

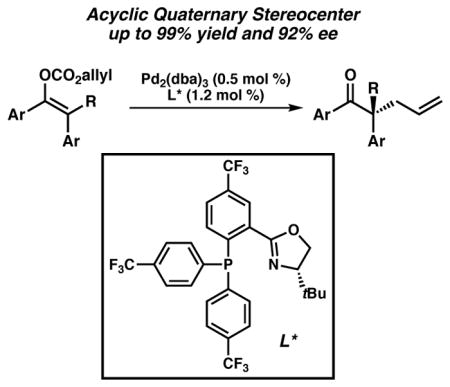
Abstract
The first enantioselective palladium-catalyzed decarboxylative allylic alkylation of fully substituted acyclic enol carbonates providing linear α-quaternary ketones is reported. Investigation into the reaction revealed that the use of an electron-deficient phosphinooxazoline ligand renders the enolate geometry of the starting material inconsequential, with the same enantiomer of product obtained in the same level of selectivity regardless of the starting ratio of enolates. As a result, a general method toward acyclic all-carbon quaternary stereocenters has been developed.
Graphical Abstract
All-carbon quaternary stereocenters are prominent features in many natural products and can provide beneficial biochemical stability and three-dimensionality to molecules for medicinal chemistry applications. As a result, a number of methods to address their synthesis have been developed, particularly in cyclic systems.1 In acyclic systems, however, the synthesis of this motif is less explored.2 This is in part due to additional problems that often arise in acyclic systems such as reduced rigidity of particular substrates leading to lower levels of selectivity. Additionally, selectively controlling the formation of fully substituted olefins/enolates is required for high selectivity in many catalytic processes. In the case of electrophilic functionalization of fully substituted enolates, the general and selective formation of such enolates as pure geometric isomers is highly challenging and has slowed progress. Although selective enolizations have been reported, these typically require highly specialized substrates, often incorporating chiral auxiliaries to impart selectivity in the enolate formation step.3
Recently, our group has become interested in strategies for preparing fully substituted acyclic enolates and applying them toward the synthesis of acyclic quaternary stereocenters, particularly via transition-metal catalyzed allylic alkylation (Figure 1). While palladium-catalyzed allylic alkylation has been employed in the synthesis of acyclic tertiary stereocenters,4 the formation of all-carbon quaternary stereocenters has proven more challenging. Utilizing chemistry developed by the Marek group6 for the synthesis of stereodefined acyclic enolates, we demonstrated the palladium-catalyzed enantioselective decarboxylative allylic alkylation of acyclic amide enolates utilizing an electron deficient C2-symmetric bisphosphine ligand (Figure 1A).7 Not surprisingly, it was observed that opposite enolate geometries of the substrate favored formation of the opposite enantiomers of the product, indicating a lack of significant dynamic kinetic resolution of the enolates during the reaction, and different facial selectivity between the two enolates. In fact, an analogous phenomenon regarding different enolate geometries was observed by the Trost lab when forming acyclic tertiary stereocenters via palladium-catalyzed allylic alkylation.8 Additionally, the Evans lab has reported the use of rhodium-catalyzed allylic alkylation to furnish acyclic α-quaternary nitriles and aldehydes (Figure 1B).9 In the latter work, they showed that both enolate geometries form the same enantiomer of product in essentially the same level of selectivity, which they believe is a consequence of substrate selectivity and not the result of an enolate equilibration event. In contrast, a dynamic equilibration of tributyltin enolates has been invoked by the Jacobsen group to directly alkylate fully substituted acyclic enolates with alkyl halides catalyzed by a [Cr(salen)] complex, affording α-quaternary ketones in high enantioselectivity starting with mixtures of enolates (Figure 1C).10 Recently, Shimizu and Kanai demonstrated the preparation of acyclic α-quaternary carboxylic acids via allylic alkylation utilizing a hybrid boron-palladium catalytic system (Figure 1D).11
Figure 1.
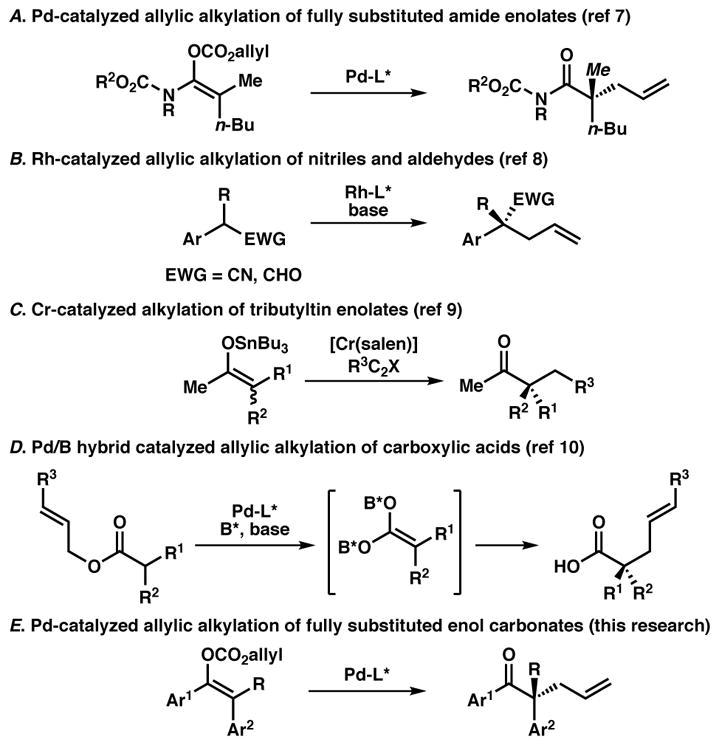
Preparation of acyclic α-quaternary carbonyl derivatives5
While acyclic ketone products can be obtained from the derivatization of α-quaternary nitriles and other carbonyl derivatives, we sought to prepare stereodefined acyclic ketone enolates and demonstrate their utility in palladium-catalyzed allylic alkylation (Figure 1E). To accomplish this goal, we utilized recently developed chemistry toward the synthesis of tetrasubstituted acyclic all-carbon olefins via sequential selective enoltosylate formation and cross-coupling.12 We were pleased to find that the reported enolization conditions could be modified to trap the resultant enolate as the allyl enol carbonate with excellent E/Z ratios (eq 1), thereby allowing us to investigate palladium-catalyzed conditions toward furnishing the acyclic quaternary stereocenter.
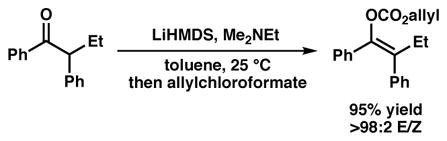 |
(1) |
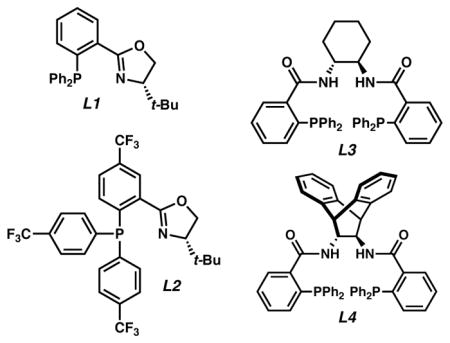
We began our studies with an initial ligand screen using 2.5 mol % Pd2(dba)3 and 6 mol % of a variety of common ligands, finding that phosphinooxazoline ligand L1 gave high conversion of the substrate albeit with very low enantioselectivity (Table 1, entry 1). This result is unsurprising since ligand L1 has previously been shown to perform poorly with α-aryl stabilized enolates, albeit on cyclic motifs.1a We were pleased to find, however, that electron-deficient phosphinooxazoline L2 gave the product in similarly excellent conversion with a promising enantiomeric excess of 54% (entry 2). Bisphosphine ligand L3 gave high conversion of the substrate but only a moderate 45% ee (entry 3), whereas L4 gave a more promising 73% ee but in poor conversion (entry 4). To our delight, we found that non-polar solvents were superior for the transformation (entries 5–9) with a 3:1 mixture of hexane/toluene providing the highest and most reproducible yields. In this solvent system, L4 provided the product in 89% isolated yield and 86% ee (entry 7). Gratifyingly, using ligand L2 gave enhanced results with the desired product obtained in 98% isolated yield and 90% ee. Moreover, the catalyst loading could be reduced to 0.5 mol % Pd2(dba)3 and 1.2 mol % of L2 (entry 9) without altering the yield and selectivity or significantly increasing reaction time.
Table 1.
Optimization of acyclic, decarboxylative allylic alkylationa
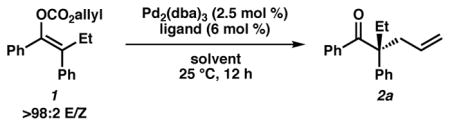
| ||||
|---|---|---|---|---|
| entry | ligand | solvent | yieldb | % eec |
| 1 | L1 | THF | – | 6 |
| 2 | L2 | THF | – | 54 |
| 3 | L3 | THF | – | −45 |
| 4 | L4 | THF | 42 | −73 |
| 5 | L2 | MeCyd | 83 | 89 |
| 6 | L2 | 3:1 MeCy/PhMe | 90 | 89 |
| 7 | L4 | 3:1 hexane/PhMe | 89 | −86 |
| 8 | L2 | 3:1 hexane/PhMe | 98 | 90 |
| 9e | L2 | 3:1 hexane/PhMe | 97 | 91 |
Conditions: 0.1 mmol 1, 2.5 mol % Pd2(dba)3, 6 mol % ligand, 1.0 mL solvent.
Yield of isolated product.
Determined by chiral SFC analysis.
methylcyclohexane
Reaction performed with 0.2 mmol 1, 0.5 mol % Pd2(dba)3, 1.2 mol % ligand, and 2.0 mL solvent.
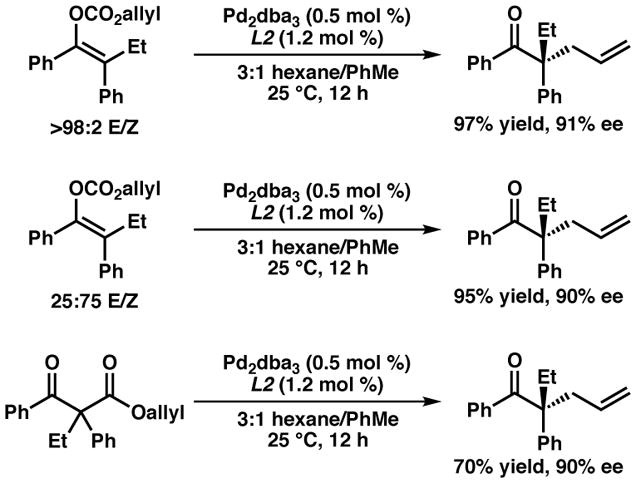
With optimal conditions identified, we next sought to examine the importance of enolate geometry on the transformation (Figure 2). Under the standard conditions, the desired product is obtained in 97% isolated yield and 91% ee favoring the S-absolute stereochemistry when the starting material consists of a >98:2 E/Z ratio of enolates. When the same starting material is prepared in a 25:75 E/Z ratio via a non-selective enolization, the alkylation product is obtained in 95% isolated yield and 90% ee, forming the same enantiomer of product (i.e. S) as in the standard case. Furthermore, the corresponding β-ketoester substrate again provides the same enantiomer (i.e. S) of product in high selectivity, albeit with a diminished yield (70% yield, 90% ee). This surprising result is exclusive to the electron-deficient phosphinooxazoline L2. With bisphosphine L4, the 25:75 E/Z ratio of enolates provides the product in a diminished 64% yield and 50% ee (compared to 89% yield and 86% ee). Additionally, when the 25:75 E/Z ratio substrate is used with L2, the ee of the product remains the same throughout the time course of the reaction (see supporting information). Based on these findings and prior examples of differing selectivities obtained with different enolate geometries in palladium-catalyzed allylic alkylation, we believe that a dynamic kinetic resolution of the two enolate geometries occurs in the reaction when L2 is used as the ligand, possibly due to facile equilibration between O-bound and C-bound palladium enolates.
Figure 2.
Importance of enolate geometry
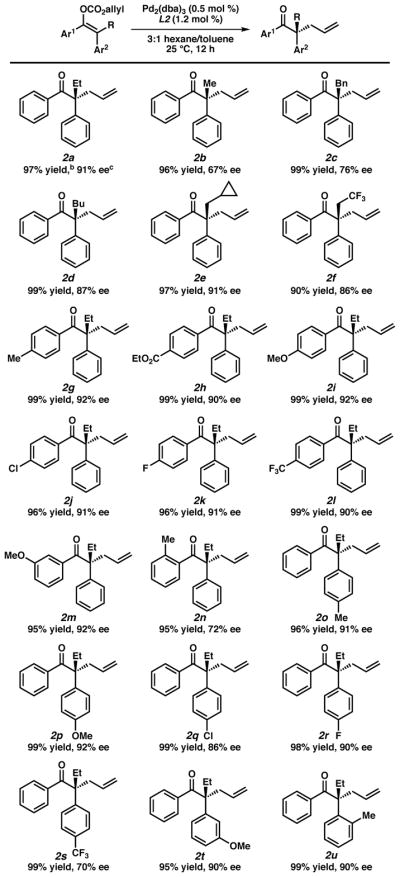
While β-ketoesters and enolate mixtures can be used to furnish the desired acyclic ketones in high enantioselectivity, due to ease of preparation and characterization, the substrate scope of the reaction was investigated with allyl enol carbonates formed via the aforementioned selective enolization. Substrates were enolized in generally high levels of selectivity (see supporting information for details). A variety of substrates containing different alkyl and aryl substituents were examined, with all alkylated products obtained in excellent yields (Table 2). Smaller α-alkyl groups such as methyl (2b) yielded a lower 67% ee, along with the α-benzyl substrate (2c) providing the alkylated product in only 76% ee. Longer alkyl chains such as n-butyl (2d) and trifluoroethyl (2f) as well as cyclopropyl methyl substrate (2e) all provided the desired alkylation products in high enantioselectivity. Examining electronic effects on the acetophenone (i.e., Ar1) portion of the substrate revealed that a variety of electron-withdrawing and donating substituents were well tolerated. Substitution at the para-position led to consistently high yields and selectivities with the p-Me (2g), p-ethyl ester (2h), p-OMe (2i), p-chloro (2j), p-fluoro (2k), and p-CF3 (2i) substrates providing products in 96–99% yield and 90–92% ee. The m-OMe (2m) substrate also performed well yielding the product in 96% yield and 91% ee. Ortho-substitution on this aryl ring, however, resulted in lower enantioselectivity with the o-Me (2n) substrate obtained excellent yield but only 72% ee. Lastly, α-aryl substitutions were surveyed (i.e. Ar2) with electron donating groups such as p-Me (2o) and p-OMe (2p) proceeding with excellent yields and 91% and 92% ee, respectively. While a p-fluoro (2r) substituent was well tolerated (90% ee), other electron-withdrawing substituents led to lower selectivity with the p-chloro (2q) substrate at 86% ee and the p-CF3 (2s) substrate significantly lower at 70% ee. Again, meta-substitution was tolerated with the m-OMe (2t) substrate delivering the product in 95% yield and 90% ee. Interestingly, ortho substituion is tolerated on the α-aryl substituent with o-Me (2u) substrate proceeding with excellent yield and 90% ee. Unfortunately, attempts to form fully alkyl quaternary stereocenters has proven challenging (see supporting information).
Table 2.
Substrate scope of asymmetric, decarboxylative allylic alkylationa
|
Reactions performed on 0.2 mmol scale.
Yield of isolated product.
Determined by chiral SFC analysis.
Absolute configuration of 2b determined by comparison to literature value,13 all other compounds are assigned by analogy.
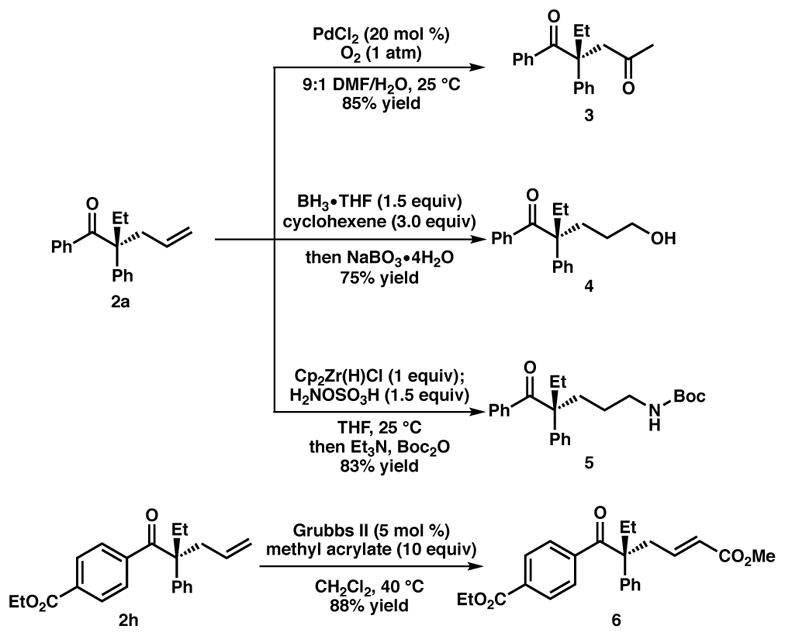
With the scope of the transformation established, we sought to demonstrate the utility of the produced acyclic α-quaternary ketones toward further functionalization (Figure 3). Treatment of the standard alkylation product 2a to Wacker oxidation conditions14 employing catalytic PdCl2 under O2 (1 atm) provided methyl ketone 3 in 85% isolated yield. In addition, treatment of ketone 2a to a hydroboration-oxidation protocol with BH3•THF and cyclohexene, followed by sodium perborate provided primary alcohol 4 in 75% isolated yield. A hydrozir-conation/amination protocol using conditions developed by the Hartwig group15 allows access to anti-Markovnikov amination product 5 in 83% yield following in situ Boc protection. Lastly, olefin metathesis of alkylation product 2h with methyl acrylate and Grubbs’ second generation catalyst6 yields α,β-unsaturated methyl ester 6 in 88% yield.
Figure 3.
Derivatization of alkylation products
In conclusion, we have developed the first enantioselective palladium-catalyzed decarboxylative allylic alkylation toward the synthesis of chiral acyclic α-quaternary ketones. The use of electron-deficient phosphinooxazoline ligand L2 is critical to achieve high yields and enantioselectivity. The allyl enol carbonate substrates could be prepared in high levels of E/Z geometrical selectivity, however a proposed dynamic kinetic enolate equilibration during the palladium catalyzed reaction allows the use of enolate mixtures and racemic β-ketoesters as substrates as well. Further exploration into the scope, mechanism, and applications of this process are underway.
Supplementary Material
Acknowledgments
We thank NIH-NIGMS (R01GM080269), the Gordon and Betty Moore Foundation, and Caltech for financial support. E.J.A. thanks the National Science Foundation for a predoctoral fellowship. We thank Dr. David VanderVelde (Caltech) for NMR expertise. Dr. Scott Virgil (Caltech) is thanked for instrumentation and SFC assistance.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and characterization data. (PDF)
References
- 1.(a) Martin SF. Tetrahedron. 1980;36:419–460. [Google Scholar]; (b) Corey EJ, Guzman-Perez A. Angew Chem, Int Ed. 1998;37:388–401. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Marek I, Sklute G. Chem Commun. 2007:1683–1691. doi: 10.1039/b615042j. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Jiang C. Synthesis. 2006:369–396. [Google Scholar]; (d) Liu Y, Han SJ, Liu WB, Stoltz BM. Acc Chem Res. 2015;48:740–751. doi: 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Behenna DC, Mohr JT, Sherden NH, Marunescu SC, Harned AM, Tani K, Seto M, Ma S, Novak Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr, White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem –Eur J. 2011;17:14199–14223. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Feng J, Holmes M, Krische MJ. Chem Rev. 2017;117:12564–12580. doi: 10.1021/acs.chemrev.7b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Das JP, Marek I. Chem Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; (c) Minko Y, Marek I. Chem Commun. 2014;50:12597–12611. doi: 10.1039/c4cc04391j. [DOI] [PubMed] [Google Scholar]; (d) Marek I, Minko Y, Pasco M, Mejuch T, Gilboa N, Chechik H, Das JP. J Am Chem Soc. 2014;136:2682–2694. doi: 10.1021/ja410424g. [DOI] [PubMed] [Google Scholar]
- 3.Selected examples: Meyer AI, Seefeld MA, Lefker BA, Blake JF, Williard PG. J Am Chem Soc. 1998;120:7429–7438.Boeckman RB, Boehmler DJ, Musselman RA. Org Lett. 2001;3:3777–3780. doi: 10.1021/ol016738i.Kummer DA, Chain WJ, Morales MR, Quiroga O, Myers AG. J Am Chem Soc. 2008;130:13231–13233. doi: 10.1021/ja806021y.Morales MR, Mellem KT, Myers AG. Angew Chem, Int Ed. 2012;51:4568–4571. doi: 10.1002/anie.201200370.Kummer DA, Chain WJ, Morales MR, Quiroga O, Myers AG. J Am Chem Soc. 2008;130:13231–13233. doi: 10.1021/ja806021y.Manthorpe JM, Gleason JL. J Am Chem Soc. 2001;123:2091–2092. doi: 10.1021/ja0058280.
- 4.a) Hannesian S, Chénard E. Org Lett. 2012;14:3222–3225. doi: 10.1021/ol301332f. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Michaelis DJ, Charpentier J, Xu J. Angew Chem Int Ed. 2011;51:204–208. doi: 10.1002/anie.201105801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang K, Peng Q, Hou XL, Wu YD. Angew Chem, Int Ed. 2008;47:1741–1744. doi: 10.1002/anie.200704629. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Lehr K, Michaelis DJ, Xu J, Buckl AK. J Am Chem Soc. 2010;132:8915–8917. doi: 10.1021/ja103771w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen JP, Ding CH, Liu W, Hou XL, Dai LX. J Am Chem Soc. 2010;132:15493–15495. doi: 10.1021/ja106703y. [DOI] [PubMed] [Google Scholar]; (f) Schwarz KJ, Amos JL, Klein JC, Do DT, Snaddon TN. J Am Chem Soc. 2016;138:5214–5217. doi: 10.1021/jacs.6b01694. [DOI] [PubMed] [Google Scholar]
- 5.Iridium has been used to make acyclic vinyl quaternary stereocenters as well as vicinal stereocenters: Liu WB, Reeves CM, Stoltz BM. J Am Chem Soc. 2013;135:17298–17301. doi: 10.1021/ja4097829.Shockley S, Hethcox JC, Stoltz BM. Angew Chem, Int Ed. 2017;56:10196–10199. doi: 10.1002/anie.201707015.Hethcox JC, Shockley S, Stoltz BM. Angew Chem, Int Ed. 2018;57:8664–8667. doi: 10.1002/anie.201804820.
- 6.(a) Minko Y, Pasco M, Lercher L, Botoshansky M, Marek I. Nature. 2012;490:522–526. doi: 10.1038/nature11569. [DOI] [PubMed] [Google Scholar]; (b) Nairoukh Z, Marek I. Angew Chem, Int Ed. 2015;54:14393–14397. doi: 10.1002/anie.201507209. [DOI] [PubMed] [Google Scholar]; (c) Nairoukh Z, Kumar GKSN, Minko Y, Marek I. Chem Sci. 2017;8:627–630. doi: 10.1039/c6sc03036j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Haimov E, Nairoukh Z, Shterenberg A, Berkovitz T, Jamison TF, Marek I. Angew Chem, Int Ed. 2016;55:5517–5520. doi: 10.1002/anie.201601883. [DOI] [PubMed] [Google Scholar]
- 7.Starkov P, Moore JT, Duquette DC, Stoltz BM, Marek I. J Am Chem Soc. 2017;139:9615–9620. doi: 10.1021/jacs.7b04086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turnbull BWH, Evans PA. J Am Chem Soc. 2015;137:6156–6159. doi: 10.1021/jacs.5b02810.Wright TB, Evans PA. J Am Chem Soc. 2016;138:15303–15306. doi: 10.1021/jacs.6b10099.See also: Evans PA, Oliver S, Chae J. J Am Chem Soc. 2012;134:19314–19317. doi: 10.1021/ja306602g.Evans PA, Oliver S. Org Lett. 2013;15:5626–5629. doi: 10.1021/ol402336u.Turnbull BWH, Oliver S, Evans PA. J Am Chem Soc. 2015;137:15374–15377. doi: 10.1021/jacs.5b10270.
- 10.Doyle AG, Jacobsen EN. Angew Chem, Int Ed. 2007;46:3701–3705. doi: 10.1002/anie.200604901. [DOI] [PubMed] [Google Scholar]
- 11.Fujita T, Yamamoto T, Morita Y, Chen H, Shimizu Y, Kanai M. J Am Chem Soc. 2018;140:5899–5903. doi: 10.1021/jacs.8b02783. [DOI] [PubMed] [Google Scholar]
- 12.(a) Li BX, Le DN, Mack KA, McClory A, Lim NK, Cravillion T, Savage S, Han C, Collum DB, Zhang H, Gosselin F. J Am Chem Soc. 2017;139:10777–10783. doi: 10.1021/jacs.7b05071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mack KA, McClory A, Zhang H, Gosselin F, Collum DB. J Am Chem Soc. 2017;139:12182–12189. doi: 10.1021/jacs.7b05057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonawane RP, Jheengut V, Rabalakos C, Larouch-Gauthier R, Scott HK, Aggarwal VK. Angew Chem, Int Ed. 2011;50:3760–3763. doi: 10.1002/anie.201008067. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji J, Shimizu I, Yamamoto K. Tetrahedron Lett. 1976;17:2975–2976. [Google Scholar]
- 15.Strom AE, Hartwig JF. J Org Chem. 2013;78:8909–8914. doi: 10.1021/jo401498w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.