


Abstract
Antibody engineering is often performed to improve therapeutic properties by directed evolution, usually by high-throughput screening of phage or yeast display libraries. Engineering antibodies in mammalian cells offer advantages associated with expression in their final therapeutic format (full-length glycosylated IgG); however, the inability to express large and diverse libraries severely limits their potential throughput. To address this limitation, we have developed homology-directed mutagenesis (HDM), a novel method which extends the concept of CRISPR/Cas9-mediated homology-directed repair (HDR). HDM leverages oligonucleotides with degenerate codons to generate site-directed mutagenesis libraries in mammalian cells. By improving HDR to a robust efficiency of 15–35% and combining mammalian display screening with next-generation sequencing, we validated this approach can be used for key applications in antibody engineering at high-throughput: rational library construction, novel variant discovery, affinity maturation and deep mutational scanning (DMS). We anticipate that HDM will be a valuable tool for engineering and optimizing antibodies in mammalian cells, and eventually enable directed evolution of other complex proteins and cellular therapeutics.
INTRODUCTION
Following their initial discovery, antibody drug candidates typically require further engineering to increase target affinity or improve a number of other characteristics associated with therapeutic developability (e.g. immunogenicity, stability, solubility) (1). This is independent of the original source of the antibody (i.e. immunized animals, recombinant or synthetic libraries) (2). Even with a lead candidate to start from, the potential protein sequence space to explore and optimize for all the relevant drug parameters expands astronomically. Therefore, antibody engineering is done at high-throughput by library mutagenesis and directed evolution using surface display screening, most notably phage and yeast display (3–6). With some exceptions (7,8), these display systems typically express antibody proteins as fragments [e.g. single-chain fragment variable (scFv) and fragment antigen binding (Fab)] and without certain post-translational modifications (i.e. glycosylation). However, for therapeutic production, scFvs and Fabs require conversion into full-length glycosylated IgG molecules which consequentially leads to a final optimization phase of evaluating and modifying drug candidates directly in mammalian cells. This step is performed at low-throughput due to the challenges associated with generating libraries in mammalian systems (i.e. inability to stably retain and replicate plasmids).
When engineering candidate antibodies, libraries are often constructed by polymerase chain reaction (PCR) mutagenesis (e.g. error-prone PCR and site-directed mutagenesis with degenerate primers), followed by cloning into expression plasmids, making them compatible for screening by phage and yeast display. With the motivation of being able to screen antibodies in their native context as full-length IgGs with proper glycosylation, attempts have also been made to incorporate libraries into mammalian cells using episomal-, viral- or transposon-mediated gene transfer (9–11). However, relative to phage (>1010) and yeast (>107), these mammalian display systems are substantially challenged by small library size (∼104 variants for genome-integrated libraries) and polyclonality (multiple antibody variants per cell). Therefore, in order to truly have a competitive platform for mammalian antibody engineering, an alternative method which overcomes these limitations is essential.
With the rapid advancements in genome editing technologies, most notably the CRISPR/Cas9 system (Cas9), it is now possible to easily make targeted genomic modifications in mammalian cells (12). While Cas9 is most widely used for gene knock-out (via non-homologous end joining, NHEJ) or gene knock-in (via homology-directed repair (HDR)), it also enables the generation of libraries in mammalian cells. For example, Cas9 has been used to promote HDR with degenerate templates, resulting in a library of genomic variants; this has been applied to both coding and non-coding regions, providing insight into gene regulation, expression and even drug resistance (13,14). In a recent study, Cas9 was also used to integrate a genomic landing pad containing a recombination site, which allowed for the introduction of a library of transgene variants (15). Although these studies illustrate the potential to integrate libraries into specific genomic regions of mammalian cells, transfection of genome editing reagents combined with low HDR efficiencies limit the scalability and ease-of-use required to generate libraries capable of exploring sufficient protein sequence space, which is crucial for directed evolution and protein engineering.
In this study, we have established the method of homology-directed mutagenesis (HDM), which relies on high-efficiency HDR by Cas9 to generate site-directed mutagenesis libraries in mammalian cells. We use as our mammalian antibody display platform, a recently developed hybridoma cell line, where antibody variable regions can be exchanged by Cas9-driven HDR, referred to as plug-and-(dis)play hybridomas (PnP) (16). A critical feature of our HDM method is that it utilizes single-stranded oligonucleotides (ssODNs) as the donor template, which relative to double-stranded DNA, drastically increase HDR integration efficiencies (17–19) and also reduce off-target integration events (20, BioRxiv: https://doi.org/10.1101/178905). By using a cellular genotype-phenotype assay, we optimized a series of parameters, allowing us to achieve a robust HDR integration efficiency of 15–35%, thus reaching up to a 35-fold improvement. Next, starting with an antibody specific for the model antigen, hen egg lysozyme (HEL), we introduce a library into the variable heavy chain (VH) complementarity determining region 3 (CDRH3) by using ssODN templates with commonly used NNK and NNB degenerate codon regions (where N=A/C/G/T, K=G/T, and B=C/G/T). Following HDM, we perform next-generation sequencing (NGS) on the entire VH region to quantitatively assess the library diversity and distribution. We further maximize the efficacy of HDM libraries by implementing an optimized gRNA target sequence and rationally selecting degenerate nucleotides to match target amino acid (a.a.) frequencies of CDRH3 regions found in the antibody repertoire of murine naïve B cells (21,22). With HDM, we were able to achieve a library size of >105 variants validated by NGS. We then screen this library for specificity towards our model antigen, which led to recovery of a novel variant with a unique CDRH3. We also show that by using HDM to generate saturation libraries across the CDRH3, we could perform directed evolution and affinity maturation. Finally, we apply HDM to the recently established method of deep mutational scanning (DMS) (23,24), which allowed us to deconstruct the antigen-binding sequence landscape of our antigen-specific antibodies. Through HDM we have successfully developed a rapid and facile method for the generation of site-directed mutagenesis libraries in mammalian cells, which represents a versatile approach for high-throughput antibody engineering.
MATERIALS AND METHODS
Hybridoma cell culture conditions
All PnP hybridoma cell lines were cultivated in high-glucose Dulbecco’s Modified Eagle Medium [Thermo Fisher Scientific (Thermo), 11960-044] supplemented with 10% fetal bovine serum [Thermo, 16000-044], 100 U/ml Penicillin/Streptomycin (Thermo, 15140-122), 2 mM Glutamine (Sigma-Aldrich, G7513), 10 mM HEPES buffer (Thermo, 15630-056) and 50 μM 2-mercaptoethanol (Sigma-Aldrich, M3148). All hybridoma cells were maintained in incubators at a temperature of 37°C and 5% CO2. Hybridomas were typically maintained in 6 ml of culture in T-25 flasks (TPP, TPP90026), and passaged every 48/72 h. All hybridoma cell lines were confirmed annually to be negative for Mycoplasma contamination. A list of all PnP cell lines is provided in Supplementary Table S4.
Guide RNA design and ssODN synthesis
Both on-target and off-target activities were assessed to identify optimal guide RNA (gRNA) sequences. Geneious version 9 (25) was utilized to locate and score gRNA sequences in the vicinity of the target site of mutagenesis. On-target activity scores were calculated according to ‘sgRNA Designer’ (26). Off-target analysis was performed by comparing the gRNA sequences to the Mus musculus genome (GRCm38.p3) downloaded from NCBI. A maximum mismatch of 3 bps was allowed and any potential off-target sites were scored by ‘CRISPR Design’ (27). When designing an optimal gRNA target sequence (Figure 2B), nucleotide propensities for highly active gRNA sequences were selected per position as proposed by Doench et al. (26), while still including in-frame stop codons and minimizing overlapping homology on the 5′ and 3′ ends of the cleavage site.
Figure 2.

Generating unbiased libraries with HDM. (A) Amino acid frequencies of observed HDM events when generating NNK/B libraries resembles the theoretical amino acid frequencies, indicating unbiased integration. (B) A PnP hybridoma cell line was engineered with an optimized gRNA target sequence to reduce bias arising from NHEJ/MMEJ events. The gRNA sequence adheres to nucleotide propensities observed to increase Cas9 activity (26) while minimizing potential off-target sequences. Following Cas9 cleavage, stop codons encoded on both the 5′ and 3′ ends of the Cas9 cut-site promote recombination of in-frame stop codons following repair via NHEJ/MMEJ. These in-frame stop codons show reduced library bias arising from non-random repair. (C) NGS analysis of the top 10 CDRH3 clones for NNK/B libraries generated from either the frameshift-indel cell line (PnP-HEL23.FI) or the frameshift-stop cell line (PnP-HEL23.FS). In addition to all in-frame NHEJ/MMEJ events containing a stop codon, a much lower percentage of cells matching the originating CDRH3 nucleotide sequence ( ) was observed. (D) Diversity profiles calculated from NGS data for NNK/B libraries generated from the PnP-HEL23.FI or the PnP-HEL23.FS cell lines. The α values represent different weights to the clonal frequency, where α = 0 is the species richness, and α = 1 and α = 2 are two common measures for library diversity known as the Shannon entropy and Simpson's index, respectively. Both the Shannon entropy (α = 1) and Simpson’s Index (α = 2) are higher in the case for libraries generated from the PnP-HEL23.FS cell line, indicating a higher degree of unbiased diversity. Data presented (mean ± sd) is representative of n = 4 experiments.
) was observed. (D) Diversity profiles calculated from NGS data for NNK/B libraries generated from the PnP-HEL23.FI or the PnP-HEL23.FS cell lines. The α values represent different weights to the clonal frequency, where α = 0 is the species richness, and α = 1 and α = 2 are two common measures for library diversity known as the Shannon entropy and Simpson's index, respectively. Both the Shannon entropy (α = 1) and Simpson’s Index (α = 2) are higher in the case for libraries generated from the PnP-HEL23.FS cell line, indicating a higher degree of unbiased diversity. Data presented (mean ± sd) is representative of n = 4 experiments.
For the optimization of parameters or the incorporation of genetic diversity, ssODNs complementary to the non-target strand were ordered directly from Integrated DNA Technologies (IDT) along with any PCR primers or gRNAs used in this study. A recent study has suggested that ssODNs complementary to the non-target strand, and subsequently also complementary to the gRNA, do not compete for Cas9 binding, but instead anneal to the non-target strand further enhancing HDR events (28). Modified ssODNs had phosphorothioate (PS) bonds between the three terminal nucleotides (nt) of the 5′ and 3′ ends. A list of all gRNA, primer and ssODN donor sequences are provided in Supplementary Tables S5 and S6.
Hybridoma transfection
PnP hybridoma cells were electroporated with the 4D-Nucleofector™System (Lonza) using the SF Cell Line 4D-Nucleofector® X Kit L (Lonza, V4XC-2024, V4XC-2032) with the program CQ-104. Cells were prepared as follows: cells were isolated and centrifuged at 125 × G for 10 min, washed with Opti-MEM® I Reduced Serum Medium (Thermo, 31985-062), and centrifuged again with the same parameters. The cells were finally resuspended in SF buffer (per kit manufacturer guidelines), after which Cas9 plasmid (PX458), Alt-R Cas9 RNP (IDT) or Alt-R gRNA (IDT) and ssODN donor were added. All experiments performed utilize Cas9 from Streptococcus pyogenes (SpCas9). Transfections for optimization of parameters were performed by transfecting 2 × 105 cells with either 1 μg Cas9 plasmid, 100 pmol Alt-R Cas9 RNP or 115 pmol Alt-R gRNA and 100 pmol ssODN donor in 20 μl, 16-well Nucleocuvette™ strips. All other transfections up to 5 × 106 cells were performed in 100 μl single Nucleocuvettes™ and reagents were scaled accordingly. Transfections of 107 cells were performed under identical conditions as transfections for 5 × 106 cells.
Flow cytometry analysis and sorting
Flow cytometry-based analysis and cell isolation were performed using the BD LSR Fortessa™ and BD FACS Aria™ III (BD Biosciences), respectively. When labeling was required, cells were washed with phosphate-buffered saline (PBS), incubated with the labeling antibody or antigen for 30 min on ice, protected from light, washed again with PBS and analyzed or sorted. The labeling reagents and working concentrations are described in Supplementary Table S7. For cell numbers different from 106, the antibody/antigen amount and incubation volume were adjusted proportionally.
Cell isolation by MACS
Magnetic assisted cell sorting (MACS) isolation of cells was performed using the OctoMACS™ Separator (Miltenyi, 130-042-109) in combination with MS columns (Miltenyi, 130-042-201) for cell counts up to 2 × 108 cells. Cells were washed with PBS, incubated with the biotinylated antibody or antigen for 30 min on ice, washed twice with PBS, resuspended in PBS and Streptavidin Microbeads (Miltenyi, 130-048-102), and incubated in the refrigerator for 15 min. Following incubation, cells were washed with additional PBS, and resuspended in 1 ml PBS. The resuspended cells were added to a pre-rinsed magnetic column, washed twice with 500 μl PBS, and then once with 500 μl growth media. At last, the column was removed from the magnetic separator and cells were flushed directly into a collection plate with 1 ml of growth media.
Measurement of antibody secretion and affinity by ELISA
Sandwich ELISAs were used to measure the secretion of IgG from hybridoma cell lines. Plates were coated with capture polyclonal antibodies specific for Vk light chains (goat anti-mouse, Jackson ImmunoResearch, 115-005-174) concentrated at 4 μg/ml in PBS (Thermo, 10010-015). Plates were then blocked with PBS supplemented with 2% m/v milk (AppliChem, A0830) and 0.05% V/V Tween®-20 (AppliChem, A1389) (PBSMT). Supernatants from cell culture (106 cells/sample, volume normalized to least concentrated samples) were then serially diluted (at 1:3 ratio) in PBS supplemented with 2% m/v milk (PBSM). After blocking, supernatants and positive controls were incubated for 1 hour at RT or O/N at 4°C, followed by three washing steps with PBS supplemented with Tween-20 0.05% V/V (PBST). A secondary HRP-conjugated antibody specific for mouse Fc region was used (goat anti-mouse, Sigma-Aldrich, A2554), concentrated at 1.7 μg/ml in PBSM, followed by three wash steps with PBST. ELISA detection was performed using a 1-Step™ Ultra TMB-ELISA Substrate Solution (Thermo, 34028) as the HRP substrate, and the reaction was terminated with H2SO4 (1M). Absorbance at 450 nm was read with Infinite® 200 PRO NanoQuant (Tecan).
For antigen specificity measurements, plates were coated with purified HEL protein (Sigma-Aldrich, 62971-10G-F) concentrated at 4 μg/ml in PBS. Blocking, washing and supernatant incubation steps were made analogously to the previously described procedure. A secondary HRP-conjugated antibody was used specific for Vk light chain (rat anti-mouse, Abcam, AB99617) concentrated at 0.7 μg/ml. ELISA detection by HRP substrate and absorbance reading was performed as previously stated. ELISA data was analyzed with the software GraphPad Prism.
Sample preparation for NGS
Sample preparation for NGS was performed similar to the antibody library generation protocol of the primer extension method described previously (29). Genomic DNA was extracted from 1–5 × 106 cells using the Purelink™ Genomic DNA Mini Kit (Thermo, K182001). All extracted genomic DNA was subjected to a first PCR step. Amplification was performed using a forward primer binding to the beginning of the VH framework region and a reverse primer specific to the intronic region immediately 3′ of the J segment. PCRs were performed with Q5® High-Fidelity DNA polymerase (NEB, M0491L) in parallel reaction volumes of 50 μl with the following cycle conditions: 98°C for 30 s; 16 cycles of 98°C for 10 s, 70°C for 20 s, 72°C for 30 s; final extension 72°C for 1 min; 4°C storage. PCR products were concentrated using DNA Clean and Concentrator (Zymo, D4013) followed by 0.8X SPRIselect (Beckman Coulter, B22318) left-sided size selection. Total PCR1 product was amplified in a PCR2 step, which added extension-specific full-length Illumina adapter sequences to the amplicon library. Individual samples were Illumina-indexed by choosing from 20 different multiplex identifier index reverse primers. Cycle conditions were as follows: 98°C for 30 s; 2 cycles of 98°C for 10 s, 40°C for 20 s, 72°C for 1 min; 6 cycles of 98°C for 10 s, 65°C for 20 s, 72°C for 1 min; 72°C for 5 min; 4°C storage. PCR2 products were concentrated again with DNA Clean and Concentrator and run on a 1% agarose gel. Bands of appropriate size (∼550 bp) were gel-purified using the Zymoclean™ Gel DNA Recovery kit (Zymo, D4008). Concentration of purified libraries were determined by a Nanodrop 2000c spectrophotometer and pooled at concentrations aimed at optimal read return. The quality of the final sequencing pool was verified on a fragment analyzer (Advanced Analytical Technologies) using DNF-473 Standard Sensitivity NGS fragment analysis kit. All samples passing quality control were sequenced. Antibody library pools were sequenced on the Illumina MiSeq platform using the reagent kit v3 (2 × 300 cycles, paired-end) with 10% PhiX control library. Base call quality of all samples was in the range of a mean Phred score of 34.
Bioinformatics analysis and graphics
The MiXCR v2.0.3 program was used to perform data pre-processing of raw FASTQ files (30). Sequences were aligned to a custom germline gene reference database containing the known sequence information of the V and J regions for the variable heavy chain of the HEL23-2A synthetic antibody gene. Clonotype formation by CDRH3 and error correction were performed as described by Bolotin et al. (30). Functional clonotypes were discarded if: (i) a duplicate CDRH3 a.a. sequence arising from MiXCR uncorrected PCR errors, or (ii) a clone count equal to one. Downstream analysis was performed using R v3.2.2 (31) and Python v2.7.13 (32). Graphics were generated using the R packages ggplot2 (33), RColorBrewer (34) and ggseqlogo (35).
Codon selection for library design
We aimed at designing a library of immunological relevance by investigating all 3,375 degenerate codon schemes with regard to every possible combination described by the IUPAC nucleotide codes. To this end, the a.a. frequencies of each degenerate codon scheme were calculated by dividing the number of codons that encode for a specific a.a. by the total number of codons encoded for a given degenerate scheme. The a.a. frequencies per position of the CDRH3 found in the murine naïve antibody repertoire were based on NGS datasets of VH genes from sorted naïve B cells, 433,618 unique CDHR3 sequences from 19 C57BL/6 mice, described previously in Greiff et al. (22). Due to the canonically high frequencies of ‘CAR’ and ‘YW’ residues at the beginning and end of the CDRH3 (Kabat numbering positions 104–106 and 117–118), these positions were kept constant. Utilizing Equation (1), an optimized degenerate codon per a.a. position can be determined by calculating the mean-squared error (MSE) of each degenerate codon relative to the naïve repertoire and then selecting the scheme with the minimum MSE.
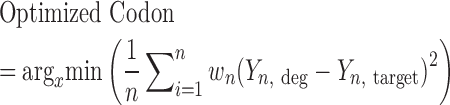 |
(1) |
Where x is the degenerate codon, n is the number of a.a. (20), wn is the a.a. weighting factor depending on each a.a.’s frequency in the target, Yn,deg is the a.a. frequency of the degenerate codon and Yn,target is the a.a. frequency of the target, in this case, the mouse naïve antibody repertoire. To obtain a measure of the overall similarity for the combination of degenerate codons for a CDRH3 of a given length, the mean of each position’s MSE was also calculated.
Calculating the diversity profiles of libraries
The diversity profile of a given library can be calculated using the Hill diversity, as previously described (36). Briefly, we calculated Hill diversity for α = 0 to α = 10 by steps of 0.2 according to Equation (2),
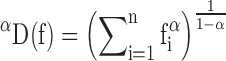 |
(2) |
where fi is the clonal frequency of clone i, n is the total number of clones and α-values represent weights. Diversities calculated at alpha values of 0, 1 and 2 represent the species richness, Shannon Diversity and the exponential inverse of Simpson’s Index, respectively. Clonal sequences were excluded from the diversity calculation if: (i) a stop codon was present, or (ii) the coding sequence was out of frame. A clone was defined based on the exact a.a. sequence of the CDRH3.
Calculating Levenshtein (edit) distances
From each of the NGS datasets of naïve repertoire optimized (NRO), NNK and NNB libraries, 5,000 CDRH3 sequences of length 14 a.a were randomly sampled. Subsequently, for each sampled sequence, the naïve repertoire utilized to develop the NRO degenerate codon scheme was searched for the presence of CDRH3 a.a. sequences of Levenshtein (edit) distances 0–6 (Figure 3C).
Figure 3.

Naive repertoire optimized (NRO) library design and analysis. (A) Side-by-side comparisons of the CDRH3 a.a. frequencies found in the naïve antibody repertoire (NGS data of naïve B cells from mice) and the theoretical NNB and NRO randomization schemes. (B) The theoretical and observed averages (from NGS) of the positional MSEs evaluated between the naïve repertoire and degenerate codon libraries across various CDRH3 lengths. The NRO scheme more closely resembles the amino acid frequencies observed in the naïve antibody repertoire compared to both NNK and NNB schemes. Equivalent average MSEs for both theoretical and observed amino acid frequencies reflect the accuracy of ssODN synthesis and unbiased HDM integration preferences. (C) Levenshtein distances for CDRH3 sequences of length 14 between the HDM libraries generated with degenerate codon schemes and the naïve antibody repertoire. The NRO scheme has a higher percentage of CDRH3 sequences with shorter edit distances indicating a higher degree of overall sequence similarity with the naïve repertoire. (D) Results from NGS data displays the ability to control the CDRH3 length distribution of the HDM library from a single transfection. By accounting for differences in integration efficiencies (Supplementary Figure S5), ssODNs of various lengths were pooled at varying ratios to mimic the length distribution of the naïve antibody repertoire.
Calculation of enrichment ratios (ERs) in DMS
The enrichment ratios (ERs) of a given variant was calculated according to previous methods (37). Clonal frequencies of variants enriched for antigen specificity (by FACS), fi,Ag+, were divided by the clonal frequencies of the variants present in the original library, fi,Ab+, according to Equation (3).
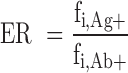 |
(3) |
A minimum value of −2 was designated to variants with log[ER] values less than or equal −2 and variants not present in the dataset were disregarded in the calculation. A clone was defined based on the exact a.a. sequence of the CDRH3.
RESULTS
Optimizing HDR efficiency
The degree of genetic diversity and library size that can be introduced using Cas9 is dependent on HDR efficiency. Previously, when establishing our PnP hybridoma platform, we observed an HDR efficiency of less than 1.0% when exchanging a fluorescent reporter protein with antibody variable regions encoded on a double-stranded DNA cassette (∼1.5 kb donor regions, ∼0.7 kb each for left and right homology arms) (16). Previous studies have shown that despite having only (micro)homology arms, much higher HDR efficiencies are observed when using ssODN donor templates (17,28,38). Due to the length limitation of commercially synthesized ssODNs, the target region of mutation is typically 50–80 nt, with ∼50 nt for each homology arm, which is highly compatible for targeting antibody CDRs for mutagenesis. However, in contrast to the previous approach, where loss of reporter protein and gain of antibody expression could easily be used to detect HDR, detection of HDR with ssODNs templates is more challenging. Therefore, to quantify and subsequently optimize ssODN-based HDR, we first developed a cellular phenotype assay based on antibody expression and antigen binding.
Starting with the PnP-HEL23 hybridoma cell line, which expresses a murine antibody sequence with specificity towards the model antigen HEL (16), we used Cas9 and gRNA targeting CDRH3 to introduce a frameshift mutation by NHEJ, resulting in the knockout of antibody expression (PnP-HEL23.FI cell line) (Figure 1A). We then designed an ssODN template that encoded the original CDRH3 a.a. sequence, but contained silent mutations, thus Cas9-driven HDR could be detected if both antibody expression and antigen specificity for HEL were restored. If only antibody expression was detected without binding to HEL, we presumed that an insertion/deletion (indel) via NHEJ or micro-homology mediated end joining (MMEJ) had occurred that knocked the antibody sequence back in-frame. Editing efficiencies were measured by flow cytometry following labeling of cells with fluorescently-tagged HEL and anti-IgH (Figure 1B).
Figure 1.

Optimizing parameters for homology directed repair (HDR). (A) An experimental assay developed to measure HDR efficiencies by flow cytometry. Cas9 is used to knockout antibody expression in the CDRH3 of a PnP hybridoma cell line which originally expressed a functional antibody targeting the model antigen HEL. A monoclonal population is then isolated and used for all other downstream experiments. Cas9 promotes HDR at the CDRH3 in the presence of a ssODN donor template which contains silent mutations. Cells which undergo HDR will therefore regain antibody expression specific for HEL. (B) Flow cytometry plots of the original cell line, PnP-HEL23 (left), the CDRH3 knockout cell line, PnP-HEL23.FI (middle) and a representative flow cytometry plot demonstrating the ability of the experimental assay to measure HDR integration efficiencies (right). (C) Optimal conditions for HDR revealed by results from assays examining the parameters: (i) Cas9 delivery method, (ii) ssODN length and (iii) PS modifications to ssODNs, data presented (mean ± sd) is representative of n = 2 independent experiments. (D) The maximum achievable HDR efficiency of ∼36% was obtained after combining the optimal parameters (constitutive Cas9, ssODN length 120 with PS modifications) and knocking out expression of the 53BP1 protein (PnP-HEL23.FS cells used here, see Figure 2).
To maximize HDR efficiency in PnP cells, several parameters regarding Cas9, gRNA, and donor templates were evaluated by performing separate and parallel transfections (2 × 105 cells). To resolve the effect of homology arm length on HDR, ssODNs ranged between 60 and 200 nt. To determine if an increase in resistance to nuclease degradation of ssODNs improves HDR, we included PS bonds in the 5′ and 3′ ends. Cas9 and gRNA was delivered to cells by transfection (electroporation) with either plasmid or ribonucleoprotein (RNP) complexes (18,39). We also generated a PnP stable cell line which constitutively expresses Cas9 from the Rosa26 safe harbor locus (Supplementary Figure S1a), as it permits the transfection of just pre-formed gRNA and ssODN donor (constant expression of Cas9 has been shown to have no toxic side effects on cells or in vivo (40)). Across all conditions, we observed the most notable increase in HDR efficiency when using the constitutive Cas9-expressing cell line, thus all subsequent experiments were performed with constitutive Cas9 expression (Figure 1C and Supplementary Figure S2). The highest HDR efficiency of 26% was observed when ssODNs with PS modifications and 120 nt length (homology arms equal to 46 nt) were used. This HDR rate remained stable when scaling up the number of cells transfected and only decreased slightly to ∼20% following transfection of 1 × 107 cells (Supplementary Figure S3). Recently, it has been shown that HDR efficiency can be improved by inhibiting the DNA repair regulator protein 53BP1, which promotes NHEJ over HDR (41,42). Therefore, we used a gRNA to target and knockout 53BP1 in our Cas9-expressing cell line (Supplementary Figure S1c); whereby subsequent testing of HDR in these cells showed an improved HDR efficiency of ∼36% (120 nt and PS-modified ssODNs), a >35-fold improvement relative to 1% (120 nt, Cas9 plasmid, unmodified ssODNs) (Figure 1D).
Assessing HDM library diversity and eliminating bias by gRNA design
Targeting the CDRs for site-directed mutagenesis has proven to be an effective means of building antibody libraries for improved properties (43,44). Introducing sequence diversity into CDRH3 alone has shown to be sufficient for a wide range of antigen-binding specificities (45). Thus, initial libraries were generated using ssODN homology templates (126 nt) that contained nine consecutive degenerate codons of NNK or NNB within the 14 a.a. long CDRH3 (e.g. ‘CAR(NNK)9YW’). These degenerate ssODN donors were transfected with gRNA into PnP-HEL23.FI cells (2 × 105 cells, two replicates per codon scheme). Here, we define the difference between HDR and HDM, in that the latter uses ssODN templates with degenerate codons. In order to precisely quantify library diversity introduced by HDM, following transfection and cell recovery and expansion, genomic DNA was isolated and targeted PCR was performed on the VH region to amplify libraries for NGS (using a previously established protocol for Illumina paired-end sequencing) (29). Across the four samples of NNK and NNB libraries, sequencing depth ranged between 577,915–847,687 read counts, with an ∼95% alignment success, resulting in 12,773–14,842 unique CDRH3 a.a. sequences (Supplementary Table S1). CDRH3 sequences with a length of 14 a.a. were classified as HDM events. Analysis of the HDM sequences revealed nearly an unbiased a.a. usage: positional a.a. usage in HDM libraries was nearly identical to what is theoretically predicted using NNK/B codon scheme (MSE = 3.07 ± 0.32) (Figure 2A). Furthermore, overlap analysis of sequences revealed almost no common CDRH3 sequences across all replicates, indicating that each HDM experiment results in a unique library (Supplementary Table S2).
In addition to HDM, a subpopulation of cells can restore antibody expression through NHEJ or MMEJ. Consistent with previous reports (46), we observed that the non-randomness of these pathways created a bias in the library: there were several grossly overrepresented variants (Figure 2C). This presence of high-frequency, redundant variants may limit library diversity and impede screening and selection steps. In order to address this issue, we used the information from the highest frequency NHEJ or MMEJ events to rationally design a gRNA target sequence which would promote either a frameshift mutation or in-frame stop codon following Cas9 cleavage and DNA repair. Using this approach, we drastically reduced the probability that an NHEJ or MMEJ event would result in a functionally expressed antibody by ensuring in-frame stop codons would appear on the 5′ and 3′ sides of the cleavage site (Figure 2B). Next, we used this new, frameshift-stop cell line (PnP-HEL23.FS) to generate HDM libraries, using once again NNK/B ssODN templates. NGS on VH genes was performed as before and similar sequencing depth and quality was obtained (Supplementary Table S1). Close analysis of the CDRH3 distribution revealed a substantial decrease in the frequencies of overrepresented (biased) variants and the generation of a more desirable uniformly diverse library. Diversity was quantified using the Hill diversity (Figure 2D and Equation 2) (36), where the diversity (αD) for each α-value represents an equivalent library in which all variants are equally present; Shannon diversity (α = 1) and Simpson's index (α = 2) are widely used for diversity comparisons.
Design, generation, and analysis of an HDM library that mimics the naïve antibody repertoire
While we have shown that with optimized parameters we can achieve considerable HDM efficiency, the growth rate and number of mammalian cells that can be handled in a typical experiment still pose challenges in achieving large library sizes (>105). With this in mind, we looked to maximize the functional quality of HDM libraries through the rational selection of degenerate codons. A major limitation in standard NNK/B codon schemes is the compounding probability of introducing a premature stop codon as the number of degenerate codons increases (Supplementary Figure S4a). However, when considering every possible combination of nucleotides, there exist 3,375 different degenerate codon schemes. Thus, as a first approach towards optimizing ssODNs, we designed degenerate codons that would most closely mimic the CDRH3 a.a. frequencies found in the antibody repertoire of murine naïve B cells (433,618 unique CDHR3 sequences) (22) (Figure 3A). It is well established that CDRH3 libraries with a more natural sequence landscape offer favorable properties (i.e. reduced immunogenicity, improved protein stability and folding) (47). Therefore, to generate the NRO library, for each position the degenerate codon was selected that produced the minimal MSE value relative to the naïve repertoire, while also using weights to punish degenerate codons that result in cysteines or stop codons (Equation 1). Following HDM and NGS, we observed that relative to the naïve repertoire, NRO libraries had substantially lower MSE values (a.a. length 14, average MSE = 47.1) compared to that of NNK/B libraries (a.a. length 14, average MSE = 85.7 ± 0.85) (Figure 3B). Notably, the possibility of introducing a premature stop codon or cysteine residue was eliminated while still maintaining adequate levels of total diversity (Supplementary Figure S4b) (48). Using another indication of sequence similarity to the naive B cell antibody repertoire, we determined based on the Levenshtein (edit) distance that the NRO library had a much higher percentage of sequences with short edit distances compared to NNK/B libraries (Figure 3C).
The CDRH3 length distribution of natural antibody repertoires also represents another important aspect to recapitulating functional diversity. Therefore, in order to assess the impact of CDRH3 length on HDM efficiency, separate transfections of 2 × 105 cells were performed with ssODN donors containing various lengths of degenerate codon regions, while keeping homology regions constant. As hypothesized, a minor decrease in integration efficiency was observed as the a.a. degenerate region increases (Supplementary Figure S5). This relationship between degenerate codon region length and integration efficiency can be taken into consideration when building libraries that aim to resemble the natural CDRH3 length distribution.
Next, by combining the previously acquired knowledge, an HDM library of unique CDRH3 sequences was constructed by transfecting 107 cells with gRNA and a pool of NRO ssODNs with varying degenerate codon lengths, which corresponded to CDRH3 lengths of 10, 12, 14, 15, 16, 18, 20, 22 a.a., thus mimicking the diversity and length distribution found in naïve repertoires. The ssODNs were pooled to mimic the naïve repertoire length distribution at weight-adjusted ratios to account for the expected decrease in integration efficiencies for longer degenerate regions. Following transfection, cells were allowed to divide for >72 hours in order to reach a total cell count >108 cells, at which point antibody expressing cells (Ab+) were labelled with biotinylated anti-IgH and isolated using MACS. NGS libraries were prepared from genomic DNA isolated from cells pre- and post-selection of antibody expression. Since the number of cells transfected was scaled up, NGS sequencing depth was correspondingly increased to 5.1 × 106 and 1.4 × 106 reads for pre- and post-selection libraries, respectively, with an ∼94% alignment success (Supplementary Table S1). Analysis of the NGS data revealed exceptional agreement between the theoretically predicted and observed a.a. frequencies across all CDRH3 lengths (MSE = 2.95 ± 1.14) (Figure 3B). Furthermore, the observed CDRH3 length distribution recapitulated what is observed in naïve repertoires (Figure 3D). There were 9.9 × 104 and 1.47 × 105 unique CDRH3 sequences identified in the samples pre- and post-selection of Ab+ cells, respectively. A possible explanation for this inconsistency can be attributed to under-sampling due to genomic DNA, where copy numbers of a given sequence are expectedly low.
Antibody screening and affinity maturation with HDM libraries
The HDM library described in the previous section was next used for antibody discovery using a directed evolution and high-throughput screening approach. Most recombinant antibody libraries, not derived from an immunized animal, require substantially more diversity than our NRO library in PnP cells (1.47 × 105 unique CDRH3s), hence phage and yeast display are often used because of their increased throughput. In order to compensate for this, we opted to screen our library against the model antigen HEL; this served as a reasonable proof-of-concept because the rest of the antibody scaffold (excluding CDRH3) was derived from a HEL-specific sequence (HEL23) (16). Thus, even with a relatively small library, we aimed to determine if we could discover antigen-specific variants possessing unique CDRH3 sequences when compared to HEL23. First, we enriched PnP cells from the library by two rounds of MACS, using a biotinylated-HEL and streptavidin conjugated magnetic beads. For each round of MACS, the library was expanded to a minimum of 5 × 107 cells in order to contain multiple copies per variant. Following MACS, a definitive HEL-specific population was visible by flow cytometry (Figure 4A). A subsequent two rounds of FACS enrichment were then performed, followed by a single-cell sort. Antigen specificity for the monoclonal populations was verified by flow cytometry (Figure 4A and Supplementary Figure S8b) and ELISA (Supplementary Figure S8a). Genotyping identified a novel clone (HEL24), which had a unique and longer CDRH3 sequence when compared to the original HEL23 (Levenshtein distance = 8) (Figure 4D). Although the newly identified variant had a lower binding affinity for the target antigen, it serves as a potential candidate for downstream engineering and optimization.
Figure 4.

HDM library screening for antibody discovery and affinity maturation. (A) Library generation by HDM coupled with mammalian cell display screening leads to the discovery of a novel CDRH3 variant, HEL24, for the target antigen, HEL. Following HDM library generation with NRO ssODNs, antibody expressing cells (Ab+) are isolated by magnetic activated cell sorting (MACS) and expanded. The library is then screened by MACS for specificity to antigen (Ag+), followed by two rounds of screening by fluorescence activated cell sorting (FACS). A monoclonal population for downstream characterization was isolated by single-cell sorting, revealing the novel variant, HEL24. (B) Single-site saturation HDM libraries for affinity maturation are created by pooling ssODNs that tile the mutagenesis site along the CDRH3. (C) Screening of saturation HDM libraries of HEL23 by FACS for higher antigen affinity variants, while using IgG staining to normalize for antibody expression. Monoclonal populations for downstream characterization were isolated by single-cell sorting. (D) The original HEL23 and HEL24 sequences and their higher affinity variants. ELISA data confirm secretion of antigen-specific antibodies into the hybridoma supernatant and antigen affinities relative to one another (Supplementary Figure S8a).
In addition to novel antibody discovery, directed evolution for affinity maturation is also an important step to engineering antibodies. Thus, we also aimed to demonstrate that HDM could be used to improve antibody affinity toward existing antigen-binding clones. To increase the affinity of the previously known HEL23 and newly discovered HEL24 clones, saturation mutagenesis libraries were generated along the CDRH3: HDM was performed with a pool of ssODNs with a single NNK codon tiled across CDRH3 (Figure 4B). With this approach, point mutation libraries containing a total of 134 and 191 variants were created based on the sequences for HEL23 and HEL24 respectively. Higher affinity variants were then enriched by FACS, where antibody avidity was normalized by simultaneous labeling for IgG surface expression (Figure 4C). Following one to two rounds of FACS selection, whereby libraries were pressured with decreasing concentrations of antigen labeling (Supplementary Table S7b), monoclonal populations were isolated and eight clonal variants were characterized. PnP cells have the advantage of simultaneously surface expressing and secreting IgG (16), therefore ELISAs were performed on these variants to confirm that relative to the original sequence, they had similar or improved antigen affinity (Supplementary Figure S8a).
Antigen specificity-sequence landscapes uncovered by HDM-mediated DMS
DMS is a new method in protein engineering, which combines directed evolution with NGS to assess the functional impact of mutations across the protein sequence landscape (37,49). In a typical DMS experiment with antibodies, saturation mutagenesis is performed on a single position at a time, followed by screening for functional antibody expression and then again for antigen binding (Supplementary Figure S9). NGS is performed along the various screening and selection steps, thus providing substantial insight into sequence-specificity relationships. Because of the need for large libraries and high-throughput screening, DMS has most often been performed using phage or yeast display systems. With the ability to generate and screen libraries in our mammalian display system, we therefore aimed to perform DMS on our HEL23 and HEL24 binding variants. We generated HDM single-position saturation libraries of CDRH1, CDRH2 and CDRH3 (pooled ssODNs for each CDR, separate transfection for each CDR). Each CDR library was then first selected on the basis of antibody expression and then screened for variants that retained binding to antigen. We extracted genomic DNA and NGS was performed on the VH genes of antibody expressing cells at both pre- and post-antigen selection. Sequencing depth of all libraries ranged from ∼215,000–890,000 reads, with an alignment success of >90% (Supplementary Table S3). NGS data was analyzed by determining the ER of each mutant, which was calculated by examining the clonal frequencies between the pre- and post-antigen selection libraries (37) (Equation 3). Heatmaps representing ER data were constructed for each CDR (Figure 5). Variants with ERs > 1 were then normalized per position and transformed into the corresponding sequence logo plots. These profiles of DMS data clearly show residues critical or detrimental for antigen binding and others which are more amenable to mutations. For example, in both HEL23 and HEL24 CDRH3 sequences, there are two a.a. positions confined to single residue to maintain antigen binding. Also, interestingly, even though HEL23 and HEL24 have identical CDRH1 and CDRH2 sequences, the DMS profiles show substantial variation and dependencies on different positions and residues. Where all positions along the CDRH1 of HEL23 appear more receptive to mutations, the stringency of certain residues found along the CDRH1 of HEL24 implies it has a greater influence on and contribution to antigen binding.
Figure 5.

Determining antigen-specificity sequence landscapes by deep mutational scanning (DMS). Heat maps and their corresponding sequence logo plots from the results of DMS of all variable heavy chain CDRs in both the HEL23 (A) and HEL24 (B) variants reveals positional variance for antigen-binding is clone specific. Libraries of single point-mutations across all heavy chain CDRs were integrated into hybridomas by HDM. NGS was then performed on libraries pre- and post-screening for antigen specificity (Supplementary Figure S9 and Table S3). Clonal (CDRH3) frequencies of variants post-screening were divided by clonal frequencies of variants pre-screening to calculate ERs. Sequence logo plots are generated by normalizing all variants with an ER > 1 (or log[ER] > 0).
DISCUSSION
Paramount to any directed evolution and protein engineering strategy is the ability to generate sufficiently sized libraries of variant clones. Since we rely here on Cas9 to introduce libraries directly in the genome of mammalian cells, we first aimed to optimize a range of parameters associated with HDR by developing an experimental assay that coupled antibody genotype to phenotype using PnP cells (Figure 1A). This allowed us to evaluate a series of parameters through which we determined constitutive Cas9 expression within the host cell played the most important role in improving HDR efficiency (Figure 1C). This is likely due to availability and abundance of Cas9 protein already localized within the nucleus at the time ssODN donors become available for DNA repair. We also resolved the optimal homology arm length, which unexpectedly did not correspond to the longest ssODN tested (200 nt), but rather to an intermediate ssODN length of 120 nt. The precise reason why such a decrease in integration occurs for longer ssODN donors is not known, but it is hypothesized that longer ssODNs are less accessible, or may even interfere with DNA repair proteins (38) and could also be attributed to a decrease in transfection efficiency. We also determined that chemical modification of the 5′ and 3′ ends of ssODNs with PS bonds led to higher HDR due to their increased stability and nuclease resistance (19). Since several studies have recently shown that inhibition of the NHEJ pathway regulator 53BP1 can improve HDR, we also knocked out 53BP1 and saw an additional improvement in HDR, reaching a maximum in this study of ∼36% (Figure 1D). In the future, it may be possible to further improve HDR by incorporating additional techniques such as the use of chemically modified gRNAs (50) and suppression of other NHEJ molecules (e.g. KU70 and DNA ligase IV) (51), or by exploiting newly engineered variants of Cas9 or other programmable nucleases (e.g. Cpf1) (52,53).
With parameters optimized for HDR, we next constructed initial HDM libraries targeting CDRH3 through the incorporation of degenerate codons (NNK and NNB) present between the homology arms of the ssODN donors. Following HDM and NGS analysis, we were able to quantitatively elucidate that the a.a. frequencies present in genomic libraries were almost exactly what would have been predicted based on the degenerate codon scheme (Figure 2A). This implies that ssODN sequences that had additional homology through similarity with the original CDRH3 were not selectively integrated at higher frequencies, suggesting that HDM is unbiased. NGS analysis did however reveal that repair of double-stranded breaks via the NHEJ/MMEJ pathways occurred in a non-random manner, resulting in several highly abundant variants that were disproportionately present in the library (Figure 2C). This phenomenon of Cas9 repair bias has been reported previously (46). While the possibility to use the error-prone NHEJ mechanism to introduce additional mutagenesis in our library could be considered a benefit, as it has even been used for discovering novel variants of cell signaling pathways (54) and dissecting enhancer regions (55); in the context of antibody engineering, the presence of highly redundant variants would have a detrimental effect on library distribution and screening (56). Therefore, we reduced the propensity of these events by using a ‘frameshift-stop’ gRNA target sequence, where following Cas9-cleavage, NHEJ/MMEJ events that would normally result in in-frame antibody sequences instead resulted in antibody sequences with premature stop codons (Figure 2B and C). HDM libraries constructed using this frameshift-stop gRNA sequence (PnP-HEL23.FS cells) showed a much more uniform distribution, thus greatly reducing overrepresented variants (Figure 2D). Although gRNA target optimization is not always necessary, as was displayed when building DMS libraries in the CDRH1 and 2, we expect that any future studies that aim to engineer antibodies (or other proteins) in mammalian cells may also benefit from careful design of gRNA sequences to ensure that NHEJ/MMEJ-biased repair does not compromise library distribution.
Even with the improved HDM efficiencies observed here, our mammalian cell libraries (105) are still substantially smaller relative to what can be routinely achieved in phage (>1010) and yeast (>107) (2). While scale-up with specialized devices capable of large-scale electroporation (>2.0 × 1010 cells) can partially compensate for this (Supplementary Figure S3), the slower growth rate and throughput associated with mammalian cell culture necessitates a strategy to maximize the functional quality of libraries. To this end, we designed ssODNs by rationally selecting degenerate codon schemes to mimic the a.a. frequencies of a particular diversity space, in this instance the naïve repertoire of mice. Although there are other approaches to build genetic diversity which very closely recapitulate the a.a. frequencies of a given repertoire (e.g. trinucleotide phosphoramidate-based oligonucleotide synthesis) (57–60), these methods are expensive and require sophisticated gene assembly and cloning strategies. In contrast, HDM relies on commercially synthesizable degenerate codon schemes and requires no gene assembly or cloning, thus representing substantial savings in time, effort and cost for library generation. Furthermore, we have shown that when compared to standard NNK and NNB degenerate codon schemes, the rationally selected codon schemes of ssODNs were able to more closely resemble the a.a. frequencies of the mouse naïve repertoire (Figure 3A and B). To generate the NRO library, we used a very minimal approach of a single degenerate codon ssODN (per CDRH3 length); however with advances in the large-scale synthesis of oligonucleotide arrays (61), ssODN pools of custom defined sequences could be included to generate more precise HDM libraries.
Next, we constructed a larger library, based primarily on increasing the number of cells transfected. Following HDM and NGS analysis, we identified that there was a minimum of 1.47 × 105 variants (unique CDRH3 a.a. sequences). Due to the fact that samples were prepared for NGS from genomic DNA with low copy numbers per variant, we believe the library size generated was actually in the range of 5 × 105 variants according to live cell counts post-transfection and flow cytometry data. Subjecting the library to antigen selection by MACS and FACS resulted in the discovery of a novel CDRH3 sequence (HEL24), which had a different length and large edit distance from the original sequence (HEL23). While our mammalian cell screening was successful at identifying a single new antigen-specific CDRH3 variant, we understand that such an approach is not in itself competitive with more robust synthetic antibody libraries screened by phage and yeast display, which often recover panels of unique CDRH3 clones (3,43,44,62). This is most assuredly due to lower library size and diversity in our mammalian system. Although, the diversity and library size is larger than what is commonly observed in mammalian-based platforms, we do not envision our approach will have a primary application in the discovery of novel antibody clones, but rather we expect the main application will be for engineering and optimizing antibodies, starting from a suitable lead candidate (which can be from in vivo selection or phage and yeast display screening). To this end, we showed that by pooling ssODNs with degenerate codons tiled along the CDRH3, that HDM could be used to rapidly engineer our ‘lead candidate’ HEL23 and HEL24 antibodies for higher affinity (Figure 4C and D; Supplementary Figure S8).
One of the most exciting applications of HDM is the ease at which we can perform DMS, a new technique in protein engineering that uncovers sequence–function relationships. DMS relies on the construction of saturation mutagenesis libraries, which requires PCR mutagenesis or synthetic genes and cloning into plasmid expression vectors, thus most DMS studies have been performed in bacteria, phage and yeast (24,37,63,64). One previous example performed DMS on an antibody clone using plasmid transfection in mammalian cells (65); however, the transient presence and polyclonality of plasmids make this a challenging approach. In the case of HDM, we were able to build saturation mutagenesis libraries for DMS in a similar manner in which we did for affinity maturation, by simply pooling ssODNs with single degenerate codons tiled along CDRH1, 2, 3. Because the library sizes required for DMS scale linearly with the number of target positions to investigate, the total library sizes were easily achievable in our hybridoma cells. Furthermore, because no cloning or plasmid transfection was required, DMS libraries could be built rapidly and easily by HDM and genomic integration ensured cellular monoclonality. Analysis of the NGS data produced by DMS revealed residues both critical and detrimental for functional antibody expression and antigen-specificity (Figure 5). This information could be used to rationally select degenerate codons that mimic the functional sequence landscape, which in turn can be used to produce combinatorial HDM libraries that can be screened for antibodies with improved properties (i.e. affinity, specificity, developability). At last, while in this study we exclusively focus on antibodies, HDM offers the potential to investigate genomic regulator sites (e.g. promoters, enhancers) or engineer other valuable biological and cellular therapeutics that rely on mammalian expression, such as chimeric antigen-receptors, T-cell receptors, cytokine receptors and intracellular signaling domains (66–69).
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the ETH Zurich D-BSSE Single Cell Unit and the ETH Zurich D-BSSE Genomics Facility for support, in particular, T. Lopes, V. Jäggin, E. Burcklen and C. Beisel. We also thank P. Heuberger for assistance with optimization.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swiss National Science Foundation [31003A_170110 to S.T.R.]; European Research Council Starting Grant [679403 to S.T.R.]; National Center of Competence in Research (NCCR) Molecular Systems Engineering (to S.T.R.); S. Leslie Misrock Foundation Professorship (to S.T.R.). Funding for open access charge: NCCR.
Conflict of interest statement. ETH Zurich has filed for patent protection on the technology described herein, and D.M.M., C.P., W.J.K. and S.T.R. are named as co-inventors on this patent (European Patent Application: 16163734.3-1402).
REFERENCES
- 1. Jain T., Sun T., Durand S., Hall A., Houston N.R., Nett J.H., Sharkey B., Bobrowicz B., Caffry I., Yu Y. et al. . Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoogenboom H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005; 23:1105–1116. [DOI] [PubMed] [Google Scholar]
- 3. Feldhaus M.J., Siegel R.W., Opresko L.K., Coleman J.R., Feldhaus J.M.W., Yeung Y.A., Cochran J.R., Heinzelman P., Colby D., Swers J. et al. . Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol. 2003; 21:163–170. [DOI] [PubMed] [Google Scholar]
- 4. Köhler G., Milstein C.. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975; 256:495–497. [DOI] [PubMed] [Google Scholar]
- 5. McCafferty J., Griffiths A.D., Winter G., Chiswell D.J.. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990; 348:552–554. [DOI] [PubMed] [Google Scholar]
- 6. Hanes J., Jermutus L., Weber-Bornhauser S., Bosshard H.R., Plückthun A.. Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14130–14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doerner A., Rhiel L., Zielonka S., Kolmar H.. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett. 2014; 588:278–287. [DOI] [PubMed] [Google Scholar]
- 8. Mazor Y., Blarcom T.V., Mabry R., Iverson B.L., Georgiou G.. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli. Nat. Biotechnol. 2007; 25:563–565. [DOI] [PubMed] [Google Scholar]
- 9. Beerli R.R., Bauer M., Buser R.B., Gwerder M., Muntwiler S., Maurer P., Saudan P., Bachmann M.F.. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Waldmeier L., Hellmann I., Gutknecht C.K., Wolter F.I., Cook S.C., Reddy S.T., Grawunder U., Beerli R.R.. Transpo-mAb display: transposition-mediated B cell display and functional screening of full-length IgG antibody libraries. MAbs. 2016; 8:726–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bowers P.M., Horlick R.A., Neben T.Y., Toobian R.M., Tomlinson G.L., Dalton J.L., Jones H.A., Chen A., Altobell L., Zhang X. et al. . Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20455–20460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Black J.B., Perez-Pinera P., Gersbach C.A.. Mammalian synthetic Biology: Engineering biological systems. Annu. Rev. Biomed. Eng. 2017; 19:249–277. [DOI] [PubMed] [Google Scholar]
- 13. Findlay G.M., Boyle E.A., Hause R.J., Klein J.C., Shendure J.. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014; 513:120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma L., Boucher J.I., Paulsen J., Matuszewski S., Eide C.A., Ou J., Eickelberg G., Press R.D., Zhu L.J., Druker B.J. et al. . CRISPR-Cas9–mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matreyek K.A., Stephany J.J., Fowler D.M.. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 2017; 45:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pogson M., Parola C., Kelton W.J., Heuberger P., Reddy S.T.. Immunogenomic engineering of a plug-and-(dis)play hybridoma platform. Nat. Commun. 2016; 7:12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen F., Pruett-Miller S.M., Huang Y., Gjoka M., Duda K., Taunton J., Collingwood T.N., Frodin M., Davis G.D.. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods. 2011; 8:753–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Renaud J.-B., Boix C., Charpentier M., De Cian A., Cochennec J., Duvernois-Berthet E., Perrouault L., Tesson L., Edouard J., Thinard R. et al. . Improved genome editing efficiency and flexibility using modified oligonucleotides with TALEN and CRISPR-Cas9 nucleases. Cell Rep. 2016; 14:2263–2272. [DOI] [PubMed] [Google Scholar]
- 20. Yoshimi K., Kunihiro Y., Kaneko T., Nagahora H., Voigt B., Mashimo T.. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016; 7:10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mena M.A., Daugherty P.S.. Automated design of degenerate codon libraries. Protein Eng. Des. Sel. 2005; 18:559–561. [DOI] [PubMed] [Google Scholar]
- 22. Greiff V., Menzel U., Miho E., Weber C., Riedel R., Cook S., Valai A., Lopes T., Radbruch A., Winkler T.H. et al. . Systems analysis reveals high genetic and Antigen-Driven predetermination of antibody repertoires throughout B cell development. Cell Rep. 2017; 19:1467–1478. [DOI] [PubMed] [Google Scholar]
- 23. Fowler D.M., Fields S.. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014; 11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whitehead T.A., Chevalier A., Song Y., Dreyfus C., Fleishman S.J., Mattos C.D., Myers C.A., Kamisetty H., Blair P., Wilson I.A. et al. . Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012; 30:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C. et al. . Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012; 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E.. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014; 32:1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. et al. . DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richardson C.D., Ray G.J., DeWitt M.A., Curie G.L., Corn J.E.. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016; 34:339–344. [DOI] [PubMed] [Google Scholar]
- 29. Menzel U., Greiff V., Khan T.A., Haessler U., Hellmann I., Friedensohn S., Cook S.C., Pogson M., Reddy S.T.. Comprehensive evaluation and optimization of amplicon library preparation methods for High-Throughput antibody sequencing. PLoS One. 2014; 9:e96727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolotin D.A., Poslavsky S., Mitrophanov I., Shugay M., Mamedov I.Z., Putintseva E.V., Chudakov D.M.. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods. 2015; 12:380–381. [DOI] [PubMed] [Google Scholar]
- 31. R Development Core Team R:A Language and Environment for Statistical Computing. 2008; Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 32. van Rossum G., Drake F.L.. The Python Language Reference Manual. 2011; UK: Network Theory Ltd. [Google Scholar]
- 33. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2009; NY: Springer-Verlag. [Google Scholar]
- 34. Brewer C.A., Hatchard G.W., Harrower M.A.. ColorBrewer in print: a catalog of color schemes for maps. Cartogr. Geogr. Inf. Sci. 2003; 30:5–32. [Google Scholar]
- 35. Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017; 33:3645–3647. [DOI] [PubMed] [Google Scholar]
- 36. Greiff V., Bhat P., Cook S.C., Menzel U., Kang W., Reddy S.T.. A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status. Genome Med. 2015; 7:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fowler D.M., Araya C.L., Fleishman S.J., Kellogg E.H., Stephany J.J., Baker D., Fields S.. High-resolution mapping of protein sequence-function relationships. Nat. Methods. 2010; 7:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang L., Guell M., Byrne S., Yang J.L., De Los Angeles A., Mali P., Aach J., Kim-Kiselak C., Briggs A.W., Rios X. et al. . Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013; 41:9049–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim S., Kim D., Cho S.W., Kim J., Kim J.-S.. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014; 24:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M. et al. . CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014; 159:440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Canny M.D., Moatti N., Wan L.C.K., Fradet-Turcotte A., Krasner D., Mateos-Gomez P.A., Zimmermann M., Orthwein A., Juang Y.-C., Zhang W. et al. . Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR–Cas9 genome-editing efficiency. Nat. Biotechnol. 2017; 36:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fasth A., Boyraz B., Paulsen B.S., Rossi D.J., Alt F.W., Talkowski M.E., Mandal P.K., Gutierrez-Martinez P., Yadav R., Frock R.L. et al. . Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR–Cas9 genome editing. Nat. Biomed. Eng. 2017; 1:878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fellouse F.A., Esaki K., Birtalan S., Raptis D., Cancasci V.J., Koide A., Jhurani P., Vasser M., Wiesmann C., Kossiakoff A.A. et al. . High-throughput generation of synthetic antibodies from highly functional minimalist Phage-displayed libraries. J. Mol. Biol. 2007; 373:924–940. [DOI] [PubMed] [Google Scholar]
- 44. Hoet R.M., Cohen E.H., Kent R.B., Rookey K., Schoonbroodt S., Hogan S., Rem L., Frans N., Daukandt M., Pieters H. et al. . Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 2005; 23:344–348. [DOI] [PubMed] [Google Scholar]
- 45. Xu J.L., Davis M.M.. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity. 2000; 13:37–45. [DOI] [PubMed] [Google Scholar]
- 46. van Overbeek M., Capurso D., Carter M.M., Thompson M.S., Frias E., Russ C., Reece-Hoyes J.S., Nye C., Gradia S., Vidal B. et al. . DNA repair profiling reveals nonrandom outcomes at Cas9-Mediated breaks. Mol. Cell. 2016; 63:633–646. [DOI] [PubMed] [Google Scholar]
- 47. Zhai W., Glanville J., Fuhrmann M., Mei L., Ni I., Sundar P.D., Van Blarcom T., Abdiche Y., Lindquist K., Strohner R. et al. . Synthetic antibodies designed on natural sequence landscapes. J. Mol. Biol. 2011; 412:55–71. [DOI] [PubMed] [Google Scholar]
- 48. Makowski L., Soares A.. Estimating the diversity of peptide populations from limited sequence data. Bioinformatics. 2003; 19:483–489. [DOI] [PubMed] [Google Scholar]
- 49. Araya C.L., Fowler D.M.. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 2011; 29:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B. et al. . Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015; 33:985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R.. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015; 33:543–548. [DOI] [PubMed] [Google Scholar]
- 52. Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z. et al. . Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018; 556:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A. et al. . Cpf1 is a single RNA-Guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Donovan K.F., Hegde M., Sullender M., Vaimberg E.W., Johannessen C.M., Root D.E., Doench J.G.. Creation of novel protein variants with CRISPR/Cas9-mediated Mutagenesis: Turning a screening By-Product into a discovery tool. PLOS One. 2017; 12:e0170445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S., Garcia S.P. et al. . BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015; 527:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Glanville J., D’Angelo S., Khan T., Reddy S., Naranjo L., Ferrara F., Bradbury A.. Deep sequencing in library selection projects: what insight does it bring. Curr. Opin. Struct. Biol. 2015; 33:146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Knappik A., Ge L., Honegger A., Pack P., Fischer M., Wellnhofer G., Hoess A., Wölle J., Plückthun A., Virnekäs B.. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides11Edited by I. A. Wilson. J. Mol. Biol. 2000; 296:57–86. [DOI] [PubMed] [Google Scholar]
- 58. Ashraf M., Frigotto L., Smith M.E., Patel S., Hughes M.D., Poole A.J., Hebaishi H.R.M., Ullman C.G., Hine A.V.. ProxiMAX randomization: a new technology for non-degenerate saturation mutagenesis of contiguous codons. Biochem. Soc. Trans. 2013; 41:1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frigotto L., Smith M.E., Brankin C., Sedani A., Cooper S.E., Kanwar N., Evans D., Svobodova S., Baar C., Glanville J. et al. . Codon-Precise, synthetic, antibody fragment libraries built using automated hexamer codon additions and validated through next generation sequencing. Antibodies. 2015; 4:88–102. [Google Scholar]
- 60. Virnekäs B., Ge L., Plückthun A., Schneider K.C., Wellnhofer G., Moroney S.E.. Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994; 22:5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kosuri S., Church G.M.. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods. 2014; 11:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee C.V., Liang W.-C., Dennis M.S., Eigenbrot C., Sidhu S.S., Fuh G.. High-affinity human antibodies from Phage-displayed synthetic fab libraries with a single framework scaffold. J. Mol. Biol. 2004; 340:1073–1093. [DOI] [PubMed] [Google Scholar]
- 63. Koenig P., Lee C.V., Walters B.T., Janakiraman V., Stinson J., Patapoff T.W., Fuh G.. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E486–E495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wrenbeck E.E., Klesmith J.R., Stapleton J.A., Adeniran A., Tyo K.E.J., Whitehead T.A.. Plasmid-based one-pot saturation mutagenesis. Nat. Methods. 2016; 13:928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Forsyth C.M., Juan V., Akamatsu Y., DuBridge R.B., Doan M., Ivanov A.V., Ma Z., Polakoff D., Razo J., Wilson K. et al. . Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. MAbs. 2013; 5:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sadelain M., Rivière I., Riddell S.. Therapeutic T cell engineering. Nature. 2017; 545:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Spangler J.B., Moraga I., Mendoza J.L., Garcia K.C.. Insights into cytokine–receptor interactions from cytokine engineering. Annu. Rev. Immunol. 2015; 33:139–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roybal K.T., Lim W.A.. Synthetic Immunology: Hacking immune cells to expand their therapeutic capabilities. Annu. Rev. Immunol. 2017; 35:229–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kariolis M.S., Kapur S., Cochran J.R.. Beyond antibodies: using biological principles to guide the development of next-generation protein therapeutics. Curr. Opin. Biotechnol. 2013; 24:1072–1077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.