-4-one (4g)
Brown powder (yield 73%), m.p. > 300 °C; IR (KBr, ν cm−1): 3328 (NH) and 1682 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.17 (s, 3H, -CH3), 2.23 (s, 3H, -CH3), 6.84 (s, 1H, Ar-H), 6.97 (d, J = 7.6 Hz, 2H, Ar-H), 7.19 (d, J = 7.6 Hz, 2H, Ar-H), 7.41 (t, J = 7.6 Hz, 2H, Ar-H), 7.49–7.58 (m, 4H, Ar-H), 8.43 (s, 1H, Ar-H), 11.07 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 425 [M+, 100]; Anal. Calcd. for C25H19N3O2S: C, 70.57; H, 4.50; N, 9.88; found C, 70.79; H, 4.61; N, 10.15.
5-(2-Oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4h)
Red powder (yield 70%), m.p. > 300 °C; IR (KBr, ν cm−1): 3271 (NH) and 1687 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.33 (s, 3H, -CH3), 2.41 (s, 3H, -CH3), 6.93 (d, J = 7.6 Hz, 1H, Ar-H), 6.98 (d, J = 7.2 Hz, 2H, Ar-H), 7.06 (t, J = 7.3 Hz, 1H, Ar-H), 7.21 (d, J = 8.0 Hz, 2H, Ar-H), 7.32–76 (m, 3H, Ar-H), 7.42 (d, J = 8.0 Hz, 2H, Ar-H), 8.75 (d, J = 7.6 Hz, 1H, Ar-H), 11.27 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C25H19N3O2S: C, 70.57; H, 4.50; N, 9.88; found C, 70.64; H, 4.64; N, 10.06.
5-(5-Fluoro-2-oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4i)
Orange powder (yield 75%), m.p. > 300 °C; IR (KBr, ν cm−1): 3290 (NH) and 1686 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.32 (s, 3H, -CH3), 2.41 (s, 3H, -CH3), 6.87 (d, J = 8.2 Hz, 2H, Ar-H), 6.92–6.95 (m, 1H, Ar-H), 7.26–7.15 (m, 3H, Ar-H), 7.38 (d, J = 8.3 Hz, 2H, Ar-H), 7.44 (d, J = 8.3 Hz, 2H, Ar-H), 8.57 (dd, J = 10.4, 2.7 Hz, 1H, Ar-H), 11.23 (s, 1H, NH isatin, D2O exchangeable); 13C-NMR (DMSO-d6, 100 MHz) δ ppm: 31.16 (2C), 118.67, 121.16 (2C), 121.30, 124.30, 128.77 (2C), 130.00 (2C), 130.33 (2C), 131.84, 132.32, 134.11, 134.28, 138.76, 140.06, 145.54, 153.37, 155.40, 156.73, 165.92, 168.63; MS, m/z [%]: 443 [M+, 100]; Anal. Calcd. for C25H18FN3O2S: C, 67.71; H, 4.09; N, 9.47; found C, 68.03; H, 4.35; N, 9.29; HRMS m/z 444.11771 M+ + 1, calcd for C25H18FN3O2S: 444.11765.
5-(5-Chloro-2-oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4j)
Orange powder (yield 73%), m.p. > 300 °C; IR (KBr, ν cm−1): 3223 (NH) and 1691 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.32 (s, 3H, -CH3), 2.41 (s, 3H, -CH3), 6.87 (d, J = 8.2 Hz, 2H, Ar-H), 6.96 (d, J = 8.4 Hz, 1H, Ar-H), 7.21 (d, J = 8.1 Hz, 2H, Ar-H), 7.50–7.35 (m, 5H, Ar-H), 8.82 (d, J = 2.2 Hz, 1H, Ar-H), 11.34 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C25H18ClN3O2S: C, 65.28; H, 3.94; N, 9.14; found C, 65.41; H, 4.12; N, 9.32; HRMS m/z 460.08807 M+ + 1, Calcd. for C25H18ClN3O2S: 460.08810.
5-(5-Bromo-2-oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4k)
Red powder (yield 86%), m.p. > 300 °C; IR (KBr, ν cm−1): 3296 (NH) and 1690 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.33 (s, 3H, -CH3), 2.43 (s, 3H, -CH3), 6.91 (d, J = 8.0 Hz, 2H, Ar-H), 7.01 (d, J = 8.0 Hz, 1H, Ar-H), 7.25 (d, J = 7.6 Hz, 2H, Ar-H), 7.39–7.51 (m, 5H, Ar-H), 9.03 (s, 1H, Ar-H), 11.41 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C25H18BrN3O2S: C, 59.53; H, 3.60; N, 8.33; found C, 59.80; H, 3.78; N, 8.59.
5-(5-Methoxy-2-oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4l)
Red powder (yield 75%), m.p. > 300 °C; IR (KBr, ν cm−1): 3256 (NH) and 1689 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.32 (s, 3H, -CH3), 2.40 (s, 3H, -CH3), 3.71 (s, 3H, -OCH3), 6.83–6.88 (m, 3H, Ar-H), 6.95 (d, J = 7.8 Hz, 1H, Ar-H), 7.19 (d, J = 8.0 Hz, 2H, Ar-H), 7.37 (d, J = 8.0 Hz, 2H, Ar-H), 7.42 (d, J = 8.0 Hz, 2H, Ar-H), 8.47 (s, 1H, Ar-H), 11.01 (s, 1H, NH isatin, D2O exchangeable); 13C-NMR (DMSO-d6, 100 MHz) δ ppm: 20.97, 21.27, 55.99, 111.14, 113.89, 118.27, 121.18, 121.22, 125.59, 128.62, 128.86, 129.00, 130.02 (2C), 130.30 (2C), 132.35, 132.56, 134.29, 137.53, 138.92, 145.49, 153.83, 155.02, 166.06, 169.03; MS, m/z [%]: 455 [M+, 100]; Anal. Calcd. for C26H21N3O3S: C, 68.55; H, 4.65; N, 9.22; found C, 68.47; H, 4.60; N, 9.53; HRMS m/z 456.13754 M+ + 1, calcd for C26H21N3O3S: 456.13764.
5-(5-Methyl-2-oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4m)
Red powder (yield 80%), m.p. > 300 °C; IR (KBr, ν cm−1): 3358 (NH) and 1693 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.26 (s, 3H, -CH3), 2.32 (s, 3H, -CH3), 2.41 (s, 3H, -CH3), 6.82 (d, J = 8.0 Hz, 1H, Ar-H), 6.91 (d, J = 7.2 Hz, 2H, Ar-H), 7.19–7.23 (m, 3H, Ar-H), 7.35 (d, J = 7.6 Hz, 2H, Ar-H), 7.43 (d, J = 7.6 Hz, 2H, Ar-H), 8.54 (s, 1H, Ar-H), 11.19 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 439 [M+, 100]; Anal. Calcd. for C26H21N3O2S: C, 71.05; H, 4.82; N, 9.56; found C, 70.89; H, 4.96; N, 9.74.
5-(5,7-Dimethyl-2-oxoindolin-3-ylidene)-3-(p-tolyl)-2-(p-tolylimino)thiazolidin-4-one (4n)
Brown powder (yield 71%), m.p. > 300 °C; IR (KBr, ν cm−1): 3319 (NH) and 1685 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.19 (s, 3H, -CH3), 2.24 (s, 3H, -CH3), 2.32 (s, 3H, -CH3), 2.41 (s, 3H, -CH3), 6.87 (d, J = 8.2 Hz, 2H, Ar-H), 7.02 (s, 1H, Ar-H), 7.21 (d, J = 8.1 Hz, 2H, Ar-H), 7.37 (d, J = 8.3 Hz, 2H, Ar-H), 7.43 (d, J = 8.3 Hz, 2H, Ar-H), 8.49 (s, 1H, Ar-H), 11.11 (s, 1H, NH isatin, D2O exchangeable); 13C-NMR (DMSO-d6, 100 MHz) δ ppm: 18.90, 21.35, 28.18 (2C), 119.54, 120.90 (2C), 126.31, 128.86 (2C), 129.87 (2C), 130.30 (2C), 130.93, 131.46, 132.59, 134.14, 138.97, 139.89, 143.02, 145.42, 153.76, 157.33, 163.30, 165.80, 169.33; MS, m/z [%]: 453 [M+, 100]; Anal. Calcd. for C27H23N3O2S: C, 71.50; H, 5.11; N, 9.26; found C, 71.67; H, 5.04; N, 9.51; HRMS m/z 454.15833 M+ + 1, calcd for C27H23N3O2S: 454.15837.
4.1.3. General Procedure for the Preparation of Target Compounds 7a–d
Route A:
A mixture of isatins 1c, e–g (1 mmoL) with an equimolar amount of 2-mercaptobenzimidazole 6 (0.15 gm, 1 mmoL) and bromoacetic acid 3 (0.14 g, 1 mmoL) in glacial acetic acid (10 mL) in the presence of sodium acetate (0.16 g, 2 mmoL), was heated under reflux for 2 h. The obtained solid was filtered off while hot, washed with hot ethanol, dried and recrystallized from DMF to afford the target hybrids 7a–d.
Route B:
To a stirred solution of 2-mercaptobenzimidazole 6 (0.15 gm, 1 mmoL) and KOH (0.06 gm, 1.1 mmoL) in ethanol (10 mL), bromoacetic acid 3 (0.14 gm, 1 mmoL) was added, and then the reaction mixture was refluxed for 1 h. The formed solid was filtered off, washed with ethanol to give compound 8, which cyclized into intermediate 9 via heating with acetic anhydride in pyridine medium at 100 °C. Intermediate 9 (0.19 g, 1 mmoL) was added to a stirred solution of the appropriate isatin derivative 1c, e–g (1 mmoL) and sodium acetate (0.16 g, 2 mmoL) in glacial acetic acid (5 mL), then the reaction mixture was refluxed for 2 h. The formed solid was filtered off, washed with ethanol and recrystallized from DMF to furnish compounds 7a–d.
2-(5-Chloro-2-oxoindolin-3-ylidene)benzo[4,5]imidazo[2,1-b]thiazol-3(2H)-one (7a)
Red powder (yield 80%), m.p. > 300 °C; IR (KBr, ν cm−1): 3325 (NH) and 1689 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 7.04 (d, J = 8.3 Hz, 1H), 7.36–7.48 (m, 1H), 7.52 (dd, J = 8.4, 2.0 Hz, 1H), 7.69 (d, J = 6.6 Hz, 1H), 8.03 (d, J = 6.5 Hz, 1H), 8.93 (d, J = 2.1 Hz, 1H), 11. 51 (s, 1H, NH isatin, D2O exchangeable); Anal. Calcd. for C17H8ClN3O2S: C, 57.72; H, 2.28; N, 11.88; found C, 58.06; H, 2.41; N, 12.16.
2-(5-Methoxy-2-oxoindolin-3-ylidene)benzo[4,5]imidazo[2,1-b]thiazol-3(2H)-one (7b)
Red powder (yield 85%), m.p. > 300 °C; IR (KBr, ν cm−1): 3296 (NH) and 1692 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 3.83 (s, 3H, -OCH3), 6.93 (d, J = 8.5 Hz, 1H, Ar-H), 7.08 (dd, J = 8.5, 2.6 Hz, 1H, Ar-H), 7.42 (m, 2H, Ar-H), 7.69 (d, J = 7.0 Hz, 1H, Ar-H), 8.01 (d, J = 6.6 Hz, 1H, Ar-H), 8.59 (d, J = 2.6 Hz, 1H, Ar-H), 11.18 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 349 [M+, 21.03]; Anal. Calcd. for C18H11N3O3S: C, 61.88; H, 3.17; N, 12.03; found C, 62.14; H, 3.42; N, 12.31.
2-(5-Methyl-2-oxoindolin-3-ylidene)benzo[4,5]imidazo[2,1-b]thiazol-3(2H)-one (7c)
Red powder (yield 87%), m.p. > 300 °C; IR (KBr, ν cm−1): 3405 (NH) and 1693 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.37 (s, 3H, -CH3), 6.90 (d, J = 7.6 Hz, 1H, Ar-H), 7.27 (d, J = 8.5, 1H, Ar-H), 7.33–7.43 (m, 2H, Ar-H), 7.68 (d, J = 7.2 Hz, 1H, Ar-H), 8.01 (d, J = 7.6 Hz, 1H, Ar-H), 8.75 (s, 1H, Ar-H), 11.26 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 333 [M+, 23.48]; Anal. Calcd. for C18H11N3O2S: C, 64.85; H, 3.33; N, 12.61; found C, 64.97; H, 3.50; N, 12.89.
2-(5,7-Dimethyl-2-oxoindolin-3-ylidene)benzo[4,5]imidazo[2,1-b]thiazol-3(2H)-one (7d)
Red powder (yield 88%), m.p. > 300 °C; IR (KBr, ν cm−1): 3382 (NH) and 1689 (C=O); 1H-NMR (DMSO-d6, 400 MHz) δ ppm: 2.20 (s, 3H, -CH3), 2.29 (s, 3H, -CH3), 7.09 (s, 1H, Ar-H), 7.34–7.48 (m, 2H, Ar-H), 7.68 (d, J = 7.0 Hz, 1H), 8.02 (d, J = 7.0 Hz, 1H), 8.62 (s, 1H, Ar-H), 11.28 (s, 1H, NH isatin, D2O exchangeable); MS, m/z [%]: 347 [M+, 100]; Anal. Calcd. for C19H13N3O2S: C, 65.69; H, 3.77; N, 12.10; found C, 65.91; H, 3.86; N, 11.97.
4.2. Biological Evaluation
4.2.1. Antiproliferative Activity Against Breast Cancer Cell Lines
Breast cancer cell lines MDA-MB-231 and MCF-7 cells were obtained from VACSERA Tissue Culture Unit. The cells were propagated in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 1% l-glutamine (2.5 mM), HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (10 mM) and 50 µg/mL gentamycin. All cells were maintained at 37 °C in a humidified atmosphere with 5% CO2 and were sub-cultured two times a week. Cytotoxicity was determined following the Crystal Violet (CV) cell cytotoxicity assay [41].
Test samples were dissolved in DMSO and kept at a stock concentration of 100 mM. Exponentially growing cells were collected using 0.25% Trypsin-EDTA and seeding was done at a density of 3000 cells/well in 96-well plates. After 24 h, the monolayer cultured cells were washed with sterile phosphate buffered saline (0.01 M pH 7.2) and simultaneously the cells were treated with 100 mL from 10 different dilutions of the test compounds as well as Doxorubicin was used as a standard compound in fresh maintenance medium and incubated at 37 °C for 24 h. Control wells were exposed to the same concentration of vehicle, DMSO (1%) used in the highest concentration of the test samples. Cell viability was >99% compared to untreated cells. The number of the surviving cells was determined by staining the cells with crystal violet followed by cell lysing using 33% glacial acetic acid. The absorbance was measured at 490 nm using ELISA reader (Thermo Fisher Scientific, Waltham, MA, USA) after gentle shaking. The absorbance values from untreated cells were considered as 100% proliferation.
The number of viable cells was determined using ELISA reader as previously mentioned and the percentage of viability was calculated as [1−(ODt/ODc)] × 100% where ODt is the mean optical density of wells treated with the test sample and ODc is the mean optical density of untreated cells. The 50% inhibitory concentration (IC50) was estimated from graphic plots of the dose response curve for each conc. using GraphPad Prism software, Version 5 (San Diego, CA, USA). The data presented are the mean of at least three separate experiments.
4.2.2. In Vitro Cytotoxic Activity towards Nontumorigenic Human WI-38 and MCF-10A Cells
The cytotoxic activity of the active conjugates towards nontumorigenic human lung fibroblast cell line (WI-38) and human breast epithelial cell line (MCF-10A) was determined using Crystal violet (CV) cell cytotoxicity assay according to the previously published procedures [42,55].
4.2.3. ELISA Immunoassay
The levels of the apoptotic markers (Bax, caspase-3) as well as the antiapoptotic marker Bcl-2 were determined using ELISA colorimetric kits per the manufacturer’s instructions, as reported earlier [41]. MDA-MB-231 cells were cultured as a monolayer in T-25 flasks and were seeded to attain 30% confluency prior to treatment. Cells were then treated separately with compounds 4m and 7b at their IC50 concentrations for 48 h. At the end of treatment, cells were collected via trypsinization and centrifuged at 10,000 rpm. The pellet was then rinsed with PBS and lysed in RIPA lysis buffer at 4 °C for 45 min, then centrifuged at 14,000 rpm for 20 min to remove the cellular debris. Lysates were then collected and stored at −80 °C for later protein determination using Pierce BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer’s recommendations.
The cell lysate was diluted 10 times, and 100 μL (50 mg protein) was added to the wells of four separate microtiter plates for the four ELISA kits that were pre-coated with primary antibodies specific to Bax, Bcl-2 and caspase-3 proteins, respectively. A secondary biotin-linked antibody specific to the protein captured by the primary antibody was further added to bind the captured protein, forming a “sandwich” of specific antibodies around the desired protein in the cell lysate. The streptavidin-HRP complex was then used to bind the biotin-linked secondary antibody through its streptavidin portion. The HRP domain reacted with the added TMB substrate to form a colored product that measured at 450 nm by a plate reader (ChroMate-4300, Orlando, FL, USA) after the reaction was terminated via the addition of stop solution.
4.2.4. Cell Cycle Analysis
TNBC MDA-MB-231 cells were treated with compound 4m for 24 h (at its IC50 concentration), and then cells were washed twice with ice-cold phosphate buffered saline (PBS). Subsequently, the treated cells were collected by centrifugation, fixed in ice-cold 70% (v/v) ethanol, washed with PBS, re-suspended with 100 μg/mL RNase, stained with 40 μg/mL PI, and analyzed by flow cytometry using FACS Calibur (Becton Dickinson, BD, Franklin Lakes, NJ, USA). The cell cycle distributions were calculated using CellQuest software 5.1 (Becton Dickinson) [35,49].
4.2.5. Annexin V-FITC Apoptosis Assay
Phosphatidylserine externalization was assayed using Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions, as reported earlier [35,49]. MDA-MB-231 cells were cultured to a monolayer then treated with conjugate 4m at its IC50 concentration as described earlier. Cells were then harvested via trypsinization, and rinsed twice in PBS followed by binding buffer. Moreover, cells were re-suspended in 100 μL of binding buffer with the addition of 1 μL of FITC-Annexin V (Becton Dickinson BD PharmingenTM, Heidelberg, Germany) followed by an incubation period of 30 min at 4 °C. Cells were then rinsed in binding buffer and resuspended in 150 μL of binding buffer with the addition of 1 μL of DAPI (1 μg/μL in PBS) (Invitrogen, Life Technologies, Darmstadt, Germany). Cells were then analyzed using the flow cytometer BD FACS Canto II (BD Biosciences, San Jose, CA, USA) and the results were interpreted with FlowJo7.6.4 software (Tree Star, Ashland, OR, USA).
4.2.6. Statistical Analysis
Data are presented as means ± S.D. Individual groups were compared using the two-tailed independent Student’s t-test. Multiple group comparisons were carried out using one-way analysis of variance (ANOVA) followed by the Tukey–Kramer test for post-hoc analysis. Statistical significance was accepted at a level of p < 0.05. All statistical analyses were performed using GraphPad InStat software, version 3.05 (GraphPad Software, Inc., La Jolla, CA, USA). Graphs were sketched using GraphPad Prism software, version 5.00 (GraphPad Software, Inc., La Jolla, CA, USA).
Acknowledgments
Patrick Maccosker, University of Wollongong, Australia, is highly appreciated for his efforts in language editing of the manuscript.
Author Contributions
Wagdy M. Eldehna and Hatem A. Abdel-Aziz have formulated the research idea; Wagdy M. Eldehna, Mohamed H. El-Naggar, Hadia Almahli and Mohaned Fares carried out the experimental, interpreted the data and discussion, and prepared the manuscript; Mahmoud M. Elaasser performed the cytotoxicity assay; Amr Elgez carried out the biological work and discussed its results. All the authors reviewed and approved the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Sample Availability: Samples of the compounds 4a–n and 7a–d are available from the authors.
References
- 1.DeSantis C., Ma J., Bryan L., Jemal A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 2.International Agency for Research on Cancer . IGlobocan: Estimated Cancer Incidence, Mortality, and Prevalence Worldwide in 2012. IARC; Lyon, France: 2014. [Google Scholar]
- 3.Siegel R., Ma J., Zou Z., Jemal A. Cancer statistics, 2014. CA Cancer J. Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Adami H., Hunter D., Trichopoulos D. Textbook of Cancer Epidemiology. Oxford University Press; New York, NY, USA: 2002. pp. 301–373. [Google Scholar]
- 5.Barnard M.E., Boeke C.E., Tamimi R.M. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim. Biophys. Acta. 2015;1856:73–85. doi: 10.1016/j.bbcan.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Rahman M., Davis S.R., Pumphrey J.G., Bao J., Nau M.M., Meltzer P.S., Lipkowitz S. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res. Treat. 2009;113:217–230. doi: 10.1007/s10549-008-9924-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdel-Aziz H.A., Eldehna W.M., Ghabbour H., Al-Ansary G.H., Assaf A.M., Al-Dhfyan A. Synthesis, crystal study, and antiproliferative activity of some 2-benzimidazolylthioacetophenones towards triple-negative breast cancer MDA-MB-468 cells as apoptosis-inducing agents. Int. J. Mol. Sci. 2016;17:1221. doi: 10.3390/ijms17081221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smid M., Wang Y., Zhang Y., Sieuwerts A.M., Yu J., Klijn J.G., Foekens J.A., Martens J.W. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68:3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 9.Medvedev A., Buneeva O., Gnedenko O., Ershov P., Ivanov A. Isatin, an endogenous nonpeptide biofactor: A review of its molecular targets, mechanisms of actions, and their biomedical implications. BioFactors. 2018;44:95–108. doi: 10.1002/biof.1408. [DOI] [PubMed] [Google Scholar]
- 10.Kidwai M., Jahan A., Mishra N.K. Isatins: A diversity orientated biological profile. Med. Chem. 2014;4:451–468. doi: 10.4172/2161-0444.1000179. [DOI] [Google Scholar]
- 11.Eldehna W.M., Al-Ansary G.H., Bua S., Nocentini A., Gratteri P., Altoukhy A., Ghabbour H., Ahmed H.Y., Supuran C.T. Novel indolin-2-onebased sulfonamides as carbonic anhydrase inhibitors: Synthesis, in vitro biological evaluation against carbonic anhydrases isoforms I, II, IV and VII and molecular docking studies. Eur. J. Med. Chem. 2017;127:521–530. doi: 10.1016/j.ejmech.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Eldehna W.M., Fares M., Abdel-Aziz M.M., Abdel-Aziz H.A. Design, synthesis and antitubercular activity of certain nicotinic Acid hydrazides. Molecules. 2015;20:8800–8815. doi: 10.3390/molecules20058800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sridhar S.K., Ramesh A. Synthesis and pharmacological activities of hydrazones, Schiff and Mannich bases of isatin derivatives. Biol. Pharm. Bull. 2001;24:1149–1152. doi: 10.1248/bpb.24.1149. [DOI] [PubMed] [Google Scholar]
- 14.Vine K.L., Matesic L., Locke J.M., Skropeta D. Recent highlights in the development of isatin-based anticancer agents. Adv. Anticancer Agents Med. Chem. 2013;2:254–312. [Google Scholar]
- 15.Goodman V.L., Rock E.P., Dagher R., Ramchandani R.P., Abraham S., Gobburu J.V., Booth B.P., Verbois S.L., Morse D.E., Liang C.Y., et al. Approval summary: Sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin. Cancer Res. 2007;13:1367–1373. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 16.McCormack P.L. Nintedanib: First global approval. Drugs. 2015;75:129–139. doi: 10.1007/s40265-014-0335-0. [DOI] [PubMed] [Google Scholar]
- 17.Lemmens L. Nintedanib in advanced NSCLC: Management of adverse events. Lung Cancer Manag. 2016;5:29–41. doi: 10.2217/lmt.15.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagarsenkar A., Guntuku L., Guggilapu S.D., Gannoju S., Naidu V.G.M., Bathini N.B. Synthesis and apoptosis inducing studies of triazole linked 3-benzylidene isatin derivatives. Eur. J. Med. Chem. 2016;124:782–793. doi: 10.1016/j.ejmech.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Karthikeyan C., Solomon V.R., Lee H., Trivedi P. Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: A molecular hybridization approach. Biomed. Prev. Nutr. 2013;3:325–330. doi: 10.1016/j.bionut.2013.04.001. [DOI] [Google Scholar]
- 20.Ke S., Shi L., Yang Z. Discovery of novel isatin–dehydroepiandrosterone conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015;25:4628–4631. doi: 10.1016/j.bmcl.2015.08.041. [DOI] [PubMed] [Google Scholar]
- 21.Solomon V.R., Hu C., Lee H. Hybrid pharmacophore design and synthesis of isatin–benzothiazole analogs for their anti-breast cancer activity. Bioorg. Med. Chem. 2009;17:7585–7592. doi: 10.1016/j.bmc.2009.08.068. [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Aziz H.A., Elsaman T., Al-Dhfyan A., Attia M.I., Al-Rashood K.A., Al-Obaid A.M. Synthesis and Anticancer Potential of Certain Novel 2-Oxo-N′-(2-oxoindolin-3-ylidene)-2H-chromene-3-carbohydrazides. Eur. J. Med. Chem. 2013;70:358–363. doi: 10.1016/j.ejmech.2013.09.060. [DOI] [PubMed] [Google Scholar]
- 23.Gudipati R., Anreddy R.N.R., Manda S. Synthesis, anticancer and antioxidant activities of some novel N-(benzo[d]oxazol-2-yl)-2-(7- or 5-substituted-2-oxoindolin-3-ylidene) hydrazinecarboxamide derivatives. J. Enzyme Inhib. Med. Chem. 2011;26:813–818. doi: 10.3109/14756366.2011.556630. [DOI] [PubMed] [Google Scholar]
- 24.Aboul-Fadl T., Kadi A., Abdel-Aziz H.A. Novel N,N′-Hydrazino-bis-isatin Derivatives with Selective Activity Against Multidrug-Resistant Cancer Cells. 20120252860. U.S. Patent. 2012 Oct 4;
- 25.Havrylyuk D., Kovach N., Zimenkovsky B., Vasylenko O., Lesyk R. Synthesis and Anticancer Activity of Isatin-Based Pyrazolines and Thiazolidines Conjugates. Arch. Pharm. Chem. Life Sci. 2011;344:514–522. doi: 10.1002/ardp.201100055. [DOI] [PubMed] [Google Scholar]
- 26.Ramshid P.K., Jagadeeshan S., Krishnan A., Mathew M., Nair S.A., Pillai M.R. Synthesis and in vitro Evaluation of Some Isatin-Thiazolidinone Hybrid Analogues as Anti-Proliferative Agents. Med. Chem. 2010;6:306–312. doi: 10.2174/157340610793358909. [DOI] [PubMed] [Google Scholar]
- 27.Kaminskyy D., Khyluk D., Vasylenko O., Zaprutko L., Lesky R. A Facile Synthesis and Anticancer Activity Evaluation of Spiro[Thiazolidinone-Isatin] Conjugates. Sci. Pharm. 2011;79:763–777. doi: 10.3797/scipharm.1109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havrylyuk D., Zimenkovsky B., Vasylenko O., Gzella A., Lesyk R. Synthesis of New 4-Thiazolidinone-, Pyrazoline-, and Isatin-Based Conjugates with Promising Antitumor Activity. J. Med. Chem. 2012;55:8630–8641. doi: 10.1021/jm300789g. [DOI] [PubMed] [Google Scholar]
- 29.Wang S., Zhao Y., Zhang G., Lv Y., Zhang N., Gong P. Design, synthesis and biological evaluation of novel 4-thiazolidinones containing indolin-2-one moiety as potential antitumor agent. Eur. J. Med. Chem. 2011;46:3509–3518. doi: 10.1016/j.ejmech.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 30.Taher A.T., Khalil N.A., Ahmed E.M. Synthesis of Novel Isatin-Thiazoline and Isatin-Benzimidazole Conjugates as Anti-Breast Cancer Agents. Arch. Pharm. Res. 2011;34:1615–1621. doi: 10.1007/s12272-011-1005-3. [DOI] [PubMed] [Google Scholar]
- 31.Eldehna W.M., Almahli H., Al-Ansary G.H., Ghabbour H.A., Aly M.H., Ismael O.E., Al-Dhfyan A., Abdel-Aziz H.A. Synthesis and in vitro antiproliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents. J. Enz. Inhib. Med. Chem. 2017;32:600–613. doi: 10.1080/14756366.2017.1279155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdel-Aziz H.A., Eldehna W.M., Keeton A.B., Piazza G.A., Kadi A.A., Attwa M.W., Abdelhameed A.S., Attia M.I. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: Synthesis and in vitro pharmacological profiling. Drug Des. Dev. Ther. 2017;11:2333. doi: 10.2147/DDDT.S140164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eldehna W.M., Fares M., Ibrahim H.S., Alsherbiny M.A., Aly M.H., Ghabbour H.A., Abdel-Aziz H.A. Synthesis and cytotoxic activity of biphenylurea derivatives containing indolin-2-one moieties. Molecules. 2016;21:762. doi: 10.3390/molecules21060762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eldehna W.M., EL-Naggar D.H., Hamed A.R., Ibrahim H.S., Ghabbour H.A., Abdel-Aziz H.A. One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDA-MB-231 cells. J. Enz. Inhib. Med. Chem. 2018;33:309–318. doi: 10.1080/14756366.2017.1417276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eldehna W.M., Abo-Ashour M.F., Ibrahim H.S., Al-Ansary G.H., Ghabbour H.A., Elaasser M.M., Ahmed H.Y., Safwat N.A. Novel [(3-indolylmethylene)hydrazono]indolin-2-ones as apoptotic antiproliferativeagents: Design, synthesis and in vitro biological evaluation. J. Enz. Inhib. Med. Chem. 2018;33:686–700. doi: 10.1080/14756366.2017.1421181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Attia M.I., Eldehna W.M., Afifi S.A., Keeton A.B., Piazza G.A., Abdel-Aziz H.A. New hydrazonoindolin-2-ones: Synthesis, exploration of the possible antiproliferative mechanism of action and encapsulation into PLGA microspheres. PLoS ONE. 2017;12:e0181241. doi: 10.1371/journal.pone.0181241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eldehna W.M., Altoukhy A., Mahrous H., Abdel-Aziz H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential antiproliferative agents. Eur. J. Med. Chem. 2015;90:684–694. doi: 10.1016/j.ejmech.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 38.Abdel-Aziz H.A., Ghabbour H.A., Eldehna W.M., Qabeel M.M., Fun H.-K. Synthesis, crystal structure and biological activity of cis/trans amide rotomers of (Z)-N′-(2-oxoindolin-3-ylidene)formohydrazide. J. Chem. 2014;2014:760434. doi: 10.1155/2014/760434. [DOI] [Google Scholar]
- 39.Eldehna W.M., Fares M., Ceruso M., Ghabbour H.A., Abou-Seri S.M., Abdel-Aziz H.A., El Ella D.A.A., Supuran C.T. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur. J. Med. Chem. 2016;110:259–266. doi: 10.1016/j.ejmech.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 40.Eldehna W.M., Fares M., Ibrahim H.S., Aly M.H., Zada S., Ali M.M., Abou-Seri S.M., Abdel-Aziz H.A., El Ella D.A.A. Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: Synthesis, in vitro biological evaluation and molecular docking. Eur. J. Med. Chem. 2015;100:89–97. doi: 10.1016/j.ejmech.2015.05.040. [DOI] [PubMed] [Google Scholar]
- 41.Eldehna W.M., Abo-Ashour M.F., Nocentini A., Gratteri P., Eissa I.H., Fares M., Ismael O.E., Ghabbour H.A., Elaasser M.M., Abdel-Aziz H.A., et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene) thiazolidin-2-ylidene) amino) benzenesulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur. J. Med. Chem. 2017;139:250–262. doi: 10.1016/j.ejmech.2017.07.073. [DOI] [PubMed] [Google Scholar]
- 42.Eldehna W.M., Al-Wabli R.I., Almutairi M.S., Keeton A.B., Piazza G.A., Abdel-Aziz H.A., Attia M.I. Synthesis and biological evaluation of certain hydrazonoindolin-2-one derivatives as new potent antiproliferative agents. J. Enz. Inhib. Med. Chem. 2018;33:867–878. doi: 10.1080/14756366.2018.1462802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delbridge A.R., Grabow S., Strasser A., Vaux D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer. 2016;16:99. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 44.Hu W., Kavanagh J.J. Anticancer therapy targeting the apoptotic pathway. Lancet Oncol. 2003;4:721–729. doi: 10.1016/S1470-2045(03)01277-4. [DOI] [PubMed] [Google Scholar]
- 45.Lopez J., Tait S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer. 2015;112:957. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youle R.J., Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 47.Jiang H., Zhao P.J., Su D., Feng J., Ma S.L. Paris saponin I induces apoptosis via increasing the Bax/Bcl-2 ratio and caspase-3 expression in gefitinib-resistant non-small cell lung cancer in vitro and in vivo. Mol. Med. Rep. 2014;9:2265–2272. doi: 10.3892/mmr.2014.2108. [DOI] [PubMed] [Google Scholar]
- 48.McIlwain D.R., Berger T., Mak T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015;7:a026716. doi: 10.1101/cshperspect.a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eldehna W.M., Ibrahim H.S., Abdel-Aziz H.A., Farrag N.N., Youssef M.M. Design, synthesis and in vitro antitumor activity of novel N-substituted-4-phenyl/benzylphthalazin-1-ones. Eur. J. Med. Chem. 2015;89:549–560. doi: 10.1016/j.ejmech.2014.10.064. [DOI] [PubMed] [Google Scholar]
- 50.Accelrys Co. Ltd. Discovery Studio 4.0. [(accessed on 1 June 2018)]; Available online: http://www.accelrys.com/
- 51.Fares M., Said M.A., Alsherbiny M.A., Eladwy R.A., Almahli H., Abdel-Aziz M.M., Ghabbour H.A., Eldehna W.M., Abdel-Aziz H.A. Synthesis, biological evaluation and molecular docking of certain sulfones as potential nonazole antifungal agents. Molecules. 2016;21:114. doi: 10.3390/molecules21010114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng G., Zheng M., Wang M., Tong J., Ge W., Zhang J., Zheng A., Li J., Gao L., Li J. Design and synthesis of new potent PTP1B inhibitors with the skeleton of 2-substituted imino-3-substituted-5-heteroarylidene-1, 3-thiazolidine-4-one: Part I. Eur. J. Med. Chem. 2016;122:756–769. doi: 10.1016/j.ejmech.2016.05.060. [DOI] [PubMed] [Google Scholar]
- 53.Mavrova A.T., Anichina K.K., Vuchev D.I., Tsenov J.A., Denkova P.S., Kondeva M.S., Micheva M.K. Antihelminthic activity of some newly synthesized 5 (6)-(un) substituted-1H-benzimidazol-2-ylthioacetylpiperazine derivatives. Eur. J. Med. Chem. 2006;41:1412–1420. doi: 10.1016/j.ejmech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Hu J., Wang Y., Wei X., Wu X., Chen G., Cao G., Shen X., Zhang X., Tang Q., Liang G., et al. Synthesis and biological evaluation of novel thiazolidinone derivatives as potential anti-inflammatory agents. Eur. J. Med. Chem. 2013;64:292–301. doi: 10.1016/j.ejmech.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 55.Soliman D.H., Eldehna W.M., Ghabbour H.A., Kabil M.M., Abdel-Aziz M.M., Abdel-Aziz H.A.K. Novel 6-Phenylnicotinohydrazide Derivatives: Design, Synthesis and Biological Evaluation as a Novel Class of Antitubercular and Antimicrobial Agents. Biol. Pharm. Bull. 2017;40:1883–1893. doi: 10.1248/bpb.b17-00361. [DOI] [PubMed] [Google Scholar]