

Abstract
Species are fundamental units in biological research and can be defined on the basis of various operational criteria. There has been growing use of molecular approaches for species delimitation. Among the most widely used methods, the generalized mixed Yule-coalescent (GMYC) and Poisson tree processes (PTP) were designed for the analysis of single-locus data but are often applied to concatenations of multilocus data. In contrast, the Bayesian multispecies coalescent approach in the software Bayesian Phylogenetics and Phylogeography (BPP) explicitly models the evolution of multilocus data. In this study, we compare the performance of GMYC, PTP, and BPP using synthetic data generated by simulation under various speciation scenarios. We show that in the absence of gene flow, the main factor influencing the performance of these methods is the ratio of population size to divergence time, while number of loci and sample size per species have smaller effects. Given appropriate priors and correct guide trees, BPP shows lower rates of species overestimation and underestimation, and is generally robust to various potential confounding factors except high levels of gene flow. The single-threshold GMYC and the best strategy that we identified in PTP generally perform well for scenarios involving more than a single putative species when gene flow is absent, but PTP outperforms GMYC when fewer species are involved. Both methods are more sensitive than BPP to the effects of gene flow and potential confounding factors. Case studies of bears and bees further validate some of the findings from our simulation study, and reveal the importance of using an informed starting point for molecular species delimitation. Our results highlight the key factors affecting the performance of molecular species delimitation, with potential benefits for using these methods within an integrative taxonomic framework.
Keywords: Molecular species delimitation, speciation, multispecies coalescent, simulation, generalized mixed Yule-coalescent, Poisson tree processes, Bayesian phylogenetics
Species identification is critical to a wide range of biological research, including studies of evolution, conservation, and biodiversity. However, various operational criteria are used for species identification, depending on the species concept that is being invoked (de Queiroz 2007). Among the most widely used are the biological species concept, which is based on reproductive isolation (Mayr 1942; Dobzhansky 1950), and the phylogenetic species concept, which is based on reciprocal monophyly (Rosen 1979; Baum and Shaw 1995). In contrast, morphology-based taxonomy usually appeals to the phenetic species concept (Michener 1970; Sokal and Crovello 1970), which remains a key framework for species identification in practice.
The last decade has witnessed the growing availability of genetic methods for species identification, providing a valuable complement to morphological taxonomy. Some of the widely used approaches for validating putative species are based on comparison of intra- and interspecific genetic distances (Hebert et al. 2003, 2004; Mallo and Posada 2016). These methods are contentious, however, partly because they do not appeal to an explicit species concept (Rubinoff et al. 2006a, 2006b; Waugh 2007). By contrast, the goal of molecular species delimitation is to build a taxonomic scheme for a set of samples and to infer a de novo delimitation of operational taxonomic units (OTUs) (Tautz et al. 2003; Vogler and Monaghan 2007; Mallo and Posada 2016). Within this burgeoning field, most methods appeal to the phylogenetic species concept and identify minimal phylogenetic units as the OTUs (Goldstein et al. 2000). These methods include the generalized mixed Yule-coalescent (GMYC) model (Pons et al. 2006; Fujisawa and Barraclough 2013), Poisson tree processes (PTP) model (Zhang et al. 2013; Kapli et al. 2017), Bayes factor delimitation (Grummer et al. 2014; Leaché et al. 2014), Bayesian coalescent method in the software Bayesian Phylogenetics and Phylogeography (BPP) (Yang 2015), and phylogeographic inference using approximate likelihoods (Jackson et al. 2017). Molecular species delimitation has been employed either as a stand-alone method or as part of an integrative taxonomic approach to species identification (e.g., Bond and Stockman 2008; Hotaling et al. 2016; Mason et al. 2016).
Methods of molecular species delimitation differ from each other in a number of respects. Among the widely used methods, Automatic Barcode Gap Discovery (ABGD) is one of the most computationally efficient. It requires the a priori specification of an intraspecific distance threshold, and the method is based on genetic distances computed from a single locus rather than an explicit species concept (Puillandre et al. 2012). The GMYC method also analyzes data from a single locus, but requires an ultrametric estimate of the gene tree. Studies have found that its performance is affected primarily by the ratio of population sizes to species divergence times, but also by varying population sizes, number of species involved, and number of sampling singletons (Fujisawa and Barraclough 2013; Dellicour and Flot 2015; Ahrens et al. 2016). Empirical studies have shown that ABGD and GMYC tend to under- and oversplit species, respectively (e.g., Paz and Crawford 2012; Pentinsaari et al. 2017; Renner et al. 2017). As with GMYC, PTP requires an estimate of the gene tree, but with branch lengths proportional to the amount of genetic change rather than to time. It tends to outperform GMYC when interspecific distances are small (Zhang et al. 2013), though the two methods often produce similar estimates of species limits (e.g., Lang et al. 2015; Arrigoni et al. 2016; Wang et al. 2016). GMYC and PTP were originally designed for the analysis of single-locus data, but are often applied to concatenated multilocus data by postulating a shared genealogical history (e.g., Arrigoni et al. 2016; Nieto-Montes de Oca et al. 2017; Renner et al. 2017).
In contrast with the methods described above, the Bayesian method in BPP was designed to analyze multiple loci but is much more computationally intensive (Yang 2015). It performs well when appropriate priors are chosen, with low rates of false positives and false negatives under most evolutionary scenarios (Yang and Rannala 2010, 2014; Zhang et al. 2011, 2014). Although the biological species concept provides the motivation for assuming limited gene flow between species, BPP appears to be robust to low levels of gene flow (Zhang et al. 2011). Studies with both simulated and empirical data have shown that BPP is more accurate than other multilocus coalescent methods (such as the information-theoretic and approximate Bayesian frameworks), while being somewhat sensitive to the number of loci and to the information content (Ence and Carstens 2011; Camargo et al. 2012; Hime et al. 2016). Empirical studies have also shown that BPP can produce delimitations that are consistent with those from other widely used methods such as GMYC and PTP (e.g., Hoppeler et al. 2016; Previšić et al. 2016; Nieto-Montes de Oca et al. 2017). Species delimitations by BPP are used widely not only for explicit taxon identification, but also as an important procedure in analyses of taxon evolution and divergence (e.g., Ruane et al. 2014; Moritz et al. 2018).
Most genetic methods for species identification, especially those based on the phylogenetic species concept, do not explicitly account for the mode of speciation. There are three main modes of speciation that differ in terms of the assumed degree of gene flow: allopatric, parapatric, and sympatric speciation (Gavrilets 2003). In each case, the formation of incipient species is related to a reduction in gene flow, which is at the core of the biological species concept. Some view speciation as a gradual and protracted process independent of any species concept (Rosindell et al. 2010; Etienne et al. 2014). For these reasons, there appears to be a gap between what we would consider to be “good” species and the taxonomic units inferred by coalescent-based methods of molecular species delimitation. This can be addressed by examining the congruence between genetic divergence and speciation. Some methods, such as GMYC and PTP, assume that gene trees accurately reflect the diversification of species, whereas BPP acknowledges the possibility of discordance between the two (Yang and Rannala 2010). This discordance is presumed to be caused primarily by among-gene differences in lineage sorting, but it can also be due to systematic error (e.g., model misspecification) and stochastic error (e.g., sampling scheme) (Mallo et al. 2014; Mallo and Posada 2016).
In this study, we compare the performance of three widely used methods of species delimitation, GMYC, PTP, and BPP, using both single-locus and multilocus sequence data generated by simulation under various speciation scenarios. We characterize the behavior of these methods, their delimitation efficacy, and their sensitivity to potential confounding factors. In addition, we validate some of the features of these methods in case studies involving sequence data from bears and bees. Our results provide practical guidelines for using molecular methods of species delimitation.
Materials and Methods
Models and Assumptions
To examine the performance of species delimitation using GMYC, PTP, and BPP, we simulated the evolution of sequence data under five speciation scenarios (Fig. 1): 1) no speciation; 2) speciation into two species with cessation of gene flow; 3) speciation into two species with ongoing gene flow; 4) speciation into five species with cessation of gene flow; and 5) speciation into four species with ongoing gene flow. In each case, we assumed Wright–Fisher panmixia within each species. Scenario I is treated as the null case in this study. Scenario II can represent either allopatric or peripatric speciation, depending on the combination of population sizes between the two species. Scenario III involves reduced but ongoing gene flow, so it can be taken to represent either parapatric or sympatric speciation. Scenarios IV and V are extensions of Scenarios II and III, respectively; we chose to model the evolution of five and four species in these scenarios, to allow variation in the tree topology and for practical convenience. In these scenarios, speciation can also be regarded as the formation of separate populations, and migration as being interchangeable with other forms of gene flow (e.g., introgression). We assumed that all speciation events were bifurcating, and that all genes evolved neutrally without gene conversion, gene duplication, or horizontal transfer.
Figure 1.
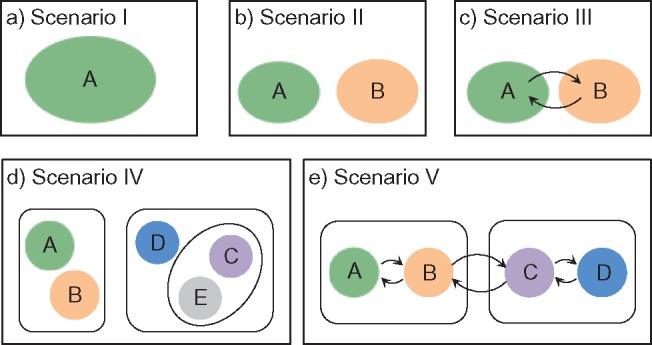
Five speciation models used for simulations in this study. a) Scenario I: a single species without population structure. b) Scenario II: speciation into two species, with cessation of gene flow. c) Scenario III: speciation into two species, with ongoing gene flow indicated by arrows. d) Scenario IV: speciation into five species, with cessation of gene flow. e) Scenario V: speciation into four species, with ongoing gene flow between adjacent species indicated by arrows.
Evolutionary Simulations
Nucleotide sequence evolution was simulated under each of the five scenarios (Table 1), with assumptions of a constant rate of 10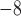 mutations per site per generation and a generation time of 1 year. Owing to its convenience and versatility, we preferred to use SimPhy (Mallo et al. 2016) to generate the trees, where possible. The species tree was simulated first, and then we simulated the evolution of 100 independent gene trees conditioned on the species tree. For simulations that could not be performed using SimPhy, we used makesamples (Hudson 2002) to simulate the evolution of 100 gene trees based on each species tree that we specified.
mutations per site per generation and a generation time of 1 year. Owing to its convenience and versatility, we preferred to use SimPhy (Mallo et al. 2016) to generate the trees, where possible. The species tree was simulated first, and then we simulated the evolution of 100 independent gene trees conditioned on the species tree. For simulations that could not be performed using SimPhy, we used makesamples (Hudson 2002) to simulate the evolution of 100 gene trees based on each species tree that we specified.
Table 1.
Genealogical simulation software and parameter settings for Scenarios I–V
| Scenario | Software | Population size 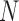
|
Crown age 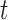 (years) (years) |
Number of loci 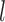
|
Sample size 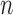
|
Migration rate 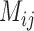 (individuals per generation) (individuals per generation) |
Speciation rate 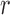 (speciation events per generation) (speciation events per generation) |
|---|---|---|---|---|---|---|---|
| I | makesamples | 10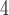 , 10 , 10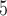 , 10 , 10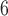
|
— | 1, 2, 5, 10, 20 | 2, 4, 10, 20, 40 | — | — |
| II | SimPhy | 10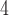 , 10 , 10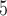 , 10 , 10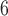
|
10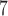 , 10 , 10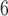 , 10 , 10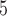
|
1, 2, 5, 10, 20 | 1, 2, 5, 10, 20 | — | — |
| III | makesamples | 10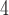
|
10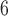
|
1, 2, 5, 10, 20 | 10 | 0.1, 1, 10, 10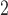 , 10 , 10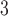
|
— |
| IV | SimPhy | 10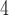
|
10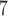
|
1, 2, 5, 10, 20 | 10 | — | 10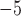 , 10 , 10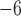 , 10 , 10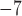 , 10 , 10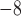
|
| V | makesamples | 10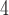
|
10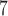
|
1, 2, 5, 10, 20 | 10 | 0.1, 1, 10, 10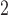 , 10 , 10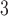
|
— |
Under each set of simulation conditions (see below; Table 1), we randomly subsampled varying numbers of gene trees (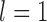 , 2, 5, 10, and 20) from the 100 generated by SimPhy or makesamples. These correspond to varying numbers of loci, because each gene tree corresponds to the evolutionary history of a single locus. We performed the jackknifing procedure 10 times for each number of loci. For each sampled gene tree, we then used Seq-Gen v1.3.2 (Rambaut and Grassly 1997) to simulate the evolution of a sequence alignment of length 1000 bp, using the Jukes–Cantor model of nucleotide substitution (Jukes and Cantor 1969). An outgroup sequence was added to the sequence alignment for each of the five scenarios during simulation, but was removed for the species-delimitation analyses. All data generated by our simulations are available in Supplementary Material of this article available on Dryad at http://dx.doi.org/10.5061/dryad.739bs, Appendix S1.
, 2, 5, 10, and 20) from the 100 generated by SimPhy or makesamples. These correspond to varying numbers of loci, because each gene tree corresponds to the evolutionary history of a single locus. We performed the jackknifing procedure 10 times for each number of loci. For each sampled gene tree, we then used Seq-Gen v1.3.2 (Rambaut and Grassly 1997) to simulate the evolution of a sequence alignment of length 1000 bp, using the Jukes–Cantor model of nucleotide substitution (Jukes and Cantor 1969). An outgroup sequence was added to the sequence alignment for each of the five scenarios during simulation, but was removed for the species-delimitation analyses. All data generated by our simulations are available in Supplementary Material of this article available on Dryad at http://dx.doi.org/10.5061/dryad.739bs, Appendix S1.
Scenario I
We began simulations with the null scenario of a single, unstructured population or species (Fig. 1a), under the classical Wright–Fisher model in makesamples. We set 15 combinations of sample sizes (i.e., the number of sampled individuals per species) and population sizes. Sample sizes (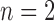 , 4, 10, 20, and 40) were double those used in Scenario II (as appropriate for some methods of species delimitation), whereas population sizes (
, 4, 10, 20, and 40) were double those used in Scenario II (as appropriate for some methods of species delimitation), whereas population sizes (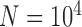 , 10
, 10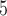 , and 10
, and 10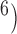 were consistent with the general settings used in Scenario II. After producing 10 replicates for each number of loci (
were consistent with the general settings used in Scenario II. After producing 10 replicates for each number of loci (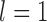 , 2, 5, 10, and 20), we had a total of 750 datasets.
, 2, 5, 10, and 20), we had a total of 750 datasets.
Scenario II
This scenario involves two reproductively isolated species with equal population sizes and with equal numbers of sampled individuals (Fig. 1b). In SimPhy, we chose: 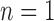 , 2, 5, 10, and 20 samples per species (as appropriate for the relevant delimitation methods; Luo et al. 2015); population sizes of
, 2, 5, 10, and 20 samples per species (as appropriate for the relevant delimitation methods; Luo et al. 2015); population sizes of 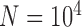 , 10
, 10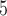 , and 10
, and 10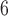 for each species; and divergence times of
for each species; and divergence times of 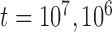 , and 10
, and 10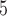 years between the two species. These yielded a total of 45 combinations of parameters. Larger population sizes and divergence times lead to greater genetic variation within species and between species, respectively; the ranges of values investigated in our study are generally consistent with the features of a broad range of eukaryotic species (Zhang et al. 2011; Fujisawa and Barraclough 2013). Taking into account the variation in the number of loci, our simulations produced a total of 2250 datasets. Based on the results from our analyses of these datasets, we chose the basic settings for the remaining simulations (including supplementary settings for BPP and variations of Scenario II). These results, together with those from Scenario I, provided benchmarks for interpreting the results from the other Scenarios and also informed the best strategy in PTP for analyzing our remaining data.
years between the two species. These yielded a total of 45 combinations of parameters. Larger population sizes and divergence times lead to greater genetic variation within species and between species, respectively; the ranges of values investigated in our study are generally consistent with the features of a broad range of eukaryotic species (Zhang et al. 2011; Fujisawa and Barraclough 2013). Taking into account the variation in the number of loci, our simulations produced a total of 2250 datasets. Based on the results from our analyses of these datasets, we chose the basic settings for the remaining simulations (including supplementary settings for BPP and variations of Scenario II). These results, together with those from Scenario I, provided benchmarks for interpreting the results from the other Scenarios and also informed the best strategy in PTP for analyzing our remaining data.
For species delimitation using BPP in particular, supplementary settings included: extreme population sizes for each species (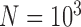 and 10
and 10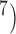 , with species divergence time
, with species divergence time 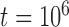 and
and 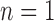 , 2, 5, 10, and 20 samples per species; and larger sample size
, 2, 5, 10, and 20 samples per species; and larger sample size 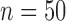 with
with 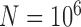 and
and 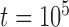 . Unless otherwise noted, the simulations for Scenario II described below exclude the supplementary settings used for BPP.
. Unless otherwise noted, the simulations for Scenario II described below exclude the supplementary settings used for BPP.
In addition, we considered a series of variations of Scenario II, involving potential confounding factors that might influence species delimitations inferred by the three methods. These included: simulating sequence evolution using makesamples vs. SimPhy for the core settings in Scenario II, to evaluate the consistency of our methods; differing vs. equal population sizes for the two species; exponentially growing vs. constant-size populations; uneven sampling vs. equal sample sizes from the two species; and substitution rate heterogeneity across species, across loci, or across lineages (see Supplementary Appendix S2 available on Dryad for details).
Scenario III
This scenario involves two sister species with ongoing gene flow (Fig. 1c). Gene flow is specified by the migration rate 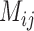 . In makesamples,
. In makesamples, 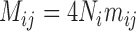 (
(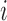 ,
, 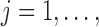 number of populations), where
number of populations), where 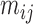 is the fraction of population
is the fraction of population 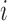 that is made up of migrants from population
that is made up of migrants from population 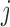 each generation, and
each generation, and 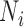 is the size of population
is the size of population 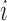 . To test the effect of ongoing gene flow on species delimitation, we set
. To test the effect of ongoing gene flow on species delimitation, we set 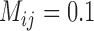 , 1, 10, 10
, 1, 10, 10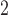 , and 10
, and 10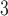 , and assumed equal amounts of reciprocal gene flow. In light of the results from Scenario II and general applicability to most empirical studies, we set the population size of each species to
, and assumed equal amounts of reciprocal gene flow. In light of the results from Scenario II and general applicability to most empirical studies, we set the population size of each species to 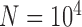 , the divergence time of the two species to
, the divergence time of the two species to 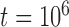 , and the sample size per species to
, and the sample size per species to 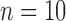 . These settings were combined with variation in the number of loci, as described above for Scenarios I and II.
. These settings were combined with variation in the number of loci, as described above for Scenarios I and II.
Scenario IV
We examined a five-species case in which speciation followed a Yule process (i.e., birth–death process with extinction rate zero; Yule 1924) (Fig. 1d), and considered a range of speciation rates (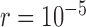 , 10
, 10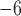 , 10
, 10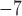 , and 10
, and 10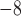 speciation events per year) for five species with the relationship ((A,B),(D,(C,E))). The most recent common ancestor of these species was set to 10
speciation events per year) for five species with the relationship ((A,B),(D,(C,E))). The most recent common ancestor of these species was set to 10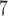 years before present, with each species having a population size
years before present, with each species having a population size 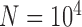 and with
and with 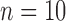 samples per species. We used SimPhy to simulate speciation with a pure-birth process, conditioned on the crown age and the number of extant species (Hartmann et al. 2010). Under these constraints, higher speciation rates (e.g.,
samples per species. We used SimPhy to simulate speciation with a pure-birth process, conditioned on the crown age and the number of extant species (Hartmann et al. 2010). Under these constraints, higher speciation rates (e.g., 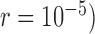 tended to produce trees with internal nodes closer to the tips (Supplementary Appendix S3 available on Dryad). Based on these settings, we performed simulations with various numbers of loci.
tended to produce trees with internal nodes closer to the tips (Supplementary Appendix S3 available on Dryad). Based on these settings, we performed simulations with various numbers of loci.
Scenario V
We examined a four-species case in which gene flow is conditioned on the geographic proximity of the species (Fig. 1e). The species have the relationship ((A,B),(C,D)), and gene flow occurs reciprocally between A and B, between B and C, and between C and D. Rates of gene flow match those in Scenario III. In makesamples, we assumed that divergences between sister species A and B and between C and D occurred 10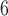 years ago, and that the four species had their most recent common ancestor 10
years ago, and that the four species had their most recent common ancestor 10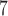 years ago. Each species was assumed to have a population size of
years ago. Each species was assumed to have a population size of 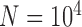 , with
, with 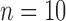 samples per species. Under each set of conditions, we performed simulations with various numbers of loci. In addition to the complete datasets, sequences of species pairs (A, B), (A, C), (A, D), and (B, C) were extracted separately so that we could explicitly test the effect of geographic distance on species delimitation.
samples per species. Under each set of conditions, we performed simulations with various numbers of loci. In addition to the complete datasets, sequences of species pairs (A, B), (A, C), (A, D), and (B, C) were extracted separately so that we could explicitly test the effect of geographic distance on species delimitation.
Species Delimitation
We performed species delimitation using three different methods: GMYC, PTP, and BPP. With the assumption that species are reciprocally monophyletic, GMYC uses a likelihood approach to identify the boundary between a Yule speciation process and intraspecific coalescence. This is done with reference to the relative node times in an ultrametric tree. To obtain these trees, we first inferred phylograms with maximum likelihood in RAxML v8.2.9 (Stamatakis 2014). We pruned non-unique sequences prior to phylogenetic inference, and conducted rapid full analyses with the Jukes–Cantor substitution model and 1000 bootstrap replicates. The phylograms were then made ultrametric through computing relative evolutionary times using the penalized-likelihood method in r8s v1.7 (Sanderson 2003), with a smoothing parameter of 10. Although a multiple-threshold version of GMYC has been developed (Monaghan et al. 2009), we only used the single-threshold version of GMYC because it has been shown to outperform the multiple-threshold version (Fujisawa and Barraclough 2013; Talavera et al. 2013). Species delimitation was done using the package splits v1.0-19 (Ezard et al. 2009) in R (R Core Team 2016).
PTP identifies the transition points between inter- and intraspecific branching events. The method postulates that the number of substitutions between species is significantly higher than that within species, with any individual substitution having a low probability of causing speciation. The mean numbers of substitutions until speciation events and until coalescent events are expected to follow exponential distributions, forming two independent Poisson processes on the tree. Since PTP does not require an ultrametric tree, we directly used the phylograms inferred in RAxML as described above. We employed three strategies in PTP: PTP heuristic (PTP-h) v2.2, Bayesian PTP maximum likelihood (bPTP-ML) v0.51, and Bayesian PTP heuristic (bPTP-h) v0.51, but did not consider the most recently proposed strategy, multi-rate PTP (Kapli et al. 2017; but see Discussion). For bPTP-ML and bPTP-h, we carried out two independent Bayesian Markov chain Monte Carlo (MCMC) analyses, with each chain having a length of 10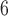 steps and the first 25% discarded as burn-in. After checking for convergence between chains, we reported the results from one of the two chains. For PTP-h, the minimal branch length was set to 10
steps and the first 25% discarded as burn-in. After checking for convergence between chains, we reported the results from one of the two chains. For PTP-h, the minimal branch length was set to 10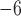 , with other parameters left at their default values.
, with other parameters left at their default values.
BPP delimits species in the multispecies coalescent framework, which assumes that the gene trees evolve within the constraints of the species tree (Rannala and Yang 2003). It uses reversible-jump MCMC to move between delimitation models while calculating posterior probabilities. We used BPP v3.3a to analyze our simulated single- and multilocus datasets based on guide trees that matched those used for simulation. Algorithms 0 and 1 (Yang and Rannala 2010) were used in two independent runs with default parameters, with each having 10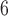 MCMC iterations following a discarded burn-in of 10,000 iterations. We checked for convergence between the two runs and combined the MCMC samples to produce estimates of the posterior probabilities of various delimitation models. For all datasets, we placed diffuse gamma priors
MCMC iterations following a discarded burn-in of 10,000 iterations. We checked for convergence between the two runs and combined the MCMC samples to produce estimates of the posterior probabilities of various delimitation models. For all datasets, we placed diffuse gamma priors 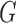 (1,500) on all
(1,500) on all 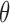 parameters and
parameters and 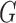 (1,100) on the root age
(1,100) on the root age  of species trees. These values are generally consistent with the settings in our simulations. For datasets from Scenario I, we randomly assigned equal numbers of sequences to each of two arbitrary species. For datasets with rate heterogeneity among loci, species delimitation was performed with a model of variable rates among loci, specified using a Dirichlet distribution with
of species trees. These values are generally consistent with the settings in our simulations. For datasets from Scenario I, we randomly assigned equal numbers of sequences to each of two arbitrary species. For datasets with rate heterogeneity among loci, species delimitation was performed with a model of variable rates among loci, specified using a Dirichlet distribution with 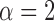 (Burgess and Yang 2008; Zhang et al. 2011).
(Burgess and Yang 2008; Zhang et al. 2011).
Evaluation of Performance
The three methods examined in this study produce species delimitations in different forms. For species delimitation using BPP, we recorded the posterior probabilities of different delimitation models (e.g., 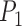 for the one-species model and
for the one-species model and 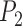 for the two-species model), and considered the rates of false positives and false negatives. For the results from GMYC and PTP, we based our evaluations on the most well supported species delimitations to capture the broad patterns. However, we did not take into account the support values for delimited entities, which should be considered when the focus of the investigation is on particular taxa. We calculated the number of delimited OTUs for each simulated species where delimitations were available, although this measure lacks the capacity to distinguish between certain outcomes (e.g., it cannot differentiate between the situations depicted in Fig. 2a,b, as illustrated below).
for the two-species model), and considered the rates of false positives and false negatives. For the results from GMYC and PTP, we based our evaluations on the most well supported species delimitations to capture the broad patterns. However, we did not take into account the support values for delimited entities, which should be considered when the focus of the investigation is on particular taxa. We calculated the number of delimited OTUs for each simulated species where delimitations were available, although this measure lacks the capacity to distinguish between certain outcomes (e.g., it cannot differentiate between the situations depicted in Fig. 2a,b, as illustrated below).
Figure 2.

An illustration of four categories used to classify the results of our species delimitations, for every species pair, by the GMYC and the PTP methods. Boxes with 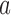 and
and 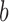 represent individuals of simulated species A and B, respectively, with additional individuals implied by the ellipses. Black bars above the boxes denote delimitation results; each bar indicates an OTU. Two illustrative examples each are given for false positives and for complex false positives.
represent individuals of simulated species A and B, respectively, with additional individuals implied by the ellipses. Black bars above the boxes denote delimitation results; each bar indicates an OTU. Two illustrative examples each are given for false positives and for complex false positives.
To enable the results to be described in finer detail, we used five categories to summarize the delimitations of GMYC and PTP for every species pair (Fig. 2). Because our focus is on the overall quality of delimitation rather than the number of delimited OTUs, these categories are largely intended to evaluate the performance of GMYC and PTP. First, two species might be correctly identified as two distinct OTUs (“correct delimitation”). Second, two species might be delimited to be a single OTU (“false negative”). Third, at least one of the two species might be inferred to be two or more OTUs that are also distinct from the OTU(s) identified for the other species (“false positive”). Fourth, at least one of the two species is inferred to be two or more OTUs, but with partial or total overlap with the OTU(s) identified for the other species (“complex false positive”). These four categories are similar, but not identical, to those defined by Ratnasingham and Hebert (2013). A fifth category comprises the cases in which we were unable to obtain a species delimitation (“not available” or “NA”).
GMYC and PTP can fail to yield a species delimitation under various circumstances. Because the trees were pruned so that they only contained unique sequences prior to phylogenetic inference, maximum-likelihood trees were unavailable for some datasets. This was problematic for both GMYC and PTP. In addition, PTP-h can fail to yield a definite species delimitation due to the failure of the likelihood-ratio test, because we used the default cut-off of 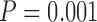 (Zhang 2013). In some cases, GMYC encountered computing errors, particularly for datasets comprising fewer sequences. Trees with zero branch lengths are actually compatible with PTP, but for convenience we preferred to include NAs for bPTP-h and bPTP-ML. For delimitations inferred for data from Scenario I, which involves a single species, we did not consider false negatives and complex false positives.
(Zhang 2013). In some cases, GMYC encountered computing errors, particularly for datasets comprising fewer sequences. Trees with zero branch lengths are actually compatible with PTP, but for convenience we preferred to include NAs for bPTP-h and bPTP-ML. For delimitations inferred for data from Scenario I, which involves a single species, we did not consider false negatives and complex false positives.
Case Studies
Our simulation study was designed to provide an insight into the performance of species-delimitation methods with data generated under known conditions. However, the idealized settings of our simulations might not adequately reflect the complex conditions under which real sequences have evolved. Therefore, based on the results from our simulation study, we carried out additional comparisons of species-delimitation methods using empirical datasets of bears and bees. These datasets comprise sequences of mitochondrial genes, which are expected to have congruent gene trees because of the absence of recombination in the mitochondrial genome.
Bears
The genus Ursus (Carnivora: Ursidae) comprises both extant and extinct species of bears, for which the taxonomy is relatively uncontroversial. For this group, we obtained a total of 172 complete or partial mitochondrial genomes from GenBank (retrieved on 20 February, 2017) and extracted the 12 protein-coding genes (excluding ND6). The sloth bear (Melursus ursinus) was added as an outgroup to allow estimation of a rooted tree. After removing duplicate sequences and/or sequences containing large proportions of missing data, we aligned the sequences while maintaining the reading frames. Our dataset then comprised concatenated sequences of the 12 protein-coding genes from 89 taxa (Supplementary Appendix S4 available on Dryad).
We inferred the gene tree using maximum likelihood in RAxML, with a separate GTR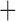 G
G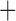 I substitution model applied to each gene. We ran rapid full analyses with 1000 bootstrap replicates. After rooting the tree and removing the outgroup sequence from the sloth bear, we used the inferred tree for species delimitation by GMYC, bPTP-h, bPTP-ML, and PTP-h. For BPP, the maximum-likelihood tree was simply treated as the guide tree for species delimitation with diffuse gamma priors
I substitution model applied to each gene. We ran rapid full analyses with 1000 bootstrap replicates. After rooting the tree and removing the outgroup sequence from the sloth bear, we used the inferred tree for species delimitation by GMYC, bPTP-h, bPTP-ML, and PTP-h. For BPP, the maximum-likelihood tree was simply treated as the guide tree for species delimitation with diffuse gamma priors 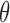 s
s 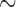
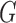 (1,500) and
(1,500) and 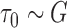 (1,100).
(1,100).
Bees
We tested the effects of locus number and sample size on species delimitation using sequence data from bees. This is a group of insects with important pollination services, but for which species diversity is relatively unclear. To test the impact of the number of loci, we downloaded a total of 38 complete or partial mitochondrial genomes of apid bees (Apidae; Hymenoptera: Apoidea) from GenBank (retrieved on 23 May, 2017). After removing duplicates and aligning the sequences, we used three datasets comprising 20 sequences of COI, 20 concatenated sequences of COI and CYTB, and 18 concatenated sequences of ATP6, COI, COII, COIII, CYTB, and ND1. Data are available in Supplementary Appendix S5 available on Dryad.
To test the impact of sample size, we downloaded sequences of the canonical barcode region (i.e., the 5’ terminus of the COI gene) from the mason bees of the genus Osmia (Apoidea: Megachilidae). After deleting sequences containing large proportions of missing data, pruning non-unique sequences, aligning the sequences, and removing species represented by fewer than five sequences, we were left with 69 sequences. In terms of species annotations, we randomly deleted some sequences to obtain two additional datasets: one comprising two sequences per species, and another comprising a single sequence per species (Supplementary Appendix S6 available on Dryad).
To allow the position of the root to be inferred, corresponding genes or regions from Megachile sculpturalis (GenBank accession NC_028017) and/or Megachile strupigera (GenBank accession KT346366) were used as the outgroup for both apid bees and mason bees (Hedtke et al. 2013). Species delimitations with GMYC, bPTP-ML, and BPP were implemented in a similar manner to our analyses of bear sequences. For our BPP analyses, the maximum-likelihood tree inferred from the 69 sequences was used as the guide tree for the other two datasets of mason bees. To achieve MCMC convergence, we drew samples from 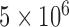 MCMC steps rather than the 10
MCMC steps rather than the 10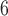 steps used in our other BPP analyses.
steps used in our other BPP analyses.
Results
Simulation Scenario I: One Species
For the null case in which sequences were sampled from a single species, BPP generally yielded the correct delimitation with high posterior probabilities (median 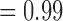 and mean
and mean 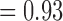 ) for the one-species model (
) for the one-species model (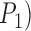 , across various parameter combinations (Table 2; Supplementary Appendix S7 available on Dryad). In some instances, however,
, across various parameter combinations (Table 2; Supplementary Appendix S7 available on Dryad). In some instances, however, 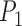 has extremely low values (min.
has extremely low values (min. 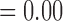 ), whereas posterior probabilities for the two-species model (
), whereas posterior probabilities for the two-species model (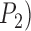 are relatively high. To avoid species inflation (Carstens et al. 2013), we consider that BPP supports the existence of two species only if
are relatively high. To avoid species inflation (Carstens et al. 2013), we consider that BPP supports the existence of two species only if 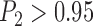 (Zhang et al. 2011). Therefore, the false-positive error rate is 1.73% for BPP, but is high for both GMYC and PTP (Tables 2 and 3; Supplementary Appendix S7 available on Dryad).
(Zhang et al. 2011). Therefore, the false-positive error rate is 1.73% for BPP, but is high for both GMYC and PTP (Tables 2 and 3; Supplementary Appendix S7 available on Dryad).
Table 2.
Descriptive statistics of the evaluation indices for delimitation results from Scenarios I to V
| Scenario | Statistics | BPP | No. of delimited OTUs | |||
|---|---|---|---|---|---|---|
| PP | GMYC | bPTP-h | bPTP-ML | PTP-h | ||
| I | Median | 0.99 | 2 | 8 | 5 | 11 |
| Mean | 0.93 | 3.38 | 11.14 | 8.44 | 12.78 | |
| Sd. | 0.19 | 2.73 | 9.49 | 8.12 | 7.99 | |
| Min. | 0.00 | 2 | 2 | 2 | 2 | |
| Max. | 1.00 | 32 | 37 | 39 | 34 | |
| II | Median | 1.00 | 1 | 1 | 1 | 1 |
| Mean | 0.91 | 1.71 | 3.18 | 2.39 | 1.49 | |
| Sd. | 0.24 | 1.08 | 4.00 | 3.17 | 1.91 | |
| Min. | 0.00 | 1 | 1 | 1 | 1 | |
| Max. | 1.00 | 13 | 20 | 20 | 18 | |
| III | Median | 0.02 | 2 | — | 3 | — |
| Mean | 0.38 | 2.14 | — | 4.38 | — | |
| Sd. | 0.46 | 1.50 | — | 3.09 | — | |
| Min. | 0.00 | 1 | — | 1 | — | |
| Max. | 1.00 | 10 | — | 10 | — | |
| IV | Median | 1.00 | 1 | — | 1 | — |
| Mean | 0.97 | 1.04 | — | 1.00 | — | |
| Sd. | 0.15 | 0.30 | — | 0 | — | |
| Min. | 0.01 | 1 | — | 1 | — | |
| Max. | 1.00 | 7 | — | 1 | — | |
| V | Median | 0.25 | 2 | — | 5 | — |
| Mean | 0.42 | 2.14 | — | 5.06 | — | |
| Sd. | 0.44 | 1.74 | — | 3.40 | — | |
| Min. | 0.00 | 1 | — | 1 | — | |
| Max. | 1.00 | 10 | — | 10 | — | |
Note: Posterior probabilities for correct delimitation models are given for the results from BPP: 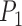 of the one-species model for Scenario I;
of the one-species model for Scenario I; 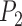 of the two-species model for Scenarios II and III;
of the two-species model for Scenarios II and III; 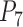 of the five-species model for Scenario IV; and
of the five-species model for Scenario IV; and 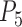 of the four-species model for Scenario V. The numbers of delimited OTUs for each simulated species are reported for both GMYC and PTP.
of the four-species model for Scenario V. The numbers of delimited OTUs for each simulated species are reported for both GMYC and PTP.
BPP  Bayesian Phylogenetics and Phylogeography; GMYC
Bayesian Phylogenetics and Phylogeography; GMYC  generalized mixed Yule-coalescent; PTP
generalized mixed Yule-coalescent; PTP  Poisson tree processes; bPTP-h
Poisson tree processes; bPTP-h  Bayesian PTP heuristic; bPTP-ML
Bayesian PTP heuristic; bPTP-ML  Bayesian PTP maximum likelihood; PTP-h
Bayesian PTP maximum likelihood; PTP-h  PTP heuristic; PP
PTP heuristic; PP  posterior probability; OTU
posterior probability; OTU  operational taxonomic unit.
operational taxonomic unit.
Table 3.
Percentages of categories with delimitation results from Scenarios I to V by GMYC and PTP
| Scenario | Category | Percent (%) | |||
|---|---|---|---|---|---|
| GMYC | bPTP-h | bPTP-ML | PTP-h | ||
| I | CD | 0 | 0 | 0 | 0 |
| FP | 76.50 | 91.00 | 91.00 | 8.33 | |
| NA | 23.50 | 9.00 | 9.00 | 91.67 | |
| II | CD | 21.89 | 48.89 | 60.33 | 39.83 |
| FN | 0 | 0 | 0 | 0 | |
| FP | 52.61 | 36.39 | 21.67 | 4.22 | |
| CFP | 13.06 | 9.28 | 12.56 | 1.89 | |
| NA | 12.44 | 5.44 | 5.44 | 54.06 | |
| III | CD | 5.60 | — | 10.80 | — |
| FN | 0 | — | 0 | — | |
| FP | 19.20 | — | 24.80 | — | |
| CFP | 45.60 | — | 56.00 | — | |
| NA | 29.60 | — | 8.40 | — | |
| IV | CD | 88.10 | — | 87.55 | — |
| FN | 5.85 | — | 11.70 | — | |
| FP | 4.70 | — | 0 | — | |
| CFP | 0.10 | — | 0 | — | |
| NA | 1.25 | — | 0.75 | — | |
| V | CD | 7.53 | — | 6.67 | — |
| FN | 10.00 | — | 4.60 | — | |
| FP | 16.47 | — | 33.00 | — | |
| CFP | 40.53 | — | 48.20 | — | |
| NA | 25.47 | — | 7.53 | — | |
Note: Values are given based on single species in Scenario I, one species pair in Scenarios II and III, ten species pairs in Scenario IV, and six species pairs in Scenario V.
CD  correct delimitation; FP
correct delimitation; FP  false positive; NA
false positive; NA  not available; FN
not available; FN  false negative; CFP
false negative; CFP  complex false positive.
complex false positive.
After excluding NAs, all of the delimitations by GMYC and PTP yielded false positives, indicating that both oversplit species under our null simulation scenario. However, they delimited varying numbers of OTUs. Given the available delimitations, GMYC inferred fewer OTUs (median 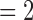 and mean
and mean 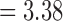 ) than PTP across all of the relevant simulation conditions. Among the three strategies of PTP, bPTP-h and PTP-h gave rise to smaller maximum numbers of delimited OTUs than did bPTP-ML, but bPTP-ML yielded fewer OTUs overall (median
) than PTP across all of the relevant simulation conditions. Among the three strategies of PTP, bPTP-h and PTP-h gave rise to smaller maximum numbers of delimited OTUs than did bPTP-ML, but bPTP-ML yielded fewer OTUs overall (median 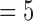 and mean
and mean 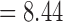 ).
).
Simulation Scenario II: Two Species
With diffuse priors in BPP, 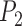 for the two-species delimitation model approaches 1.00 under most conditions, yielding an average false-negative error rate of 14.40% (Fig. 3 and Table 2). With large divergence times (
for the two-species delimitation model approaches 1.00 under most conditions, yielding an average false-negative error rate of 14.40% (Fig. 3 and Table 2). With large divergence times (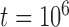 or 10
or 10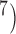 ,
, 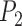 is greater than 0.95 for almost all population sizes. When
is greater than 0.95 for almost all population sizes. When 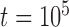 , the
, the 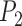 values show complex patterns: with smaller population sizes (
values show complex patterns: with smaller population sizes (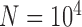 and 10
and 10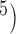 ,
, 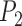 is below 0.95 especially for smaller sample sizes (
is below 0.95 especially for smaller sample sizes (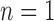 and 2), but tends to increase with number of loci
and 2), but tends to increase with number of loci 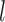 ; with a larger population size (
; with a larger population size (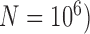 ,
, 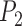 first decreases, then approaches 1.00 with larger values of
first decreases, then approaches 1.00 with larger values of 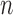 and
and 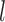 .
.
Figure 3.

Species delimitations estimated by the Bayesian coalescent method in BPP. Boxplots are shown for posterior probabilities of the two-species delimitation model (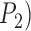 , across every 10 replicates under each set of conditions for Scenario II. Nine combinations (
, across every 10 replicates under each set of conditions for Scenario II. Nine combinations (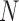 ,
, 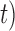 of population size
of population size 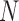 and divergence time
and divergence time 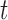 are shown along the top, together with five values of sample size
are shown along the top, together with five values of sample size 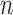 on the right. The
on the right. The 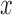 -axis represents the number of loci, while probabilities are given on the
-axis represents the number of loci, while probabilities are given on the 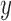 -axis.
-axis.
The results from our BPP analyses of supplementary (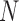 ,
, 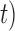 combinations (10
combinations (10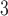 , 10
, 10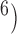 are similar to those from (10
are similar to those from (10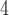 , 10
, 10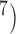 . Results from (10
. Results from (10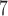 , 10
, 10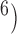 are broadly consistent with those from (10
are broadly consistent with those from (10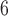 , 10
, 10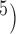 , except for large values of
, except for large values of 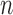 and
and 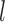 and some failures in MCMC convergence. Given that MCMC convergence was generally good in our BPP delimitations, here we only note the cases in which MCMC convergence was not achieved. With sample size
and some failures in MCMC convergence. Given that MCMC convergence was generally good in our BPP delimitations, here we only note the cases in which MCMC convergence was not achieved. With sample size 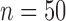 and (
and (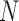 ,
, 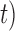 as (10
as (10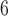 , 10
, 10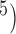 ,
, 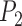 values are 1.00 across different values of
values are 1.00 across different values of 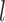 (Supplementary Appendix S8 available on Dryad).
(Supplementary Appendix S8 available on Dryad).
The number of delimited OTUs for each simulated species (median 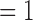 and mean
and mean 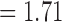 ) gives the impression that GMYC might correctly delimit species under this scenario (Table 2). However, GMYC actually produced a low rate of correct delimitations (21.89%) with no false negatives. False positives (52.61%) and complex false positives (13.06%) occurred quite often, with the latter limited to certain combinations of (
) gives the impression that GMYC might correctly delimit species under this scenario (Table 2). However, GMYC actually produced a low rate of correct delimitations (21.89%) with no false negatives. False positives (52.61%) and complex false positives (13.06%) occurred quite often, with the latter limited to certain combinations of (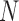 ,
, 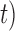 (Fig. 4 and Table 3). GMYC failed to yield a species delimitation with small sample size
(Fig. 4 and Table 3). GMYC failed to yield a species delimitation with small sample size 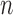 and/or number of loci
and/or number of loci 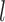 , and encountered computing errors with some (
, and encountered computing errors with some (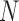 ,
, 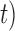 combinations. Although correct delimitations generally accompany false positives, the percentage of the latter is significantly higher than that of the former (
combinations. Although correct delimitations generally accompany false positives, the percentage of the latter is significantly higher than that of the former (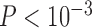 , non-parametric paired Wilcoxon test). The test also reveals that larger numbers of loci did not improve the chance of obtaining correct delimitations (
, non-parametric paired Wilcoxon test). The test also reveals that larger numbers of loci did not improve the chance of obtaining correct delimitations (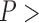 0.05 for
0.05 for 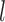 comparisons of 1∼2, 1∼5, 1∼10, and 1∼20), but larger samples sizes did (
comparisons of 1∼2, 1∼5, 1∼10, and 1∼20), but larger samples sizes did (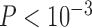 for
for 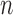 comparisons 2∼5, 2∼10, and 2∼20). In contrast, analyzing larger numbers of loci resulted in more false-positive errors, whereas sample size did not have a significant influence on this error rate.
comparisons 2∼5, 2∼10, and 2∼20). In contrast, analyzing larger numbers of loci resulted in more false-positive errors, whereas sample size did not have a significant influence on this error rate.
Figure 4.

Species delimitations estimated by GMYC for datasets from Scenario II. Panels show nine combinations of population size 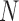 and divergence time
and divergence time 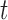 along the top, and four values of sample size
along the top, and four values of sample size 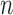 on the right. The
on the right. The 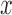 -axis represents the number of loci. The
-axis represents the number of loci. The 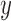 -axis represents the number of cases classified by correct delimitation (CD), false positive (FP), complex false positive (CFP), and not available (NA) among the 10 replicates under each set of conditions, which are denoted by different shades according to the legend in the bottom-right. No false negatives occurred in the results from Scenario II.
-axis represents the number of cases classified by correct delimitation (CD), false positive (FP), complex false positive (CFP), and not available (NA) among the 10 replicates under each set of conditions, which are denoted by different shades according to the legend in the bottom-right. No false negatives occurred in the results from Scenario II.
PTP-h produced many NA results (54.06%), with most due to unclear species delimitations. However, correct delimitations dominate the remaining available results (Table 3; Supplementary Appendix S9 available on Dryad). The other two methods, bPTP-h and bPTP-ML, performed similarly to each other, with far fewer NA results. However, bPTP-h oversplit species more frequently (Table 3; Supplementary Appendix S10 available on Dryad). Because we penalize species overestimation more greatly than underestimation, we only provide the detailed results from bPTP-ML here (Fig. 5). This method produced correct species delimitations in the majority of cases (60.33%; Table 3). However, it had variable rates of success under different conditions, as also shown by the larger standard deviation (3.17) of the numbers of delimited OTUs for each simulated species (Table 2). Under some conditions (e.g., the combination of 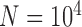 and
and 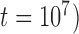 , the performance of bPTP-ML generally increases with number of loci
, the performance of bPTP-ML generally increases with number of loci 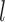 and sample size
and sample size 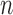 . It did not produce any false negatives, whereas false positives mainly occur in the (
. It did not produce any false negatives, whereas false positives mainly occur in the (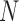 ,
, 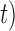 combinations of (10
combinations of (10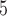 , 10
, 10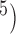 and (10
and (10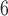 , 10
, 10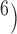 . Complex false positives tend to dominate the results when
. Complex false positives tend to dominate the results when 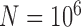 and
and 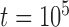 .
.
Figure 5.

Species delimitations estimated by the Bayesian PTP maximum likelihood (bPTP-ML) for datasets from Scenario II. Panels show nine combinations of population size 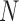 and divergence time
and divergence time 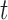 along the top, and four values of sample size
along the top, and four values of sample size 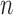 on the right. The
on the right. The 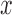 -axis represents the number of loci. The
-axis represents the number of loci. The 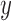 -axis represents the number of cases classified by correct delimitation (CD), false positive (FP), complex false positive (CFP), and not available (NA) among the 10 replicates under each set of conditions, which are denoted by different shades according to the legend in the bottom-right. No false negatives occurred in the results from Scenario II.
-axis represents the number of cases classified by correct delimitation (CD), false positive (FP), complex false positive (CFP), and not available (NA) among the 10 replicates under each set of conditions, which are denoted by different shades according to the legend in the bottom-right. No false negatives occurred in the results from Scenario II.
We found that species delimitations are not significantly different whether we performed simulations using makesamples or SimPhy. Population growth also had no significant influence on the performance of the species-delimitation methods. However, having one species of population size larger than that of the other species (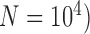 led to false positives dominating the delimitations by GMYC and bPTP-ML. Both unbalanced sampling and mutation rate heterogeneity at different levels were found to have small effects on the performance of these methods (see Supplementary Appendix S2 available on Dryad for details).
led to false positives dominating the delimitations by GMYC and bPTP-ML. Both unbalanced sampling and mutation rate heterogeneity at different levels were found to have small effects on the performance of these methods (see Supplementary Appendix S2 available on Dryad for details).
Simulation Scenario III: Two Species with Ongoing Gene Flow
With the basic settings informed by Scenario II (population size 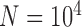 , divergence time
, divergence time 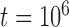 , and sample size
, and sample size 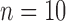 ) and with reference to delimitation results under these conditions in Scenario II, we found that varying degrees of gene flow did influence the performance of these methods. BPP identified the correct species delimitation when the migration rate was very low (
) and with reference to delimitation results under these conditions in Scenario II, we found that varying degrees of gene flow did influence the performance of these methods. BPP identified the correct species delimitation when the migration rate was very low (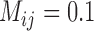 ) (Supplementary Appendix S11 available on Dryad). When
) (Supplementary Appendix S11 available on Dryad). When 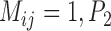 approaches 1.00 only with larger numbers of loci. For higher migration rates, nearly all
approaches 1.00 only with larger numbers of loci. For higher migration rates, nearly all 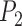 values approach 0.00, indicating the presence of false negatives.
values approach 0.00, indicating the presence of false negatives.
Compared with BPP, both GMYC and bPTP-ML appear to be more sensitive to the presence of gene flow. A large proportion of false positives was produced by GMYC when 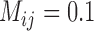 , and complex false positives dominate the available results of GMYC when
, and complex false positives dominate the available results of GMYC when 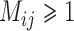 . Of the different methods in PTP, we only consider bPTP-ML here because of its superior performance in Scenarios I and II. It produced a variety of results, including correct delimitations, when
. Of the different methods in PTP, we only consider bPTP-ML here because of its superior performance in Scenarios I and II. It produced a variety of results, including correct delimitations, when 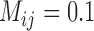 and 1. For higher migration rates, however, bPTP-ML tended to produce complex false positives. Neither GMYC nor bPTP-ML yielded false negatives under this scenario. The numbers of delimited OTUs for each simulated species in this scenario are also higher than those in Scenario II for both GMYC and bPTP-ML (Table 2).
and 1. For higher migration rates, however, bPTP-ML tended to produce complex false positives. Neither GMYC nor bPTP-ML yielded false negatives under this scenario. The numbers of delimited OTUs for each simulated species in this scenario are also higher than those in Scenario II for both GMYC and bPTP-ML (Table 2).
Simulation Scenario IV: Five Species
We use the values of the speciation rates, which controlled the relative depths of the internal nodes in our simulated trees, to refer to the five chronograms in this scenario (Supplementary Appendix S3 available on Dryad). When speciation rate 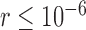 (i.e., relatively deep speciation events), BPP generally obtained the correct species delimitation model with posterior probabilities (
(i.e., relatively deep speciation events), BPP generally obtained the correct species delimitation model with posterior probabilities (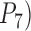 approaching 1.00 (Fig. 6a; Supplementary Appendix S12 available on Dryad). This model is denoted as “1111”, where the numbers “1” indicate the correct resolutions of (A,B) from (D,(C,E)), A from B, D from (C,E), and C from E, respectively. Failure to identify each of these distinctions is denoted by “0”. When
approaching 1.00 (Fig. 6a; Supplementary Appendix S12 available on Dryad). This model is denoted as “1111”, where the numbers “1” indicate the correct resolutions of (A,B) from (D,(C,E)), A from B, D from (C,E), and C from E, respectively. Failure to identify each of these distinctions is denoted by “0”. When 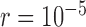 (i.e., shallow speciation events), posterior probabilities are spread across all delimitation models except 0000, given small numbers of loci (i.e.,
(i.e., shallow speciation events), posterior probabilities are spread across all delimitation models except 0000, given small numbers of loci (i.e., 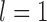 or 2); for larger
or 2); for larger 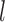 , however,
, however, 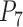 always approaches 1.00.
always approaches 1.00.
Figure 6.

a) Posterior probabilities of the correct species delimitation model 1111 (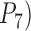 by BPP in Scenario IV. Probabilities are along the
by BPP in Scenario IV. Probabilities are along the 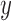 -axis for every 10 replicates of each number of loci (
-axis for every 10 replicates of each number of loci (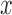 -axis) combined with the speciation rates (indicated by text above the boxplots). b) Symbol plots of correct delimitations by GMYC in Scenario IV. Yellow (light grey in printed version) circles represent results from the four species pairs along the two basal branches, while blue (dark grey in printed version) circles represent results from the six species pairs across the two basal branches. Relative areas of circles correspond to the percentages of correct delimitations in the respective full delimitations (Supplementary Appendix S12 available on Dryad), with the maximum area indicating 100%. Symbols in (c) have the same meaning as in (b), but show correct delimitations by bPTP-ML in Scenario IV. (d) Correlogram of posterior probabilities inferred by BPP across 250 datasets in Scenario V. Diagonal lines running from top-left to bottom-right in the red panels below the diagonal and red pies above the diagonal denote negative correlation, whereas diagonal lines running from bottom-left to top-right in the blue panels below the diagonal and blue pies above the diagonal denote positive correlation (the blue and red colors are only shown in online version). Darker colors indicate stronger relationships. Delimitation models are denoted by the character “m” and numbers along the diagonal, such as “m111” for delimitation model 111.
-axis) combined with the speciation rates (indicated by text above the boxplots). b) Symbol plots of correct delimitations by GMYC in Scenario IV. Yellow (light grey in printed version) circles represent results from the four species pairs along the two basal branches, while blue (dark grey in printed version) circles represent results from the six species pairs across the two basal branches. Relative areas of circles correspond to the percentages of correct delimitations in the respective full delimitations (Supplementary Appendix S12 available on Dryad), with the maximum area indicating 100%. Symbols in (c) have the same meaning as in (b), but show correct delimitations by bPTP-ML in Scenario IV. (d) Correlogram of posterior probabilities inferred by BPP across 250 datasets in Scenario V. Diagonal lines running from top-left to bottom-right in the red panels below the diagonal and red pies above the diagonal denote negative correlation, whereas diagonal lines running from bottom-left to top-right in the blue panels below the diagonal and blue pies above the diagonal denote positive correlation (the blue and red colors are only shown in online version). Darker colors indicate stronger relationships. Delimitation models are denoted by the character “m” and numbers along the diagonal, such as “m111” for delimitation model 111.
GMYC yielded the correct delimitation under some circumstances in the five-species scenario (Fig. 6b; Supplementary Appendix S12 available on Dryad). When 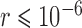 , GMYC correctly delimited all of the species pairs, though with a few false negatives and false positives. When
, GMYC correctly delimited all of the species pairs, though with a few false negatives and false positives. When 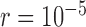 and with smaller
and with smaller 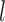 , it mainly produced false negatives for the four species pairs along the two basal branches, but correct delimitations for the other six pairs. For
, it mainly produced false negatives for the four species pairs along the two basal branches, but correct delimitations for the other six pairs. For 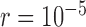 and large
and large 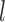 , some false positives appear together with correct delimitations for all ten species pairs, making GMYC delimitations complicated. Conversely, bPTP-ML produced results with some clear patterns (Fig. 6c; Supplementary Appendix S12 available on Dryad). When
, some false positives appear together with correct delimitations for all ten species pairs, making GMYC delimitations complicated. Conversely, bPTP-ML produced results with some clear patterns (Fig. 6c; Supplementary Appendix S12 available on Dryad). When 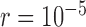 , it typically yielded false negatives and correct delimitations for the above four species pairs and the six species pairs, respectively. When
, it typically yielded false negatives and correct delimitations for the above four species pairs and the six species pairs, respectively. When 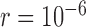 together with
together with 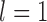 and
and 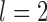 , some false negatives accompany correct delimitations. Under the remaining conditions, bPTP-ML almost always yielded correct delimitations. Numbers of delimited OTUs for GMYC and bPTP-ML have lower mean and median values (Table 2), but these can mask the presence of false negatives.
, some false negatives accompany correct delimitations. Under the remaining conditions, bPTP-ML almost always yielded correct delimitations. Numbers of delimited OTUs for GMYC and bPTP-ML have lower mean and median values (Table 2), but these can mask the presence of false negatives.
Simulation Scenario V: Four Species with Ongoing Gene Flow between Adjacent Species
For our scenario with four species experiencing ongoing gene flow between geographically adjacent species, BPP yielded posterior probabilities similar to those from Scenario III (Supplementary Appendix S13 available on Dryad). Only with a low migration rate (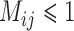 ; and
; and 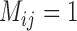 with larger
with larger 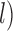 were high probabilities obtained for the correct delimitation model. This model is referred to as “111”, where the numbers “1” denote correct resolutions of (A,B) from (C,D), A from B, and C from D, respectively. Failure to identify each of these distinctions is denoted by “0”. Otherwise, the model 000 has a high posterior probability (Fig. 6d), though it is relatively low when
were high probabilities obtained for the correct delimitation model. This model is referred to as “111”, where the numbers “1” denote correct resolutions of (A,B) from (C,D), A from B, and C from D, respectively. Failure to identify each of these distinctions is denoted by “0”. Otherwise, the model 000 has a high posterior probability (Fig. 6d), though it is relatively low when 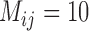 .
.
As with Scenario III, GMYC and bPTP-ML show stronger sensitivity to the presence of gene flow, but they both yielded a few false negatives with every level of gene flow (Tables 2 and 3; Supplementary Appendix S13 available on Dryad). A variety of results containing some correct delimitations (mainly with smaller numbers of loci) and many false positives were obtained for GMYC when 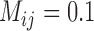 and for bPTP-ML when
and for bPTP-ML when 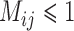 . Otherwise, complex false positives dominate their delimitations.
. Otherwise, complex false positives dominate their delimitations.
Separate analyses of the four species pairs from Scenario V (Supplementary Appendix S14 available on Dryad) show that when 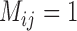 ,
, 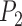 values from BPP for (A, C) and (A, D) are higher than those for (A, B) and (B, C). Even with a very high migration rate (
values from BPP for (A, C) and (A, D) are higher than those for (A, B) and (B, C). Even with a very high migration rate (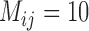 ),
), 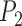 for (A, D) approaches 1.00 for large numbers of loci
for (A, D) approaches 1.00 for large numbers of loci 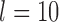 and
and 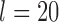 . For bPTP-ML, correct delimitations for (A, C) and (A, D) were obtained more frequently than those for (A, B) and (B, C) when
. For bPTP-ML, correct delimitations for (A, C) and (A, D) were obtained more frequently than those for (A, B) and (B, C) when 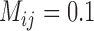 . For GMYC, which produced limited correct delimitations, there are no clear differences in the numbers of correct delimitations among the four species pairs. However, for
. For GMYC, which produced limited correct delimitations, there are no clear differences in the numbers of correct delimitations among the four species pairs. However, for 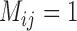 , more false positives were produced for (A, C) and (A, D), whereas complex false positives dominate the results from (A, B) and (B, C). For all three species-delimitation methods, delimitation differences among the species pairs, indicating the effects of geographic distance, become negligible when the migration rate is very small or very large (as appropriate for each method).
, more false positives were produced for (A, C) and (A, D), whereas complex false positives dominate the results from (A, B) and (B, C). For all three species-delimitation methods, delimitation differences among the species pairs, indicating the effects of geographic distance, become negligible when the migration rate is very small or very large (as appropriate for each method).
Species Delimitation in Bears
The maximum-likelihood estimate of the mitochondrial tree of Ursus shows that the brown bear (U. arctos) is paraphyletic with respect to the polar bear (U. maritimus) (Fig. 7). The sequences from the cave bear (U. spelaeus) are also not monophyletic, but this is corrected with the recent designation of sequence NC_011112 as belonging to U. ingressus (Stiller et al. 2014). With the species annotation and reciprocal monophyly as references, a guide tree comprising 10 taxa (Fig. 7) aided BPP in identifying 9 OTUs with a posterior probability of 0.84. The three strategies in PTP, bPTP-ML, bPTP-h, and PTP-h, estimated 17, 22, and 20 OTUs, respectively. GMYC analysis using a single threshold identified the presence of 20 OTUs, matching the result obtained using PTP-h.
Figure 7.

Species delimitations estimated for a dataset comprising 89 sequences from bears (genus Ursus). The maximum-likelihood tree is shown on the left. The vertical bars, from left to right, indicate the OTUs inferred by BPP, bPTP-ML, bPTP-h, PTP-h, and GMYC, respectively. Clades (of different colors in online version) in the tree indicate the 10 taxa in the guide tree for BPP delimitation, and a collapsed clade at the bottom with the label “HQ6859.._ Ursus_ arctos” represents 34 sequences of Ursus arctos with accession numbers beginning with “HQ6859” (Supplementary Appendix S4 available on Dryad).
Species Delimitation in Bees
Species included in our datasets account for a small portion of the described species of apid bees and mason bees, so we do not focus on the inferred relationships here. We tested the effects of the number of loci 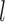 and sample size
and sample size 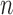 on the performance of BPP, GMYC, and bPTP-ML with the available species annotations. For Apidae (Supplementary Appendix S15 available on Dryad), our duplicate BPP analyses with
on the performance of BPP, GMYC, and bPTP-ML with the available species annotations. For Apidae (Supplementary Appendix S15 available on Dryad), our duplicate BPP analyses with 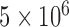 MCMC steps failed to converge when
MCMC steps failed to converge when 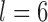 , resulting in posterior probabilities approaching 1.00 for two different delimitation models. One of these models, along with the species delimitations with the highest posterior probabilities when
, resulting in posterior probabilities approaching 1.00 for two different delimitation models. One of these models, along with the species delimitations with the highest posterior probabilities when 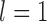 and
and 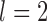 , are consistent with the species annotations. Both GMYC and bPTP-ML produced estimates congruent with the species annotations, except that GMYC delineated two subspecies of Apis mellifera from other subspecies when
, are consistent with the species annotations. Both GMYC and bPTP-ML produced estimates congruent with the species annotations, except that GMYC delineated two subspecies of Apis mellifera from other subspecies when 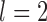 .
.
For Osmia bees (Supplementary Appendix S16 available on Dryad), when 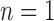 , GMYC and bPTP-ML mixed the annotated species to varying degrees. When
, GMYC and bPTP-ML mixed the annotated species to varying degrees. When 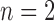 , the two methods generally identified the annotated species, but with one more OTU for some species. When
, the two methods generally identified the annotated species, but with one more OTU for some species. When 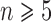 , the methods delimited more OTUs for the annotated species. Delimitations from our BPP analyses are consistent with the 10 Osmia species whenever
, the methods delimited more OTUs for the annotated species. Delimitations from our BPP analyses are consistent with the 10 Osmia species whenever 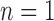 , 2, or
, 2, or 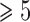 .
.
Discussion
Performance of Species-Delimitation Methods
We have presented a comprehensive comparison of the performance of three widely used molecular species-delimitation methods, based on five different simulation scenarios. The Bayesian coalescent method in BPP, designed for multiple loci, was found to yield high posterior probabilities for correct species delimitations under a variety of conditions. It was relatively robust to the influence of unequal population sizes, population growth, unbalanced sampling, and mutation rate heterogeneity. Some of these findings are consistent with those of previous studies (e.g., Zhang et al. 2011; Barley et al. 2018). However, we note that our use of BPP was carried out under favorable conditions. For example, we used diffuse gamma priors that were compatible with the population sizes and divergence times in our simulations (Leaché and Fujita 2010; Yang 2015). We also specified the true species tree as the guide tree, although the species tree can be jointly estimated with species delimitation by BPP or independently inferred by BPP or other software such as *BEAST (Heled and Drummond 2010; Yang and Rannala 2014; Caviedes-Solis et al. 2015; Yang 2015).
We confirmed that BPP encountered problems when the migration rate between species was relatively high (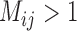 ), and that its delimitation efficacy was somewhat affected by geographic distance (Zhang et al. 2011). These results are not surprising, given the assumption of BPP that no gene flow exists between species. We also found that when the ratio of population size to divergence time (
), and that its delimitation efficacy was somewhat affected by geographic distance (Zhang et al. 2011). These results are not surprising, given the assumption of BPP that no gene flow exists between species. We also found that when the ratio of population size to divergence time (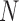 /
/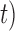 was relatively high, BPP had a high probability of underestimating the number of species, although this could potentially be overcome by analyzing larger numbers of loci and/or using larger samples. Therefore, recently diverged species (as shown by our Scenarios II and IV) pose a challenge to BPP, especially when they have larger population sizes. This outcome is consistent with a previous finding that more loci are needed when analyzing species that have a shallow evolutionary history (Hime et al. 2016).
was relatively high, BPP had a high probability of underestimating the number of species, although this could potentially be overcome by analyzing larger numbers of loci and/or using larger samples. Therefore, recently diverged species (as shown by our Scenarios II and IV) pose a challenge to BPP, especially when they have larger population sizes. This outcome is consistent with a previous finding that more loci are needed when analyzing species that have a shallow evolutionary history (Hime et al. 2016).
We obtained different species delimitations across the three PTP strategies and even the newly developed multi-rate PTP method, which did not perform better than bPTP-ML according to our evaluation criteria (results not shown). However, our focus is on comparison of GMYC, PTP, and BPP. In contrast with BPP, the first two methods aim to delimit species efficiently with data from a single locus, but are increasingly being applied to multilocus datasets. However, our results highlight some differences between the methods. First, unlike the single-threshold GMYC, the best PTP strategy bPTP-ML did not encounter computing errors. Second, bPTP-ML correctly delimited species in Scenario II more frequently than did GMYC, contributing to the better overall performance of bPTP-ML compared with GMYC. Third, larger 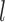 and
and 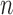 generally enhanced correct delimitations in bPTP-ML, whereas the effect of the former was more modest for GMYC. Fourth, where species were not delimited correctly, the results often differed between GMYC and bPTP-ML. Last, the numbers of delimited OTUs for simulated species indicate that GMYC and PTP can infer different numbers of OTUs in practice.
generally enhanced correct delimitations in bPTP-ML, whereas the effect of the former was more modest for GMYC. Fourth, where species were not delimited correctly, the results often differed between GMYC and bPTP-ML. Last, the numbers of delimited OTUs for simulated species indicate that GMYC and PTP can infer different numbers of OTUs in practice.
There are also considerable similarities in the performance of GMYC and bPTP-ML. Both methods were sensitive to the ratio 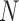 /
/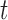 . Large values of
. Large values of 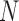 /
/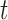 led to complex false positives involving polyphyletic species and false positives involving oversplitting (e.g., Scenario II). Both GMYC and bPTP-ML were also very sensitive to ongoing gene flow, with negative impacts seen even with very low levels of gene flow. Further, both were generally robust to the effects of potential confounding factors (e.g., unbalanced sampling and mutation rate heterogeneity), but to a lesser extent than BPP overall (especially to differing population sizes; Supplementary Appendix S2 available on Dryad). Additionally, our results support the suggestion that GMYC should not be used when the dataset consists of very few putative species, and we extend this suggestion to include bPTP-ML. This is because of the imbalance between speciation and coalescence (Talavera et al. 2013; Dellicour and Flot 2015), which poses a challenge to identifying transition points between inter- and intraspecific processes. In our results, problems appeared in the forms of no correct delimitations (Scenario I), no false negatives (Scenario II, Scenario III, and individual pairs from Scenario V), and even computing errors in GMYC.
led to complex false positives involving polyphyletic species and false positives involving oversplitting (e.g., Scenario II). Both GMYC and bPTP-ML were also very sensitive to ongoing gene flow, with negative impacts seen even with very low levels of gene flow. Further, both were generally robust to the effects of potential confounding factors (e.g., unbalanced sampling and mutation rate heterogeneity), but to a lesser extent than BPP overall (especially to differing population sizes; Supplementary Appendix S2 available on Dryad). Additionally, our results support the suggestion that GMYC should not be used when the dataset consists of very few putative species, and we extend this suggestion to include bPTP-ML. This is because of the imbalance between speciation and coalescence (Talavera et al. 2013; Dellicour and Flot 2015), which poses a challenge to identifying transition points between inter- and intraspecific processes. In our results, problems appeared in the forms of no correct delimitations (Scenario I), no false negatives (Scenario II, Scenario III, and individual pairs from Scenario V), and even computing errors in GMYC.
On the whole, our results indicate that both GMYC and bPTP-ML are able to perform well in the absence of gene flow between species; the latter method tends to perform better overall, although in some cases it produced larger numbers of inferred OTUs. Nevertheless, GMYC and bPTP-ML have a number of important shortcomings. First, they need gene trees to be specified and they treat these as being equivalent to species trees. This assumption is problematic when there is strong discordance between the gene trees and the species tree (Degnan and Rosenberg 2006, 2009). Consequently, concatenation of multiple loci requires caution when using methods designed to analyze single-locus data. Second, when 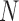 /
/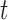 was relatively high, the performance of both GMYC and bPTP-ML was poor regardless of the number of loci or the sample size. Third, GMYC and bPTP-ML rely on the accuracy of the input trees, and upstream errors can result in misleading species delimitations (Tang et al. 2014). GMYC seems to be generally robust to the choice of method used to infer the ultrametric input tree (Talavera et al. 2013; da Cruz and Weksler 2018), but this result is in contrast with the known sensitivity of node-time estimates to the choice of clock model and tree prior (e.g., Duchêne et al. 2014).
was relatively high, the performance of both GMYC and bPTP-ML was poor regardless of the number of loci or the sample size. Third, GMYC and bPTP-ML rely on the accuracy of the input trees, and upstream errors can result in misleading species delimitations (Tang et al. 2014). GMYC seems to be generally robust to the choice of method used to infer the ultrametric input tree (Talavera et al. 2013; da Cruz and Weksler 2018), but this result is in contrast with the known sensitivity of node-time estimates to the choice of clock model and tree prior (e.g., Duchêne et al. 2014).
Factors Affecting Molecular Species Delimitation
The combination of population size 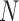 and divergence time
and divergence time 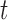 has differing impacts on the three species-delimitation methods. When the ratio
has differing impacts on the three species-delimitation methods. When the ratio 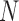 /
/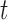 was relatively high for two species, BPP tended to produce false-negative errors. To obtain a correct species delimitation with a small number of loci and small sample size (e.g., 2 for both), the maximum of the ratio
was relatively high for two species, BPP tended to produce false-negative errors. To obtain a correct species delimitation with a small number of loci and small sample size (e.g., 2 for both), the maximum of the ratio 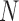 /
/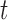 for BPP analyses should be around 1 (Fig. 3). With higher
for BPP analyses should be around 1 (Fig. 3). With higher 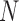 /
/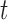 , GMYC and bPTP-ML increasingly produced false positives and complex false positives in Scenario II. With shallow speciation events in Scenario IV, GMYC mainly yielded false negatives with false positives for closely related species, whereas bPTP-ML increased the number of false negatives. These results demonstrate the important influence of incomplete lineage sorting on species delimitation, which becomes more probable with higher
, GMYC and bPTP-ML increasingly produced false positives and complex false positives in Scenario II. With shallow speciation events in Scenario IV, GMYC mainly yielded false negatives with false positives for closely related species, whereas bPTP-ML increased the number of false negatives. These results demonstrate the important influence of incomplete lineage sorting on species delimitation, which becomes more probable with higher 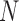 /
/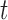 .
.
There is growing use of multi-locus datasets for molecular species delimitation, but this often involves a trade-off between the number of loci and sample size for each species (Fujisawa and Barraclough 2013; Hime et al. 2016). We investigated the possible effects of this trade-off, particularly in Scenario II of our simulations, finding that the effects were outweighed by those of the population size and divergence time. This was previously demonstrated for GMYC (Fujisawa and Barraclough 2013), but we have found that it also holds true for BPP and bPTP-ML. However, the effects of the number of loci  and sample size
and sample size  cannot be negligible. Although both
cannot be negligible. Although both  and
and  appeared to have no impact with lower
appeared to have no impact with lower  /
/ (somewhat consistent with the results of Yang and Rannala 2017), increasing both
(somewhat consistent with the results of Yang and Rannala 2017), increasing both  and
and  improved correct delimitations of BPP with higher
improved correct delimitations of BPP with higher 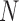 /
/ (Figs. 3 and 6a). Under some conditions in Scenario II, increasing
(Figs. 3 and 6a). Under some conditions in Scenario II, increasing  and/or
and/or  improved the performance of bPTP-ML and GMYC.
improved the performance of bPTP-ML and GMYC.
Our concatenations of larger numbers of loci generally did not have negative effects on the performance of bPTP-ML and GMYC, indicating that the impacts of violating the assumption of gene-tree discordance are modest. However, the situation might be considerably more complex for real data. The performance of BPP with concatenations of 20 loci was equal to or worse than that with single locus (results not shown), suggesting that concatenating independent loci can have a negative impact on methods that have been designed to analyze multiple loci.
The presence of gene flow had negative impacts on the three methods examined in our study, particularly on GMYC and bPTP-ML. These two methods were also substantially affected by differing population sizes resulting in higher 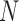 /
/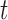 , along with the number of putative species. In contrast, unbalanced sampling, population growth, and mutation rate heterogeneity appeared to have limited impacts on species delimitation using these methods (Supplementary Appendix S2 available on Dryad).
, along with the number of putative species. In contrast, unbalanced sampling, population growth, and mutation rate heterogeneity appeared to have limited impacts on species delimitation using these methods (Supplementary Appendix S2 available on Dryad).
Species Delimitation, Population Delimitation, or a Mosaic?
Our simulation study has shed light on the performance of species-delimitation methods across a range of speciation scenarios, but its implications apply equally to studies of highly structured populations (Sukumaran and Knowles 2017). For example, BPP has been variously used to delimit populations (e.g., Potter et al. 2016), to delimit species (e.g., Mason et al. 2016), to delimit populations with the potential to elevate them to species (e.g., Oliveira et al. 2015), and to delimit evolving lineages within and across species (e.g., Moritz et al. 2018). Our speciation scenarios can be interpreted as being analogous to specific models of population structure. For example, two species with unequal population sizes (Supplementary Appendix S2 available on Dryad) can be treated as a simple case of the continent-island model without migration. Our simulation Scenario V is a one-dimensional stepping-stone model of population structure.
The delimited OTUs can represent species, populations, or even a mosaic of species and populations. In this study, we have simply treated speciation events as being instantaneous, while assuming Wright–Fisher panmixia for each species. In contrast, both the protracted speciation model and the viewpoint that a species is a separately evolving metapopulation lineage (de Queiroz 2007) treat speciation as an extended process. However, these two treatments are not strictly contradictory. Our modelling of speciation as an instantaneous event can be interpreted as the initiation of speciation (i.e., lineage separation), with the delimited OTUs then representing populations or a mosaic of species and populations. If the extended process is relatively short and the newly formed species do not have pronounced structure, then species divergence is effectively an instantaneous event. If there is any continuation of gene flow, the effects are captured in our Scenarios III and V.
The results of molecular species delimitation should be interpreted alongside other lines of evidence, such as comparative morphology, population genetics, and ecology (Sukumaran and Knowles 2017). The importance of using such an integrative approach to taxonomy is underscored by our finding that PTP and GMYC yielded high rates of false positives and complex false positives in some circumstances. If interpreted naively, the results of these analyses would lead to an artificial inflation in the number of species.
Implications of Case Studies
Our case studies, based on species of bears and bees, confirmed some of the findings from our simulation study. These included differences in the performance of the three species-delimitation methods, along with the modest effects of increasing the number of loci and sample size. To interpret these results, we referred to the species annotations accompanying the sequence data from bears and bees, most of which are attributed to traditional morphological taxonomy.
Like the species annotations in our cases, additional information from morphological characters, behavioral traits, and geographic distributions would be needed to provide informed staring points for species delimitation. Accordingly, molecular species delimitation is either implicitly or explicitly carried out as part of an integrative taxonomy approach (Dayrat 2005; Will et al. 2005). In terms of other lines of evidence as above, the informed starting points provide important background for interpreting the presence of population structure.
Our analysis of mitochondrial genomes from bears enhances our understanding of these vulnerable, endangered, or extinct animals. For example, our results point to various delimitations of the brown bear, a species that has been the subject of numerous mitochondrial studies (Davison et al. 2011). Its mitochondrial paraphyly with respect to the polar bear has been recognized as an instance of introgression due to past hybridization between the two species (Hailer et al. 2012).
Compared with bears, many bee species remain undescribed, despite their ecological and economic importance. Currently, pollinator bees fundamental to agricultural productivity are declining towards extinction (Ollerton et al. 2014; Carswell 2015). Our delimitation results are broadly consistent with the species annotations, indicating that molecular species delimitation complements rather than replaces the traditional taxonomy. With molecular species delimitation, the first step to protecting species diversity of bees can be accelerated.
Conclusions
We have compared the performance of three widely used methods of species-delimitation across a range of simulation scenarios and evolutionary parameters. Our results have drawn attention to the better accuracy and robustness of the Bayesian coalescent method in BPP, along with the performance of the GMYC model and the PTP model under a range of conditions. All three methods are negatively influenced by gene flow and are sensitive to the ratio of population size to divergence time, reflecting the important impact of incomplete lineage sorting on species delimitation. Unexpectedly, we found only a modest benefit in increasing the number of loci and the sample size per species. Future studies of molecular species delimitation, particularly focusing on a range of empirical datasets, will provide further insights into the relative impacts of different confounding factors. With a greater understanding of the behavior of molecular species-delimitation methods, genetic data will increasingly contribute to integrative taxonomy and other areas of biological research.
Acknowledgments
We thank the reviewers and the editors for their constructive comments, and Dr Qingsong Zhou, Huanxi Cao, and Dr Pengfei Gao for advice on software. We thank those who generated, edited, and submitted the sequences analyzed in our case studies. We acknowledge the University of Sydney HPC service for providing computing resources for some of our analyses.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.739bs.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 31201701; the Youth Innovation Promotion Association of the Chinese Academy of Sciences [2017118]; and the Ministry of Environmental Protection of China [grant number 2111101]. A.L. was funded by a visiting scholarship from the Chinese Academy of Sciences to carry out research at the University of Sydney. S.Y.W.H. was supported by a Future Fellowship from the Australian Research Council. We acknowledge the support of the National Science Fund for Distinguished Young Scholars [grant number 31625024 to C.Z.].
References
- Ahrens D.,, Fujisawa T.,, Krammer H.,, Eberle J.,, Fabrizi S.,, Vogler A.P. 2016. Rarity and incomplete sampling in DNA-based species delimitation. Syst. Biol. 65:478–494. [DOI] [PubMed] [Google Scholar]
- Arrigoni R.,, Berumen M.L.,, Chen C.A.,, Terraneo T.I.,, Baird A.H.,, Payri C.,, Benzoni F. 2016. Species delimitation in the reef coral genera Echinophyllia and Oxypora (Scleractinia, Lobophylliidae) with a description of two new species. Mol. Phylogenet. Evol. 105:146–159. [DOI] [PubMed] [Google Scholar]
- Barley A.J.,, Brown J.M.,, Thomson R.C. 2018. Impact of model violations on the inference of species boundaries under the multispecies coalescent. Syst. Biol. 67:269–284. [DOI] [PubMed] [Google Scholar]
- Baum D.A.,, Shaw K.L. 1995. Genealogical perspectives on the species problem. In: Hoch P.C.,, Stephenson A.G., editors. Experimental and molecular approaches to plant biosystematics.St. Louis: Missouri Botanical Garden; p. 289–303. [Google Scholar]
- Bond J.E.,, Stockman A.K. 2008. An integrative method for delimiting cohesion species: finding the population-species interface in a group of Californian trapdoor spiders with extreme genetic divergence and geographic structuring. Syst. Biol. 57:628–646. [DOI] [PubMed] [Google Scholar]
- Burgess R.,, Yang Z. 2008. Estimation of hominoid ancestral population sizes under Bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol. Biol. Evol. 25:1979–1994. [DOI] [PubMed] [Google Scholar]
- Camargo A.,, Morando M.,, Avila L.J.,, Sites J.W., Jr. 2012. Species delimitation with ABC and other coalescent-based methods: a test of accuracy with simulations and an empirical example with lizards of the Liolaemus darwinii complex (Squamata: Liolaemidae). Evolution 66:2834–2849. [DOI] [PubMed] [Google Scholar]
- Carstens B.C.,, Pelletier T.A.,, Reid N.M.,, Satler J.D. 2013. How to fail at species delimitation? Mol. Ecol. 22:4369–4383. [DOI] [PubMed] [Google Scholar]
- Carswell C. 2015. Bumblebees aren’t keeping up with a warming planet. Science 349:126–127. [DOI] [PubMed] [Google Scholar]
- Caviedes-Solis I.W.,, Bouzid N.M.,, Banbury B.L.,, Leaché A.D. 2015. Uprooting phylogenetic uncertainty in coalescent species delimitation: a meta-analysis of empirical studies. Curr. Zool. 61:866–873. [Google Scholar]
- da Cruz M.D.,, Weksler M. 2018. Impact of tree priors in species delimitation and phylogenetics of the genus Oligoryzomys (Rodentia: Cricetidae). Mol. Phylogenet. Evol. 119:1–12. [DOI] [PubMed] [Google Scholar]
- Davison J.,, Ho S.Y.W.,, Bray S.C.,, Korsten M.,, Tammeleht E.,, Hindrikson M.,, Østbye K.,, Østbye E.,, Lauritzen S-E.,, Austin J.,, Cooper A.,, Saarma U. 2011. Late-Quaternary biogeographic scenarios for the brown bear (Ursus arctos), a wild mammal model species. Quat. Sci. Rev. 30:418–430. [Google Scholar]
- Dayrat B. 2005. Towards integrative taxonomy. Biol. J. Linn. Soc. 85:407–415. [Google Scholar]
- de Queiroz K. 2007. Species concepts and species delimitation. Syst. Biol. 56:879–886. [DOI] [PubMed] [Google Scholar]
- Degnan J.H.,, Rosenberg N.A. 2006. Discordance of species trees with their most likely gene trees. PLOS Genet. 2:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H.,, Rosenberg N.A. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Dellicour S.,, Flot J. 2015. Delimiting species-poor data sets using single molecular markers: a study of Barcode Gaps, Haplowebs and GMYC. Syst. Biol. 64:900–908. [DOI] [PubMed] [Google Scholar]
- Dobzhansky T. 1950. Mendelian populations and their evolution. Am. Nat. 84:401–418. [Google Scholar]
- Duchêne S.,, Lanfear R.,, Ho S.Y.W. 2014. The impact of calibration and clock-model choice on molecular estimates of divergence times. Mol. Phylogenet. Evol. 78:277–289. [DOI] [PubMed] [Google Scholar]
- Ence D.D.,, Carstens B.C. 2011. SpedeSTEM: a rapid and accurate method for species delimitation. Mol. Ecol. Resour. 11:473–480. [DOI] [PubMed] [Google Scholar]
- Etienne R.S.,, Morlon H.,, Lambert A. 2014. Estimating the duration of speciation from phylogenies. Evolution 68:2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezard T.,, Fujisawa T.,, Barraclough T.G. 2009. Splits: species limits by threshold statistics (version 1.0-19). Available from: URL http://R-Forge.R-project.org/projects/splits/. [Google Scholar]
- Fujisawa T.,, Barraclough T.G. 2013. Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: a revised method and evaluation on simulated data sets. Syst. Biol. 62:707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilets S. 2003. Models of speciation: what have we learned in 40 years? Evolution 57:2197–2215. [DOI] [PubMed] [Google Scholar]
- Goldstein P.Z.,, DeSalle R.,, Amato G.,, Vogler A.P. 2000. Conservation genetics at the species boundary. Conserv. Biol. 14:120–131. [Google Scholar]
- Grummer J.A.,, Bryson R.W., Jr.,, Reeder T.W. 2014. Species delimitation using Bayes factors: simulations and application to the Sceloporus scalaris species group (Squamata: Phrynosomatidae). Syst. Biol. 63:119–133. [DOI] [PubMed] [Google Scholar]
- Hailer F.,, Kutschera V.E.,, Hallström B.M.,, Klassert D.,, Fain S.R.,, Leonard J.A.,, Arnason U.,, Janke A. 2012. Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science 336:344–347. [DOI] [PubMed] [Google Scholar]
- Hartmann K.,, Wong D.,, Stadler T. 2010. Sampling trees from evolutionary models. Syst. Biol. 59:465–476. [DOI] [PubMed] [Google Scholar]
- Hebert P.D.,, Cywinska A.,, Ball S.L.,, deWaard J.R. 2003. Biological identifications through DNA barcodes. Proc. Biol. Sci. 270:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert P.D.,, Penton E.H.,, Burns J.M.,, Janzen D.H.,, Hallwachs W. 2004. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 101:14812–14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedtke S.M.,, Patiny S.,, Danforth B.N. 2013. The bee tree of life: a supermatrix approach to apoid phylogeny and biogeography. BMC Evol. Biol. 13:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J.,, Drummond A.J. 2010. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hime P.M.,, Hotaling S.,, Grewelle R.E.,, O’Neill E.M.,, Voss S.R.,, Shaffer H.B.,, Weisrock D.W. 2016. The influence of locus number and information content on species delimitation: an empirical test case in an endangered Mexican salamander. Mol. Ecol. 25:5959–5974. [DOI] [PubMed] [Google Scholar]
- Hoppeler F.,, Shah R.D.T.,, Shah D.N,, Jähnig S.C.,, Tonkin J.D.,, Sharma S.,, Pauls S.U. 2016. Environmental and spatial characterization of an unknown fauna using DNA sequencing—an example with Himalayan Hydropsychidae (Insecta: Trichoptera). Freshw. Biol. 61:1905–1920. [Google Scholar]
- Hotaling S.,, Foley M.E.,, Lawrence N.M.,, Bocanegra J.,, Blanco M.B.,, Rasoloarison R.,, Kappeler P.M.,, Barrett M.A.,, Yoder A.D.,, Weisrock D.W. 2016. Species discovery and validation in a cryptic radiation of endangered primates: coalescent-based species delimitation in Madagascar’s mouse lemurs. Mol. Ecol. 25:2029–2045. [DOI] [PubMed] [Google Scholar]
- Hudson R.R. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338. [DOI] [PubMed] [Google Scholar]
- Jackson N.D.,, Carstens B.C.,, Morales A.E.,, O’Meara B.C. 2017. Species delimitation with gene flow. Syst. Biol. 66:799–812. [DOI] [PubMed] [Google Scholar]
- Jukes T.H.,, Cantor C.R. 1969. Evolution of protein molecules. In: Munro H.N., editor. Mammalian protein metabolism New York: Academic Press; p. 21–123. [Google Scholar]
- Kapli P.,, Lutteropp S.,, Zhang J.,, Kobert K.,, Pavlidis P.,, Stamatakis A.,, Flouri T. 2017. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang A.S.,, Bocksberger G.,, Stech M. 2015. Phylogeny and species delimitations in European Dicranum (Dicranaceae, Bryophyta) inferred from nuclear and plastid DNA. Mol. Phylogenet. Evol. 92:217–225. [DOI] [PubMed] [Google Scholar]
- Leaché A.D.,, Fujita M.K. 2010. Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proc. Biol. Sci. 277:3071–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaché A.D.,, Fujita M.K.,, Minin V.N.,, Bouckaert R.R. 2014. Species delimitation using genome-wide SNP data. Syst. Biol. 63:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo A.,, Lan H.,, Ling C.,, Zhang A.,, Shi L.,, Ho S.Y.W.,, Zhu C. 2015. A simulation study of sample size for DNA barcoding. Ecol. Evol. 5:5869–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo D.,, De Oliveira Martins L.,, Posada D. 2014. Unsorted homology within locus and species trees. Syst. Biol. 63:988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo D.,, De Oliveira Martins L.,, Posada D. 2016. SimPhy: phylogenomic simulation of gene, locus, and species trees. Syst. Biol. 65:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo D.,, Posada D. 2016. Multilocus inference of species trees and DNA barcoding. Phil. Trans. R. Soc. Lond B Biol. Sci. 371:20150335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason V.C.,, Li G.,, Minx P.,, Schmitz J.,, Churakov G.,, Doronina L.,, Melin A.D.,, Dominy N.J.,, Lim N.T,, Springer M.S.,, Wilson R.K.,, Warren W.C.,, Helgen K.M.,, Murphy W.J. 2016. Genomic analysis reveals hidden biodiversity within colugos, the sister group to primates. Science 2:e1600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr E. 1942. Systematics and the origin of species. New York: Columbia University Press. [Google Scholar]
- Michener C.D. 1970. Diverse approaches to systematics. Evol. Biol. 4:1–38. [Google Scholar]
- Monaghan M.T.,, Wild R.,, Elliot M.,, Fujisawa T.,, Balke M.,, Inward D.J.G.,, Lees D.C.,, Ranaivosolo R.,, Eggleton P.,, Barraclough T.G.,, Vogler A.P. 2009. Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Syst. Biol. 58:298–311. [DOI] [PubMed] [Google Scholar]
- Moritz C.,, Pratt R.C.,, Bank S.,, Bourke G.,, Bragg J.G.,, Doughty P.,, Keogh J.S.,, Laver R.J.,, Potter S.,, Teasdale L.C.,, Tedeschi L.G.,, Oliver P.M. 2018. Cryptic lineage diversity, body size divergence and sympatry in a species complex of Australian lizards (Gehyra). Evolution 72:54–66. [DOI] [PubMed] [Google Scholar]
- Nieto-Montes de Oca A.,, Barley A.J.,, Meza-Lázaro R.N.,, García-Vázquez U.O.,, Zamora-Abrego J.G.,, Thomson R.C.,, Leaché A.D. 2017. Phylogenomics and species delimitation in the knob-scaled lizards of the genus Xenosaurus (Squamata: Xenosauridae) using ddRADseq data reveal a substantial underestimation of diversity. Mol. Phylogenet. Evol. 106:241–253. [DOI] [PubMed] [Google Scholar]
- Oliveira E.F.,, Gehara M.,, São-Pedro V.A.,, Chen X.,, Myers E.A.,, Burbrink F.T.,, Mesquita D.O.,, Garda A.A.,, Colli G.R.,, Rodrigues M.T.,, Arias F.J.,, Zaher H.,, Santos R.M.L.,, Costa G.C. 2015. Speciation with gene flow in whiptail lizards from a Neotropical xeric biome. Mol. Ecol. 24:5957–5975. [DOI] [PubMed] [Google Scholar]
- Ollerton J.,, Erenler H.,, Edwards M.,, Crockett R. 2014. Extinctions of aculeate pollinators in Britain and the role of large-scale agricultural changes. Science 346:1360–1362. [DOI] [PubMed] [Google Scholar]
- Paz A.,, Crawford A.J. 2012. Molecular-based rapid inventories of sympatric diversity: a comparison of DNA barcode clustering methods applied to geography-based vs clade-based sampling of amphibians. J. Biosci. 37:887–896. [DOI] [PubMed] [Google Scholar]
- Pentinsaari M.,, Vos R.,, Mutanen M. 2017. Algorithmic single-locus species delimitation: effects of sampling effort, variation and nonmonophyly in four methods and 1870 species of beetles. Mol. Ecol. Resour. 17:393–404. [DOI] [PubMed] [Google Scholar]
- Pons J.,, Barraclough T.G.,, Gomes-Zurita J.,, Cardoso A.,, Duran D.P.,, Hazell S.,, Kamoun S.,, Sumlin W.D.,, Vogler A.P. 2006. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 55:595–609. [DOI] [PubMed] [Google Scholar]
- Potter S.,, Bragg J.G.,, Peter B.M.,, Bi K.,, Moritz C. 2016. Phylogenomics at the tips: inferring lineages and their demographic history in a tropical lizard, Carlia amax. Mol. Ecol. 25:1367–1380. [DOI] [PubMed] [Google Scholar]
- Previšić A.,, Gelemanović A.,, Urbanič G.,, Ternjej I. 2016. Cryptic diversity in the Western Balkan endemic copepod: four species in one? Mol. Phylogenet. Evol. 100:124–134. [DOI] [PubMed] [Google Scholar]
- Puillandre N.,, Lambert A.,, Brouillet S.,, Achaz G. 2012. ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21:1864–1877. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. v3.3.2. Available from: URL https://www.R-project.org/. [Google Scholar]
- Rambaut A.,, Grassly N.C. 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 13:235–238. [DOI] [PubMed] [Google Scholar]
- Rannala B.,, Yang Z. 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164:1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnasingham S.,, Hebert P.D.N. 2013. A DNA-based registry for all animal species: the barcode index number (BIN) system. PLOS ONE 8:e66213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M.A.,, Heslewood M.M.,, Patzak S.D.,, Schäfer-Verwimp A.,, Heinrichs J. 2017. By how much do we underestimate species diversity of liverworts using morphological evidence? An example from Australasian Plagiochila (Plagiochilaceae: Jungermanniopsida). Mol. Phylogenet. Evol. 107:576–593. [DOI] [PubMed] [Google Scholar]
- Rosen D.E. 1979. Fishes from the uplands and intermontane basins of Guatemala: revisionary studies and comparative geography. Bull. Am. Mus. Nat. Hist. 162:267–376. [Google Scholar]
- Rosindell J.,, Cornell S.J.,, Hubbell S.P.,, Etienne R.S. 2010. Protracted speciation revitalizes the neutral theory of biodiversity. Ecol. Lett. 13:716–727. [DOI] [PubMed] [Google Scholar]
- Ruane S.,, Bryson R.W., Jr.,, Pyron R.A.,, Burbrink F.T. 2014. Coalescent species delimitation in milksnakes (Genus Lampropeltis) and impacts on phylogenetic comparative analyses. Syst. Biol. 63:231–250. [DOI] [PubMed] [Google Scholar]
- Rubinoff D.,, Cameron S.,, Will K. 2006a. A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J. Hered. 97:581–594. [DOI] [PubMed] [Google Scholar]
- Rubinoff D.,, Cameron S.,, Will K. 2006b. Are plant DNA barcodes a search for the Holy Grail? Trends Ecol. Evol. 21:1–2. [DOI] [PubMed] [Google Scholar]
- Sanderson M.J. 2003. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19:301–302. [DOI] [PubMed] [Google Scholar]
- Sokal R.R.,, Crovello T.J. 1970. The biological species concept: a critical evaluation. Am. Nat. 104:127–153. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiller M.,, Molak M.,, Prost S.,, Rabeder G.,, Baryshnikov G.,, Rosendahl W.,, Münzel S.,, Bocherens H.,, Grandal-d’Anglade A.,, Hilpert B.,, Germonpré M.,, Stasyk O.,, Pinhasi R.,, Tintori A.,, Rohland N.,, Mohandesan E.,, Ho S.Y.W.,, Hofreiter M.,, Knapp M. 2014. Quat. Int. 339–340:224–231. [Google Scholar]
- Sukumaran J.,, Knowles L.L. 2017. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. USA 114:1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G.,, Dincă V.,, Vila R. 2013. Factors affecting species delimitations with the GMYC model: insights from a butterfly survey. Methods Ecol. Evol. 4:1101–1110. [Google Scholar]
- Tang C.Q.,, Humphreys A.M.,, Fontaneto D.,, Barraclough T.G. 2014. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 5:1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D.,, Arctander P.,, Minelli A.,, Thomas R.H.,, Vogler A.P. 2003. A plea for DNA taxonomy. Trends Ecol. Evol. 18:70–74. [Google Scholar]
- Vogler A.P.,, Monaghan M.T. 2007. Recent advances in DNA taxonomy. J. Zool. Syst. Evol. Res. 45:1–10. [Google Scholar]
- Wang C.,, Agrawal S.,, Laudien J.,, Häussermann V.,, Held C. 2016. Discrete phenotypes are not underpinned by genome-wide genetic differentiation in the squat lobster Munida gregaria (Crustacea: Decapoda: Munididae): a multi-marker study covering the Patagonian shelf. BMC Evol. Biol. 16:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh J. 2007. DNA barcoding in animal species: progress, potential and pitfalls. BioEssays 29:188–197. [DOI] [PubMed] [Google Scholar]
- Will K.W.,, Mishler B.D.,, Wheeler Q.D. 2005. The perils of DNA barcoding and the need for integrative taxonomy. Syst. Biol. 54:844–851. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2015. The BPP program for species tree estimation and species delimitation. Curr. Zool. 61:854–865. [Google Scholar]
- Yang Z.,, Rannala B. 2010. Bayesian species delimitation using multilocus sequence data. Proc. Natl. Acad. Sci. USA 107:9264–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.,, Rannala B. 2014. Unguided species delimitation using DNA sequence data from multiple loci. Mol. Biol. Evol. 31:3125–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.,, Rannala B. 2017. Bayesian species identification under the multispecies coalescent provides significant improvements to DNA barcoding analyses. Mol. Ecol. 26:3028–3036. [DOI] [PubMed] [Google Scholar]
- Yule G.U. 1924. A mathematical theory of evolution, based on the conclusions of Dr. J. C. Willis, F.R.S. Phil.Trans. R. Soc. Lond. B. 213:21–87. [Google Scholar]
- Zhang C.,, Rannala B.,, Yang Z. 2014. Bayesian species delimitation can be robust to guide-tree inference errors. Syst. Biol. 63:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.,, Zhang D.,, Zhu T.,, Yang Z. 2011. Evaluation of a Bayesian coalescent method of species delimitation. Syst. Biol. 60:747–761. [DOI] [PubMed] [Google Scholar]
- Zhang J.,, Kapli P.,, Pavlidis P.,, Stamatakis A. 2013. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]