

Abstract
The insoluble salt Cs15K[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3] (CsCo9) is tested as heterogeneous oxygen evolution catalyst in light-induced experiments, when combined with the homogeneous photosensitizer [Ru(bpy)3]2+ and the oxidant Na2S2O8 in neutral pH. Oxygen evolution occurs in parallel to a solid transformation. Post-catalytic essays indicate that the CsCo9 salt is transformed into the corresponding [Ru(bpy)3]2+ salt, upon cesium loss. Remarkably, analogous photoactivated oxygen evolution experiments starting with the [Ru(bpy)3](5+x)K(6−2x)[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]·(39+x)H2O (RuCo9) salt demonstrate much higher efficiency and kinetics. The origin of this improved performance is at the cation-anion, photosensitizer-catalyst pairing in the solid state. This is beneficial for the electron transfer event, and for the long-term stability of the photosensitizer. The latter was confirmed as the limiting process during these oxygen evolution reactions, with the polyoxometalate catalyst exhibiting robust performance in multiple cycles, upon addition of photosensitizer, and/or oxidant to the reaction mixture.
Keywords: water splitting, oxygen evolution, polyoxometalates, photosensitizer, cobalt
Introduction
Sunlight is the preferred carbon-neutral energy source for competing with fossil fuels for energy production, because solar radiation is readily accessible at almost any location on the surface of the Earth (Cook et al., 2010). Artificial photosynthesis aims to mimic natural photosynthesis, where sunlight is stored in the form of chemical bonds through reduction of CO2 into sugars, employing H2O as the ultimate source of electrons (Mcevoy and Brudvig, 2006). Therefore, an artificial photosynthesis device would convert sunlight into spatially separated electron/hole pairs and store its energy subsequently into chemical bonds by means of water splitting, obtaining hydrogen as a clean fuel together with oxygen as the only side product (Lewis and Nocera, 2006; Balzani et al., 2008; Barber, 2009). Unfortunately, the market introduction of commercial artificial photosynthesis devices is still hampered by the lack of robust, inexpensive and efficient water oxidation catalysts (WOCs) (Dau et al., 2010; Seh et al., 2017).
Over the last decades, scientists have reported a wide variety of new WOCs. Homogeneous organometallic compounds work at fast oxygen evolution rates and offer good processability (Concepcion et al., 2008, 2009; Bozoglian et al., 2009; Blakemore et al., 2010; Xu et al., 2010; Lloret-Fillol et al., 2011; Mccool et al., 2011; Barnett et al., 2012; Duan et al., 2012; Liu and Wang, 2012; Zhang et al., 2013; Goberna-Ferrón et al., 2014). However, they often suffer from limited long-term stability due to oxidative degradation of the organic ligands in the harsh working conditions needed for water oxidation. Precious-metal-based WOCs, for instance Ir-, and Ru-based materials, have shown superior performance and stability for water oxidation catalysis (Pillai et al., 2000; Youngblood et al., 2009; Blakemore et al., 2010; Duan et al., 2012). Unfortunately, the high production price due to metal scarcity questions their viable implementation into commercial devices. Earth abundant transition metal oxides and perovskites are a robust alternative, but exclusively in alkaline media (Galán-Mascarós, 2015). Therefore, alternatives to the current state-of-the-art catalysts are needed.
Polyoxometalates (POMs) have recently appeared as a promising new catalyst class (Geletii et al., 2008; Sartorel et al., 2008). When employed as WOCs, they combine the most appealing features of homogeneous and heterogeneous materials, and many of them can be obtained from inexpensive raw materials. They are all-inorganic molecular clusters with high stability under strongly oxidizing conditions. At the same time, their molecular nature provides access to the tunability and superior processing capabilities of homogeneous catalysts for their easier implementation into devices (Pope, 1983; Pope and Müller, 2001). POMs have shown high catalytic activity in water oxidation over a remarkable pH range (0-10), and they retain their catalytic activity under heterogeneous conditions as their corresponding insoluble salts, or when anchored onto solid supports (Wu et al., 2012; Guo et al., 2013; Quintana et al., 2013; Soriano-López et al., 2013).
Among polyoxometalates, the cobalt-containing POMs (Co-POMs) have emerged as the most promising WOCs due to their high efficiency and kinetics (Goberna-Ferrón et al., 2012; Lv et al., 2012, 2014; Evangelisti et al., 2013). After Hill et al. reported the OER activity of the [Co4(H2O)2(PW9O34)2]10− polyanion (Yin et al., 2010; Huang et al., 2011; Stracke and Finke, 2011, 2013, 2014), we turned our attention to the high nuclearity [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16− (Goberna-Ferrón et al., 2012, 2015; Soriano-López et al., 2013; Co9, Figure 1). Co9 shows good activity for photo-assisted water oxidation in homogeneous conditions, exhibiting fast charge transfer kinetics with the model [Ru(bpy)3]2+ photosensitizer (bpy = 1,2-dipyridyl; Natali et al., 2017). It is also active in the solid state when processed as an insoluble salt with alkaline metal countercations (Soriano-López et al., 2013).
Figure 1.
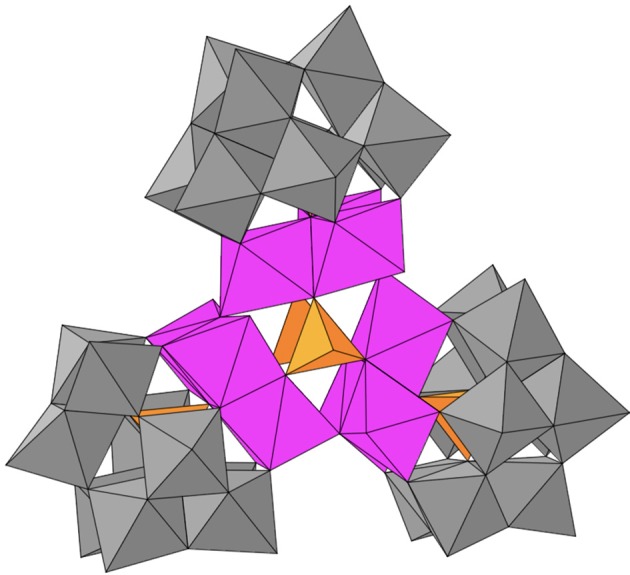
Polyhedral representation of the polyanion [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16− (Co9); WO6, gray octahedra; PO4, orange tetrahedra; CoO6, pink octahedra.
In this work we report the next required step on the road to technological applications for Co9 in an artificial photosynthesis platform, namely its combination with a photosensitizer in a light-induced process in heterogeneous conditions. These essays have been very successful with other POMs and water oxidation catalysts to assess photo-induced catalytic performance, mechanistic considerations, and stability issues (Puntoriero et al., 2010; Gao et al., 2013; Sartorel et al., 2013; Al-Oweini et al., 2014; Xiang et al., 2014; Natali et al., 2017). Our experiments confirm the efficient electron transfer between catalyst and sensitizer, even when both species are combined into an insoluble salt. The latter opens up interesting possibilities for future combinations of cationic photosensitizers with polyanionic WOCs for the construction of compact functional photoelectrodes.
Experimental section
Materials and synthesis
Tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate and sodium persulfate were purchased from TCI and Sigma-Aldrich (>99% purity) and used without further purification. The synthesis of Cs15K[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]·39H2O (CsCo9) was already reported (Soriano-López et al., 2013). [Ru(bpy)3](5+x)K(6−2x)[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]·(39+x)H2O (RuCo9) was prepared by metathesis: A stoichiometric excess of [Ru(bpy)3]Cl2 was added to a solution containing Na8K8[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]·43H2O (KCo9). RuCo9 immediately precipitated as an orange powder. It was filtered, washed with water and acetone, and air-dried.
Material characterizations
Elemental CHN analysis was performed with an Elemental Microanalyzer Flash model 1112. Detection of Co, Ru, and W was performed on an inductively coupled plasma atomic emission spectrometer iCap 6500 (Thermo Fisher Scientific), and K was detected on a 2,380 atomic absorption spectrophotometer (Perkin-Elmer), both by Mikroanalytisches Labor Pascher (Remagen/Germany). Thermogravimetric analyses were performed with powder samples using a TGA/SDTA851 Mettler Toledo with a MT1 microbalance. Dynamic light scattering was used to measure the particle size distribution employing a Malvern NanoZS analyzer. FT-IR spectra were collected in the 3600–400 cm−1 range with a Bruker Optics FTIR Alpha spectrometer equipped with a DTGS detector and a KBr beamsplitter at 4 cm−1 resolution. Raman measurements were acquired using a Renishaw inVia Reflex Raman confocal microscope (Gloucestershire, UK) equipped with a diode laser emitting at 785 nm at a nominal power of 300 mW, and a Peltier-cooled CCD detector (−70°C) coupled to a Leica DM-2500 microscope. X-ray photoelectron spectroscopy (XPS) (K-ALPHA, Thermo Scientific SSTTI at University of Alicante) was used to analyze the surface of the samples. All spectra were collected using Al-Kα radiation (1486.6 eV), monochromatized by a twin crystal monochromator, yielding a focused X-ray spot with a diameter of 400 μm, at 3 mA x 12 kV. The alpha hemispherical analyzer was operated in the whole energy band, and 50 eV in a narrow scan to selectively measure the particular elements.
Photoinduced water oxidation catalysis
Oxygen evolution experiments were performed in a 6.7 mL headspace Schlenk tube sealed with a rubber septum (PFTE). The Schlenk tube was covered with aluminum foil, in order to avoid an early light-induced reaction of the system, and filled with 1 mM (9.4 mg) [Ru(bpy)3]Cl2, 5 mM (14.9 mg) Na2S2O8, the desired amount of catalyst, and 12.5 mL of 40 mM KPi buffer solution at pH 7.0. Experiments employing the RuCo9 salt as catalyst were performed with and without addition of [Ru(bpy)3]Cl2, the former for comparison in the same conditions required for Co3O4 experiments. Suspensions were completely deaerated by purging with nitrogen. A baseline of 20 min was recorded to ensure that no oxygen leakage or side reactions took place. Next, the system was exposed to the light of a blue LED lamp (wavelength at peak emission = 465 nm; OSRAM Opto Semiconductors) working at 0.20 A and 11.4 V. The concentration of oxygen in the headspace was measured by employing a O2-sensor probe (Ocean Optics NeoFOX oxygen-sensing system equipped with a FOXY probe). Turnover number (TON) and turnover frequency (TOF) were estimated per Co9 content as obtained from chemical analyses on fresh compounds (see SI).
Results and discussion
Visible-light-driven water oxidation by CsCo9 in heterogeneous conditions
Water oxidation experiments were carried out with [Ru(bpy)3]2+ as a model photosensitizer and S2 as sacrificial electron acceptor, in a suspension of the insoluble salt Cs15K[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3] (CsCo9). Light irradiation (λ > 400 nm) of this mixture promotes oxygen evolution, which was monitored using a fluorescence O2-sensor probe for increasing amounts of CsCo9 (1–50 mg). The proposed net reaction mechanism for light-driven water oxidation catalyzed by POMs is depicted in Scheme 1. No oxygen evolution was detected in the absence of any of the components.
Scheme 1.
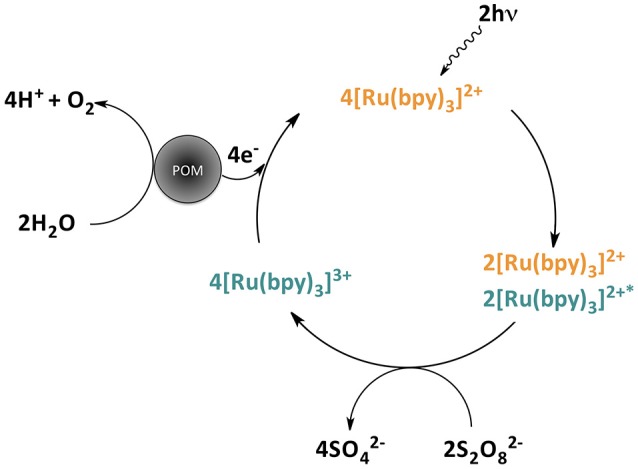
Schematic representation of the light-driven water oxidation catalysis reaction, employing [Ru(bpy)3]2+ as photosensitizer, S2 as sacrificial electron acceptor, and a polyoxometalate as water oxidation catalyst (further side reactions giving rise to sulfate radical anions were omitted for clarity).
The reaction starts with fast kinetics immediately after light irradiation, and slows down until oxygen evolution stops reaching a plateau after 2 h. We analyzed the oxygen production as a function of catalyst content (Figure 2 and Table 1). The highest values of turnover number (TON) and turnover frequency (TOF) obtained were 14.2 and 10.8 h−1, for the minimum quantity used (1 mg, ≈ 0.1 μmol). In terms of chemical yield (CY, see SI), a maximum 9.2% was reached for intermediate CsCo9 contents (10 mg, ≈ 1 μmol) in the investigated range. After oxygen evolution, CsCo9was recovered from the reaction vessel to perform structural characterization. The FT-IR spectrum shows the typical Co9 bands within the 1,100-400 cm−1 range, identical to those observed with the freshly made CsCo9. We also found additional bands in the region between 1,200 and 1,600 cm−1, which can be attributed to the bipyridyl (bpy) ligand (Figure S1). The same information is obtained from the Raman spectra (Figure S2). Moreover, comparison of the XPS spectra (Figure S3) showed the appearance of intense Ru peaks in the recovered CsCo9, and disappearance of the expected Cs peak (Figure S4). The data in their entirety suggest that cation exchange occurred under turnover conditions, i.e., Cs+ cations are replaced by [Ru(bpy)3]2+ cations. Indeed, the Raman spectrum of the recovered CsCo9 is reminiscent of the corresponding Raman spectrum of the salt obtained by addition of an excess of [Ru(bpy)3]Cl2 to an aqueous K16Co9 solution (Figure S9).
Figure 2.
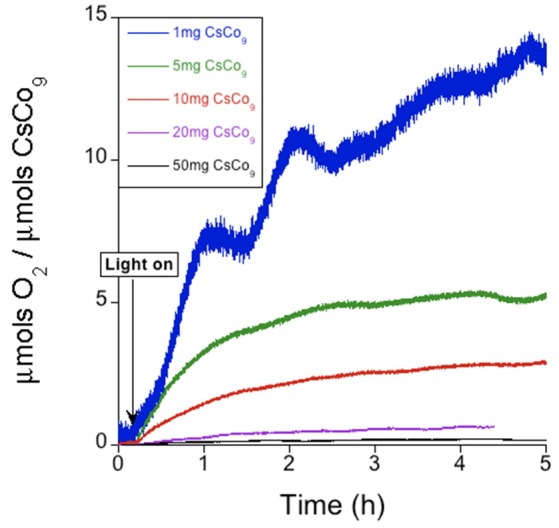
Oxygen evolution profile of the CsCo9 salt in KPi (40 mM) buffer at pH 7, with [Ru(bpy)3]2+ (1 mM) and S2 (5 mM).
Table 1.
Comparison of visible-light-driven oxygen evolution performance of CsCo9 and RuCo9 catalysts(a).
| POM | Catalyst (mg) | 1 | 5 | 10 | 20 | 50 |
|---|---|---|---|---|---|---|
| CsCo9 (first run) | TON | 14.2 | 5.3 | 2.9 | 0.7 | 0.2 |
| TOF (h−1) | 10.8 | 4.0 | 1.4 | 0.3 | 0.2 | |
| CY (%) | 4.4 | 8.4 | 9.2 | 4.0 | 3.6 | |
| RuCo9 | TON | 27.3 | 20.3 | 16.7 | 7.2 | 2.7 |
| TOF (h−1) | 19.1 | 11.9 | 17.0 | 9.0 | 4.3 | |
| CY (%) | 7.6 | 28.4 | 47.6 | 42.4 | 36.4 | |
| POM | Catalyst (μm) | 0.1 | 0.5 | 1 | 2 | 5 |
TON, total turnover number after completion of the reaction; TOF, slope of the oxygen evolution curve at the starting time; CY, total chemical yield after completion of the reaction.
Visible-light-driven water oxidation by RuCo9 in heterogeneous conditions
Addition of an excess of [Ru(bpy)3]Cl2 to an aqueous KCo9 solution forms immediately an insoluble precipitate. The presence of the [Ru(bpy)3]2+ cation and the [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16− anion were confirmed by FT-IR spectroscopy (Figure S8) with the signature bands for both molecular species. However, the exact stoichiometry was difficult to completely assess. We carried out elemental CHN analyses, and metal ICP analyses, along with thermogravimetry analyses, and they were not fully consistent (Table S1). It is worthy to note at this point that our attempts to crystallize this compound in order to accurately characterize its composition and structure failed, because slow diffusion between solutions of cation and anion produce insoluble single crystals of the compound [Ru(bpy)3]2K12[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]·xH2O (Table S2, Figure S13). This Ru/POM stoichiometry is far too low in comparison with our RuCo9 analyses (Table S1), and thus it is not representative of the RuCo9 catalyst. The obtention of this crystalline phase, though, precludes the isolation of other salts with higher [Ru(bpy)3]2+ content, closer to the present RuCo9 solid. The most plausible explanation is that RuCo9 actually consists of a mixture of different [Ru(bpy)3]/Co9 salts, and their slightly different solubility and composition gives small deviations depending on the given analytical technique. With all the analytical data taken into account (Table S1), we assign an average stoichiometry [Ru(bpy)3](5+x)K(6−2x)[Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]·(39+x)H2O (RuCo9), where −1 < x < 1 (see Table S1 and Figure S5). This powder is insoluble in water at room temperature with an average particle size of 374 nm (Figure S6).
When a suspension of RuCo9 in a solution of S2 is irradiated (λ > 400 nm), oxygen evolution starts. In this case, the proposed reaction mechanism is analogous to that depicted in Scheme 1, but with photosensitizer and catalyst bound together in the solid state through electrostatic cation-anion interactions. Remarkably, the measured oxygen evolution in these conditions (Figure 3) is significantly superior to the first run starting from photosensitizer in solution (Table 1 and Figure 4). The maximum TON (27.3) and TOF (19.1 h−1) values are doubled, and the CY showed a remarkable increase up to 47.6%. Pulsed experiments confirmed that oxygen evolves exclusively when the light source is switched on (Figure S7).
Figure 3.
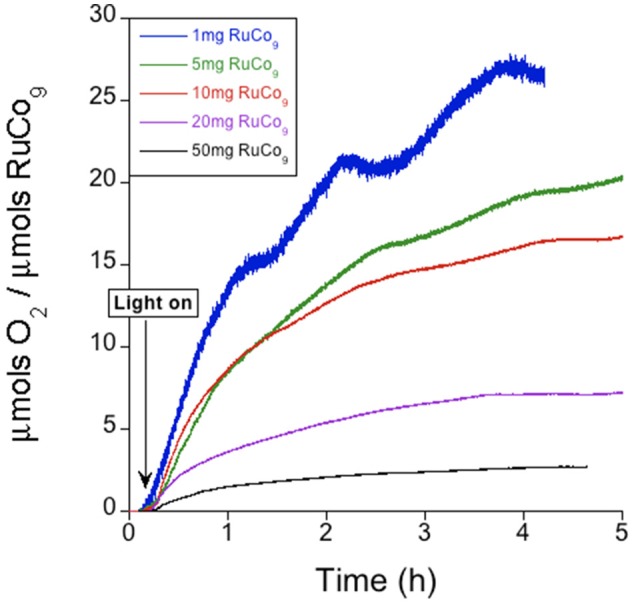
Oxygen evolution profile for solid RuCo9 in KPi (40 mM) buffer at pH 7 with S2 (5 mM).
Figure 4.
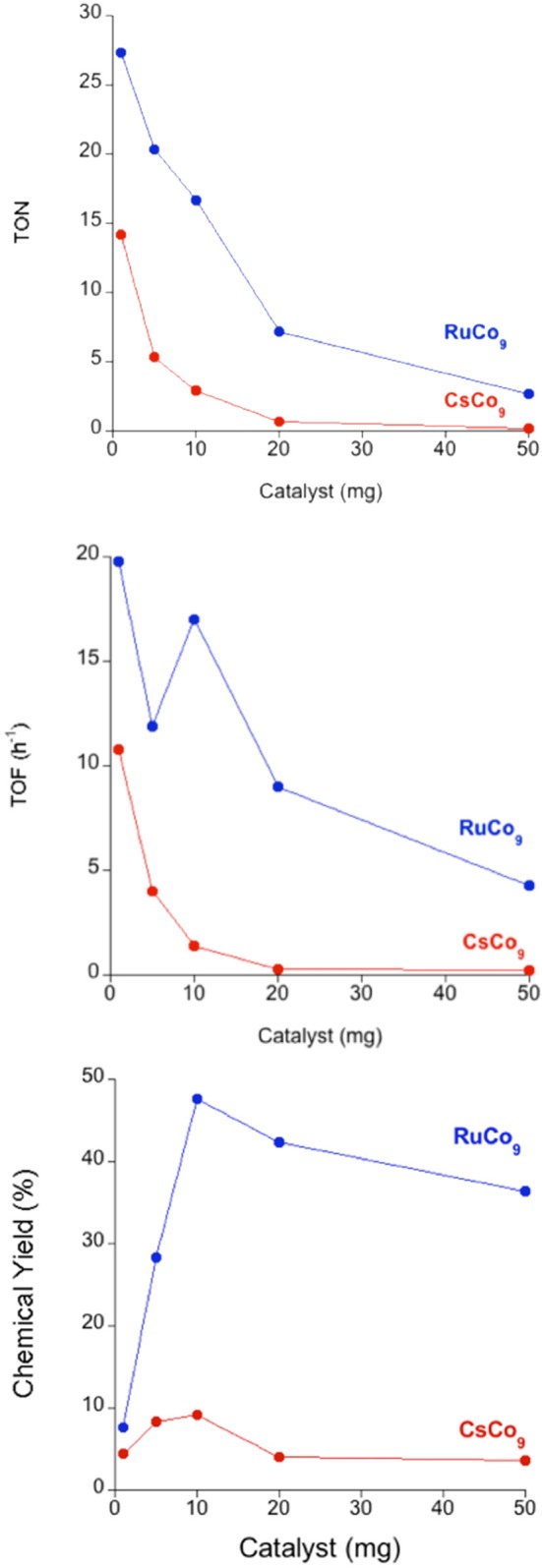
Comparison of TON, TOF, and chemical yield (CY) for RuCo9 (blue) and CsCo9 (red). Experiments were carried out in a KPi (40 mM) buffer at pH 7, with S2 (5 mM). [Ru(bpy)3]2+ (1 mM) was added to the suspension of CsCo9, whereas no homogeneous photosensitizer was added for the RuCo9 catalyst.
Stability of the RuCo9 system
In order to determine the limiting agent in the photo-assisted oxygen evolution reaction, we carried out different tests. Successive additions of S2 to the as-used RuCo9 suspension indicate that oxygen evolution activity is severely affected after each cycle (Figure 5 and Table 2), i.e., the system can barely perform three cycles before reaching complete deactivation. After deactivation, addition of an aliquot containing the photosensitizer [Ru(bpy)3]2+ and S2 to the reaction vessel restarts oxygen evolution, with rates and yields comparable to those obtained with CsCo9 (Figure 5). This behavior can only be explained with deactivation of the photosensitizer in RuCo9 recycling experiments, probably due to oxidative degradation of the organic ligands, during the harsh working conditions. The catalytic POM appears to be robust, since its performance is maintained during successive cycles.
Figure 5.
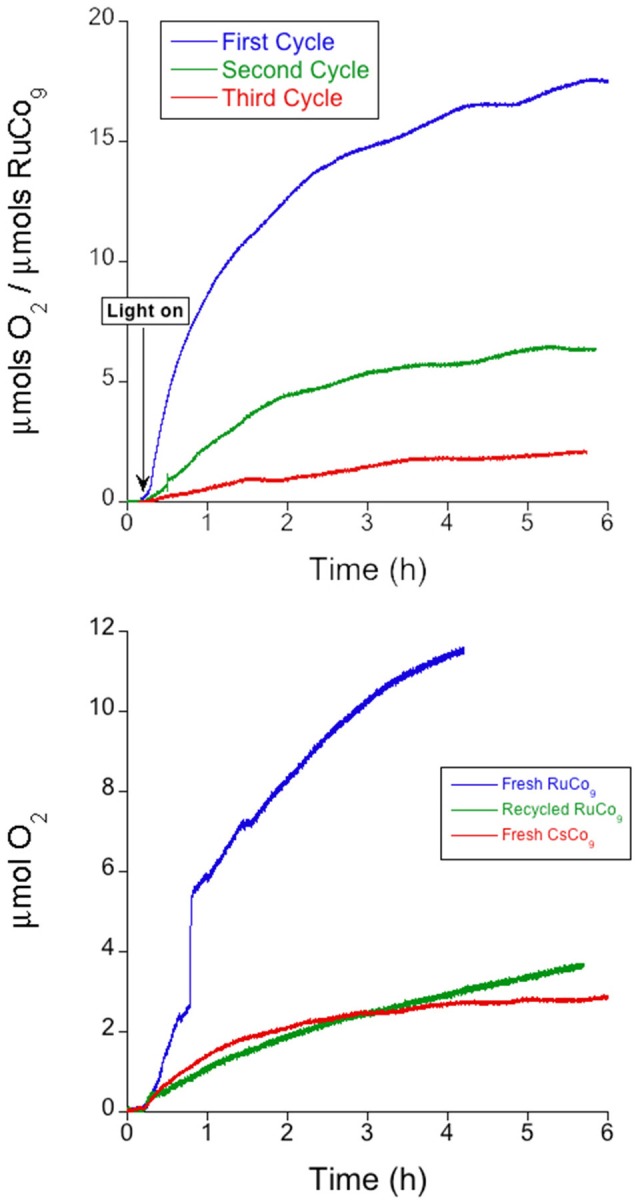
Top: Oxygen evolution with successive additions of oxidant S2 (5 mM) to a KPi (40 mM) buffered suspension of RuCo9 (10 mg, 0.9 μmol) at pH 7. Bottom: Oxygen evolution after addition of photosensitizer [Ru(bpy)3]2+ (1 mM) and oxidant S2 (5 mM) as solid reagents to a KPi (40 mM) buffered suspension of recycled RuCo9 at pH 7 compared with the same experiments starting from fresh CsCo9.
Table 2.
Comparison of the RuCo9-catalyzed light-driven oxygen evolution performance obtained for successive addition of S2 (5 mM) to the reaction vessela.
| TON | TOF (h−1) | CY (%) | |
|---|---|---|---|
| 1st cycle | 16.7 | 17.0 | 47.6 |
| 2nd cycle | 6.4 | 2.7 | 17.6 |
| 3rd cycle | 2.1 | 0.7 | 5.6 |
TON, total turnover number at the final reaction time; TOF, slope of the oxygen evolution curve at the starting time; CY, total chemical yield at the final reaction time.
Analysis of adventitious CoOx formation
In water oxidation with cobalt-based catalysts, it is fundamental to rule out the in situ formation of cobalt oxide CoOx, a competent heterogeneous WOC. This could occur through Co2+ leaching from the RuCo9 salt, and the subsequent formation of CoOx under oxidative conditions. Thus, we analyzed the as-used RuCo9 with different experimental techniques in the search for traces of CoOx.
RuCo9 was recovered from the reaction vessel after the visible-light-driven water oxidation experiments. The signature FT-IR and Raman bands of the Co9 cluster remain identical when compared with pristine RuCo9, suggesting that the bulk POM structure is maintained during the experiments. Raman spectroscopy is particularly suited to detect even traces of CoOx due to its high surface sensitivity, but no bands that could be assigned to a CoOx species are present (Figures S8–S10).
As with fresh RuCo9, the elemental and ICP analyses showed small deviations, making difficult to confirm final stoichiometry. The numbers are not too different from the original stoichiometry (Table S1). However, we need to point out that these analyses show a decrease for all elements, except for W that increases. We assign this surprising result to the deterioration of the compounds during working conditions [triggered by the [Ru(bpy)3]2+ decomposition], making them even more insoluble, and untractable.
Another powerful surface-sensitive technique is XPS. Pristine and recovered RuCo9 salts display analogous XPS spectra (Figure S11). The presence of CoOx should include the appearance of a typical Co3+ peak below 780 eV (Chuang et al., 1976; Tan et al., 1991; Hara et al., 2000). Close analysis of the Co and O edges in search of such features that could be assigned to the presence of an CoOx phase were negative. XPS spectra of RuCo9 before and after oxygen evolution show intense bands only in the 780-783 eV range, which differ from those expected for CoOx (Figure S12). This indicates that no cobalt oxide amounts are formed during turnover conditions within the detection limit of these techniques.
In order to gather additional indirect proof of the absence of the significant participation of cobalt oxide impurities, we compared the photo-induced oxygen evolution reaction starting from the RuCo9 salt to the Co3O4 catalyst (Figure 6 and Table 3). For equimolar conditions, RuCo9 displays an overall better performance, with faster onset kinetics and a higher efficiency. This is incompatible with the attribution of the catalytic activity observed for RuCo9 to very small traces of CoOx, which may be below the detection limit of Raman or XPS techniques.
Figure 6.
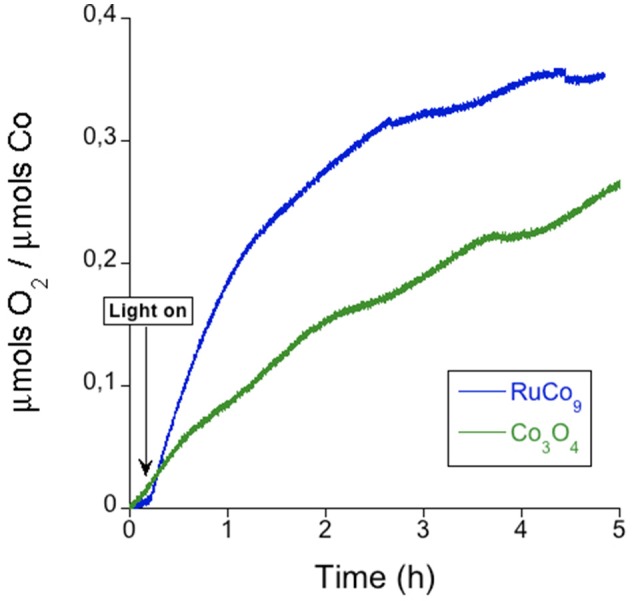
Comparison of the measured oxygen evolution employing equimolar Co amounts for RuCo9 (blue) and Co3O4 (green). The experiments were performed in a KPi (40 mM) buffer at pH 7 with [Ru(bpy)3]2+ (1 mM) as photosensitizer and S2 (5 mM).
Table 3.
Comparison of the light-driven oxygen evolution data catalyzed by RuCo9 and by Co3O4 under the same reaction conditionsa,b.
| Catalyst | TON | TOF (h−1) | CY (%) |
|---|---|---|---|
| RuCo9 | 3.2 | 2.3 | 17.2 |
| Co3O4 | 0.9 | 0.3 | 12.8 |
TON, total turnover number at the final reaction time; TOF, slope of the oxygen evolution curve at the starting time; CY, total chemical yield at the final reaction time.
The experiments were performed in a KPi (40 mM) buffer at pH 7 with [Ru(bpy)3]2+ (1 mM) as photosensitizer, S2 (5 mM) as sacrificial electron acceptor, and with 13.99 μmols of Co in the form of RuCo9 or Co3O4.
Conclusions
We compared the heterogeneous catalytic activity of two different Co9 starting materials under visible-light-driven water oxidation conditions at neutral pH. Direct combination of CsCo9 with a homogeneous photosensitizer yields a maximum turnover number (TON) of 14.2 and a maximum turnover frequency (TOF) of 10.8 h−1 with oxygen yields around 10%. Pre-catalytic incorporation of the cationic photosensitizer into the polyoxometalate salt through substitution of the alkali metal improves the oxygen evolution notably, affording chemical yields close to 50%. We associate this improvement to two beneficial effects of photosensitizer immobilization. On the one hand, the closer cation-anion (photosensitizer/catalyst) interaction in the solid state facilitates electron transfer, and therefore enhances the oxygen evolution kinetics. Additionally, the incorporation of the photosensitizer into the solid state partially improves its stability, an additional benefit to increase the efficiency of the overall process.
Our experimental data indicate that oxygen evolution eventually stops due to decomposition of the photosensitizer. Successive additions of photosensitizer re-start the water oxidation reaction at consistent rates, supporting the stable catalytic performance of Co9. We carried out careful surface analyses on the as-used catalyst in the search of traces of cobalt oxide. Neither Raman nor XPS spectroscopy showed any feature that could be associated with CoOx species. Additionally, RuCo9 exhibits superior catalytic performance than Co3O4. Thus, the hypothetical presence of undetectable CoOx traces cannot be responsible for the observed catalytic activity. This supports the genuine catalytic activity of RuCo9 for photo-induced water oxidation as the first example, to the best of our knowledge, of an effective photosensitizer/catalyst electron transfer in an ionic salt. The superior performance of this ionic composite opens up interesting perspectives for the use of such materials in the development of compact photoanodes for artificial photosynthesis.
Author contributions
GP and JRG-M proposed the concept. GP, JRG-M, and JS-L designed the experiments. JS-L and FS carried out the experiments. All authors analyzed the data and contributed to the manuscript writing.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. We would like to acknowledge the financial support from the Spanish Ministerio de Economía y Competitividad (MINECO) through project CTQ2015-71287-R and the Severo Ochoa Excellence Accreditation 2014-2018 SEV-2013-0319; and the Generalitat de Catalunya (2017-SGR-1406 and the CERCA Programme). GP and FS are grateful for financial support by the Swiss National Science Foundation (Sinergia Grant No. CRSII2_160801/1) and by the University Research Priority Program Solar Light to Chemical Energy Conversion (URPP LightChEC) of the University of Zurich. This collaboration took place in the context of the COST PoCheMoN action supported by the European Research Area.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00302/full#supplementary-material
References
- Al-Oweini R., Sartorel A., Bassil B. S., Natali M., Berardi S., Scandola F., et al. (2014). Photocatalytic water oxidation by a mixed-valent MnIVO3 manganese oxo core that mimics the natural oxygen-evolving center. Angew. Chem. Int. Ed. 53, 11182–11185. 10.1002/anie.201404664 [DOI] [PubMed] [Google Scholar]
- Balzani V., Credi A., Venturi M. (2008). Photochemical conversion of solar energy. ChemSusChem 1, 26–58. 10.1002/cssc.200700087 [DOI] [PubMed] [Google Scholar]
- Barber J. (2009). Photosynthetic energy conversion: natural and artificial. Chem. Soc. Rev. 38, 185–196. 10.1039/B802262N [DOI] [PubMed] [Google Scholar]
- Barnett S. M., Goldberg K. I., Mayer J. M. (2012). A soluble copper-bipyridine water-oxidatzion electrocatalyst Nat. Chem. 4, 498–502. 10.1038/nchem.1350 [DOI] [PubMed] [Google Scholar]
- Blakemore J. D., Schley N. D., Balcells D., Hull J. F., Olack G. W., Incarvito C. D., et al. (2010). Half-sandwich iridium complexes for homogeneous water-oxidation catalysis. J. Am. Chem. Soc. 132, 16017–16029. 10.1021/ja104775j [DOI] [PubMed] [Google Scholar]
- Bozoglian F., Romain S., Ertem M. Z., Todorova T. K., Sens C., Mola J., et al. (2009). The Ru-Hbpp water oxidation catalyst. J. Am. Chem. Soc. 131, 15176–15187. 10.1021/ja9036127 [DOI] [PubMed] [Google Scholar]
- Chuang T. J., Brundle C. R., Rice D. W. (1976). Interpretation of the x-ray photoemission spectra of cobalt oxides and cobalt oxide surfaces. Surf. Sci. 59, 413–429. 10.1016/0039-6028(76)90026-1 [DOI] [Google Scholar]
- Concepcion J. J., Jurss J. W., Brennaman M. K., Hoertz P. G., Patrocinio A. O., Murakami Iha N. Y., et al. (2009). Making oxygen with ruthenium complexes. Acc. Chem. Res. 42, 1954–1965. 10.1021/ar9001526 [DOI] [PubMed] [Google Scholar]
- Concepcion J. J., Jurss J. W., Templeton J. L., Meyer T. J. (2008). One site is enough. catalytic water oxidation by [Ru(tpy)(bpm)(OH2)]2+ and [Ru(tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc. 130, 16462–16463. 10.1021/ja8059649 [DOI] [PubMed] [Google Scholar]
- Cook T. R., Dogutan D. K., Reece S. Y., Surendranath Y., Teets T. S., Nocera D. G. (2010). Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 110, 6474–6502. 10.1021/cr100246c [DOI] [PubMed] [Google Scholar]
- Dau H., Limberg C., Reier T., Risch M., Roggan S., Strasser P. (2010). The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis. ChemCatChem 2, 724–761. 10.1002/cctc.201000126 [DOI] [Google Scholar]
- Duan L., Bozoglian F., Mandal S., Stewart B., Privalov T., Llobet A., et al. (2012). A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 4, 418–423. 10.1038/nchem.1301 [DOI] [PubMed] [Google Scholar]
- Evangelisti F., Car P.-E., Blacque O., Patzke G. R. (2013). Photocatalytic water oxidation with cobalt-containing tungstobismutates: tuning the metal core. Catal. Sci. Technol. 3, 3117–3129. 10.1039/c3cy00475a [DOI] [Google Scholar]
- Galán-Mascarós J. R. (2015). Water oxidation at electrodes modified with earth-abundant transition-metal catalysts. ChemElectroChem 2, 37–50. 10.1002/celc.201402268 [DOI] [Google Scholar]
- Gao J., Cao S., Tay Q., Liu Y., Yu L., Ye K., et al. (2013). Molecule-based water-oxidation catalysts (WOCs): Cluste-size-dependent dye-sensitized polyoxometalates for visible-light-driven O2 evolution. Sci. Rep. 3:1853. 10.1038/srep01853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geletii Y. V., Botar B., Köegerler P., Hillesheim D. A., Musaev D. G., Hill C. L. (2008). An all-inorganic, stable, and highly active tetraruthenium homogeneous catalyst for water oxidation. Angew. Chem. Int. Ed. 47, 3896–3899. 10.1002/anie.200705652 [DOI] [PubMed] [Google Scholar]
- Goberna-Ferrón S., Peña B., Soriano-López J., Carbó J. J., Zhao H., Poblet J. M., et al. (2014). A fast metal-metal bonded water oxidation catalyst. J. Catal. 315, 25–32. 10.1016/j.jcat.2014.04.010 [DOI] [Google Scholar]
- Goberna-Ferrón S., Soriano-López J., Galán-Mascarós J. R., Nyman M. (2015). Solution speciation and stability of cobalt-polyoxometalate water oxidation catalysts by X-ray scattering. Eur. J. Inorg. Chem. 2015, 2833–2840. 10.1002/ejic.201500404 [DOI] [Google Scholar]
- Goberna-Ferrón S., Vigara L., Soriano-López J., Galán-Mascarós J. R. (2012). Identification of a nonanuclear {} polyoxometalate cluster as a homogeneous catalyst for water oxidation. Inorg. Chem. 51, 11707–11715. 10.1021/ic301618h [DOI] [PubMed] [Google Scholar]
- Guo S.-X., Liu Y., Lee C.-Y., Bond A. M., Zhang J., Geletii Y. V., et al. (2013). Graphene-supported [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− for highly efficient electrocatalytic water oxidation. Energy Environ. Sci. 6, 2654–2663. 10.1039/c3ee41892h [DOI] [Google Scholar]
- Hara M., Waraksa C. C., Lean J. T., Lewis B. A., Mallouk T. E. (2000). Photocatalytic water oxidation in a buffered tris(2,2'-bipyridyl)ruthenium complex-colloidal IrO2 system. J. Phys. Chem. A 104, 5275–5280. 10.1021/jp000321x [DOI] [Google Scholar]
- Huang Z., Luo Z., Geletii Y. V., Vickers J. W., Yin Q., Wu D., et al. (2011). Efficient light-driven carbon-free cobalt-based molecular catalyst for water oxidation. J. Am. Chem. Soc. 133, 2068–2071. 10.1021/ja109681d [DOI] [PubMed] [Google Scholar]
- Lewis N. S., Nocera D. G. (2006). Powering the planet: chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. U.S.A. 103, 15729–15735. 10.1073/pnas.0603395103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wang F. Y. (2012). Transition metal complexes thast catalyze oxygen formation from water: 1979–2010. Coord. Chem. Rev. 256, 1115–1136. 10.1016/j.ccr.2012.01.015 [DOI] [Google Scholar]
- Lloret-Fillol J., Codolà Z., Garcia-Bosch I., Gómez L., Pla J. J., Costas M. (2011). Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat. Chem. 3, 807–813. 10.1038/nchem.1140 [DOI] [PubMed] [Google Scholar]
- Lv H., Geletii Y. V., Zhao C., Vickers J. W., Zhu G., Luo Z., et al. (2012). Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 41, 7572–7589. 10.1039/c2cs35292c [DOI] [PubMed] [Google Scholar]
- Lv H., Song J., Geletii Y. V., Vickers J. W., Sumliner J. M., Musaev D. G., et al. (2014). An exceptionally fast homogeneous carbon-free cobalt-based water oxidation catalyst. J. Am. Chem. Soc. 136, 9268–9271. 10.1021/ja5045488 [DOI] [PubMed] [Google Scholar]
- Mccool N. S., Robinson D. M., Sheats J. E., Dismukes G. C. (2011). A Co4O4 “cubane” water oxidation catalyst inspired by photosynthesis. J. Am. Chem. Soc. 133, 11446–11449. 10.1021/ja203877y [DOI] [PubMed] [Google Scholar]
- Mcevoy J. P., Brudvig G. W. (2006). Water-splitting chemistry of photosystem II. Chem. Rev. 106, 4455–4483. 10.1021/cr0204294 [DOI] [PubMed] [Google Scholar]
- Natali M., Bazzan I., Goberna-Ferrón S., Al-Oweini R., Ibrahim M., Bassil B. S., et al. (2017). Photo-assisted water oxidation by high-nuclearity cobalt-oxo cores: tracing the catalyst fate during oxygen evolution turnover. Green Chem. 19, 2416–2426. 10.1039/C7GC00052A [DOI] [Google Scholar]
- Pillai K. C., Kumar A. S., Zen J.-M. (2000). Nafion–RuO2-Ru composite electrodes for efficient electrocatalytic water oxidation. J. Mol. Catal. A. 160, 277–285. 10.1016/S1381-1169(00)00262-4 [DOI] [Google Scholar]
- Pope M. T. (1983). Heteropoly and Isopoly Oxometalates. Berlin: Springer. [Google Scholar]
- Pope M. T., Müller A. (2001). Polyoxometalate Chemistry From Topology via Self-Assembly to Applications. Dordrecht: Kluwer Academic Publishers. [Google Scholar]
- Puntoriero F., La Ganga G., Sartorel A., Carraro M., Scorrano G., Bonchio M., et al. (2010). Photo-induced water oxidation with tetra-nuclear ruthenium sensitizer and catalyst: a unique 4 × 4 ruthenium interplay triggering high efficiency with low-energy visible light. Chem. Commun. 46, 4725–4727. 10.1039/c0cc00444h [DOI] [PubMed] [Google Scholar]
- Quintana M., López A. M., Rapino S., Toma F. M., Iurlo M., Carraro M., et al. (2013). Knitting the catalytic pattern of artificial photosynthesis to a hybrid graphene nanotexture. ACS Nano 7, 811–817. 10.1021/nn305313q [DOI] [PubMed] [Google Scholar]
- Sartorel A., Bonchio M., Campagna S., Scandola F. (2013). Tetrametallic molecular catalysts for photochemical water oxidation. Chem. Soc. Rev. 42, 2262–2268. 10.1039/C2CS35287G [DOI] [PubMed] [Google Scholar]
- Sartorel A., Carraro M., Scorrano G., De Zorzi R., Geremia S., McDaniel N. D., et al. (2008). Polyoxometalate embedding of a tetraruthenium(IV)-oxo-core by template-directed metalation of [γ-SiW10O36]8−: a totally inorganic oxygen-evolving catalyst. J. Am. Chem. Soc. 130, 5006–5007. 10.1021/ja077837f [DOI] [PubMed] [Google Scholar]
- Seh Z. W., Kibsgaard J., Dickens C. F., Chorkendorff I., Nørskov J. K., Jaramillo T. F. (2017). Combining theory and experiment in electrocatalysis: insights into materials design. Science 355:eaad4998. 10.1126/science.aad4998 [DOI] [PubMed] [Google Scholar]
- Soriano-López J., Goberna-Ferrón S., Vigara L., Carbó J. J., Poblet J. M., Galán-Mascarós J. R. (2013). Cobalt polyoxometalates as heterogeneous water oxidation catalysts. Inorg. Chem. 52, 4753–4755. 10.1021/ic4001945 [DOI] [PubMed] [Google Scholar]
- Stracke J. J., Finke R. G. (2011). Electrocatalytic water oxidation beginning with the cobalt polyoxometalate [Co4(H2O)2(PW9O34)2]10−: identification of heterogeneous CoOx as the dominant catalyst. J. Am. Chem. Soc. 133, 14872–14875. 10.1021/ja205569j [DOI] [PubMed] [Google Scholar]
- Stracke J. J., Finke R. G. (2013). Water oxidation catalysis beginning with 2.5 μM [Co4(H2O)2(PW9O34)2]10−: investigation of the true electrochemically driven catalyst at ≥600 mV overpotential at a glassy carbon electrode. ACS Catal. 3, 1209–1219. 10.1021/cs400141t [DOI] [Google Scholar]
- Stracke J. J., Finke R. G. (2014). Distinguishing Homogeneous from heterogeneous water oxidation catalysis when beginning with polyoxometalates. ACS Catal. 4, 909–933. 10.1021/cs4011716 [DOI] [Google Scholar]
- Tan B. J., Klabunde K. J., Sherwood P. M. A. (1991). XPS studies of solvated metal atom dispersed (SMAD) catalysts. Evidence for layered cobalt-manganese particles on alumina and silica. J. Am. Chem. Soc. 113, 855–861. 10.1021/ja00003a019 [DOI] [Google Scholar]
- Wu J., Liao L., Yan W., Xue Y., Sun Y., Yan X., et al. (2012). Polyoxometalates immobilized in ordered mesoporous carbon nitride as highly efficient water oxidation catalysts. ChemSusChem 5, 1207–1212. 10.1002/cssc.201100809 [DOI] [PubMed] [Google Scholar]
- Xiang R., Ding Y., Zhao J. (2014). Visible-light-induced water oxidation mediated by a mononuclear-cobalt(II)-substituted silicotungstate. Chem. Asian J. 9, 3228–3237. 10.1002/asia.201402483 [DOI] [PubMed] [Google Scholar]
- Xu Y., Fischer A., Duan L., Tong L., Gabrielsson E., Åkermark B. (2010). Chemical and light-driven oxidation of water catalyzed by an efficient dinuclear ruthenium complex. Angew. Chem. Int. Ed. 49, 8934–8937. 10.1002/anie.201004278 [DOI] [PubMed] [Google Scholar]
- Yin Q., Tan J. M., Besson C., Geletii Y. V., Musaev D. G., Kuznetsov A. E., et al. (2010). A fast soluble carbon-free molecular water oxidation catalyst based in abundant metals. Science 328, 342–345. 10.1126/science.1185372 [DOI] [PubMed] [Google Scholar]
- Youngblood W. J., Lee S. H. A., Kobayashi Y., Hernandez-Pagan E. A., Hoertz P. G., Moore T. A., et al. (2009). Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 131, 926–927. 10.1021/ja809108y [DOI] [PubMed] [Google Scholar]
- Zhang M. T., Chen Z., Kang P., Meyer T. J. (2013). Electrocatalytic water oxidation with a copper(II) polypeptide complex. J. Am. Chem. Soc. 135, 2048–2051. 10.1021/ja3097515 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.