

Abstract
Background
Identification of gut microbiota features associated with antibiotic-resistant bacterial colonization may reveal new infection prevention targets.
Methods
We conducted a matched, case–control study of long-term acute care hospital (LTACH) patients to identify gut microbiota and clinical features associated with colonization by Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae (KPC-Kp), an urgent antibiotic resistance threat. Fecal or rectal swab specimens were collected and tested for KPC-Kp; 16S rRNA gene-based sequencing was performed. Comparisons were made between cases and controls in calibration and validation subsamples using microbiota similarity indices, logistic regression, and unit-weighted predictive models.
Results
Case (n = 32) and control (n = 99) patients had distinct fecal microbiota communities, but neither microbiota diversity nor inherent clustering into community types distinguished case and control specimens. Comparison of differentially abundant operational taxonomic units (OTUs) revealed 1 OTU associated with case status in both calibration (n = 51) and validation (n = 80) subsamples that matched the canonical KPC-Kp strain ST258. Permutation analysis using the presence or absence of OTUs and hierarchical logistic regression identified 2 OTUs (belonging to genus Desulfovibrio and family Ruminococcaceae) associated with KPC-Kp colonization. Among clinical variables, the presence of a decubitus ulcer alone was independently and consistently associated with case status. Combining the presence of the OTUs Desulfovibrio and Ruminococcaceae with decubitus ulcer increased the likelihood of KPC-Kp colonization to >38% in a unit-weighted predictive model.
Conclusions
We identified microbiota and clinical features that distinguished KPC-Kp gut colonization in LTACH patients, a population particularly susceptible to KPC-Kp infection. These features may warrant further investigation as markers of risk for KPC-Kp colonization.
Keywords: carbapenem-resistant Enterobacteriaceae, Klebsiella pneumoniae, microbiota, long-term acute care hospitals, microbiome
Health care–associated infections due to multidrug-resistant organisms (MDROs) pose an increasing public health threat. Carbapenemase-producing Enterobacteriaceae (CPE) are one of the most concerning MDROs because of their ability to spread rapidly across regions [1, 2] and to cause invasive infections associated with high mortality [3]. Although CPE control programs have been successful in some locales [4], globally the problem continues to worsen [5], necessitating more effective prevention strategies.
Colonization of the gastrointestinal tract by CPE usually precedes infection. Although a healthy, indigenous gut microbiota likely provides resistance to colonization by enteric MDROs such as CPE, exposure to antibiotics and other clinical factors can disrupt gut microbial community homeostasis and reduce “colonization resistance,” thus increasing CPE colonization risk [6]. Development of “microbiome disruption indices,” that is, measurements of microbiota characteristics that inversely associate with colonization resistance, has been proposed as an infection prevention strategy to improve identification of patients at risk for MDRO colonization, infection, or transmission [7]. A first step toward development of a microbiome disruption index for predicting CPE colonization is to determine whether the composition of the microbiota differs in CPE-colonized and -noncolonized patients. To this end, we compared the fecal microbiota and clinical care exposures of patients with and without colonization by Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae (KPC-Kp), the most common CPE in the United States, and sought features that distinguished the 2 groups.
METHODS
Study Design and Enrollment Criteria
We conducted a matched, case–control study in 1 long-term acute care hospital (LTACH) in Chicago, Illinois. Cases were defined as patients with KPC-Kp intestinal colonization identified by fecal or rectal swab screening within 3 days of admission to the LTACH and no evidence of colonization or infection with CPE other than KPC-Kp. Patients who had negative admission screening tests and no history of CPE colonization or infection were eligible to serve as controls and were matched 1:1 or 1:2 to cases by admission date (±14 days), depending on availability.
Patients were excluded from study participation if they had a history of acute bacterial or viral colitis (including Clostridium difficile infection) or active inflammatory bowel disease within 14 days before sample collection, or if they had a rectal tube, fecal incontinence device, or colostomy at the time of sample collection. These exclusion criteria were applied to reduce potential confounding effects of colonic inflammation and ensure that samples were representative of recently passed feces, respectively. Patients participated in the study only once.
The study protocol was reviewed and granted expedited approval by the institutional review board at Rush University Medical Center. Written informed consent was waived.
Specimen Collection, Processing, and Screening for CPE
Fecal or rectal swab specimens were collected from study participants, screened for KPC-Kp, and used for microbiota community analysis (Supplementary Table 1). In a previous investigation, we determined that fecal and rectal swab microbiota from the same subject were highly similar, thus justifying the combined analysis of those 2 specimen types in the current investigation [8]. Specimens were screened for bla-KPC by polymerase chain reaction (PCR) [9, 10] and plated simultaneously on MacConkey agar (Remel Inc, San Diego, CA); plates were incubated at 35ºC in ambient air for up to 48 hours. Unique colony morphologies of suspected K. pneumoniae were identified to the species level, and antibiotic susceptibility was determined using the Microscan Walkaway Plus system (Beckman Coulter, Indianapolis, IN). Carbapenem-resistant K. pneumoniae isolates underwent a second round of confirmatory PCR testing for bla-KPC.
16S rRNA Gene Sequencing
To determine if the fecal microbial community of KPC-Kp(+) patients differed from that of KPC-Kp(-) patients, we purified DNA from fecal specimens or rectal swabs using the PowerMag Soil DNA Isolation Kit (Mo Bio Laboratories, Inc., Cardlsbad, CA) on the EpMotion 5075 (Eppendorf, Hamburg, Germany). The bacterial 16S rRNA genes were amplified by PCR using dual-index primers specific to the V4 region [11] from 1 μL of the sample DNA, as described previously [12]. Amplicons were prepared for sequencing and sequenced using the 500 cycle MiSeq Reagent Kit, v. 2 (Illumina, catalog No. MS-102–2003), on a MiSeq (Illumina, San Diego, CA) by the University of Michigan Microbial Systems Molecular Biology Laboratory as described previously [12].
Sequence Analysis of Microbiota
Sequence files were deposited in the NCBI’s Sequence Read Archive (SRA; Bioproject PRJNA428477, SAMN8292036-8292166). Sequences were processed and analyzed using mothur (v. 1.39.5) (Supplementary Methods) [13]. Briefly, sequences were aligned to the recreated SILVA SEED reference alignment (release 119) and trimmed to the same start and end positions [14]. Chimeras were removed using uchime [15]. Samples with <1000 sequences after processing were not included. For all microbiota analyses, sequences were clustered into operational taxonomic units (OTUs) based on 97% sequence similarity by the average neighbor method. Shannon diversity was calculated based on OTU composition. A distance metric that takes relative abundances of both shared and nonshared OTUs into account, θYC, was calculated between samples [16], and analysis of molecular variance (AMOVA) [17] was used to test for significant differences between the communities of KPC-Kp(+) and KPC-Kp(-) patients. Principal coordinates analysis (PCoA) was used to visualize the θYC distances between samples. Linear discriminant analysis (LDA) effect size (LEfSe) was used to determine if specific OTUs were differentially abundant in KPC-Kp(+) and KPC-Kp(-) specimens [18]. We also investigated the taxonomic composition by classifying sequences within mothur using the Ribosomal Database Project (RDP) training set (v. 10) [19]. Samples were grouped into community types using partitioning around medoids (PAM), calculated from the Jensen-Shannon divergence of the taxonomic composition, as used previously to group fecal microbiota profiles into enterotypes [20]. To investigate the sequences included in each OTU, bin.seqs was used. The unique OTU003 sequence was further analyzed using RDP’s online Sequence Match against 16S rRNA gene sequences for isolates (type and nontype), size ≥1200 nucleotides, and good quality (on December 20, 2017) [9] and compared with specific genomes by BLAST in PATRIC [21]. Stepwise sequence analysis is available at https://github.com/aseekatz/Rush_CX.study.
Clinical Data
Demographic and clinical data were extracted from hospital electronic medical records. Time-dependent exposures (eg, antibiotics) were measured from LTACH admission to the time of fecal or rectal swab specimen collection and coded as present or absent.
Development of Predictive Models
KPC-Kp colonization predictive indices were developed using a 2-stage approach. First, permutation analysis was used to reduce the initial set of 300 candidate OTU predictors to a number that was more manageable for constructing a robust, linear predictive model. Each OTU was recoded from a continuous quantity (relative abundance) to a dummy-coded variable based on presence or absence in a patient’s specimen. The entire patient sample was randomly assigned to 2 groups, and the random assignment was permuted 100 times. For each permutation of the random assignments, risk ratios for each OTU were calculated for both groups. An OTU was considered validated if the risk ratio in the 2 groups both exceeded an absolute value of 2.0 and predicted KPC-Kp colonization status in the same direction. The use of this permutation approach is equivalent to a k-fold cross-validation.
Second, the patient sample was divided into calibration and validation subsamples based on date of LTACH admission: The first 51 patients who participated in the study were included in the calibration subsample, and the next 80 patients who participated in the study were included in the validation subsample. The larger number in the validation subsample was designed to increase the power of the validation analyses.
Logistic regression was used to develop optimal clinical and microbiome predictive models in the calibration subsample. The dummy-coded OTUs and clinical variables that were found to be independent predictors of KPC-Kp status by logistic regression were then combined into simple unit-weighted models to create more robust predictions. Finally, the unit-weighted models were cross-validated in the validation subsample of patients to determine if they were generalizable across the entire sample. Analyses were conducted using SPSS software, version 19 (IBM SPSS, Chicago, IL), and R, version 2.13.1 (http://CRAN.R-project.org).
RESULTS
Characteristics of Study Participants
One hundred thirty-one patients (32 KPC-Kp(+) case patients and 99 KPC-Kp(-) control patients) were included in the study between June 19, 2014, and January 15, 2016 (Figure 1). On average, patients were elderly, and most (56%) were receiving antibiotics at the time of fecal or rectal swab specimen collection. In bivariate analyses, case patients were more likely than control patients to have poor functional status, comorbid medical conditions, decubitus ulcers, and medical device use (Table 1.) In a multivariable logistic regression model, the sole clinical predictor of KPC-Kp colonization was presence of a decubitus ulcer (odds ratio [OR], 11.2; 95% confidence interval [CI], 1.3–95; P = .026).
Figure 1.

Patient participation. Patients were screened for participation between June 10, 2014, and January 17, 2016. The calibration subsample was collected between June 19, 2014, and February 6, 2015. The validation subsample was collected between March 21, 2015, and January 15, 2016. Case patients grew carbapenem-resistant, Klebsiella pneumoniae isolates from fecal or rectal swab samples that were verified to carry the bla-KPC gene by polymerase chain reaction (PCR). Control patients’ specimens were negative for bla-KPC and bla-NDM genes by screening PCR and culture-negative for carbapenem-resistant Enterobacteriaceae. Abbreviations: CPE, carbapenemase-producing Enterobacteriaceae; EIA, enzyme immunoasssay; KPC-Kp, Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae.
Table 1.
Comparison of Selected Demographic Information and Clinical Care Exposures for Cases and Controls
| Case (n = 32) | Control (n = 99) | OR (95% CI) | P Value | |
|---|---|---|---|---|
| No. (%) | No. (%) | |||
| Demographics | ||||
| Age, mean ± SD, y | 66 ± 13 | 62 ± 16 | 1.02 (0.99–1.04) | .26 |
| Female | 19 (59) | 40 (40) | 2.2 (0.96–4.8) | .061 |
| Black vs white, Asian, Hispanic, other | 23 (72) | 61 (62) | 1.59 (0.67–3.80) | .29 |
| Clinical status and comorbid conditions | ||||
| Mental statusa | 16 (50) | 26 (26) | 2.8 (1.2–6.4) | .012 |
| Activity levelb | 32 (100) | 71 (72) | — | <.001 |
| Oral feeding | 12 (38) | 57 (58) | 0.44 (0.19–1) | .048 |
| Gastronomy tube feeding | 21 (66) | 37 (37) | 3.2 (1.4–7.4) | .005 |
| Fecal incontinence | 23 (72) | 48 (48) | 2.7 (1.1–6.4) | .021 |
| Urinary bladder catheter | 28 (88) | 44 (44) | 8.8 (2.8–26.8) | <.001 |
| Mechanical ventilation | 19 (59) | 23 (23) | 4.8 (2.1–11.2) | <.001 |
| Decubitus ulcer | 30 (94) | 53 (54) | 13 (2.9–57.5) | <.001 |
| Other MDRO carriage or infectionc | 23 (72) | 26 (26) | 7.2 (2.9–17.5) | <.001 |
| Charlson Comorbidity Index, median ± IQR | 4 ± 2.5 | 3 ± 3 | 1.24 (1.04–1.49) | .017 |
| Myocardial infarction/stroke | 18 (56) | 28 (28) | 3.3 (1.4–7.4) | .004 |
| Diabetes | 19 (59) | 44 (44) | 1.8 (0.81–4.1) | .14 |
| Medicationsd | ||||
| Any antibiotic | 18 (56) | 55 (56) | 1 (0.46–2.3) | .95 |
| Antibiotic with anti-anaerobic activity | 14 (44) | 46 (46) | 0.9 (0.4–2.0) | .79 |
| Carbapenem (meropenem) | 5 (16) | 11 (11) | 1.5 (0.5–4.6) | .54 |
| Probiotic | 0 | 5 (5) | — | .33 |
| Proton pump inhibitor | 13 (41) | 32 (32) | 1.4 (0.63–3.3) | .39 |
Abbreviations: CI, confidence interval; IQR, interquartile range; MDRO, multidrug-resistant organism; OR, odds ratio.
aDisoriented or unresponsive vs awake and alert.
bBedbound or wheelchair-bound vs ambulatory (1 case patient was wheelchair-bound).
cOther MDROs included methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus spp., extended-spectrum beta-lactamase-producing Enterobacteriaceae, carbapenem-resistant Acinetobacter baumannii, and carbapenem-resistant Pseudomonas aeruginosa.
dReceipt of medication between admission and fecal specimen collection.
The Fecal Microbiota Community, but not Microbiota Diversity, Distinguished Patients With or Without KPC-Kp Colonization
The overall diversity of the fecal microbiota as measured by the Shannon diversity index (d) did not differ between specimens from KPC-Kp(+) and KPC-Kp(-) patients (Figure 2A). However, diversity was significantly decreased in patients who received antibiotics between the time of admission and fecal sample collection, independent of KPC-Kp status (Wilcoxon test, P < .01) (Figure 2B). This difference was consistent across different antibiotic classes (Supplementary Figure 1).
Figure 2.

Diversity of the fecal microbiota in patients admitted to 1 long-term acute care hospital. The diversity of the fecal microbiota (Shannon diversity index, d, y-axis) did not differ in (A) patients positive or negative for Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae (KPC-Kp) but (B) was significantly increased in patients who had not received antibiotics between admission and fecal specimen collection (Abx). (Wilcoxon test, *P < .01). Abbreviation: Abx, Antibiotics.
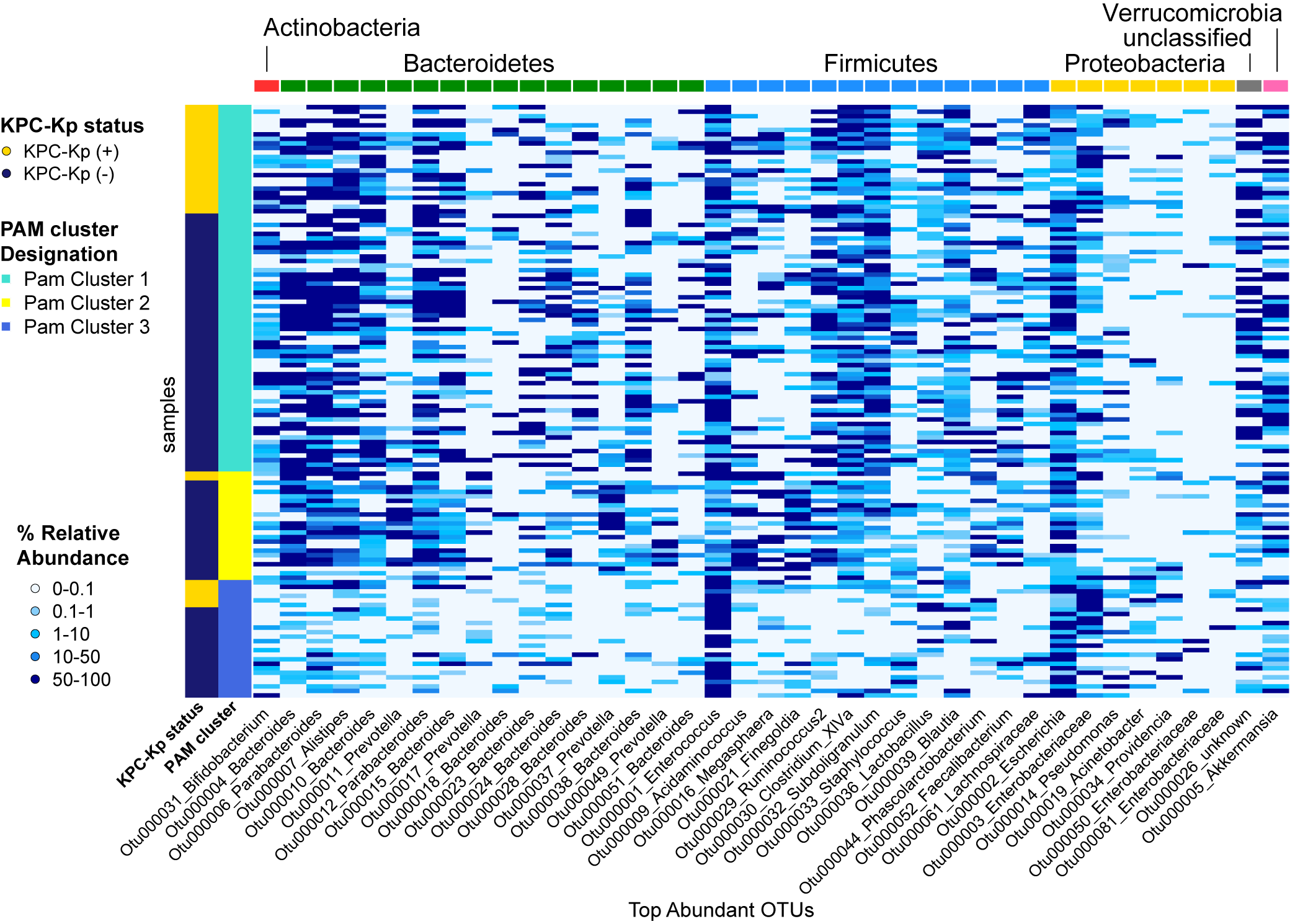
Use of AMOVA on θYC distances to compare communities from KPC-Kp(+) and KPC-Kp(-) patients indicated that the microbial communities in patients from these 2 groups were distinct (P = .017), although this relatively subtle difference was not obvious by PCoA (Figure 3A). Next, we tried community clustering using the PAM algorithm based on Jensen-Shannon divergence (Figure 3B) [20, 22]. Likely due to high microbiota variability between subjects (Supplementary Figure 2), clustering was relatively weak, with a maximum silhouette score of 0.28 for 3 clusters, each defined by different dominant genera (Figure 3B, C). PAM cluster 1 was a high-diversity cluster with several Bacteroidetes and Firmicutes OTUs, PAM cluster 2 was defined by mainly Bacteroidetes OTUs, and PAM cluster 3 was characterized by high abundances of OTU001 (Enterococcus), OTU002 (Escherichia), and OTU003 (Enterobacteriaceae). Examination of the binned OTU sequences revealed that OTU003 contained a single unique sequence, matching various Enterobacteriaceae family members, including 16S rRNA gene sequences from the canonical KPC-Kp strain ST258 (Supplementary Methods). Significant overrepresentation of specimens from KPC-Kp(+) patients was not observed in any of the clusters (chi-square test).
Figure 3.

Fecal microbiota structure and composition in long-term acute care hospital patients. A, Principal coordinates analysis (PCoA) based on the Yue & Clayton dissimilarity index suggested a difference in the microbiota structure of the fecal microbiota in Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae (KPC-Kp)(+) and KPC-Kp(-) patients (analysis of molecular variance, *P < .05). B, Biplot visualizing the operational taxonomic units (OTUs) driving the microbiota community structure in all patients, as represented by 3 community clusters calculated using partitioning around medoids (PAM) clustering (based on Jensen-Shannon divergence). C, Mean relative abundance of the 40 most abundant OTUs in each PAM cluster (n = number of samples; d = Shannon diversity).
We next sought to determine if specific features of the microbiota associated with KPC-Kp status. LEfSe evaluation revealed 12 OTUs that were differentially abundant in KPC-Kp(+) vs KPC-Kp(-) patients using the entire sample (n = 131 patients). To validate this observation, we ran LEfSe estimation independently on the calibration and validation subsamples. Different OTUs were observed to be differentially abundant in the calibration and validation subsets (Figure 4). The only OTU that cross-validated across the 2 subsamples was OTU003, which was also identified as differentially abundant in all samples, suggesting that a higher relative abundance of the OTU containing KPC-Kp was associated with KPC-Kp colonization (Figure 4).
Figure 4.

Differentially abundant operational taxonomic units (OTUs) in patients with or without Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae (KPC-Kp) colonization. Relative abundances of operational taxonomic units identified to be differentially abundant between KPC-Kp(+) and KPC-Kp(-) patients (left panel) using Linear discriminant analysis (LDA) effect size, represented as log10(relative abundance +1). Heatmap (right panel) demonstrates the LDA value of each OTU (representing magnitude of difference) in calibration, validation, or all patient samples.
In a final effort to identify features that distinguished the fecal microbiota of KPC-Kp(+) and KPC-Kp(-) patients, we applied a permutation analysis to investigate whether specific OTUs were differentially present or absent in fecal specimens from case and control patients. In the first stage of the analysis and based on cross-validated risk ratios of 2.0, the number of candidate differential OTUs was reduced from a filtered count of 302 (present in at least 0.01% of the total data set and in at least 5% of all samples) to 30. A risk ratio of 2.0 was selected because, assuming a sample size of 30 in both the calibration and validation subsamples and a base likelihood of 18% OTU relative abundance (the median OTU relative abundance for the 302 OTUs considered in the entire patient sample), there was a statistically significant likelihood of achieving a risk ratio of 2.0 (P = .014.) Identification of candidate OTUs was not ambiguous: all discarded OTUs had 0 cross-validations. Of the 30 candidate OTUs that were retained, the number of cross-validations varied from 15 to 87 out of 100 permutations, and all cross-validated agreements were in the same direction. Hierarchical logistic regression analysis was used to further reduce the number of OTUs from 30 to 2: (OTU109 [Desulfovibrio] and OTU179 [Ruminococcaceae]). The elimination process was based on Wald statistics in the logistic regression analysis. If there was a tie in the Wald statistics, the bivariate risk ratio was used as a tie-breaker.
Unit-Weighted Models to Predict KPC-Kp Colonization at the Time of LTACH Admission
Unit-weighted predictive functions (index values) were created using the 1 clinical and 2 microbiome variables that were found to be independent, statistically significant predictors of KPC-Kp status in logistic regression models, that is, decubitus ulcer, OTU109 (Desulfovibrio), and OTU179 (Ruminococcaceae). These 3 variables were dummy coded as “0” if absent and “1” if present and used to create simple clinical, microbiome, and combined index value models (Figure 5). The clinical index model and the microbiome index model were statistically significant predictors of KPC-Kp status in both the calibration and validation subsets (Figure 5A, B, D, E). The combined model, which included both clinical and microbiome predictors, performed better than either model alone (Figure 5C, F). The increase in model performance occurred for both the calibration and validation subsets, and the predictive function was a statistically significant predictor in both subsets (Figure 5C, F)
Figure 5.

Clinical, microbiome, and combined predictive models. A, Clinical calibration model, based on the presence or absence of a decubitus ulcer. In a multivariable logistic regression model, presence of a decubitus ulcer was the sole independent clinical predictor of blaKPC-positive K. pneumoniae carriage. B) Microbiome calibration model, based on the presence or absence of 2 operational taxonomic units (OTUs; OTU 109 [Desulfovibrio] and OTU 179 [Ruminococcaceae]) identified in the permutation analysis in which 30 of 302 candidate OTUs were retained. C, Combined clinical and microbiome model. D, Clinical validation model. E, Microbiome validation model. F, Combined validation model. Bars show upper bounds of standard errors, which are symmetric around the proportions (lower bounds not shown). Abbreviation: KPC, Klebsiella pneumoniae carbapenemase.
DISCUSSION
The appearance of MDROs such as KPC-Kp in healthcare settings poses a significant threat. Recently, prevention efforts have begun to target long-term care facilities such as LTACHs, which can serve as foci for the amplification and spread of KPC-Kp and other MDROs [23–29]. Improved knowledge of factors that predispose to KPC-Kp carriage in LTACH populations could lead to new, more effective prevention strategies.
A growing body of evidence suggests that the indigenous microbiota of the gastrointestinal tract plays a critical role in determining susceptibility to MDRO carriage and infection [30–32]. The prime example of this is infection with Clostridium difficile, which is associated with alteration of the gut microbiota following antibiotic administration. Antibiotics have been shown to alter the structure and function of the gut microbiota in a manner that decreases colonization resistance against C. difficile. Restoration of the gut microbiota by means of fecal microbiota transplantation further demonstrates the critical role that the gut microbiome can play in C. difficile infection [33, 34].
Our current study indicates that patients who are colonized with KPC-Kp upon admission to an LTACH have a distinct gut microbiome compared with noncolonized patients. The overall bacterial community structure of fecal specimens from KPC-Kp(+) patients was dissimilar to that of fecal specimens from KPC-Kp(-) patients (Figure 3A), but overall diversity did not differ significantly between the 2 groups. Decreased microbiota diversity has been observed in patients with recurrent C. difficile infection, which is a rationale for treating this infection with microbiota transplantation [35, 36]. In the current study, the absence of association between KPC-Kp status and microbiota diversity may be explained in part by antibiotic exposure in >50% of both cases and controls at the time of specimen collection, which in and of itself is associated with decreased microbiota diversity [37–39]. Although we did not have access to patients’ antibiotic exposure histories before LTACH admission, it is likely that many patients would have received multiple prior antibiotic courses [23], which would also have been expected to reduce the diversity of their microbiota.
We utilized 2 approaches to seek specific members of patients’ gut microbiota that associated with KPC-Kp colonization. Analysis by LEfSe was used to detect significant differential abundance of microbial community members between different groups, followed by linear discriminant analysis to evaluate the effect size of those features. We refined our use of LEfSe by performing this analysis on samples from calibration and validation subsets of patients. Whereas multiple OTUs were identified as differentially abundant in KPC-Kp(+) patients compared with KPC-Kp(-) patients, only 1 OTU (OTU003) was identified as significant in both calibration and validation subsamples. This OTU003 is predicted to be the OTU that contains the common KPC-Kp strain ST258. Although this may seem to be a trivial result, it is important to recall that LEfSe analysis takes into account relative abundance of a given OTU when determining significance. In fact, our alternate approach for examining OTUs that was based on presence or absence of all OTUs did not identify OTU003 as being significantly associated with KPC-Kp colonization. Closer examination of these data suggests that virtually all patient specimens included sequences classified as OTU003. Therefore, it appears that KPC-Kp-colonized patients are distinguished from noncolonized patients by having KPC-Kp as a relatively abundant member of the community.
In a second, independent analytic approach that subset the study sample using permutation, the association of OTU109 (Desulfovibrio) and OTU179 (Ruminococcaceae) with KPC-Kp colonization was consistent across calibration and validation subsets (Figure 5.) Assessing the generalizability of microbiota analyses across different subsets of specimens can limit spurious associations identified with these complex, heterogeneous data sets where collinearity is likely to be a problem. Although we did not investigate the biological significance of the association between OTU109 (Desulfovibrio) or OTU179 (Ruminococcaceae) and KPC-Kp colonization, the consistency of the association across all patient subsets suggests that these OTUs might be candidates for investigation in longitudinal analyses as microbiome indices of KPC-Kp acquisition risk. The association between a clinical factor—presence of a decubitus ulcer—and KPC-Kp colonization is likely an indicator of poor functional status associated with KPC-Kp carriage. As in our previous work with C. difficile [22], models that included both clinical and microbiome variables improved the prediction of KPC-Kp status compared with either model alone.
The strengths of our study include its size and use of calibration and validation subsets to investigate the generalizability of associations between the microbiome and clinical features and KPC-Kp status. Our study also has limitations. We used a cross-sectional design, which prevents us from knowing whether microbiome features that were observed to be associated with KPC-Kp positivity were present before colonization. Our ability to detect KPC-Kp is not limiting, and there is little clinical utility to using microbiota analysis for detection of KPC-Kp. However, although 16S rRNA gene sequence analysis of full bacterial communities may not be a feasible clinical diagnostic tool, such analyses are the first step toward identifying potential biomarkers for future validation and development. Additionally, studying the fecal microbiota of KPC-Kp(-) patients longitudinally over time would provide an opportunity to investigate features of the microbiota associated with acquisition of KPC-Kp colonization. Such a longitudinal design could also be used to interrogate characteristics of the microbiota of KPC-Kp(+) patients that are associated with development of clinical infection. The potential value of this approach has been suggested by work that showed that the relative abundance of vancomycin-resistant enterococci or Enterobacteriaceae was associated with an increased risk of bacteremia [40].
In summary, we observed statistically significant and consistent differences in clinical characteristics and microbiome features of fecal specimens from KPC-Kp-colonized and -noncolonized LTACH patients. These results provide support for continuing investigation of the gut microbiota as a potential diagnostic tool to identify patients at risk of KPC-Kp acquisition, providing possible targets for intervention to prevent or eliminate KPC-Kp colonization and reduce the risk of invasive infection.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
Acknowledgments
We thank the LTACH patients for participation in the study and the staff for assistance with logistical details. We thank the University of Michigan Medical School Human Microbiome Initiative and the Microbial Systems Molecular Biology lab for sample processing and logistics.
Financial support. This work was supported in part by Centers for Disease Control and Prevention Epicenters Program Cooperative Agreements U54 CK00016 03S1 and U54 CK000481. A.M.S. was supported in part by the National Center for Advancing Translational Research (UL1TR000433).
Potential conflicts of interest. M.Y.L. has received research support in the form of contributed product from OpGen and Sage Products (now part of Stryker Corporation) and has received an investigator-initiated grant from the CareFusion Foundation (now part of BD). M.K.H. has received research support in the form of contributed product from Clorox, Medline, Mölnlycke, OpGen, and Sage Products and has received an investigator-initiated grant from Clorox. R.A.W. has received research support in the form of contributed product from Clorox, Sage Products, Mölnlycke, and Medline. N.M. received research grants from Cepheid, Inc. and bioMerieux, and honoraria from Cepheid. V.B.Y. received consulting fees from MedImmune, Exarca Pharmaseuticals, Finch Therapeutics and Vedanta Biosciences. All other authors declare no conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Cantón R, Akóva M, Carmeli Y, et al. ; European Network on Carbapenemases Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin Microbiol Infect 2012; 18:413–31. [DOI] [PubMed] [Google Scholar]
- 2. Won SY, Munoz-Price LS, Lolans K, et al. ; Centers for Disease Control and Prevention Epicenter Program Emergence and rapid regional spread of Klebsiella pneumoniae carbapenemase-producing Enterobacteriaceae. Clin Infect Dis 2011; 53:532–40. [DOI] [PubMed] [Google Scholar]
- 3. Falagas ME, Tansarli GS, Karageorgopoulos DE, Vardakas KZ. Deaths attributable to carbapenem-resistant Enterobacteriaceae infections. Emerg Infect Dis 2014; 20:1170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwaber MJ, Lev B, Israeli A, et al. ; Israel Carbapenem-Resistant Enterobacteriaceae Working Group Containment of a country-wide outbreak of carbapenem-resistant Klebsiella pneumoniae in Israeli hospitals via a nationally implemented intervention. Clin Infect Dis 2011; 52:848–55. [DOI] [PubMed] [Google Scholar]
- 5. Bonomo RA, Burd EM, Conly J, et al. . Carbapenemase-producing organisms: a global scourge!Clin Infect Dis. 2018;. 66:1290–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol 2012; 20:313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Halpin AL, de Man TJ, Kraft CS, et al. . Intestinal microbiome disruption in patients in a long-term acute care hospital: a case for development of microbiome disruption indices to improve infection prevention. Am J Infect Control 2016; 44:830–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bassis CM, Moore NM, Lolans K, et al. ; CDC Prevention Epicenters Program Comparison of stool versus rectal swab samples and storage conditions on bacterial community profiles. BMC Microbiol 2017; 17:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cole JM, Schuetz AN, Hill CE, Nolte FS. Development and evaluation of a real-time PCR assay for detection of Klebsiella pneumoniae carbapenemase genes. J Clin Microbiol 2009; 47:322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangold KA, Santiano K, Broekman R, et al. . Real-time detection of blaKPC in clinical samples and surveillance specimens. J Clin Microbiol 2011; 49:3338–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kozich JJ, Westcott SL, Baxter NT, et al. . Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 2013; 79:5112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seekatz AM, Theriot CM, Molloy CT, et al. . Fecal microbiota transplantation eliminates Clostridium difficile in a murine model of relapsing disease. Infect Immun 2015; 83:3838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schloss PD, Westcott SL, Ryabin T, et al. . Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009; 75:7537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pruesse E, Quast C, Knittel K, et al. . SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 2007; 35:7188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edgar RC, Haas BJ, Clemente JC, et al. . UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011; 27:2194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yue JC, Clayton MK. A similarity measure based on species proportions. Commun Stat-Theory Methods 2005; 34: 2123–31. [Google Scholar]
- 17. Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology 2001; 26: 32–6. [Google Scholar]
- 18. Segata N, Izard J, Waldron L, et al. . Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 73:5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arumugam M, Raes J, Pelletier E, et al. ; MetaHIT Consortium Enterotypes of the human gut microbiome. Nature 2011; 473:174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wattam AR, Davis JJ, Assaf R, et al. . Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res 2017; 45:D535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schubert AM, Rogers MA, Ring C, et al. . Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio 2014; 5:e01021–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gould CV, Rothenberg R, Steinberg JP. Antibiotic resistance in long-term acute care hospitals: the perfect storm. Infect Control Hosp Epidemiol 2006; 27:920–5. [DOI] [PubMed] [Google Scholar]
- 24. Snitkin E, Won S, Pirani A, et al. . Integrated genomic and interfacility patient-transfer data reveal the transmission pathways of multidrug-resistant Klebsiella pneumoniae in a regional outbreak. Science Translational Medicine 22 Nov 2017; 9:eaan0093. [DOI] [PubMed] [Google Scholar]
- 25. Prabaker K, Lin MY, McNally M, et al. ; Centers for Disease Control and Prevention Prevention Epicenters Program Transfer from high-acuity long-term care facilities is associated with carriage of Klebsiella pneumoniae carbapenemase-producing Enterobacteriaceae: a multihospital study. Infect Control Hosp Epidemiol 2012; 33:1193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kahvecioglu D, Ramiah K, McMaughan D, et al. . Multidrug-resistant organism infections in US nursing homes: a national study of prevalence, onset, and transmission across care settings, October 1, 2010-December 31, 2011. Infect Control Hosp Epidemiol 2014; 35 (Suppl 3): S48–5. [DOI] [PubMed] [Google Scholar]
- 27. Han JH, Goldstein EJ, Wise J, et al. . Epidemiology of carbapenem-resistant Klebsiella pneumoniae in a network of long-term acute care hospitals. Clin Infect Dis 2017; 64:839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marchaim D, Chopra T, Bogan C, et al. . The burden of multidrug-resistant organisms on tertiary hospitals posed by patients with recent stays in long-term acute care facilities. Am J Infect Control 2012; 40:760–5. [DOI] [PubMed] [Google Scholar]
- 29. Lee BY, Bartsch SM, Wong KF, et al. . The importance of nursing homes in the spread of methicillin-resistant Staphylococcus aureus (MRSA) among hospitals. Med Care 2013; 51:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Araos R, Montgomery V, Ugalde JA, et al. . Microbial disruption indices to detect colonization with multidrug-resistant organisms. Infect Control Hosp Epidemiol 2017; 38:1312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Araos R, Tai AK, Snyder GM, et al. . Predominance of Lactobacillus spp. among patients who do not acquire multidrug-resistant organisms. Clin Infect Dis 2016; 63:937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee YJ, Arguello ES, Jenq RR, et al. . Protective factors in the intestinal microbiome against Clostridium difficile infection in recipients of allogeneic hematopoietic stem cell transplantation. J Infect Dis 2017; 215:1117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drekonja D, Reich J, Gezahegn S, et al. . Fecal microbiota transplantation for Clostridium difficile infection: a systematic review. Ann Intern Med 2015; 162:630–8. [DOI] [PubMed] [Google Scholar]
- 34. Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 2010; 44:354–60. [DOI] [PubMed] [Google Scholar]
- 35. Chang JY, Antonopoulos DA, Kalra A, et al. . Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis 2008; 197:435–8. [DOI] [PubMed] [Google Scholar]
- 36. Seekatz AM, Rao K, Santhosh K, Young VB. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med 2016; 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 2008; 6:e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abeles SR, Jones MB, Santiago-Rodriguez TM, et al. . Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome 2016; 4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Isaac S, Scher JU, Djukovic A, et al. . Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother 2017; 72:128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taur Y, Xavier JB, Lipuma L, et al. . Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 2012; 55:905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.