

Abstract
The tumor immune microenvironment (TIME) has recently been recognized as a critical mediator of treatment response in solid tumors, especially for immunotherapies. Recent clinical advances in immunotherapy highlight the need for reproducible methods to accurately and thoroughly characterize the tumor and its associated immune infiltrate. Tumor enzymatic digestion and flow cytometric analysis allow broad characterization of numerous immune cell subsets and phenotypes; however, depth of analysis is often limited by fluorophore restrictions on panel design and the need to acquire large tumor samples to observe rare immune populations of interest. Thus, we have developed an effective and high throughput method for separating and enriching the tumor immune infiltrate from the non-immune tumor components. The described tumor digestion and centrifugal density-based separation technique allows separate characterization of tumor and tumor immune infiltrate fractions and preserves cellular viability, and thus, provides a broad characterization of the tumor immunologic state. This method was used to characterize the extensive spatial immune heterogeneity in solid tumors, which further demonstrates the need for consistent whole tumor immunologic profiling techniques. Overall, this method provides an effective and adaptable technique for the immunologic characterization of subcutaneous solid murine tumors; as such, this tool can be used to better characterize the tumoral immunologic features and in the preclinical evaluation of novel immunotherapeutic strategies.
Keywords: This Month in JoVE, Issue 136, Tumor immune microenvironment, cancer, solid tumors, immune enrichment, tumor infiltrating leukocytes isolation, tumor immune profiling, tumor digestion.
Introduction
The suppressive TIME has recently been recognized as a hallmark of cancer, and is known to play a significant role in the development, progression, and protection of solid tumor cancers as well as mitigate their susceptibility to immunotherapy1. The TIME is composed of numerous cellular subsets and phenotypes, all of which provide critical insight into the immunologic state of the tumor. These immunologic subsets can be further stratified into cells of lymphocyte or myeloid origin, which together constitute the majority of the innate and adaptive immune responses2,3. Recent advances in the field of cancer immunotherapy have demonstrated that immunotherapeutic strategies (i.e., immune checkpoint inhibitors, chimeric antigen receptor T-cells, etc.) have the potential to induce durable cancer regressions; however, they remain relatively ineffective in solid tumor cancers4,5. Numerous groups have shown the critical hurdle that the TIME can play on treatment success6,7, and thus, there remains a need to accurately evaluate new immunotherapies in the pre-clinical setting specifically focusing on their ability to modulate or overcome the TIME8.
Current efforts to characterize the TIME typically utilize either microscopy or flow cytometry along with antibody labeling strategies to identify immune cellular subsets and their features9,10. These two strategies provide uniquely different information, as microscopy allows spatial appreciation of cellular subsets and flow cytometry provides high throughput and broader quantification of cellular changes. Despite the recent improvements in fluorescent multiplex immunohistochemistry optimizing spectral imaging systems, which now can support up to 7 parameters, limited panel size makes broad level immune profiling difficult and thus this technique is often reserved for more focused analyses. As a result, flow cytometry remains one of the most widely used immune profiling techniques. Despite its widespread use in TIME characterization, the methods used for processing and staining are quite variable. Most often protocols utilize a tumor dissociating enzyme (i.e., collagenases, DNase, etc.) and manual dissociation methods to achieve single-cell suspensions, followed by antibody staining and analysis7. Despite the benefits of each method, numerous groups have shown the extensive variability that can be induced through these techniques11. This makes cross-study comparisons of tumor microenvironment profiling extremely difficult, even when assessing the same murine tumor model. Furthermore, these methods provide limited potential to assess tumor cellular and tumor immune infiltrate components independently, since both components are interspersed after digestion. Sample quantity then limits multi-panel staining and analysis, which becomes a major issue when attempting to characterize rare immunologic subsets (i.e., tumor-specific T-cells)12. More recent techniques such as mass cytometry, or cytometry by time of flight (CyTOF), allow high-dimensional phenotypic analysis of cellular subsets with some systems supporting panel designs of greater than 42 independent parameters13. Despite the tremendous power of CyTOF technology in immune profiling, it remains limited because of the expense, analysis expertise, and the access to the equipment. In addition, many CyTOF protocols recommend purification of immune subsets to improve signal-to-noise ratios14, and thus we suggest that our enrichment method could be used upstream of CyTOF analysis to improve data quality.
Herein we describe a tumor microenvironment digestion and analysis method that incorporates tumor immune infiltrate separation. The purpose of this method is to allow independent high-throughput profiling of the tumor immune infiltrate and tumor cellular fractions for broader characterization of the tumor microenvironment. Using this method, we further demonstrate the importance of performing whole tumor analysis, as a subcutaneous solid tumor model was found to have significant spatial immunologic heterogeneity. Overall, this method can more accurately and consistently compare between samples because it enriches for immune cellular subsets within the tumor and allows for independent profiling of tumor cell and immune fractions of a tumor.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine.
1. Tumor Harvest and Digestion
NOTE: The time required for harvest is ~3 - 5 min/tumor, and the time required for processing is ~1 h + 2 - 3 min/tumor.
Euthanize mice by carbon dioxide inhalation and spray each mouse down with 70% ethanol before harvesting to prevent hair contamination. Surgically remove subcutaneous tumors from the mice and ensure that the tumors are free of any contaminating tissue (i.e., hair, skin, peritoneum, etc.). Place each tumor in 1.8 mL of base RPMI-1640 media without fetal bovine serum (FBS) in a 24-well plate. For larger harvests, keep plates cold on ice to preserve cellular viability. NOTE: If sterility is required, all tumor dissections will need to be performed in a biosafety cabinet with sterile surgical tools (i.e., scissors, forceps, etc.), as well as any further steps. Tumors with the longest diameter between 0.5 - 1 cm are optimal, and larger or smaller tumors will require scaling of reagents.
Use scissors to cut the tumors into less than 1 mm3 pieces within each well.
Prepare a 10x digestion cocktail by dissolving collagenase I at 10 mg/mL, collagenase IV at 2,500 U/mL, and DNase I at 200 U/mL in base RPMI-1640 media (without FBS). NOTE: The digestion cocktail can be prepared a day in advance and stored at -20 °C; however, prolonged storage is not recommended as enzymes will lose activity over time once dissolved.
Add 200 µL of 10x dissociation cocktail containing collagenase I, collagenase IV, and DNase I to each well with the 1 mm3 pieces.
Allow the samples to digest for 1 h at 37 °C while lightly shaking at 60 - 100 rpm using an orbital plate shaker. NOTE: The elevation of temperature to 37 °C during the enzymatic digestion may cause immune cell activation.
After 1 h, neutralize the reaction by adding 1 mL of RPMI-1640 media supplemented with 5% FBS and 2 mM EDTA to each well.
Pipette the tumor digestion and the remaining tumor pieces into a 40 µm cell strainer seated on top of a 50 mL centrifuge tube. NOTE: Cut the tip off of a 1 mL pipette tip for easier transfer of the tumor digestion to the strainer.
Use a 10 mL syringe plunger to mechanically disaggregate the tumor pieces through the strainer. Periodically rinse the strainer with 2 mL of supplemented RPMI-1640 media (containing 5% FBS) and repeat until the cell strainer is clear of tumor. NOTE: Approximately 4 - 10 mL of total media should be sufficient to adequately rinse the strainer of tumor. NOTE: Place samples on ice until this step is completed for all samples to preserve cellular viability.
Adjust each 50 mL centrifuge tube to the same volume using the supplemented RPMI-1640 media. NOTE: The adjustment of volume is critical for centrifuge balancing. Skipping this step could result in centrifuge damage or personal injury.
Centrifuge the cell suspensions (5 min, 805 x g, 4 °C), discard the supernatants, and re-suspend in 2 mL of supplemented RPMI-1640 media. NOTE: A cell aggregate may form following the centrifugation that is difficult to resuspend. Use a 5 mL serological pipette and mix rapidly to break it up before proceeding.
2. Separation of Immune and Tumor Cellular Fractions
NOTE: The time required for this step is approximately 1 h.
Add 3 mL of density gradient medium to the bottom of a 15 mL centrifuge tube. Prepare and label enough tubes for each tumor.
Layer the 2 mL of tumor cell suspension on top of the density gradient medium by pipetting slowly down the side of the tube to prevent mixing of the two layers.
Carefully place the layered tubes in the centrifuge and spin for 20 min at 805 x g, 20 °C, with no brake. NOTE: It is extremely important to verify proper balancing and ensure that no brake is used during centrifugation. Any disruption of this process will result in layer mixing and the full sample recovery will be difficult.
Carefully transfer the tumor infiltrating leukocyte (TIL) layer and the top media layer to a new 15 mL tube using a transfer pipette. Discard all remaining density gradient medium and resuspend the tumor pellet at the bottom of the tube in 2 mL of supplemented RPMI-1640. NOTE: After centrifugation there will be four visible layers from the top to bottom which respectively correspond to: media, TILs, density gradient medium, and tumor cell pellet. See Figure 2A for an example of the various layers.
Centrifuge both the TIL and tumor samples in separate tubes (5 min, 805 x g, 4 °C) and discard the supernatant.
Resuspend the TIL sample in 200 µL and the tumor pellet in 2 mL of supplemented RPMI-1640 media. Keep on ice to preserve viability.
3. Plating and Staining
Note: The time required for plating is 30 s/tumor; the time required for surface staining is 1 h; the time required for intracellular staining is 40 min; the time required for flow cytometry analysis is 1 - 4 min/tumor.
Prepare and label a 96-well U-bottom plate for each immune staining panel and an additional plate for cell counts. NOTE: Typically, three plates are prepared in total: a lymphocyte-focused panel plate, a myeloid-focused panel plate, and a cell count plate.
Aliquot the single-cell suspensions into each of the staining panel plates and a set volume into the count plate. NOTE: The count plate is used to calculate the total number of cells added to each staining panel, thereby allowing accurate cell population counts. A flow cytometer with 96-well plate sampling and volumetric analysis capabilities can provide the number of cells per µL in each sample, and therefore, calculate the number of cells added to each staining panel. Count plates can be acquired the following day after fixation overnight at 4 °C, rinsing with FACs buffer (phosphate buffered saline (PBS) with 2% FBS), and resuspending in a set volume of FACs buffer.
Wash the cells twice with 200 µL of Dulbecco's phosphate buffered saline (DPBS) per sample by centrifuging (5 min, 805 x g, 4 °C) and discarding the supernatant between each rinse to remove any free FBS.
Add 50 µL of Fc block, mix the samples using a multi-channel pipette, and incubate for 20 min at 4 °C.
Add 50 µL of 2x concentrated extracellular targeting antibody and fixable viability stain mixture, mix using a multi-channel pipette, and incubate for 30 min in the dark at RT or 4 °C. NOTE: The exact staining conditions for the antibody depend on the manufacturer's instructions. All staining dilutions must be in DPBS, with no added FBS to prevent saturation of the fixable viability dye. Please see the Table of Materials/Equipment for the optimal dilutions of the staining panel used in this manuscript. Antibody and fixable viability staining solution is prepared at a 2x concentration to provide a final 1x staining concentration in 100 µL of total staining volume when added to the Fc block.
Wash the samples twice with FACs buffer by centrifuging (5 min, 805 x g, 4 °C) and discarding the supernatants between each rinse.
Resuspend in 200 µL of 1x fixation/permeabilization solution and incubate overnight at 4 °C protected from light. NOTE: The experiment can be paused overnight at this point: keep samples at 4 °C protected from light.
The following day, centrifuge the plates (5 min, 805 x g, 4 °C) and discard the supernatant.
Rinse once with 200 µL of 1x permeabilization buffer, centrifuge (5 min, 805 x g, 4 °C), and discard the supernatant.
Add 100 µL of 1x concentrated intracellular antibody staining panel prepared in 1x permeabilization buffer and incubate for 30 min in the dark at RT or 4 °C (this depends on staining recommendations from the manufacturer).
Rinse once with 1x permeabilization buffer (5 min, 805 x g, 4 °C), discard the supernatant, and rinse a second time with FACs buffer (PBS with 2% FBS).
Resuspend the samples in 300 µL of FACs buffer (PBS with 2% FBS), transfer the samples to flow cytometry tubes, and perform flow cytometry analysis.
Representative Results
Our results demonstrate the significant benefit of TIL separation from non-immune tumor components, as explained in the protocol. Additionally, using the described method we demonstrate the significant immunologic heterogeneity of established solid tumors.
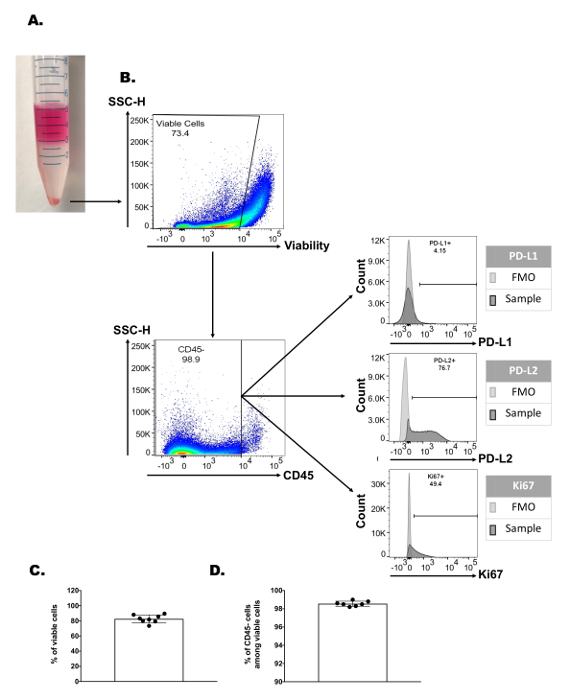
A significant problem with many tumor dissociation techniques is the loss of sample viability, most often as a result of harsh digestion conditions (i.e., elevated temperatures, inadequate nutrient supply, etc.). Use of the described digestion method, with or without density gradient medium enrichment, provides approximately 70 - 90% total cellular viability as assessed by flow cytometry (Figure 1A). Similar viabilities were observed in the TIL enriched and tumor enriched fractions, suggesting that this method causes minimal overall toxicity (Figure 1A, Figure 2C). Furthermore, the density gradient medium enrichment promoted a greater than 2-fold increase in the CD45 positive TIL fraction among total viable cells analyzed compared to non-enriched tumor digestions (Figure 1B, C). In addition, enrichment was found to enhance numerous immune cell subsets commonly assessed in immune microenvironment studies, including myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), macrophages, CD8 T-cells, and CD4 T-cells (Figure 1D, E). This suggests that the density gradient medium enrichment promotes global immunocyte enrichments and does not isolate specific lymphocyte or myeloid populations (see Supplemental Figure 1A, B for the flow cytometry gating method used to identify specific cell populations). It is worth noting however, that many commercially available density gradient separation medias allow a significant fraction of erythrocytes and granulocytes to pass through the separation phase. Thus, if these populations are of interest, further protocol optimization may be necessary.
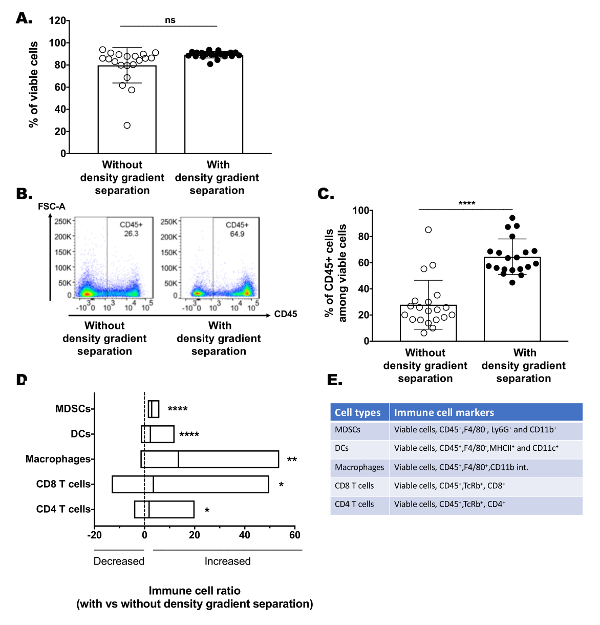
Figure 1: Gentle and effective enrichment of tumor immune infiltrate. Established MEER tumors were excised and digested using the described method. Following digestion, single-cell suspensions were divided such that half underwent density gradient medium enrichment and the other half were directly stained. (A) Cumulative flow cytometry assessment of the cellular viability for tumor digestions with or without density gradient medium enrichment. (B) Representative flow cytometry gating plots showing CD45 positive immune cell enrichment of a single tumor digestion with and without density gradient medium enrichment. (C) Cumulative flow cytometry CD45 positive cell percentages among total viable cells from tumor digestions with or without density gradient medium enrichment. (D) Enrichment ratios of various myeloid and lymphocyte immune populations. Tumors were digested into single-cell suspension before being stained directly or undergoing density gradient enrichment prior to staining and flow cytometry analysis. Each cell-type enrichment ratio was calculated by dividing a given cell-population percentage (among total viable cells assessed) from the density gradient enriched tumor digestion by its non-enriched counterpart. Bar lines represent the median and the edges of the bar represent the minimum and maximum ratios. (E) Table of immune cellular marker expression used to identify each immune cell population analyzed. Statistical significance was determined with an unpaired t-test or Mann Whitney test. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001, (n = 20 tumors, N = 2 for all graphs). Please click here to view a larger version of this figure.
One of the most significant advantages that this method provides is the ability to independently profile the non-immune tumor fraction. After isolation of the pelleted non-immune tumor fraction that passes through the density gradient medium phase, complex tumor profiling panels can be performed which previously may have been limited by fluorophore number and sample quantity limitations. Figure 2A shows an example of the tumor flow cytometry characterization with over 70% viability and 99% CD45 negative cell enrichment. The CD45 negative fraction can then be profiled for numerous tumor features such as overall tumor viability or apoptosis (i.e., caspase 3, annexin V, etc.), proliferation capacity (i.e., Ki67), and the expression of various immunosuppressive markers (i.e., programmed death-ligand 1 (PD-L1) or PD-L2). It should be noted however, that the CD45 negative fraction includes both tumor cells as well as other stromal and vascular elements of the tumor microenvironment. Thus, if direct tumor cell characterization is required, a tumor cell specific marker would need to be used (i.e., cytokeratin, specific tumor-associated antigen, etc.). Similarly, other tumor stromal elements could be easily identified and characterized after labeling with a cell-type specific marker such as tumor vascular endothelial cells (CD31) and cancer associated fibroblasts (α-smooth muscle actin). Nevertheless, the level of viability and CD45 negative cell enrichment is highly consistent across repeated samples, indicating the robustness of the density gradient separation technique for non-immune tumor component enrichment (Figure 2B, C).
Figure 2: Enrichment and profiling of the non-immune tumor component features. (A) Representative image of a sample following density gradient medium enrichment with visible media layer (pink), TIL layer (thin murky white), density gradient medium layer (clear), and tumor pellet at the bottom of the tube. (B) Representative flow cytometry gating strategy used for profiling the non-immune tumor component, with cell viability scatter plot (top), CD45 negative cell enrichment scatter plot (bottom left), and three independent markers of interest expression via histogram plots (PD-L1 top, PD-L2 middle, and Ki67 bottom). FMO histogram represents a sample stained with all other panel colors but lacking the marker of interest. Cumulative flow cytometry assessment of (C) cellular viability and (D) CD45 negative cell percentages among total viable cells of the tumor pellet fraction following density gradient medium enrichment (n = 7 - 8 tumors). Please click here to view a larger version of this figure.
Despite significant efforts to characterize the genetic heterogeneity of solid tumors, little work has detailed the spatial heterogeneity of immunologic subsets in solid tumors. We thus aimed to determine how myeloid and lymphocyte immunologic subsets drastically varied throughout a single solid tumor. Thus, an established MEER tumor was divided into four equal parts with special care to ensure each section contained approximately equal intra-tumoral regions, as well as equal dermal- and peritoneal-facing portions (Figure 3A). Each tumor piece was digested separately, and then enriched by density gradient medium and split among a myeloid and lymphocyte panel. This allowed quantification of various features including a number of commonly assessed immunocyte populations: T cells, CD4+ T cells, CD8+ T cells, macrophages, DCs, MDSCs (see Supplementary Figure 1A, B for the flow cytometry gating strategy). Within a single tumor, significant cell variability between the different tumor partitions was noted (Figure 3b). For example, both DC and T cell total cell counts were found to vary by as much as 4 to 5-fold among adjacent tumor parts. These drastic changes were also noted when analyzing cell populations as a percentage of total viable cells (Supplementary Figure 2A) and were observed at similar levels in two other independent tumors (Supplementary Figure 2B, C). These data demonstrate the importance of isolating and analyzing the entire tumor when performing immune microenvironment analyses, which may seem counterintuitive if current protocols include dividing the tumor to allow complementary analysis by independent methods (i.e., histology, RNA quantification, etc.). In addition, these data may explain discrepancies in immunologic analyses of tumor biopsies, as regional intratumoral immunologic heterogeneity could drastically skew biopsy results.
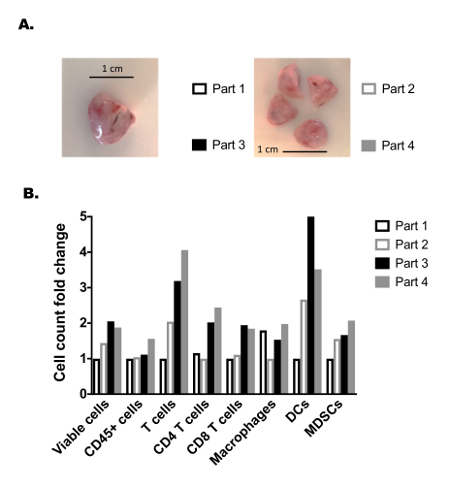
Figure 3: Assessment of established tumor immune heterogeneity. (A) Representative image of an established MEER tumor following surgical resection (left) and after equal division into 4 adjacent parts (right). Scale bar is 1 cm, shown in black on each image. (B) Fold changes of various myeloid and lymphocyte cell counts between each of the 4 adjacent tumor parts; each tumor section underwent the same digestion, density gradient medium enrichment procedure, staining, and analysis. Each tumor part is shown as a cell count fold change, as determined by dividing its cell count by the tumor part with the lowest analyzed cell count for a given cell population. Please click here to view a larger version of this figure.
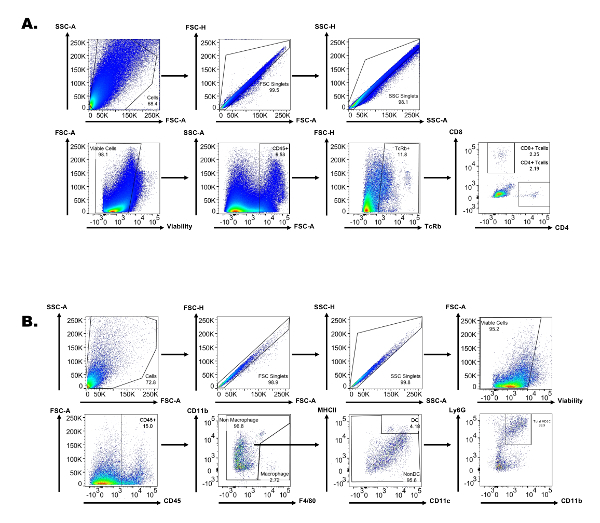 Supplemental Figure 1: Representative flow cytometry gating strategy used for tumor immune component profiling. (A) Gating strategy with respective scatter plots used for lymphocyte immune profiling. (B) Gating strategy with respective scatter plots used for tumor myeloid immune profiling. Please click here to view a larger version of this figure.
Supplemental Figure 1: Representative flow cytometry gating strategy used for tumor immune component profiling. (A) Gating strategy with respective scatter plots used for lymphocyte immune profiling. (B) Gating strategy with respective scatter plots used for tumor myeloid immune profiling. Please click here to view a larger version of this figure.
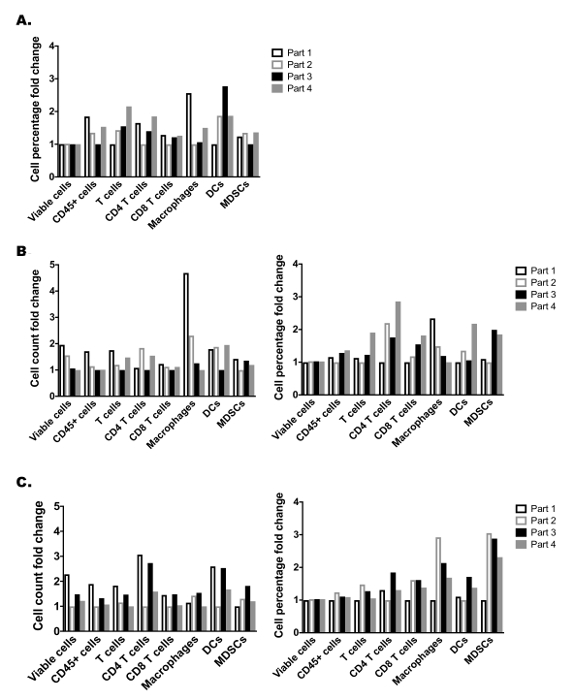 Supplemental Figure 2: Additional assessment of established tumor heterogeneity. (A) Cellular percentage among total viable cells fold change from 4 equal tumor parts shown in Figure 3. (B) and (C) are two additional established MEER tumors divided into 4 adjacent parts and analyzed for tumor immune heterogeneity. Cell count fold changes (left) and cell percentages among total viable cells fold changes (right) for each myeloid and lymphocyte cell population analyzed showing the variability of each of the 4 adjacent tumor parts. Please click here to view a larger version of this figure.
Supplemental Figure 2: Additional assessment of established tumor heterogeneity. (A) Cellular percentage among total viable cells fold change from 4 equal tumor parts shown in Figure 3. (B) and (C) are two additional established MEER tumors divided into 4 adjacent parts and analyzed for tumor immune heterogeneity. Cell count fold changes (left) and cell percentages among total viable cells fold changes (right) for each myeloid and lymphocyte cell population analyzed showing the variability of each of the 4 adjacent tumor parts. Please click here to view a larger version of this figure.
Discussion
The TIME is composed of diverse and complex cellular components and molecules. Recent evidence suggests that accurate characterization of this environment can provide a better understanding of treatment success or failure, and can even help identify mechanisms of therapeutic resistance. For example, increasing intratumoral levels of various immunosuppressive cells (i.e., MDSCs, T regulatory cells, etc.) can mitigate effector immune responses and abrogate immunotherapeutic effects. On the other hand, the increasing intratumoral presence of effector immune populations (e.g., tumor-specific CD8+ T cells, natural killer cells, T helper cells) can be powerful predictors of treatment success. Another characterization that continues to accurately represent tumor immunologic status is the phenotypic features of the tumor cells themselves. This includes expression of various immunosuppressive ligands, enzymes, or molecules aimed at downregulating effector immune responses (i.e., PD-L1, PD-L2, indolamine 2,3 dioxygenase, nitric oxide, etc.). Taken together, this demonstrates the need for accurate and broad characterization of both the immune and tumor cellular features of solid tumors.
The technique we describe herein allows separation and enrichment of both the immune and tumor cell components of solid murine subcutaneous tumors. Then, independent flow cytometric profiling can be performed so that the full TIME and interaction of tumor cells with the TIME can be appreciated. Following enrichment, extensive immune flow cytometry panels can be optimized to allow characterization of key myeloid and lymphoid cellular components, as well as a variety of features describing their phenotypic status. Similarly, extensive tumor cell focused panel can be used independently to provide broad characterization of tumor cell features as well. The relative simplicity of this technique allows for the simultaneous analysis of large numbers of murine tumors, which is critical for observing trends despite the biologic variability of murine tumor models. In addition, tumor immune enrichment provides better identification of rare immunologic subsets (i.e., tumor-specific cytotoxic T cells), which are often significantly underrepresented within the tumor compared to other non-immune cell components. Optimization of this technique can allow additional downstream analyses on these isolated subsets which require enrichment, such as coculture ex vivo assays, mRNA profiling, or adoptive cell transfer studies.
Using this technique in a subcutaneous murine tumor model, we demonstrate the significant enrichment of global and independent immune subsets. We also demonstrate the non-immune tumor profiling which can be achieved from the same tumor samples. Finally, we show the importance of whole tumor immunologic profiling, as we observed significant immunologic heterogeneity in adjacent sections of a tumor. Ideally, this technique of collagenase and DNase-based digestion and gradient-based separation of the tumor immune and non-immune fractions could be applied to the majority of murine tumor models, both orthotopic and subcutaneous. We have also observed that small non-established tumors and less collagen-dense tumor models (i.e., B16 melanoma) often do not require an enzymatic digestion step to generate single-cell suspensions15; however, gradient-based immune enrichment performs equally as well in these tumor models, further validating its versatility. Despite prior concerns about collagenase antigen digestion on specific cellular subsets11, we have yet to observe a notable loss of staining quality for any of the markers that we have profiled. Nevertheless, antigen sensitivity to these digestion conditions should be assessed using proper non-digested control samples before broad adoption of this technique.
In conclusion, our technique allows for accurate, broad, and high-throughput characterization of the tumor immune and non-immune components of murine solid tumors. By performing independent profiling of these subsets, researchers can broadly characterize the TIME and gain better mechanistic insight into solid tumor preclinical therapies. This method can be easily applied to the vast majority of murine tumor models for high throughput analysis of lymphocyte and myeloid immunocyte population counts and phenotypes via flow cytometry, or other downstream assays that require subset enrichment.
Disclosures
The authors have nothing to disclose.
Acknowledgments
JMN acknowledges financial support from National Institute of General Medical Sciences (T32GM088129) and the National Institute of Dental & Craniofacial Research (F31DE026682) both of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This project was also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom.
References
- Fouad YA, Aanei C. Revisiting the hallmarks of cancer. American Journal of Cancer Research. 2017;7(5):1016–1036. [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nature Immunology. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27(45):5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou VL, Burotto M. Pseudoprogression and Immune-Related Response in Solid Tumors. Journal of Clinical Oncology. 2015;33(31):3541–3543. doi: 10.1200/JCO.2015.61.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Shin S, Dy G. Advances in Cancer Immunotherapy in Solid Tumors. Cancers. 2016;8(12):106. doi: 10.3390/cancers8120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkert C, et al. Tumor-Associated Lymphocytes As an Independent Predictor of Response to Neoadjuvant Chemotherapy in Breast Cancer. Journal of Clinical Oncology. 2010;28(1):105–113. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- Lee Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stakheyeva M, et al. Role of the immune component of tumor microenvironment in the efficiency of cancer treatment: perspectives for the personalized therapy. Current Pharmaceutical Design. 2017;23 doi: 10.2174/1381612823666170714161703. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-Cytometry: A Method for Highly Multiplex Quantitative Tissue Imaging Analysis Applied to Dendritic Cell Subset Microanatomy in Lymph Nodes. Immunity. 2012;37(2):364–376. doi: 10.1016/j.immuni.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Vonderheide RH. Multicolor Flow Cytometric Analysis of Immune Cell Subsets in Tumor-Bearing Mice. Cold Spring Harbor Protocols. 2013;2013(10) doi: 10.1101/pdb.prot077198. [DOI] [PubMed] [Google Scholar]
- Goodyear AW, Kumar A, Dow S, Ryan EP. Optimization of murine small intestine leukocyte isolation for global immune phenotype analysis. Journal of Immunological Methods. 2014;405:97–108. doi: 10.1016/j.jim.2014.01.014. [DOI] [PubMed] [Google Scholar]
- Casadevall A, Fang FC. Rigorous Science: A How-To Guide. mBio. 2016;7(6):01902–01916. doi: 10.1128/mBio.01902-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, et al. CyTOF supports efficient detection of immune cell subsets from small samples. Journal of Immunological Methods. 2014;415:1–5. doi: 10.1016/j.jim.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AW, Strauss-Albee DM, Blish CA. Application of Mass Cytometry (CyTOF) for Functional and Phenotypic Analysis of Natural Killer Cells. Methods Mol. Biol. 2016;1441:13–26. doi: 10.1007/978-1-4939-3684-7_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachynski RK, Scholz A, Monnier J, Butcher EC, Zabel BA. Evaluation of Tumor-infiltrating Leukocyte Subsets in a Subcutaneous Tumor Model. Journal of Visualized Experiments. 2015. [DOI] [PMC free article] [PubMed]