

Abstract
Between the 1930s and 50s, evolutionary biologists developed a successful theory of why organisms age, firmly rooted in population genetic principles. By the 1980s the evolution of aging had a secure experimental basis. Since the force of selection declines with age, aging evolves due to mutation accumulation or a benefit to fitness early in life. Here we review major insights and challenges that have emerged over the last 35 years: selection does not always necessarily decline with age; higher extrinsic (i.e., environmentally caused) mortality does not always accelerate aging; conserved pathways control aging rate; senescence patterns are more diverse than previously thought; aging is not universal; trade-offs involving lifespan can be ‘broken’; aging might be ‘druggable’; and human life expectancy continues to rise but compressing late-life morbidity remains a pressing challenge.
The evolution of aging in humans
Human life expectancy worldwide has increased dramatically. During the ~ 300,000 generations since the divergence from our most recent common ancestor with the great apes, lifespan evolved to double its previous value [1]. In the last ~ 200 years there has been a further substantial increase, on average about 2.5 years per decade, attributable to environmental changes, including improved food, water, hygiene, and living conditions, reduced impact of infectious disease with immunization and antibiotics, and improved medical care at all ages [2–5]. As a result, most people are now living long beyond the ages at which most would have been dead in the past. Natural selection has therefore not had an opportunity to maintain evolutionary fitness at older ages. Presumably as a consequence, advancing age is the major risk factor for diverse types of loss of function, and for highly prevalent chronic and killer diseases, including cancer, cardiovascular disease, and dementia [6, 7]. Consequently, healthy life expectancy has not increased as much as has overall life expectancy [8, 9], and there is a growing period of late-life morbidity before death (WHO data on life expectancy [10]).
Many modern humans inhabit a very different environment from that in which their life history evolved, with both protection from many of its dangers, such as predators, infectious diseases, and harsh physical conditions, and freedom from the need to forage extensively to avoid starvation [1]. However, the ready availability of calorie-dense food, together with the low requirement for physical exercise, are resulting in a tidal wave of metabolic disease that has a major impact at all ages, but particularly on deaths from cardiovascular disease later in life [11]. Modern humans therefore often have many features in common with laboratory model organisms, which also inhabit highly protected, calorie rich, and physically restricted environments.
Aging human populations have become a grand challenge to societies worldwide. The major burden of ill health is now falling on older people. Declining birth rates, together with the population bulge in some countries from the baby boomers and generally longer lives, are increasing the ratio of dependent to independent members of society, posing major economic and social problems [12]. Current demographic trends indicate that life expectancy is likely to continue to increase in all countries for which there are good data [13], and it is unclear when any limit to human lifespan will be seen (see the recent debate in [14–16]). There is hence a pressing need to find ways of keeping people healthy for longer and hence compressing and reversing the growing period of morbidity at the end of life [9, 17]. Interestingly, it has been found that late-life disability and morbidity are lower among people living beyond 100 years [18]. Morbidity can thus be restricted, at least in principle, to the very end of life (Fig. 1). In this review, we discuss what can be learned about aging by considering its evolutionary biology, and how evolutionary thinking could help inform practical measures to ameliorate the effects of human aging.
Fig. 1.
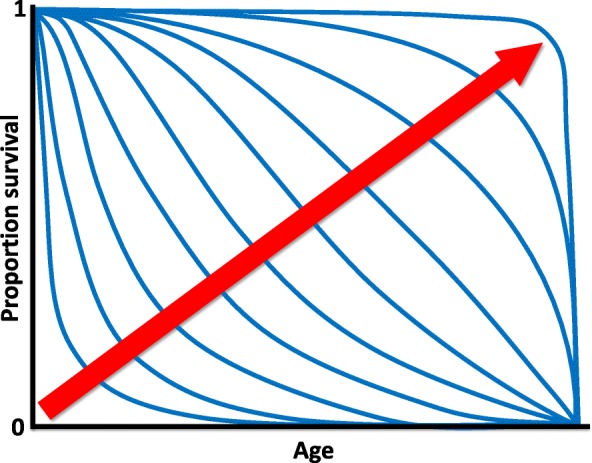
Rectangularization of survival curves. Hypothetical survival curves with different degrees of ‘rectangularization’ (as indicated by the red arrow): increasing ‘rectangularization’ implies a high, constant probability of survival to a very advanced age (i.e., a long ‘shoulder’ of the curve) and a marked compression of morbidity and death into a very narrow age range (i.e., an almost vertical drop in survival at the end of life)
Why does aging evolve?
Aging, or senescence, is characterized demographically by increasing mortality and decreasing reproductive success with advancing adult age [19–21]. These effects of aging, and other types of age-related loss of function, have been extensively documented under field conditions [22]. Aging manifests itself most clearly under benign environmental conditions in captivity, since in the wild individuals of many species are hard to track throughout life (but see [23]), and high rates of age-independent mortality (e.g., due to predators, pathogens, food shortage) can obscure the intrinsic tendency of adult survivorship and fecundity to decline with age [19, 21, 24]. The occurrence of aging in nature poses an evolutionary puzzle: why would such a deleterious, maladaptive process evolve [20]? This puzzle is deepened by the fact that aging is apparently neither inevitable nor universal: germ lines and several organisms do not exhibit senescent decline [21, 25–27].
The basic puzzle of why organisms age was addressed in a series of trail-blazing studies published between the 1930s and 1950s [19, 24, 28]: Fisher [29] and Haldane [30] were the first to realize that aging results from natural selection typically having a much larger impact on survival and reproduction early as compared to late in life, a notion further developed by Medawar [31, 32] and Williams [33]. This idea was later mathematically formalized by Hamilton in 1966 [34] (also see [25, 35, 36]) (Fig. 2). Importantly, Hamilton corrected the error of using Fisher’s so-called ‘reproductive value’ as a measure of how sensitive fitness is to age-specific changes by using the ‘intrinsic rate of increase’ (also called the ‘Malthusian parameter’) as a fitness measure [19, 34, 37, 38]. The underlying driver of the evolution of aging is that various forms of ‘extrinsic’ (i.e., environmentally caused) hazards, such as disease, predation, and accidents, largely determine the adult mortality rate and hence cause a characteristic decline with time in the numbers of surviving individuals in a cohort. Genetic variants that affect fitness at later ages will therefore encounter a weakened force of natural selection, because some of their bearers will die from extrinsic hazard, at a rate no different from non-bearers, up to the age when the variant starts to manifest its phenotypic effects and hence affect fitness. The population genetic theory of aging posits that this process leads to two non-exclusive mechanisms (Fig. 2). The first is ‘mutation accumulation’ (MA), proposed by Medawar in 1952 [32]. If the force of selection declines with age, deleterious mutations whose effects are restricted to late life can accumulate to higher frequency under mutation-selection balance, due to a progressively weakened force of natural selection. J.B.S. Haldane discussed Huntington’s disease, caused by a dominant mutation, and with an average age of onset of ~ 35 years, as an example of mutation accumulation [30]. The second mechanism is ‘antagonistic pleiotropy’ (AP), a concept proposed by Medawar [31, 32] and Williams [33]. Here, selection can favor mutations or alleles with positive effects on fitness-related traits early in life, even if these same genetic variants have negative effects late in life, because selection will act less strongly against the late-life deleterious effects if its strength declines with age. Cellular senescence, a cell cycle arrest in normally dividing cells, is a potential example of antagonistic pleiotropy. The process is important during development and wound healing, where it participates in tissue remodeling. Cellular senescence is also vital in protection against cancer, because it occurs in response to DNA damage. However, during aging senescent cells, instead of being removed by the immune system, accumulate in tissues and cause damage, by secreting inflammatory molecules, and hence are important in the etiology of many aging-related diseases [6, 39]. A physiological version of antagonistic pleiotropy, the ‘disposable soma’ (DS) hypothesis, assumes that there is a physiological (energetic) trade-off between damage repair and somatic maintenance versus reproductive investment [40–42]. The mathematical theory of MA and AP was worked out chiefly by Charlesworth [19, 37, 38, 43]. An important assumption of the theory is that aging should evolve universally whenever there is a sharp distinction between parents and their offspring (or between somatic and reproductive structures); if there is no such distinction then the theory does not apply [33, 38, 44] (also see below).
Fig. 2.
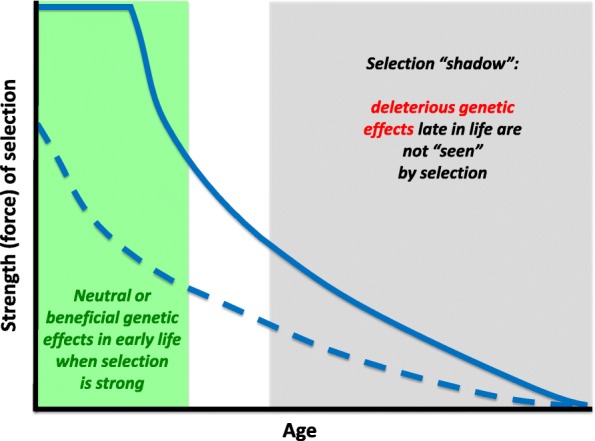
The declining force of selection. The strength (‘force’) of selection measures how strongly natural selection acts on changes in survival and/or fecundity. Often, but not always, the force of selection declines with age. If this is the case, then alleles with neutral effects on fitness early in life but with deleterious effects late in life can accumulate in a population, unchecked by selection (mutation accumulation). Similarly, alleles with positive effects on fitness components early in life can be selectively favored even if they have negative effects late in life (antagonistic pleiotropy). The late-life negative effects in the ‘selection shadow’ cannot be effectively eliminated by selection, leading to senescence. While the force acting on survival (solid line) only starts to decrease with age after the onset of reproduction, the strength of selection on fecundity (dashed line) can increase or decrease before the onset of reproduction (for details see references [36, 38])
A large body of experiments, mainly in the fruit fly Drosophila melanogaster, but also other organisms, supports the AP and MA mechanisms (reviewed in [45–53]). In particular, trade-offs between lifespan and fecundity (or other fitness components) consistent with AP have been found in artificial selection or ‘experimental evolution’ experiments performed on outbred laboratory stocks [54–59], in analyses of mutants and transgenes [46–51, 60–64], and in studies of naturally segregating polymorphisms [47, 65–67]. MA is also well supported, mainly by quantitative genetic studies [49, 68–70] (but see [71] for a critique). In humans, data from medical genetics and genome-wide association studies (GWAS) indicate that both mechanisms might play a role in explaining late-onset diseases and trade-offs between lifespan and fitness-related traits [49, 72–80]. MA is supported by a large number of dominant mutations with late age of onset, and by a recent quantitative genetic analysis of a human historical population [73, 81]. With regard to AP, for example, mutations in BRCA1/2 cause increased risk of breast and ovarian cancer yet have positive pleiotropic effects upon fertility [80]; however, since these mutations are rare, it is somewhat difficult to see how they are consistent with AP.
A classic prediction of the evolutionary theory of aging, due to Medawar [32] and Williams [33], is that low ‘extrinsic’ (i.e., environmentally imposed) adult mortality leads to the evolution of low intrinsic adult mortality (i.e., slowed aging), while the opposite is expected under high extrinsic adult mortality. This postulate was borne out in an experimental evolution experiment where fruit flies were exposed to high versus low extrinsic adult mortality [58]. Extrinsic mortality affects senescence only if it has differential effects among different age classes in an age-structured population [37, 38, 82–84], a point that was implicit in Williams’ 1957 focus on adult (as opposed to preadult) mortality [33, 52]. Indeed, extrinsic mortality often has age-dependent effects: for example, in many large mammals, juveniles and old individuals are more susceptible to extrinsic mortality than prime-aged individuals [85]. Complications can arise if extrinsic mortality affects population growth/density, or if it interacts with organismal condition; both factors can affect the rate of aging if extrinsic mortality is age-dependent. This can lead to situations where lifespan evolves to be longer, not shorter, under high extrinsic mortality [25, 86–88]. For instance, increased extrinsic mortality can select against senescence of a physiological trait that reduces the susceptibility to this source of mortality, causing the evolution of improved somatic condition and longer life; this has been confirmed in guppies and the nematode worm Caenorhabditis remanei [87, 88]. Yet, what remains true is that levels of extrinsic, adult mortality are a key driver of the evolution of aging [28].
Aging therefore evolves as a non-adaptive side effect of the declining ability of selection to maintain fitness at older ages. In humans, where age-related changes are particularly well documented, aging has proved to be a complex process of functional decline and accumulation of diverse pathologies in different tissues [6, 89]. Williams predicted in 1957 [33] that aging is likely to be a genetically complex trait, and different lineages and taxa might well exhibit different proximate mechanisms of senescence. Indeed, natural variation in the rate of aging is likely influenced by many genes [90–92], since survival and reproduction between them harness the activity of much of the genome.
Some mechanisms of aging are evolutionarily conserved
Despite Williams’ prediction that “senescence should always be a generalized deterioration, and never due largely to changes in a single system” [33], aging in laboratory animals has—initially somewhat surprisingly [93]—turned out to be highly malleable to simple genetic, environmental, and pharmacological interventions. Furthermore, similar interventions seem to ameliorate the effects of aging in distantly related organisms, suggesting evolutionary conservation of mechanisms [46, 62, 94–97]. Aging in diverse organisms has characteristic hallmarks, including genetic instability, failure of key cellular processes and components, and impairments of tissue function [6, 21]. These processes can interact within cells and tissues, through action at a distance between them, and through deterioration of the aging systemic environment [6, 98, 99]. The interventions that ameliorate the effects of aging in laboratory animals slow down or suppress at least some of these aging hallmarks.
Dietary restriction (DR) is the longest established and currently most effective means of improving health during aging and extending lifespan in the laboratory. The food intake of DR animals is experimentally reduced, while avoiding malnutrition. Various DR regimes have proved effective in diverse model and non-model invertebrates and vertebrates [100–103], as a result of either conserved mechanisms or parallel evolution. In rodents and rhesus monkeys, DR improves almost all aspects of health during aging, except wound healing and resistance to certain viruses [104–108]. Reduction in intake of specific dietary components, particularly protein, rather than of overall calories, underlies the health improvements from DR [109–115]. DR animals often gorge their daily ration in one meal, and fast until the next one, and this intermittent fasting may play a role in the health improvements [116–119]. DR has been suggested to induce evolved mechanisms for surviving food shortages in nature. Fecundity is usually reduced during DR [120], and organisms short of food might thus reallocate nutrients to somatic maintenance, and hence survive the famine to reproduce more successfully with the return of the food supply [121] (but see [122] for evidence against this hypothesis). However, these results have been obtained largely with laboratory animals, while animals in natural populations often respond to food provisioning with increases in reproduction, function, and survival [123]. In nature some degree of DR may therefore be the norm [124, 125].
Organisms sense both nutrients and their own nutritional status through multiple, parallel mechanisms. An important contributor is nutrient-sensing signaling through the highly evolutionarily conserved insulin/insulin-like growth factor signaling (IIS) and the target of rapamycin (TOR) network, which matches the costly activities of organisms, such as growth, metabolism, and reproduction, to nutrient and stress status (Fig. 3). Beginning with the isolation of the first long-lived laboratory mutants in the late 1970s and early 1980s, it was found that genetically reduced activity of IIS/TOR can increase lifespan in the nematode worm Caenorhabditis elegans, the fruit fly D. melanogaster, and the mouse Mus musculus [51, 61–64, 96, 97, 126–146]. These mutant animals show a prolonged healthspan and are protected against both natural aging-related decline and the pathology associated with genetic models of human age-related diseases [7]. Remarkably, genetic variants in, and altered expression of, the orthologues of the genes encoding components of this network are also associated with survival to advanced ages in humans [147–152]. The IIS/TOR network contains many potential drug targets, and rapamycin, a licensed drug that targets a protein complex in the TOR network, can extend lifespan in diverse laboratory organisms, including mice [153–157].
Fig. 3.
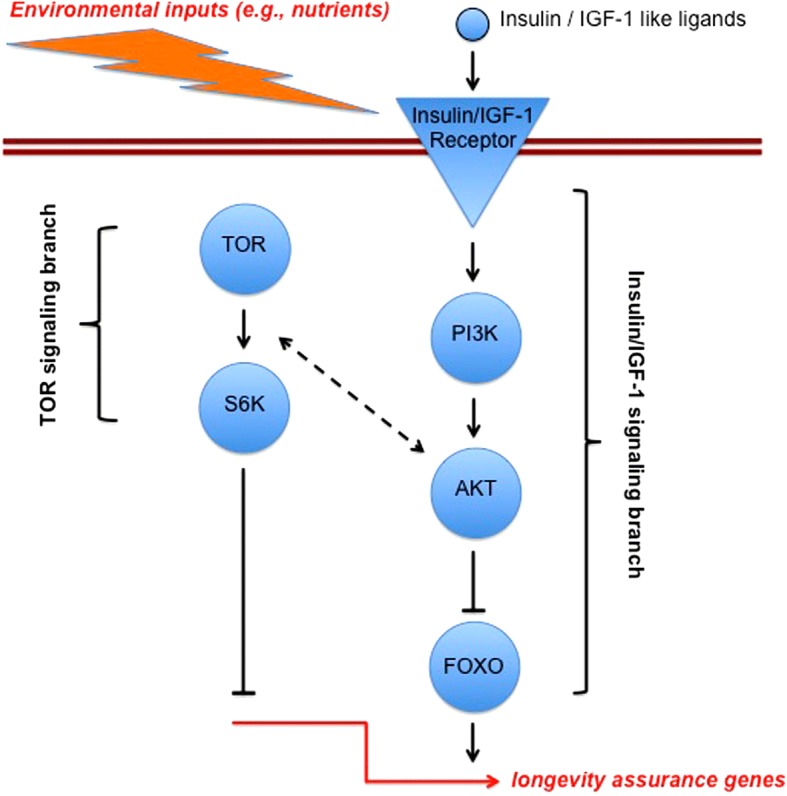
A strongly simplified representation of the evolutionarily conserved insulin/insulin-like growth factor signaling (IIS) and target of rapamycin (TOR) network which regulates lifespan in distinct organisms, from invertebrates to humans. In response to environmental inputs (e.g., nutrients) the IIS and/or TOR branches of the network become activated; reduced input (inhibition) of the signaling network leads to the activation of downstream transcription factors (such as the forkhead box O transcription factor FOXO) that regulate the expression of hundreds of target genes, many of which are involved in longevity assurance (but which also affect other life-history traits, including growth, size, and reproduction). Many of the genetically homologous components of this network have been experimentally shown to affect lifespan in C. elegans, Drosophila, and mouse; evidence from GWAS shows that genetic polymorphisms in some of these components are also associated with exceptional longevity in humans. The homologs of IIS/TOR components have different names in different species: for example, in C. elegans, the insulin-like receptor (INR) is called DAF-2, PI3K is called AGE-1, and FOXO is called DAF-16; in humans, the FOXO homolog associated with longevity is called FOXO3A. Note that humans, in contrast to invertebrates, not only have an insulin receptor but also an insulin-like growth factor 1 receptor (IGF-1); it is thought that the different physiological functions of the insulin receptor versus the IGF-1 receptor in humans are being subsumed by a single insulin receptor in invertebrates
The IIS/TOR nutrient-sensing network might also play a significant role in aging and life history in nature. For example, multiple lines of evidence suggest that insulin-like growth factor-1 (IGF-1) signaling mediates physiological life-history variation in mammalian populations [158, 159], and in D. melanogaster naturally segregating polymorphisms in IIS have been linked to latitudinal gradients (clines) in life-history traits [160–163], with some natural alleles of the insulin-like receptor (InR) gene having pleiotropic effects upon lifespan, stress resistance, fecundity, and body size [164, 66]. Analysis of quantitative trait loci affecting gene expression (so-called eQTLs) in recombinant inbred fruit fly lines has also identified some IIS variants that affect transcription in response to dietary change [165]. Moreover, in honeybees and other social insects, IIS/TOR has profound physiological and developmental effects on caste development, foraging behavior, and probably longevity itself [163, 166–173].
Does the existence of such a conserved signaling network with major effects on aging contradict Williams’ assertion that senescence should not be due to a single cause or system? Evolutionary theories of aging generally do not consider the evolution of phenotypic plasticity, the ability of a single genotype to produce different phenotypes in response to changes in the environment [163]. As individuals go through life, they can encounter widely varying environmental challenges, as can different populations. Phenotypic plasticity of life history in response to varying nutrition, infection, predation, and physical stresses is therefore widespread. The consistent role of the IIS/TOR network in aging may well represent a high degree of evolutionary conservation and optimization of the mechanisms of phenotypic plasticity in life history. Moreover, while aging does result from various forms of system failure owing to limitations of defense against aging-related damage, longevity assurance is a highly regulated process of maintenance and repair. From this point of view, it might not be so surprising then that signaling pathways have evolved that can plastically and genetically match an organism’s investment into somatic maintenance, repair, survival, growth, and reproduction with the prevailing environmental conditions [163, 174].
Although the IIS/TOR network is a major regulator of life history in many taxa [46, 64, 163, 175], several mapping studies and artificial selection experiments in D. melanogaster have failed to identify canonical IIS/TOR genes as harboring natural variation for lifespan or other life-history traits [165, 176–179], with a few exceptions [164, 66]. ‘Longevity genes’, discovered via strong loss-of-function mutations in the laboratory, might thus not always harbor variants in natural populations [47, 177], even though segregating IIS polymorphisms seem to contribute to the exceptional longevity of human centenarians [147–152]. Given the conserved role of IIS/TOR in regulating life-history physiology in response to the external and internal ‘milieu’, a possible explanation for the lack of standing variation is that the plasticity of the network has been optimized by selection but that it is now under selective constraint, with most newly arising mutations being deleterious and purged by purifying selection [180, 181]. Additionally, mapping studies often work with inbred lines and homozygous effects of recessive variants that would play little role in genetic variation in life-history phenotypes in natural, outbred populations, and the experiments are conducted in a laboratory environment that is very different from that in which the fly life history has evolved.
Genomic analyses of selection experiments in flies have also revealed other mechanisms that might be important determinants of the rate of aging. Sequencing of the genomes of flies after 50 generations of longevity selection revealed a statistical enrichment of allele frequency changes at loci involved in defenses against fungal infections [178], and a similar ‘evolve and resequence’ study also identified immunity genes as candidate loci for postponed senescence [179]. The fact that over-expression of immune genes, leading to immune hyperactivity, shortens lifespan, while reduced immune signaling can promote longevity (reviewed in [182]), suggests that allele frequency changes at these loci might underlie, to some degree, evolutionary changes in the rate of aging in these experiments. Little is understood about the mechanistic interplay between immunity and aging, but it is clear from studies of both model organisms [182] and humans [183, 184] that increased inflammation (‘inflammaging’) is a major feature of the aging process, and these artificial selection experiments could provide a powerful context for analyzing the mechanisms at work.
Is aging universal? Diverse patterns of senescence among species
Classic theories of aging pertain mainly to relatively short-lived species with increasing mortality and decreasing fertility after maturity, but patterns of aging—including reproductive senescence—are very diverse [21, 27, 185–191]. In particular, although many species do age, some appear to show ‘negligible’ senescence (i.e., only weak or no signs of aging with advancing age) [21, 27, 192–194] (Fig. 4), whereas others could—at least theoretically—exhibit ‘negative’ senescence (i.e., physiological improvement with age) [195]. In freshwater polyps of the genus Hydra (Fig. 4), for instance, survival and fertility do not decline with age [189]. Similarly, many plants (e.g., ~ 93% of angiosperms) show no signs of aging [196, 187]; some trees, for example, live thousands of years (Fig. 4). However, a caveat is that aging might in many cases exist but not be detectable because the studied individuals were not old enough [197]; for example, a recent study of turtles—typically thought of as exhibiting strongly ‘negligible’ senescence—has shown that reproduction and survival do in fact decline with age, contrary to previous expectations [198]. Many organisms, such as numerous invertebrates and fish, start to reproduce before they are fully grown. Increasing body size can then lead to increased fecundity and also to protection against size-specific predators and other sources of mortality. Under these circumstances, the force of natural selection can increase over part of adult life, because the reproductive value of the organism increases [25, 35, 38, 199]. Non- or slow-aging species, including some animals (e.g., basal metazoans such as Hydra and sea anemones) and most higher plants, are characterized by modular organization, indeterminate (including clonal) growth, and the capacity to regenerate due to stem cell activity; often such organisms start to reproduce before they have finished growing, or they can grow indefinitely [26, 200] (but see [201]). Some clones of grasses, for example, have been estimated to become 15,000 years old [202]. In addition, unlike the standard laboratory model organisms, which set aside and sequestrate their germline early in development, in organisms such as Hydra and higher plants the cells that will become the germline are only identified during adulthood, and these organisms therefore maintain cell lineages with high regenerative potential. Thus, the force of natural selection does not always decline monotonically with age [25–27, 35, 38].
Fig. 4.

Longevous organisms. Many organisms age very slowly, if at all. Top left: the freshwater polyp Hydra (top left) is potentially immortal. Bottom left: some trees like this bristlecone pine (Pinus longaeva) live for thousands of years. Top right: in the naked mole-rat (Heterocephalus glaber) mortality does not increase with age. Bottom right: the bowhead whale (Balaena mysticetus) is the longest-lived mammal, with an estimated maximum lifespan of 211 years. Images: Hydra – © Frank Fox and www.mikro-foto.de Wikimedia Commons/CC-BY-SA-3.0; bristlecone pine - J Brew/Wikimedia Commons/CC-SA-1.0; naked mole-rat ©Roman Klementschitz/Wikimedia Commons/CC-BY-SA-3.0/GFDL; bowhead whale - Olga Shpak/Wikimedia Commons/CC-BY-SA-3.0
These observations suggest that aging might not be universal [25–27], despite some claims to the contrary [203]. An obvious example of a biological system that defies senescent deterioration is the germ line itself, at least when sexual reproduction counteracts the accumulation of deleterious mutations [204–206]—indeed, already in 1885 Weismann [207] stressed that aging might be a phenomenon of the soma (also see [33]). Generally, senescence should only evolve in those organisms that have a distinction between parents and offspring, even when reproduction occurs asexually [33, 38, 44]; for example, if the parent reproduces by simple splitting or dividing symmetrically into identical offspring, then there is no clear delineation of parents versus offspring, selection cannot distinguish between them since there is no age structure, and aging is not expected to evolve [38, 44, 208]. Questions of potential immortality naturally occupy a central place in human thought: might it be possible to increase human lifespan significantly beyond the current level and in such a way that people stay healthy much longer? A recent study has claimed that such hopes are misplaced since life expectancy might reach a limit at around 115 years [14] but this analysis remains controversial [15, 16].
Organisms that have evolved extraordinary longevity are rich material for understanding how the effects of aging can be combatted, and they are usually found in nature rather than in the typical laboratory. Significant work in this direction is being carried out in several fascinating invertebrate and, importantly, vertebrate systems. In addition to the work in Hydra (mentioned above) and social insects (discussed below), studies of the naked mole rat (Heterocephalus glaber; Fig. 4), the most long-lived rodent, have revealed that it is remarkably resistant to oxidative stress and cancer [209, 210]. Intriguingly, a recent analysis based on over 3000 data points suggests that mortality rate does not increase with age in this species, even though certain physiological functions do exhibit (attenuated) senescent decline [190]. Similarly, a short-lived (median lifespan ~ 4 months) fish, the turquoise killifish (Nothobranchius furzeri), has been developed into a convenient organism for studying vertebrate aging [211–213]. This model is promising since these fish exhibit an array of aging traits, including cancer, can be easily reared and manipulated in the laboratory, are amenable to transgenesis and genomics, and possess natural populations that differ in their rates of aging [211]. Planarian flatworms have also recently been suggested as a promising and experimentally tractable model system, since they are potentially somatically immortal, possess pluripotent stem cells, have an amazing ability to regenerate all tissues and body parts, and are amenable to RNAi screens [211]. More work on aging and longevity is also needed in wild populations, especially in vertebrate populations (reviewed in [22, 214]). Importantly, a review of the evidence for aging in wild animals by Nussey and colleagues, based on 175 species (mainly birds and mammals but also in other vertebrates and insects) from 340 separate studies, has shown that aging is prevalent in natural populations [22].
Trade-offs with lifespan are pervasive but can be uncoupled
Studies of natural populations have also found support for phenotypic trade-offs consistent with the notion of AP/DS [22, 197]. In bats, for example, species that produce more offspring are shorter-lived than those that produce fewer offspring [215]. Similarly, a recent review of 26 studies of free-ranging populations of 24 vertebrate species (birds, mammals, reptiles) has identified clear-cut trade-offs between early and late fitness components [53], and data in humans have unraveled a genetically based trade-off between reproduction and lifespan [78]. Trade-offs thus seem to be pervasive: high resource allocation to growth or reproduction early in life is often associated with earlier or more rapid aging. However, there is also growing evidence that trade-offs between lifespan and other fitness components are context-dependent and can be ‘uncoupled’, as is observed in some long-lived C. elegans or Drosophila mutants (reviewed in [47, 50, 166, 175]), or upon manipulation of specific dietary amino acids in flies [109, 144] (see below), without any apparent fitness costs of longevity. In these cases, a likely explanation is the artificially benign laboratory environment occupied by these organisms, which may allow them to realize their physiologically maximal possible investments into both survival and reproduction.
The most famous example of an ‘uncoupling’ of the fecundity–longevity trade-off is seen in eusocial insects (i.e., ants, bees, termites). In many ants, for example, queens are extraordinarily long-lived and highly fertile as compared to the short-lived and sterile workers [166, 216–223], even though within the worker caste reproductive costs have been found among fertile bumblebee workers [224]. (On the other hand, in naked mole rats, which are also eusocial, queens and workers have approximately equivalent lifespans but workers do not reproduce while queens can produce up to 900 pups [225].) How can social insect queens (or kings in termites) escape this trade-off? Surprisingly little formal analysis of this problem exists; the standard explanation that has been put forward is that queens and kings live much longer because they are shielded from extrinsic mortality by the workers [216, 219]. In addition, queens or kings may defy the fecundity–longevity trade-off because of trade-offs at the colony level [226], with resources provided by workers freeing them from individual-level trade-offs; at the colony level, queens and kings might be viewed, metaphorically, as representing the ‘immortal germline’, whereas workers can be seen as representing the ‘disposable soma’ [227]. Classic theories of aging may also not fully apply to eusocial insects [226]: their populations exhibit not only age structure but also strong social structure and division of labor. Since in such a situation survival is not only age- but also state-dependent, the force of selection does not necessarily decline with age [83]. More theoretical work on aging in eusocial insects is warranted, especially the development of class-structured inclusive fitness (kin selection) models [166, 226–228].
Beyond eusocial insects, work on Volvocalean green algae (some of which are unicellular, whereas others form multicellular colonies) suggests that division of labor might be a general principle underlying the decoupling of trade-offs [229, 230]. Unicellular algae face a trade-off between flagellar locomotion and reproduction. In these small planktonic algae, survival is dependent upon locomotory ability, i.e., staying in the water column and escaping predators. The flagellum in turn depends on the centriole, yet the centriole is also required for cell division (reproduction), so that the cell has to forgo locomotion while it divides, thereby leading to a survival-reproduction trade-off. By contrast, colonial forms of these green algae have apparently managed to uncouple the trade-off by having sterile cells devoted to motility, while other cells are specialized to perform the reproductive function—a situation akin to the differentiation of the germline and soma. A convex trade-off function, i.e., an upward-bent curve that relates survival to reproduction, should favor colony formation and specialization into separate survival (somatic) versus reproductive (germline) functions, whereas such a division of labor should not evolve when the trade-off curve is concave, i.e., bent downward [229]. This principle has been generalized, in a theoretical cost–benefit analysis of accelerating and decelerating performance functions [231].
A major aim of mechanistic research into aging is to compress morbidity at the end of life, by shortening its duration and lessening its severity. Importantly, improvement of health during aging should not be associated with adverse side effects. The pleiotropy route to the evolution of aging could be taken to imply that any amelioration of the effects of aging could be achieved only at the cost of problems earlier in adult life, because of the predicted genetic correlation between early and late fitness. However, the finding that increased lifespan can be achieved in the absence of associated costs to reproduction, both in the laboratory and in nature in the case of social insects, indicates that this correlation can be broken.
An important insight into the likely explanation for the ‘breaking’ or ‘uncoupling’ of trade-offs comes from the different outcomes of attempts to measure reproductive costs by looking at natural correlations across individuals as opposed to experimental manipulation of reproductive rate. Generally, across individuals in natural populations, there is a positive phenotypic correlation between fecundity and lifespan. However, the causal connection between the two traits may be the opposite, as experimental manipulations of, for instance, increasing clutch size in birds, often lead to reduced future fecundity or survival [232]. This difference occurs because the individual variation in condition and circumstances may obscure the underlying cost of reproduction: healthy individuals in a rich environment may have high fecundity and lifespan despite the cost of reproduction, which is only revealed by experimental manipulations. This underlying cost of reproduction may then constrain the combinations of life history traits that can evolve [233, 234]. Organisms that live in an environment that is beneficial for development may indeed not experience costs of reproduction [233, 234], as often seems to be the case in laboratory animals [235]. In addition, positive correlations between fitness-related traits can also be caused by mutational variation in recessive deleterious effects [236]. This arises because such deleterious mutations can have negative pleiotropic effects on two or more traits but the extent of these negative effects varies genetically among individuals.
Prevention of late-life morbidity in humans ideally would involve interventions that could be started at the earliest in middle age. Pharmacological prevention of cardiovascular disease, with statins and blood pressure lowerers, is already routine in clinical practice [237]. Unsurprisingly, many of the proteins that have turned out to be important in aging also play prominent roles in the etiology of age-related diseases, and are already the targets of licensed drugs. Consideration is hence starting to be given to widening the preventative, pharmacological approach, for instance by repurposing drugs such as rapamycin, which inhibits TOR and is used to treat cancer and to prevent rejection of transplanted organs, and metformin, used to treat type 2 diabetes and which may have several modes of action; importantly, both drugs have been found to extend lifespan in model organisms [238–241]. Other possible approaches to emerge from experimental work with animals include removal of damaging senescent cells that accumulate during aging [242, 243], use of factors from young blood that restore the age-related loss of function of stem cells or synapses between nerve cells in the brain [100, 244], and alteration of the composition of the microorganisms in the gut to a younger profile [245–247], which has already been shown to extend lifespan in the turquoise killifish [248].
However, despite the considerable promise of these approaches, the extent to which they can yield health benefits free of side effects needs detailed study, since they could pose some new challenges for an aged system. For instance, removal of senescent cells, or restoration of stem cell function, could be beneficial in the short term, but in the longer term could lead to stem cell exhaustion and tissue dysfunction.
Key lessons from the evolution of aging
We conclude our brief ‘tour d’horizon’ of the evolution of aging with four key messages:
Everything we know about the evolution of aging tells us that it is not a programmed process, so it has often been thought of as being intractable to experimental analysis or medical intervention. But evolutionarily conserved high-level regulators of phenotypic plasticity have turned out to be able to produce a major rearrangement of physiology and to ameliorate the effects of aging.
Amelioration of aging can protect against multiple types of loss of function and age-related diseases, potentially without side effects given that we can ‘shake off’ trade-offs in some circumstances—this is, at least potentially, very good news for the compression of late-life morbidity in humans.
Some species achieve extraordinary longevity—the longest-lived vertebrate, the Greenland shark (Somniosus microcephalus), reaches maturity at 150 years and lives ~ 400 years [249], and a clam, the ocean quahog (Arcrtica islandica), probably lives up to 500 years [250]. Notably also, bats and birds have longer lifespans than mammals with similar body sizes [251]. Although a major challenge, it will be revealing to understand a lot more about how these slow-aging creatures achieve their long lives, and whether their secrets could help to improve human health late in life.
Some species (e.g., Hydra) do apparently not age. Such organisms clearly deserve much more mechanistic investigation as they might hold key lessons for regeneration and repair and thus for our understanding of how long life can be achieved.
Acknowledgements
We are grateful to Brian Charlesworth, Jean-Michel Gaillard, and two anonymous reviewers for helpful comments on a previous version of our manuscript. TF is funded by the Swiss National Science Foundation (SNSF grants PP00P3_165836 and 310030E-164207) and a DFG Mercator Fellowship held at the EvoPAD program, Institute for Evolution and Biodiversity, University of Münster. LP is funded by the Max Planck Society, the Wellcome Trust, and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement number 741989).
Authors’ contributions
TF and LP wrote the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Finch CE. Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci U S A. 2010;107(suppl 1):1718–1724. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaupel JW, Carey JR, Christensen K, Johnson TE, Yashin AI, Holm NV, et al. Biodemographic trajectories of longevity. Science. 1998;280(5365):855–860. doi: 10.1126/science.280.5365.855. [DOI] [PubMed] [Google Scholar]
- 3.Wilmoth JR. Demography of longevity: past, present, and future trends. Exp Gerontol. 2000;35(9-10):1111–1129. doi: 10.1016/S0531-5565(00)00194-7. [DOI] [PubMed] [Google Scholar]
- 4.Oeppen J, Vaupel JW. Broken limits to life expectancy. Science. 2002;296(5570):1029–1031. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- 5.Vaupel JW. Biodemography of human ageing. Nature. 2010;464(7288):536–542. doi: 10.1038/nature08984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22(17):R741–RR52. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Crimmins EM. Lifespan and healthspan: past, present, and promise. Gerontologist. 2015;55(6):901–911. doi: 10.1093/geront/gnv130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salomon JA, Wang H, Freeman MK, Vos T, Flaxman AD, Lopez AD, et al. Healthy life expectancy for 187 countries, 1990–2010: a systematic analysis for the Global Burden Disease Study 2010. Lancet. 2012;380(9859):2144–2162. doi: 10.1016/S0140-6736(12)61690-0. [DOI] [PubMed] [Google Scholar]
- 10.Global Health Observatory data repository. Life expectancy and Healthy life expectancy. Data by WHO region. http://apps.who.int/gho/data/view.main.SDG2016LEXREGv?lang=en. Accessed 18 Jul 2018.
- 11.Younis A, Goldkorn R, Goldenberg I, Geva D, Tzur B, et al. Impaired fasting glucose is the major determinant of the 20-year mortality risk associated with metabolic syndrome in nondiabetic patients with stable coronary artery disease. J Am Heart Assoc. 2017;6:e006609. doi: 10.1161/JAHA.117.006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carone G, Costello D, Diez Guardia N, Mourre G, Przywara B, Salomäki A. The economic impact of ageing populations in the Eu25 member states, Directorate general for economic and financial affairs European economy economic working paper no. 236.2005. 10.2139/ssrn.873872.
- 13.Kontis V, Bennett JE, Mathers CD, Li G, Foreman K, Ezzati M. Future life expectancy in 35 industrialised countries: projections with a Bayesian model ensemble. Lancet. 2017;389(10076):1323–1335. doi: 10.1016/S0140-6736(16)32381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong X, Milholland B, Vijg J. Evidence for a limit to human lifespan. Nature. 2016;538(7624):257–259. doi: 10.1038/nature19793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenart A, Vaupel JW. Questionable evidence for a limit to human lifespan. Nature. 2017;546(7660):E13–EE4. doi: 10.1038/nature22790. [DOI] [PubMed] [Google Scholar]
- 16.Barbi E, Lagona F, Marsili M, Vaupel JW, Wachter KW. The plateau of human mortality: demography of longevity pioneers. Science. 2018;360:1459–1461. doi: 10.1126/science.aat3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tetzlaff J, Muschik D, Epping J, Eberhard S, Geyer S. Expansion or compression of multimorbidity? 10-year development of life years spent in multimorbidity based on health insurance claims data of Lower Saxony, Germany. Intl J Pub Health. 2017;62(6):679–686. doi: 10.1007/s00038-017-0962-9. [DOI] [PubMed] [Google Scholar]
- 18.Sebastiani P, Perls T. The genetics of extreme longevity: lessons from the New England centenarian study. Front Genet. 2012;3:277. doi: 10.3389/fgene.2012.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charlesworth B, Charlesworth D. Elements of evolutionary genetics. Greenwood Village: Roberts and Company Publishers; 2010. [Google Scholar]
- 20.Rose MR. Evolutionary biology of aging. New York and Oxford: Oxford University Press; 1991. [Google Scholar]
- 21.Finch CE. Longevity, senescence, and the genome. Chicago and London: The University of Chicago Press; 1990. [Google Scholar]
- 22.Nussey DH, Froy H, Lemaitre JF, Gaillard JM, Austad SN. Senescence in natural populations of animals: widespread evidence and its implications for bio-gerontology. Ageing Res Rev. 2013;12(1):214–225. doi: 10.1016/j.arr.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clutton-Brock T, Sheldon BC. Individuals and populations: the role of long-term, individual-based studies of animals in ecology and evolutionary biology. Trends Ecol Evol. 2010;25:562–573. doi: 10.1016/j.tree.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Charlesworth B. Evolutionary mechanisms of senescence. Genetica. 1993;91:11–19. doi: 10.1007/BF01435984. [DOI] [PubMed] [Google Scholar]
- 25.Baudisch A. Inevitable aging? Berlin: Springer; 2008. [Google Scholar]
- 26.Munné-Bosch S. Senescence: is it universal or not? Trends Plant Sci. 2015;20(11):713–720. doi: 10.1016/j.tplants.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Shefferson RP, Jones OR, Salguero-Gómez R. The evolution of senescence in the tree of life. Cambridge: Cambridge University Press; 2017. [Google Scholar]
- 28.Charlesworth B. Fisher, Medawar, Hamilton and the evolution of aging. Genetics. 2000;156(3):927–931. doi: 10.1093/genetics/156.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher RA. The genetical theory of natural selection. Oxford: Oxford University Press; 1930. [Google Scholar]
- 30.Haldane JBS. New paths in genetics. London: Allen and Unwin; 1941. [Google Scholar]
- 31.Medawar PB. Old age and natural death. Mod Quart. 1946;2:30–49. [Google Scholar]
- 32.Medawar PB. An unsolved problem of biology. London: H.K. Lewis; 1952. [Google Scholar]
- 33.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. doi: 10.1111/j.1558-5646.1957.tb02911.x. [DOI] [Google Scholar]
- 34.Hamilton WD. Moulding of senescence by natural selection. J Theor Biol. 1966;12(1):12–45. doi: 10.1016/0022-5193(66)90184-6. [DOI] [PubMed] [Google Scholar]
- 35.Baudisch A. Hamilton’s indicators of the force of selection. Proc Natl Acad Sci U S A. 2005;102(23):8263–8268. doi: 10.1073/pnas.0502155102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose MR, Rauser CL, Benford G, Matos M, Mueller LD. Hamilton’s forces of natural selection after forty years. Evolution. 2007;61(6):1265–1276. doi: 10.1111/j.1558-5646.2007.00120.x. [DOI] [PubMed] [Google Scholar]
- 37.Charlesworth B. Evolution in age-structured populations. 1. Cambridge: Cambridge University Press; 1980. [Google Scholar]
- 38.Charlesworth B. Evolution in age-structured populations. 2. Cambridge: Cambridge University Press; 1994. [Google Scholar]
- 39.Partridge L, Deelen J, Slagboom PE. Facing up to the global challenges of ageing. Nature. 2018; in press [DOI] [PubMed]
- 40.Kirkwood TBL. Evolution of ageing. Nature. 1977;270:301–304. doi: 10.1038/270301a0. [DOI] [PubMed] [Google Scholar]
- 41.Kirkwood TBL. The disposable soma theory of aging. In: Harrison DE, editor. Genetic effects on aging II. Caldwell: Telford Press; 1990. pp. 9–19. [Google Scholar]
- 42.Abrams PA, Ludwig D. Optimality theory, Gompertz’ law, and the disposable soma theory of senescence. Evolution. 1995;49(6):1055–1066. doi: 10.1111/j.1558-5646.1995.tb04433.x. [DOI] [PubMed] [Google Scholar]
- 43.Charlesworth B. Patterns of age-specific means and genetic variances of mortality rates predicted by the mutation accumulation theory of aging. J Theor Biol. 2001;210:47–65. doi: 10.1006/jtbi.2001.2296. [DOI] [PubMed] [Google Scholar]
- 44.Partridge L, Barton NH. Optimality, mutation and the evolution of ageing. Nature. 1993;362:305–311. doi: 10.1038/362305a0. [DOI] [PubMed] [Google Scholar]
- 45.Partridge L, Barton NH. Evolution of aging - testing the theory using Drosophila. Genetica. 1993;91(1-3):89–98. doi: 10.1007/BF01435990. [DOI] [PubMed] [Google Scholar]
- 46.Partridge L, Gems D. Mechanisms of ageing: public or private? Nat Rev Genet. 2002;3:165–175. doi: 10.1038/nrg753. [DOI] [PubMed] [Google Scholar]
- 47.Flatt T, Schmidt PS. Integrating evolutionary and molecular genetics of aging. Biochim Biophys Acta. 2009;1790(10):951–962. doi: 10.1016/j.bbagen.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stearns SC, Partridge L. The genetics of aging in Drosophila. In: Masoro E, Austad S, editors. Handbook of the biology of aging. 5th ed. Cambridge: Academic Press; 2001. p. 353–68.
- 49.Hughes KA, Reynolds RM. Evolutionary and mechanistic theories of aging. Annu Rev Entomol. 2005;50:421–445. doi: 10.1146/annurev.ento.50.071803.130409. [DOI] [PubMed] [Google Scholar]
- 50.Flatt T. Survival costs of reproduction in Drosophila. Exp Gerontol. 2011;46(5):369–375. doi: 10.1016/j.exger.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 51.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120(4):449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Gaillard JM, Lemaître JF. The Williams’ legacy: a critical reappraisal of his nine predictions about the evolution of senescence. Evolution. 2017;71(12):2768–2785. doi: 10.1111/evo.13379. [DOI] [PubMed] [Google Scholar]
- 53.Lemaitre JF, Berger V, Bonenfant C, Douhard M, Gamelon M, Plard F, et al. Early-late life trade-offs and the evolution of ageing in the wild. Proc R Soc Lond B. 2015;282(1806):20150209. [DOI] [PMC free article] [PubMed]
- 54.Rose M, Charlesworth B. A test of evolutionary theories of senescence. Nature. 1980;287:141–142. doi: 10.1038/287141a0. [DOI] [PubMed] [Google Scholar]
- 55.Rose MR. Laboratory evolution of postponed senescence in Drosophila melanogaster. Evolution. 1984;38(5):1004–1010. doi: 10.1111/j.1558-5646.1984.tb00370.x. [DOI] [PubMed] [Google Scholar]
- 56.Partridge L, Prowse N, Pignatelli P. Another set of responses and correlated responses to selection on age at reproduction in Drosophila melanogaster. Proc R Soc Lond B. 1999;266:255–261. doi: 10.1098/rspb.1999.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sgro CM, Partridge L. A delayed wave of death from reproduction in Drosophila. Science. 1999;286:2521–2524. doi: 10.1126/science.286.5449.2521. [DOI] [PubMed] [Google Scholar]
- 58.Stearns SC, Ackermann M, Doebeli M, Kaiser M. Experimental evolution of aging, growth, and reproduction in fruitflies. Proc Natl Acad Sci U S A. 2000;97:3309–3313. doi: 10.1073/pnas.97.7.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zwaan B, Bijlsma R, Hoekstra RF. Direct selection on life span in Drosophila melanogaster. Evolution. 1995;49(4):649–659. doi: 10.1111/j.1558-5646.1995.tb02301.x. [DOI] [PubMed] [Google Scholar]
- 60.Partridge L, Gems D, Withers DJ. Sex and death: what is the connection? Cell. 2005;120(4):461. doi: 10.1016/j.cell.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 61.Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 62.Kenyon C. A conserved regulatory system for aging. Cell. 2001;105(2):165–168. doi: 10.1016/S0092-8674(01)00306-3. [DOI] [PubMed] [Google Scholar]
- 63.Tatar M, Kopelman A, Epstein D, Tu M-P, Yin C-M, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 64.Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299(5611):1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 65.Paaby AB, Schmidt PS. Dissecting the genetics of longevity in Drosophila melanogaster. Fly (Austin) 2009;3(1):29–38. doi: 10.4161/fly.3.1.7771. [DOI] [PubMed] [Google Scholar]
- 66.Paaby AB, Bergland AO, Behrman EL, Schmidt PS. A highly pleiotropic amino acid polymorphism in the Drosophila insulin receptor contributes to life-history adaptation. Evolution. 2014;68(12):3395–3409. doi: 10.1111/evo.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paaby AB, Schmidt PS. Functional significance of allelic variation at methuselah, an aging gene in Drosophila. PLoS One. 2008;3(4):e1987. doi: 10.1371/journal.pone.0001987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Charlesworth B, Hughes KA. Age-specific inbreeding depression and components of genetic variance in relation to the evolution of senescence. Proc Natl Acad Sci U S A. 1996;93(12):6140–6145. doi: 10.1073/pnas.93.12.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaw FH, Promislow DE, Tatar M, Hughes KA, Geyer CJ. Toward reconciling inferences concerning genetic variation in senescence in Drosophila melanogaster. Genetics. 1999;152(2):553–566. doi: 10.1093/genetics/152.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hughes KA, Alipaz JA, Drnevich JM, Reynolds RM. A test of evolutionary theories of aging. Proc Natl Acad Sci U S A. 2002;99(22):14286–14291. doi: 10.1073/pnas.222326199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moorad JA, Promislow DE. What can genetic variation tell us about the evolution of senescence? Proc R Soc Lond B. 2009;276(1665):2271–2278. doi: 10.1098/rspb.2009.0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodríguez JA, Marigorta UM, Hughes DA, et al. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat Ecol Evol. 2017;1(3):55. doi: 10.1038/s41559-016-0055. [DOI] [PubMed] [Google Scholar]
- 73.Wright A, Charlesworth B, Rudan I, Carothers A, Campbell H. A polygenic basis for late-onset disease. Trends Genet. 2003;19(2):97–106. doi: 10.1016/S0168-9525(02)00033-1. [DOI] [PubMed] [Google Scholar]
- 74.Charlesworth B. Evolution of senescence: Alzheimer’s disease and evolution. Curr Biol. 1996;6(1):20–22. doi: 10.1016/S0960-9822(02)00411-6. [DOI] [PubMed] [Google Scholar]
- 75.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75(1):685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Byars SG, Huang QQ, Gray L-A, Bakshi A, Ripatti S, Abraham G, et al. Genetic loci associated with coronary artery disease harbor evidence of selection and antagonistic pleiotropy. PLoS Genet. 2017;13(6):e1006328. doi: 10.1371/journal.pgen.1006328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carter AJ, Nguyen AQ. Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med Genet. 2011;12(1):1–13. doi: 10.1186/1471-2350-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Byars SG, Stearns SC. Genetic links between post-reproductive lifespan and family size in Framingham. Evol Med Pub Health. 2013;2013(1):241–253. doi: 10.1093/emph/eot013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kang H-J, Feng Z, Sun Y, Atwal G, Murphy ME, Rebbeck TR, et al. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci U S A. 2009;106(24):9761–9766. doi: 10.1073/pnas.0904280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith KR, Hanson HA, Mineau GP, Buys SS. Effects of BRCA1 and BRCA2 mutations on female fertility. Proc R Soc Lond B. 2012;279(1732):1389–1395. doi: 10.1098/rspb.2011.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moorad JA, Walling CA. Measuring selection for genes that promote long life in a historical human population. Nat Ecol Evol. 2017;1:1773–1781. doi: 10.1038/s41559-017-0329-x. [DOI] [PubMed] [Google Scholar]
- 82.Abrams PA. Does increased mortality favor the evolution of more and rapid senescence? Evolution. 1993;47(3):877–887. doi: 10.1111/j.1558-5646.1993.tb01241.x. [DOI] [PubMed] [Google Scholar]
- 83.Williams PD, Day T. Antagonistic pleiotropy, mortality source interactions, and the evolutionary theory of senescence. Evolution. 2003;57(7):1478–1488. doi: 10.1111/j.0014-3820.2003.tb00356.x. [DOI] [PubMed] [Google Scholar]
- 84.Caswell H. Extrinsic mortality and the evolution of senescence. Trends Ecol Evol. 2007;22(4):173–174. doi: 10.1016/j.tree.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 85.Gaillard J-M, Festa-Bianchet M, Yoccoz NG, Loison A, Toïgo C. Temporal variation in fitness components and population dynamics of large herbivores. Annu Rev Ecol Syst. 2000;31:367–393. doi: 10.1146/annurev.ecolsys.31.1.367. [DOI] [Google Scholar]
- 86.Williams PD, Day T, Fletcher Q, Rowe L. The shaping of senescence in the wild. Trends Ecol Evol. 2006;21(8):458–463. doi: 10.1016/j.tree.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 87.Reznick DN, Bryant MJ, Roff D, Ghalambor CK, Ghalambor DE. Effect of extrinsic mortality on the evolution of senescence in guppies. Nature. 2004;431(7012):1095–1099. doi: 10.1038/nature02936. [DOI] [PubMed] [Google Scholar]
- 88.Chen H, Maklakov AA. Longer life span evolves under high rates of condition-dependent mortality. Curr Biol. 2012;22(22):2140–2143. doi: 10.1016/j.cub.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 89.Kirkwood TBL, Martin GM, Partridge L. Evolution, senescence and health in old age. In: Stearns SC, editor. Evolution in health and disease. Oxford: Oxford University Press; 1999. pp. 219–230. [Google Scholar]
- 90.Burke MK, King EG, Shahrestani P, Rose MR, Long AD. Genome-wide association study of extreme longevity in Drosophila melanogaster. Genome Biol Evol. 2014;6(1):1–11. doi: 10.1093/gbe/evt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Highfill CA, Reeves GA, Macdonald SJ. Genetic analysis of variation in lifespan using a multiparental advanced intercross Drosophila mapping population. BMC Genet. 2016;17(1):113. doi: 10.1186/s12863-016-0419-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ivanov DK, Escott-Price V, Ziehm M, Magwire MM, Mackay TFC, Partridge L, et al. Longevity GWAS using the Drosophila genetic reference panel. J Gerontol A Biol Sci Med Sci. 2015;70(12):1470–1478. doi: 10.1093/gerona/glv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reznick DN. The genetic basis of aging: an evolutionary biologist’s perspective. Sci Aging Knowl Environ. 2005;2005(11):pe7. doi: 10.1126/sageke.2005.11.pe7. [DOI] [PubMed] [Google Scholar]
- 94.McElwee JJ, Schuster E, Blanc E, Piper MD, Thomas JH, Patel DS, et al. Evolutionary conservation of regulated longevity assurance mechanisms. Genome Biol. 2007;8(7):R132. doi: 10.1186/gb-2007-8-7-r132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith ED, Tsuchiya M, Fox LA, Dang N, Hu D, Kerr EO, et al. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 2008;18(4):564–570. doi: 10.1101/gr.074724.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kenyon CJ. The genetics of ageing. Nature. 2010;464(7288):504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 97.Fontana L, Partridge L, Longo VD. Extending healthy life span – from yeast to humans. Science. 2010;328(5976):321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539:180. doi: 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Castellano JM, Mosher KI, Abbey RJ, McBride AA, James ML, Berdnik D, et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature. 2017;544:488–492. doi: 10.1038/nature22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- 101.Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161(1):106–118. doi: 10.1016/j.cell.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anderson RM, Le Couteur DG, de Cabo R. Caloric restriction research: new perspectives on the biology of aging. J Gerontol A Biol Sci Med Sci. 2017;73(1):1–3. doi: 10.1093/gerona/glx212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kapahi P, Kaeberlein M, Hansen M. Dietary restriction and lifespan: lessons from invertebrate models. Ageing Res Rev. 2017;39:3–14. doi: 10.1016/j.arr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gardner EM. Caloric restriction decreases survival of aged mice in response to primary influenza infection. J Gerontol A Biol Sci Med Sci. 2005;60(6):688–694. doi: 10.1093/gerona/60.6.688. [DOI] [PubMed] [Google Scholar]
- 105.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557. doi: 10.1038/ncomms4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ingram DK, de Cabo R. Calorie restriction in rodents: caveats to consider. Ageing Res Rev. 2017;39:15–28. doi: 10.1016/j.arr.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grandison RC, Piper MDW, Partridge L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature. 2009;462(7276):1061–1065. doi: 10.1038/nature08619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Levine ME, Suarez Jorge A, Brandhorst S, Balasubramanian P, Cheng C-W, Madia F, et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014;19(3):407–417. doi: 10.1016/j.cmet.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mirzaei H, Suarez JA, Longo VD. Protein and amino acid restriction, aging and disease: from yeast to humans. Trends Endocrinol Metab. 2014;25(11):558–566. doi: 10.1016/j.tem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Solon-Biet Samantha M, McMahon Aisling C, Ballard JWilliam O, Ruohonen K, Wu Lindsay E, Cogger Victoria C, et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014;19(3):418–430. doi: 10.1016/j.cmet.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Simpson SJ, Le Couteur DG, Raubenheimer D, Solon-Biet SM, Cooney GJ, Cogger VC, et al. Dietary protein, aging and nutritional geometry. Ageing Res Rev. 2017;39:78–86. doi: 10.1016/j.arr.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 114.Piper MDW, Soultoukis GA, Blanc E, Mesaros A, Herbert SL, Juricic P, et al. Matching dietary amino acid balance to the in silico-translated exome optimizes growth and reproduction without cost to lifespan. Cell Metab. 2017;25(3):610–621. doi: 10.1016/j.cmet.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol. 2018;596(4):623–645. doi: 10.1113/JP275075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mattson MP, Allison DB, Fontana L, Harvie M, Longo VD, Malaisse WJ, et al. Meal frequency and timing in health and disease. Proc Natl Acad Sci U S A. 2014;111(47):16647–16653. doi: 10.1073/pnas.1413965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brandhorst S, Choi In Y, Wei M, Cheng Chia W, Sedrakyan S, Navarrete G, et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015;22(1):86–99. doi: 10.1016/j.cmet.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Longo VD, Panda S. Fasting, circadian rhythms, and time-restricted feeding in healthy lifespan. Cell Metab. 2016;23(6):1048–1059. doi: 10.1016/j.cmet.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martinez-Lopez N, Tarabra E, Toledo M, Garcia-Macia M, Sahu S, Coletto L, et al. System-wide benefits of intermeal fasting by autophagy. Cell Metab. 2017;26(6):856–71.e5. doi: 10.1016/j.cmet.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moatt JP, Nakagawa S, Lagisz M, Walling CA. The effect of dietary restriction on reproduction: a meta-analytic perspective. BMC Evol Biol. 2016;16(1):199. doi: 10.1186/s12862-016-0768-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shanley DP, Kirkwood TBL. Calorie restriction and aging: a life-history analysis. Evolution. 2000;54(3):740–750. doi: 10.1111/j.0014-3820.2000.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 122.Zajitschek F, Georgolopoulos G, Vourlou A, et al. Evolution under dietary restriction decouples survival from fecundity in Drosophila melanogaster females. J Gerontol A Biol Sci Med Sci. 2018; in press [DOI] [PubMed]
- 123.Strandin T, Babayan SA, Forbes KM. Reviewing the effects of food provisioning on wildlife immunity. Philos Trans R Soc Lond B. 2018;373(1745):20170088. doi: 10.1098/rstb.2017.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boutin S. Food supplementation experiments with terrestrial vertebrates: patterns, problems, and the future. Can J Zool. 1990;68:203–220. doi: 10.1139/z90-031. [DOI] [Google Scholar]
- 125.Ruffino L, Salo P, Koivisto E, Banks PB, Korpimäki E. Reproductive responses of birds to experimental food supplementation: a meta-analysis. Front Zool. 2014;11:80. doi: 10.1186/s12983-014-0080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Klass M, Hirsh D. Non-ageing developmental variant of Caenorhabditis elegans. Nature. 1976;260:523–525. doi: 10.1038/260523a0. [DOI] [PubMed] [Google Scholar]
- 127.Klass MR. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech Ageing Dev. 1983;22(3-4):279–286. doi: 10.1016/0047-6374(83)90082-9. [DOI] [PubMed] [Google Scholar]
- 128.Johnson TE, Mitchell DH, Kline S, Kemal R, Foy J. Arresting development arrests aging in the nematode Caenorhabditis elegans. Mech Ageing Dev. 1984;28(1):23–40. doi: 10.1016/0047-6374(84)90150-7. [DOI] [PubMed] [Google Scholar]
- 129.Johnson TE. Aging can be genetically dissected into component processes using long-lived lines of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1987;84(11):3777–3781. doi: 10.1073/pnas.84.11.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118(1):75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Johnson TE, Lithgow GJ. The search for the genetic basis of aging: the identification of gerontogenes in the nematode Caenorhabditis elegans. J Am Geriatr Soc. 1992;40(9):936–945. doi: 10.1111/j.1532-5415.1992.tb01993.x. [DOI] [PubMed] [Google Scholar]
- 132.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366(6454):461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 133.Kenyon C. The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos Trans R Soc Lond B. 2011;366(1561):9–16. doi: 10.1098/rstb.2010.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morris J, Tissenbaum H, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- 135.Kimura K, Tissenbaum H, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 136.Ogg S, Paradis S, Gottlieb S, Patterson G, Lee L, Tissenbaum H, et al. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 137.Lee S, Kennedy S, Tolonen A, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- 138.Lin K, Dorman J, Rodan A, Kenyon C. daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 139.Murphy C, McCarroll S, Bargmann C, Fraser A, Kamath R, Ahringer J, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–284. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 140.Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429(6991):562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 141.Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 2004;305(5682):361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- 142.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14(10):885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299(5606):572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 144.Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008;22(3):807–818. doi: 10.1096/fj.07-9261com. [DOI] [PubMed] [Google Scholar]
- 145.Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloen A, Even PC, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 146.Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317(5836):369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- 147.Flachsbart F, Caliebe A, Kleindorp R, Blanché H, von Eller-Eberstein H, Nikolaus S, et al. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A. 2009;106(8):2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105(37):13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, Leahy DJ, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A. 2008;105(9):3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tazearslan C, Huang J, Barzilai N, Suh Y. Impaired IGF1R signaling in cells expressing longevity-associated human IGF1R alleles. Aging Cell. 2011;10(3):551–554. doi: 10.1111/j.1474-9726.2011.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Passtoors WM, Beekman M, Deelen J, van der Breggen R, Maier AB, Guigas B, et al. Gene expression analysis of mTOR pathway: association with human longevity. Aging Cell. 2013;12(1):24–31. doi: 10.1111/acel.12015. [DOI] [PubMed] [Google Scholar]
- 152.Flachsbart F, Dose J, Gentschew L, Geismann C, Caliebe A, Knecht C, et al. Identification and characterization of two functional variants in the human longevity gene FOXO3. Nat Commun. 2017;8(1):2063. doi: 10.1038/s41467-017-02183-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11(1):35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13(3):468–477. doi: 10.1111/acel.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kennedy BK, Lamming DW. The mechanistic target of rapamycin: the grand conducTOR of metabolism and aging. Cell Metab. 2016;23(6):990–1003. doi: 10.1016/j.cmet.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Swanson EM, Dantzer B. Insulin-like growth factor-1 is associated with life-history variation across Mammalia. Proc R Soc Lond B. 2014;281(1782):20132458. doi: 10.1098/rspb.2013.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dantzer B, Swanson EM. Mediation of vertebrate life histories via insulin-like growth factor-1. Biol Rev. 2012;87(2):414–429. doi: 10.1111/j.1469-185X.2011.00204.x. [DOI] [PubMed] [Google Scholar]
- 160.De Jong G, Bochdanovits Z. Latitudinal clines in Drosophila melanogaster: body size, allozyme frequencies, inversion frequencies, and the insulin-signalling pathway. J Genet. 2003;82(3):207–223. doi: 10.1007/BF02715819. [DOI] [PubMed] [Google Scholar]
- 161.Fabian DK, Kapun M, Nolte V, Kofler R, Schmidt PS, Schlötterer C, et al. Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol Ecol. 2012;21(19):4748–4769. doi: 10.1111/j.1365-294X.2012.05731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kapun M, Fabian DK, Goudet J, Flatt T. Genomic evidence for adaptive inversion clines in Drosophila melanogaster. Mol Biol Evol. 2016;33(5):1317–1336. doi: 10.1093/molbev/msw016. [DOI] [PubMed] [Google Scholar]
- 163.Flatt T, Amdam GV, Kirkwood TBL, Omholt SW. Life-history evolution and the polyphenic regulation of somatic maintenance and survival. Q Rev Biol. 2013;88(3):185–218. doi: 10.1086/671484. [DOI] [PubMed] [Google Scholar]
- 164.Paaby AB, Blacket MJ, Hoffmann AA, Schmidt PS. Identification of a candidate adaptive polymorphism for Drosophila life history by parallel independent clines on two continents. Mol Ecol. 2010;19(4):760–774. doi: 10.1111/j.1365-294X.2009.04508.x. [DOI] [PubMed] [Google Scholar]
- 165.Stanley PD, Ng’oma E, O’Day S, King EG. Genetic dissection of nutrition-induced plasticity in insulin/insulin-like growth factor signaling and median life span in a Drosophila multiparent population. Genetics. 2017;206(2):587–602. doi: 10.1534/genetics.116.197780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rodrigues MA, Flatt T. Endocrine uncoupling of the trade-off between reproduction and somatic maintenance in eusocial insects. Curr Opin Insect Sci. 2016;16:1–8. doi: 10.1016/j.cois.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 167.Ament SA, Corona M, Pollock HS, Robinson GE. Insulin signaling is involved in the regulation of worker division of labor in honey bee colonies. Proc Natl Acad Sci U S A. 2008;105(11):4226–4231. doi: 10.1073/pnas.0800630105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Corona M, Velarde RA, Remolina S, Moran-Lauter A, Wang Y, Hughes KA, et al. Vitellogenin, juvenile hormone, insulin signaling, and queen honey bee longevity. Proc Natl Acad Sci U S A. 2007;104(17):7128–7133. doi: 10.1073/pnas.0701909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang Y, Mutti NS, Ihle KE, Siegel A, Dolezal AG, Kaftanoglu O, et al. Down-regulation of honey bee IRS gene biases behavior toward food rich in protein. PLoS Genet. 2010;6(4):e1000896. doi: 10.1371/journal.pgen.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mutti NS, Wang Y, Kaftanoglu O, Amdam GV. Honey bee PTEN - description, developmental knockdown, and tissue-specific expression of splice-variants correlated with alternative social phenotypes. PLoS One. 2011;6(7):e22195. doi: 10.1371/journal.pone.0022195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang Y, Azevedo SV, Hartfelder K, Amdam G. Insulin-like peptides (AmILP1 and AmILP2) differentially affect female caste development in the honey bee (Apis mellifera) J Exp Biol. 2013;216:4347–4357. doi: 10.1242/jeb.085779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Patel A, Fondrk MK, Kaftanoglu O, Emore C, Hunt G, Frederick K, et al. The making of a queen: TOR pathway is a key player in diphenic caste development. PLoS One. 2007;2(6):e509. doi: 10.1371/journal.pone.0000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Libbrecht R, Corona M, Wende F, Azevedo DO, Serrão JE, Keller L. Interplay between insulin signaling, juvenile hormone, and vitellogenin regulates maternal effects on polyphenism in ants. Proc Natl Acad Sci U S A. 2013;110(27):11050–11055. doi: 10.1073/pnas.1221781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kirkwood TBL. Understanding the odd science of aging. Cell. 2005;120(4):437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 175.Flatt T, Heyland A. Mechanisms of life history evolution - the genetics and physiology of life history traits and trade-offs. Oxford: Oxford University Press; 2011. [Google Scholar]
- 176.Geiger-Thornsberry GL, Mackay TF. Quantitative trait loci affecting natural variation in Drosophila longevity. Mech Ageing Dev. 2004;125(3):179–189. doi: 10.1016/j.mad.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 177.Flatt T. Assessing natural variation in genes affecting Drosophila lifespan. Mech Ageing Dev. 2004;125(3):155–159. doi: 10.1016/j.mad.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 178.Remolina SC, Chang PL, Leips J, Nuzhdin SV, Hughes KA. Genomic basis of aging and life-history evolution in Drosophila melanogaster. Evolution. 2012;66(11):3390–3403. doi: 10.1111/j.1558-5646.2012.01710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Carnes MU, Campbell T, Huang W, Butler DG, Carbone MA, Duncan LH, et al. The genomic basis of postponed senescence in Drosophila melanogaster. PLoS One. 2015;10(9):e0138569. doi: 10.1371/journal.pone.0138569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Minghui W, Qishan W, Zhen W, Qingping W, Xiangzhe Z, Yuchun P. The molecular evolutionary patterns of the insulin/FOXO signaling pathway. Evol Bioinforma. 2013;9:1–16. doi: 10.6026/97320630009001. [DOI] [Google Scholar]
- 181.Alvarez-Ponce D, Aguade M, Rozas J. Comparative genomics of the vertebrate insulin/TOR signal transduction pathway: a network-level analysis of selective pressures. Genome Biol Evol. 2011;3:87–101. doi: 10.1093/gbe/evq084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Garschall K, Flatt T. The interplay between immunity and aging in Drosophila. F1000Res. 2018;7:160. doi: 10.12688/f1000research.13117.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sanada F, Taniyama Y, Muratsu J, Otsu R, Shimizu H, Rakugi H, Morishita R. Source of chronic inflammation in aging. Front Cardiovasc Med. 2018;5:12. doi: 10.3389/fcvm.2018.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bektas A, Schurman SH, Sen R, Ferrucci L. Aging, inflammation and the environment. Exp Gerontol. 2018;105:10–18. doi: 10.1016/j.exger.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Comfort A. The biology of senescence. London: Routledhe & Kegan Paul; 1956. [Google Scholar]
- 186.Jones OR, Scheuerlein A, Salguero-Gomez R, Camarda CG, Schaible R, Casper BB, et al. Diversity of ageing across the tree of life. Nature. 2014;505(7482):169–173. doi: 10.1038/nature12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Baudisch A, Salguero-Gómez R, Jones OR, Wrycza T, Mbeau-Ache C, Franco M, et al. The pace and shape of senescence in angiosperms. J Ecol. 2013;101(3):596–506. doi: 10.1111/1365-2745.12084. [DOI] [Google Scholar]
- 188.Garcia MB, Dahlgren JP, Ehrlén J. No evidence of senescence in a 300-year-old mountain herb. J Ecol. 2011;99(6):1424–1430. doi: 10.1111/j.1365-2745.2011.01871.x. [DOI] [Google Scholar]
- 189.Schaible R, Scheuerlein A, Dańko MJ, Gampe J, Martínez DE, Vaupel JW. Constant mortality and fertility over age in Hydra. Proc Natl Acad Sci U S A. 2015;112(51):15701–15706. doi: 10.1073/pnas.1521002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ruby JG, Smith M, Buffenstein R. Naked mole-rat mortality rates defy Gompertzian laws by not increasing with age. elife. 2018;7:e31157. doi: 10.7554/eLife.31157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lemaître J-F, Gaillard J-M. Reproductive senescence: new perspectives in the wild. Biol Rev. 2017;92:2182–2199. doi: 10.1111/brv.12328. [DOI] [PubMed] [Google Scholar]
- 192.Finch CE, Austad SN. History and prospects: symposium on organisms with slow aging. Exp Gerontol. 2001;36(4):593–597. doi: 10.1016/S0531-5565(00)00228-X. [DOI] [PubMed] [Google Scholar]
- 193.Finch CE. Update on slow aging and negligible senescence - a mini-review. Gerontology. 2009;55(3):307–313. doi: 10.1159/000215589. [DOI] [PubMed] [Google Scholar]
- 194.Finch CE. Variations in senescence and longevity include the possibility of negligible senescence. J Gerontol A Biol Sci Med Sci. 1998;53(4):B235–B239. doi: 10.1093/gerona/53A.4.B235. [DOI] [PubMed] [Google Scholar]
- 195.Vaupel JW, Baudisch A, Dolling M, Roach DA, Gampe J. The case for negative senescence. Theor Popul Biol. 2004;65(4):339–351. doi: 10.1016/j.tpb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 196.Salguero-Gómez R, Shefferson RP, Hutchings MJ. Plants do not count … or do they? New perspectives on the universality of senescence. J Ecol. 2013;101(3):545–554. doi: 10.1111/1365-2745.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Peron G, Gimenez O, Charmantier A, Gaillard JM, Crochet PA. Age at the onset of senescence in birds and mammals is predicted by early-life performance. Proc R Soc Lond B. 2010;277:2849–2856. doi: 10.1098/rspb.2010.0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Warner DA, Miller DAW, Bronikowski AM, Janzen FJ. Decades of field data reveal that turtles senesce in the wild. Proc Natl Acad Sci U S A. 2016;113:6502–6507. doi: 10.1073/pnas.1600035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Partridge L, Barton NH. On measuring the rate of ageing. Proc R Soc Lond B. 1996;263(1375):1365–1371. doi: 10.1098/rspb.1996.0200. [DOI] [Google Scholar]
- 200.Petralia RS, Mattson MP, Yao PJ. Aging and longevity in the simplest animals and the quest for immortality. Ageing Res Rev. 2014;16:66–82. doi: 10.1016/j.arr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bythell JC, Brown BE, Kirkwood TBL. Do reef corals age? Biol Rev. 2017;93(2):1192–1102. doi: 10.1111/brv.12391. [DOI] [PubMed] [Google Scholar]
- 202.Noodén LD. Whole plant senescence. In: Thiman KV, Leopold AC, editors. Senescence and aging in plants. Cambridge: Academic Press; 1988. p. 391–439.
- 203.Nelson P, Masel J. Intercellular competition and the inevitability of multicellular aging. Proc Natl Acad Sci U S A. 2017;114(49):12982–12987. doi: 10.1073/pnas.1618854114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bell G. Sex and death in protozoa. Cambridge: Cambridge University Press; 1988. [Google Scholar]
- 205.Kirkwood TBL. Immortality of the germ-line versus disposability of the soma. In: Woodhead AD, Thompson KH, editors. Evolution of longevity in animals: a comparative approach. Boston: Springer US; 1987. pp. 209–218. [Google Scholar]
- 206.Jones DL. Aging and the germ line: where mortality and immortality meet. Stem Cell Rev. 2007;3(3):192–200. doi: 10.1007/s12015-007-0009-3. [DOI] [PubMed] [Google Scholar]
- 207.Weissmann A. Die Kontinuität des Keimplasmas als Grundlage einer Theorie der Vererbung. Jena: Gustav Fischer; 1885. [Google Scholar]
- 208.Ackermann M, Chao L, Bergstrom CT, Doebeli M. On the evolutionary origin of aging. Aging Cell. 2007;6(2):235–244. doi: 10.1111/j.1474-9726.2007.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Buffenstein R. Negligible senescence in the longest living rodent, the naked mole-rat: insights from a successfully aging species. J Comp Physiol B. 2008;178(4):439–445. doi: 10.1007/s00360-007-0237-5. [DOI] [PubMed] [Google Scholar]
- 210.Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature. 2011;479(7372):223–227. doi: 10.1038/nature10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Valenzano DR, Aboobaker A, Seluanov A, Gorbunova V. Non-canonical aging model systems and why we need them. EMBO J. 2017;36(8):959–963. doi: 10.15252/embj.201796837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kim Y, Nam HG, Valenzano DR. The short-lived African turquoise killifish: an emerging experimental model for ageing. Dis Model Mech. 2016;9(2):115–129. doi: 10.1242/dmm.023226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Valenzano Dario R, Benayoun Bérénice A, Singh Param P, Zhang E, Etter Paul D, Hu C-K, et al. The African turquoise killifish genome provides insights into evolution and genetic architecture of lifespan. Cell. 2015;163(6):1539–1554. doi: 10.1016/j.cell.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Monaghan P, Charmantier A, Nussey DH, Ricklefs RE. The evolutionary ecology of senescence. Funct Ecol. 2008;22(3):371–378. doi: 10.1111/j.1365-2435.2008.01418.x. [DOI] [Google Scholar]
- 215.Wilkinson GS, South JM. Life history, ecology and longevity in bats. Aging Cell. 2002;1(2):124–131. doi: 10.1046/j.1474-9728.2002.00020.x. [DOI] [PubMed] [Google Scholar]
- 216.Keller L, Genoud M. Extraordinary lifespans in ants: a test of evolutionary theories of ageing. Nature. 1997;389:958–960. doi: 10.1038/40130. [DOI] [Google Scholar]
- 217.Keller L, Jemielity S. Social insects as a model to study the molecular basis of ageing. Exp Gerontol. 2006;41:553–556. doi: 10.1016/j.exger.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 218.Kuhn JMM, Korb J. Editorial overview: social insects: aging and the re-shaping of the fecundity/longevity trade-off with sociality. Curr Opin Insect Sci. 2016;16:vii–vix. doi: 10.1016/j.cois.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 219.Heinze J, Schrempf A. Aging and reproduction in social insects – a mini-review. Gerontology. 2008;54:160–167. doi: 10.1159/000122472. [DOI] [PubMed] [Google Scholar]
- 220.Schrempf A, Giehr J, Röhrl R, Steigleder S, Heinze J. Royal Darwinian demons: enforced changes in reproductive efforts do not affect the life expectancy of ant queens. Am Nat. 2017;189(4):436–442. doi: 10.1086/691000. [DOI] [PubMed] [Google Scholar]
- 221.von Wyschetzki K, Rueppell O, Oettler J, Heinze J. Transcriptomic signatures mirror the lack of the fecundity/longevity trade-off in ant queens. Mol Biol Evol. 2015;32:3173–3185. doi: 10.1093/molbev/msv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hartmann A, Heinze J. Lay eggs, live longer: division of labor and life span in a clonal ant species. Evolution. 2003;57:2424–2429. doi: 10.1111/j.0014-3820.2003.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 223.Kramer BH, Schrempf A, Scheuerlein A, Heinze J. Ant colonies do not trade-off reproduction against maintenance. PLoS One. 2015;10:e0137969. doi: 10.1371/journal.pone.0137969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Blacher P, Huggins TJ, Bourke AFG. Evolution of ageing, costs of reproduction and the fecundity–longevity trade-off in eusocial insects. Proc R Soc Lond B. 2017;284:20170380. doi: 10.1098/rspb.2017.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Buffenstein R, Jarvis JUM. The naked mole rat - a new record for the oldest living rodent. Sci Aging Knowl Environ. 2002;2002(21):pe7. doi: 10.1126/sageke.2002.21.pe7. [DOI] [PubMed] [Google Scholar]
- 226.Kramer BH, van Doorn GS, Weissing FJ, Pen I. Lifespan divergence between social insect castes: challenges and opportunities for evolutionary theories of aging. Curr Opin Insect Sci. 2016;16:76–80. doi: 10.1016/j.cois.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 227.Kramer BH, Schaible R. Life span evolution in eusocial workers - a theoretical approach to understanding the effects of extrinsic mortality in a hierarchical system. PLoS One. 2013;8:e61813. doi: 10.1371/journal.pone.0061813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bourke AFG. Kin selection and the evolutionary theory of aging. Annu Rev Ecol Evol Syst. 2007;38:103–128. doi: 10.1146/annurev.ecolsys.38.091206.095528. [DOI] [Google Scholar]
- 229.Michod RE. Evolution of individuality during the transition from unicellular to multicellular life. Proc Natl Acad Sci U S A. 2007;104:8613–8618. doi: 10.1073/pnas.0701489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Nedelcu A, Michod RE. Molecular mechanisms of life history trade-offs and the evolution of multicellular complexity in volvocalean green algae. In: Flatt T, Heyland A, editors. Mechanisms of life history evolution – the genetics and physiology of life history traits and trade-offs. Oxford: Oxford University Press; 2011. p. 271–83.
- 231.Rueffler C, Hermisson J, Wagner GP. Evolution of functional specialization and division of labor. Proc Natl Acad Sci U S A. 2012;109(6):1830–1831. doi: 10.1073/pnas.1110521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Partridge L. Measuring reproductive costs. Trends Ecol Evol. 1992;7(3):99–100. doi: 10.1016/0169-5347(92)90250-F. [DOI] [PubMed] [Google Scholar]
- 233.van Noordwijk A, de Jong G. Acquisition and allocation of resources: their influence on variation in life history tactics. Am Nat. 1986;128(1):137–142. doi: 10.1086/284547. [DOI] [Google Scholar]
- 234.Metcalf CJE. Invisible trade-offs: Van Noordwijk and de Jong and life-history evolution. Am Nat. 2016;187(4):iii–iiv. doi: 10.1086/685487. [DOI] [PubMed] [Google Scholar]
- 235.Klepsatel P, Gáliková M, Maio N, Ricci S, Schlötterer C, Flatt T. Reproductive and post-reproductive life history of wild-caught Drosophila melanogaster under laboratory conditions. J Evol Biol. 2013;26(7):1508–1520. doi: 10.1111/jeb.12155. [DOI] [PubMed] [Google Scholar]
- 236.Charlesworth B. Optimization models, quantitative genetics, and mutation. Evolution. 1990;44:520–538. doi: 10.1111/j.1558-5646.1990.tb05936.x. [DOI] [PubMed] [Google Scholar]
- 237.Sundström J, Gulliksson G, Wirén M. Synergistic effects of blood pressure-lowering drugs and statins: systematic review and meta-analysis. BMJ Evid Based Med. 2018;23(2):64–69. doi: 10.1136/bmjebm-2017-110888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Blenis J. TOR, the gateway to cellular metabolism, cell growth, and disease. Cell. 2017;171(1):10–13. doi: 10.1016/j.cell.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 239.Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6(268):268ra179. doi: 10.1126/scitranslmed.3009892. [DOI] [PubMed] [Google Scholar]
- 240.Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab. 2016;23(6):1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Johnson SC, Kaeberlein M. Rapamycin in aging and disease: maximizing efficacy while minimizing side effects. Oncotarget. 2016;7(29):44876–44878. doi: 10.18632/oncotarget.10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Childs BG, Gluscevic M, Baker DJ, Laberge R-M, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16:718–735. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rando Thomas A, Chang Howard Y. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148(1):46–57. doi: 10.1016/j.cell.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Clark RI, Walker DW. Role of gut microbiota in aging-related health decline: insights from invertebrate models. Cell Mol Life Sci. 2018;75(1):93–101. doi: 10.1007/s00018-017-2671-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kundu P, Blacher E, Elinav E, Pettersson S. Our gut microbiome: the evolving inner self. Cell. 2017;171(7):1481–1493. doi: 10.1016/j.cell.2017.11.024. [DOI] [PubMed] [Google Scholar]
- 247.Schmidt TSB, Raes J, Bork P. The human gut microbiome: from association to modulation. Cell. 2018;172(6):1198–1215. doi: 10.1016/j.cell.2018.02.044. [DOI] [PubMed] [Google Scholar]
- 248.Smith P, Willemsen D, Popkes M, Metge F, Gandiwa E, Reichard M, Valenzano DR. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. elife. 2017;6:e27014. doi: 10.7554/eLife.27014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Nielsen J, Hedeholm RB, Heinemeier J, Bushnell PG, Christiansen JS, Olsen J, Ramsey CB, Brill RW, Simon M, Steffensen KF, et al. Eye lens radiocarbon reveals centuries of longevity in the Greenland shark (Somniosus microcephalus) Science. 2016;353(6300):702–704. doi: 10.1126/science.aaf1703. [DOI] [PubMed] [Google Scholar]
- 250.AnAge: The Animal Ageing and Longevity Database. Arctica islandica. http://genomics.senescence.info/species/entry.php?species=Arctica_islandica. Accessed 18 Jul 2018.
- 251.Prothero J, Jürgens KD. Scaling of lifespan in mammals. Basic Life Sci. 1987;42:49–74. doi: 10.1007/978-1-4613-1939-9_4. [DOI] [PubMed] [Google Scholar]