

ABSTRACT
Impaired NK cell cytotoxic activity contributes to the local dysfunctional immune environment in endometriosis (EMS), which is an estrogen-dependent gynecological disease that affects the function of ectopic endometrial tissue clearance. The reason for the impaired cytotoxic activity of NK cells in an ectopic lesion microenvironment (ELM) is largely unknown. In this study, we show that the macroautophagy/autophagy level of endometrial stromal cells (ESCs) from EMS decreased under negative regulation of estrogen. The ratio of peritoneal FCGR3− NK to FCGR3+ NK cells increases as EMS progresses. Moreover, the autophagy suppression results in the downregulation of HCK (hematopoietic cellular kinase) by inactivating STAT3 (signal transducer and activator of transcription 3), as well as the increased secretion of the downstream molecules CXCL8/IL8 and IL23A by ESCs, and this increase induced the upregulation of FCGR3− NK cells and decline of cytotoxic activity in ELM. This process is mediated through the depression of microRNA MIR1185-1-3p, which is associated with the activation of the target gene PTGS2 in NK cells. FCGR3− NK with a phenotype of PTGS2/COX2high IFNGlow PRF1low GZMBlow induced by hck knockout (hck−/-) or 3-methyladenine (3-MA, an autophagy inhibitor)-stimulated ESCs accelerates ESC’s growth both in vitro and in vivo. These results suggest that the estrogen-autophagy-STAT3-HCK axis participates in the differentiation of PTGS2high IFNGlow PRF1low GZMBlow FCGR3− NK cells in ELM and contributes to the development of EMS. This result provides a scientific basis for potential therapeutic strategies to treat diseases related to impaired NK cell cytotoxic activity.
Abbreviations: anti-FCGR3: anti-FCGR3 with neutralizing antibody; Ctrl-ESC: untreated ESCs; CXCL8: C-X-C motif chemokine ligand 8; ectoESC: ESCs from ectopic lesion; ELM: ectopic lesion microenvironment; EMS: endometriosis; ESCs: endometrial stromal cells; eutoESC:eutopic ESCs; HCK: hematopoietic cellular kinase; HCK(OE): overexpression of HCK; IFNG: interferon gamma; IL23A (OE): overexpression of IL23A; KLRK1: Killer cell lectin like receptor K1; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; 3 -MA: 3-methyladenine; 3-MA-ESC: 3-MA-treated ESCs; MIR1185-1-3p+: overexpression of HsMIR1185-1-3p; NK: natural killer; normESCs: normal ESCs; Rap-ESC:rapamycin-treated ESCs; PCNA: proliferating cell nuclear antigen; PF: peritoneal fluid; SFKs: SRC family of cytoplasmic tyrosine kinases; si-HCK: silencing of HCK; siIL23A: silencing of IL23A; USCs: uterus stromal cells.
KEYWORDS: Autophagy, endometrial stromal cells, FCGR3, hematopoietic cellular kinase, NK cells, PTGS2
Introduction
Natural killer (NK) cells are important components of the innate immune system and play a central role in the defense against viral infections, as well as tumor surveillance [1]. The effector functions of NK cells include cytotoxicity and production of cytokines and chemokines. In humans, more than 90% of the NK cells are NCAM1/CD56dimNK cells (in these cases, ‘bright’ and ‘dim’ are used to denote the relative expression of NCAM1/CD56) in the peripheral blood that can induce apoptosis of virus-infected cells through the release of granzymes and PRF1 or binding of ligands to their death receptors. This subset of NK cells usually expresses high levels of the low-affinity FCGR3 (Fc fragment of IgG receptor IIIa). Engagement of FCGR3 is necessary for antibody-dependent cell-mediated cytotoxicity (ADCC) and sufficient to induce IFNG (interferon gamma) and TNF (tumor necrosis factor) secretion in addition to chemokine secretion [2,3]. NK cell function is controlled by the integration of signals from various activation and inhibitory receptors, which bind to components of pathogens and tumoral antigens [4–6]. The most potent activation receptors of NK cells are the ADCC-mediating molecule FCGR3 and KLRK1/NKG2D (killer cell lectin like receptor K1) [3–6]. Moreover, NK cells mediate ‘natural cytotoxicity’ through a set of activating natural cytotoxicity receptors, e.g. NCR1/NKp46 (natural cytotoxicity triggering receptor 1), NCR2/NKp44, and NCR3/NKp30, which recognize their ligands in tumor or virus-infected cells [3,4,7]. In contrast, NCAM1bright FCGR3− NK cells are poorly cytotoxic and are major cytokine producers that respond to cytokines, such as IL12, IL18, or IL15. Although this subset of NK cells constitutes the minority of peripheral blood NK cells, it is primarily in secondary lymphoid organs or other tissues [2,3]. Accumulating evidence indicates that the imbalance of NCAM1dim FCGR3+ NK and NCAM1bright FCGR3− NK ratio and impairment of NK cells cytotoxic activity are associated with several physiological and pathological processes, including normal pregnancy, infectious diseases, malignancies, and endometriosis (EMS). However, the mechanisms for the imbalance of NK cell subsets and the impaired cytotoxic activity remain largely unclear in the local tissue and organ microenvironment.
Under the influence of various factors, shed endometrial-like tissue in retrograde menstruation reaches the peritoneal cavity, adheres to endoabdominal structures, proliferates and implants to form ectopic lesions that lead to dysmenorrhea, chronic pelvic pain and infertility, which is referred to as EMS [8]. Although the majority of women have retrograde menstruation during their reproductive years, only about one in ten women develop EMS. Therefore, the pathogenesis of EMS still remains controversial despite extensive research. Today, EMS is considered to be an estrogen-dependent benign disease with malignancy-like behavior (e.g. unrestrained proliferation, decreased apoptosis and aggressive invasion as well as the potential for recurrence). A large body of evidence suggests that immune system alterations play critical roles in the initiation and progression of this enigmatic disorder in addition to hormonal and intrinsic abnormalities of the endometrium [9,10].
The distorted immune response against endometrial cells is responsible for the poor response to treatment, and poor clearance of the ectopic endometrium. Several studies have shown that the levels of activated macrophages, T cells, B cells, and inflammatory cytokines are increased in women with EMS [9–11]. Specifically, reductions in NK cell cytotoxicity (such as low levels of GZMB, PRF1, TRAIL, and LAMP1/CD107a) have been observed in the peritoneal fluid (PF) of patients with EMS [12,13]. Moreover, the levels of most cell-activating receptors decreased when NK cells are downregulated, whereas the levels of most inhibitory receptors are upregulated. However, the underlying mechanisms remain unknown.
MicroRNAs are small, non-coding RNAs that regulate target genes though degradation or the inhibition of post-transcriptional gene expression [14]. Recently, the role of microRNAs in the management of NK cell developmental and functional programs have been suggested [15–17]. MIR1185–1-3p (accession number: MIMAT0022838; miRBase ID: hsa-miR-1185-1-3p) was identified in mammalian genomes in 2008 [18]. It has been reported that MIR1185 can induce endothelial cell apoptosis by targeting UVRAG (UV radiation resistance associated gene) and KRIT1 (krev1 interaction trapped gene 1) [19], and promotes arterial stiffness by modulating VCAM1 (vascular cell adhesion molecule 1) and SELE/selectin E expression [20]. However, expression and function of MIR1185-1-3p in NK cells is still unclear.
Of note, autophagy has been linked to various pathophysiological processes, including tumorigenesis [21], development [21], cell death [21], and immunity [22]. Our previous study shows that the autophagy of ectopic endometrial stromal cells (ESCs) is significantly decreased, and this status is possibly mediated by the estrogen-SDF1/CXCL12-CXCR4 axis [23]. However, whether and how the change in the autophagy level of ESC is associated with the functional defects and impaired cytotoxicity of NK cells in PF from EMS are still unknown.
Therefore, the aim of this study was to investigate whether ESC autophagy regulates the balance of FCGR3+ NK to FCGR3− NK cells, as well as the levels of cytotoxicity-related molecules in the endometriotic milieu, and to explore the effects of these ESCs (abnormal autophagy)-educated NK cells on the growth of ectopic ESCs and lesions in vitro and in vivo.
Results
The low cytotoxic FCGR3− NK cells increase in ELM with disease progression
To investigate the change pattern of the NK cell ratio and phenotype with disease progression, we first collected the PF from women with or without EMS. As shown in Figure 1, the percentage of NCAM1+ NK cells in PTPRC/CD45+ total leukocytes of PF increased from patients in the stage I-II disease, whereas it further declined in patients with stage III-IV disease (Figure 1(a,b). The decrease in the advanced-stage patients possibly resulted from the sharp rise in macrophages (the highest population, ~ 60%) [24], and regulatory T cells [25,26]. In contrast, the ratio of FCGR3− NK to FCGR3+ NK cells (FCGR3−:FCGR3+ NK) declined in the early stages and then peaked in patients with advanced EMS (Figure 1(a,c)). These results indicated the dominant subset of NK cells is FCGR3+ NK in the initial stage of this disease. However, such an advantage tends to favor FCGR3− NK during the advanced stage.
Figure 1.
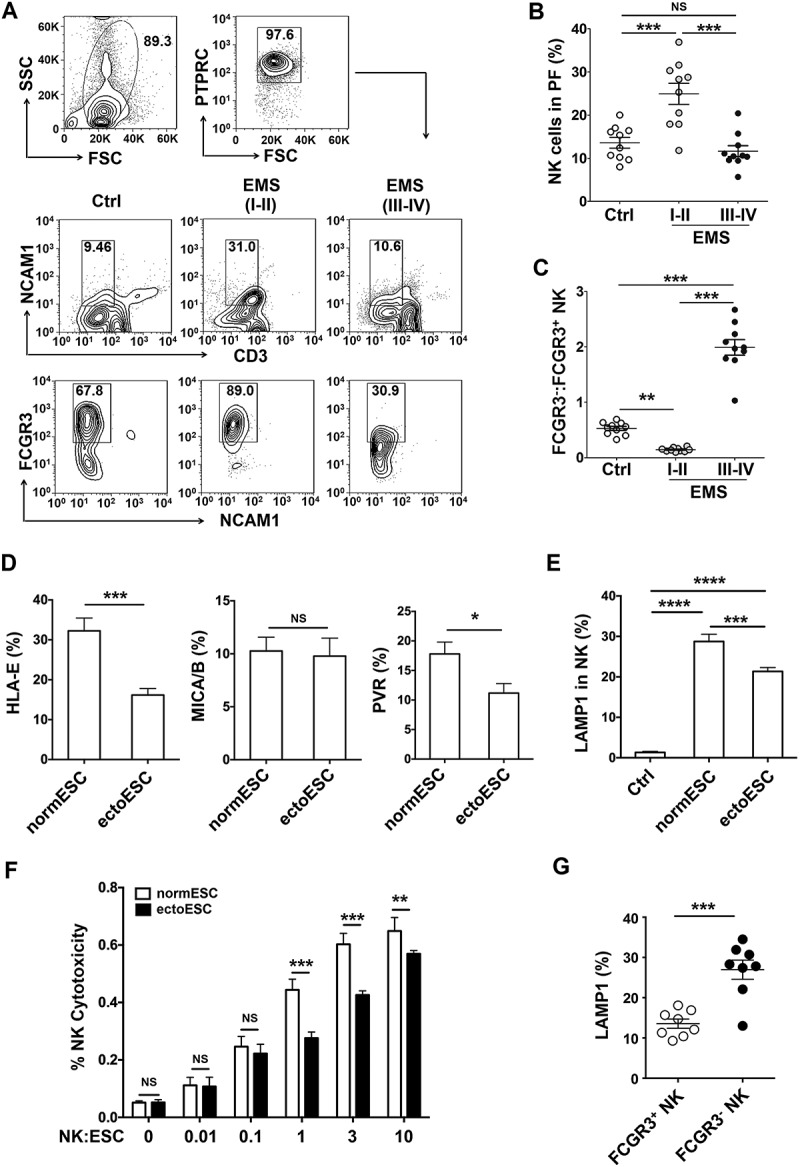
The low cytotoxic FCGR3− NK cells increase in ELM with disease progression. (a-c) The percentage and the ratio of FCGR3−:FCGR3+ NK cells in PF from women with or without EMS were analyzed by FCM (one-way ANOVA). (d) The expression of HLA-E, MICA/B and PVR on normESCs (n = 6) and ectoESCs (n = 6) was detected by FCM (Student t test). (e) After coculture with normESCs or ectoESCs for 48 h, the expression of LAMP1 on NK cells (n = 6) was detected by FCM (one-way ANOVA). (f) NK cells (n = 6) were cocultured with normESCs or ectoESCs for the cytotoxicity assay at different T/E (target cells:effector cells) ratios (1:100, 1:10, 1:1, 3:1 or 10:1) for 4 h (Student t test). (g) After coculture with ectoESCs for 48 h, the expression of LAMP1 on FCGR3+ NK cells and FCGR3− NK cells (n = 6) was analyzed by FCM (Student t test). Ctrl, PF from women without endometriosis (n = 10); EMS (I-II), PF from women with endometriosis were in early stages (stage I and II, n = 10); EMS (III-IV), PF from women with endometriosis were in advanced stages (stage III and IV, n = 10). normESC, normal ESCs from healthy women; ectoESC, ectopic ESCs from women with EMS. Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. NS, no statistical difference.
Compared with normal ESCs (normESCs), ESCs from ectopic lesions (ectoESCs) of patients with stage III-IV disease expressed lower levels of activating NK cell ligands HLA-E and PVR/CD155 (Figure 1(d)). ESCs led to the elevation of LAMP1 on NK cells, especially normESCs (Figure 1(e)). Compared with ectoESCs, NK cells displayed a more powerful cytotoxic activity in response to normESCs (Figure 1(f)). After coculture with ectoESC, the level of LAMP1 on FCGR3+ NK cells was higher than that on FCGR3− NK cells (Figure 1(g)). These results above suggest that the decrease of cytotoxic activity of NK cells in PF should be closely related to ectoESC, and FCGR3+ NK cells preferentially degranulate in response to ectoESC.
The low autophagy of ESCs leads to a high level of FCGR3− NK cells in ELM
Before analyzing the relationship between ESCs and the accumulation of FCGR3− NK cells, we observed that the autophagy-related molecules MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) and BECN1/Beclin 1 of ectoESCs were significantly decreased compared to normESCs (Figure 2(a)), and stimulation with 17-β estradiol also markedly led to the suppression of autophagy grade (Figure 2(b)). Then we established a coculture system to mimic the microenvironment of the peritoneal cavity and detected the ratio of FCGR3− NK cells from peripheral blood when the cells were cultured alone or cocultured with 3-methyladenine (3-MA, an autophagy inhibitor)-treated ESCs (3-MA-ESC), rapamycin (an autophagy inducer)-treated ESCs (Rap-ESC), or untreated, control ESCs (Ctrl-ESC). After coculture with ESC, the ratio of FCGR3− NK cells and the levels of KIR2DL1 and KIR3DL1 perceptibly increased, but NCR3, NCR2, IFNG, PRF1 and GZMB of NK cells significantly decreased (Figure 2(c,d and Fig. S1). The treatments with 3-MA-ESC and Rap-ESC further enhanced and restricted these effects, respectively (Figure 2(c,d) and Fig. S1). Coculturing with ATG5-silenced ESCs (siATG5-ESC) also led to a higher ratio of FCGR3− NK cells and a less cytotoxic activity compared with control ESCs (NC-ESC) (Figure 2(e-g).
Figure 2.
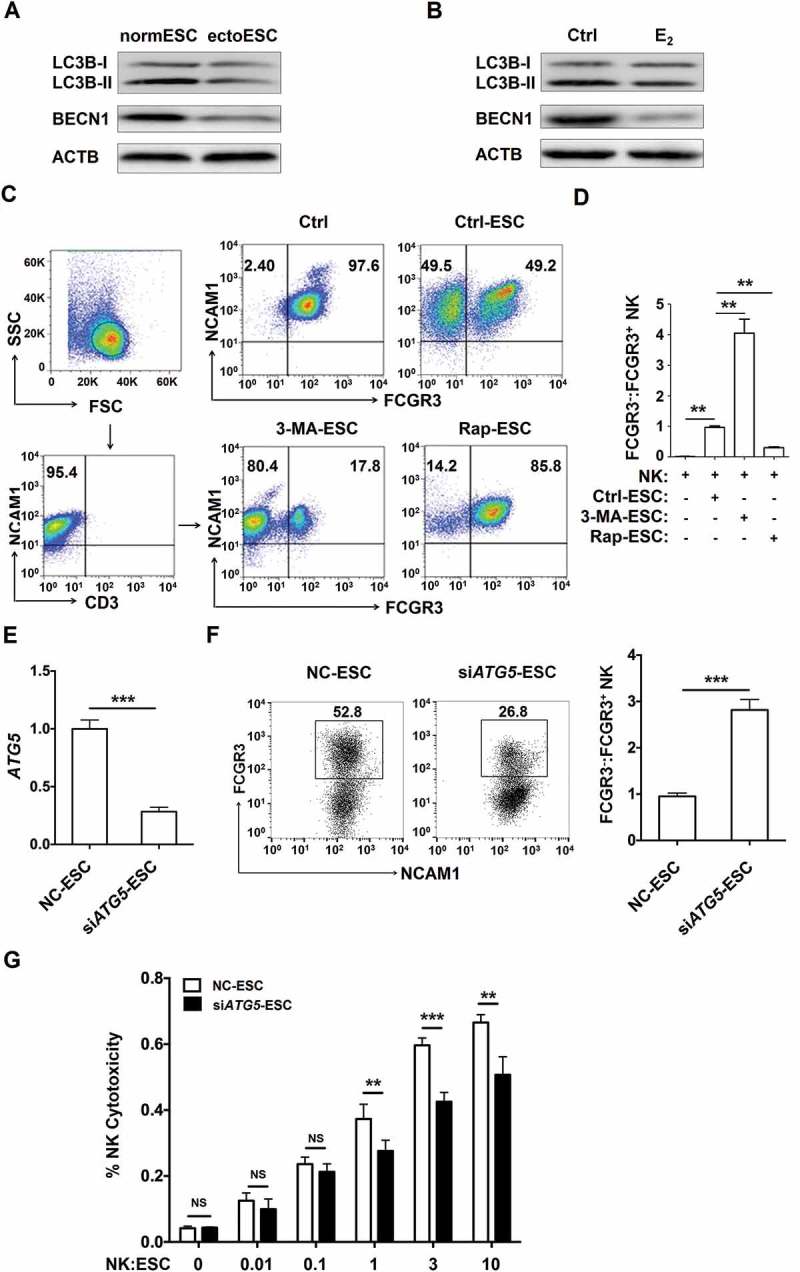
The low autophagy of ESCs leads to the high level of FCGR3− NK cells in vitro. (a) The expression of LC3B and BECN1 in normESC (n = 6) and ectoESC (n = 6) was analyzed by western blotting. (b) The expression of LC3B and BECN1 in normal ESC (n = 6) treated with or without 17β-estrogen (10−8 M) was analyzed by western blotting. (c,d) After coculture with Ctrl-ESC, 3-MA-ESC or Rap-ESC for 48 h, the ratio of FCGR3−:FCGR3+ NK cells (n = 6) was analyzed by FCM (one-way ANOVA). (e) The ATG5 mRNA levels in NC-ESC and siATG5-ESC by RT-PCR (Student t test). (f) After coculture with NC-ESC and siATG5-ESC for 48 h, the ratio of FCGR3−:FCGR3+ NK cells (n = 6) was analyzed by FCM (Student t test). (g) NK cells (n = 6) were cocultured with NC-ESC or siATG5-ESC for the cytotoxicity assay at different T/E ratios (1:100, 1:10, 1:1, 3:1 or 10:1) for 4 h (Student t test). E2, 17β-estrogen; Ctrl-ESC, normal ESCs; 3-MA-ESC, normal ESCs pretreated with 3-MA (10 mM) for 4 h; Rap-ESC, normal ESCs pretreated with rapamycin (1 mM) for 4 h. NC-ESC, Control ESCs transfected with control lentivirus; siATG5-ESC, ATG5-silenced ESCs transfected with ATG5 lentivirus. Data are expressed as the mean± SEM. **P < 0.01 and ***P < 0.001. NS, no statistical difference.
To further explore the reasons of FCGR3− NK cells accumulation, as depicted in Figure 3A, PKH26-labeled FCGR3+ NK and PKH67-labled FCGR3− NK cells were mixed and further cocultured with NC-ESC or siATG5-ESC for 48 h. As shown, there was no different of PKH67+ NK cells proliferation (Figure 3(b)) and dead PKH26+ NK cells (Figure 3(c,d)) between NC-ESC culture group and siATG5-ESC culture group. However, there was a subset of FCGR3− PKH26+ NK cells after coculture with NC-ESC or siATG5-ESC (Figure 3(c,e)), suggesting ESCs can trigger the differentiation of PKH26+ FCGR3− NK from PKH26+ FCGR3+ NK. Moreover, the ratio of FCGR3− PKH26+ NK cells in group of culture with siATG5-ESC was higher than that of culture with NC-ESC (Figure 3(c,e)). These data above suggest that the low autophagy of ESC led to more differentiation of FCGR3− NK cells from FCGR3+ NK cells in vitro.
Figure 3.
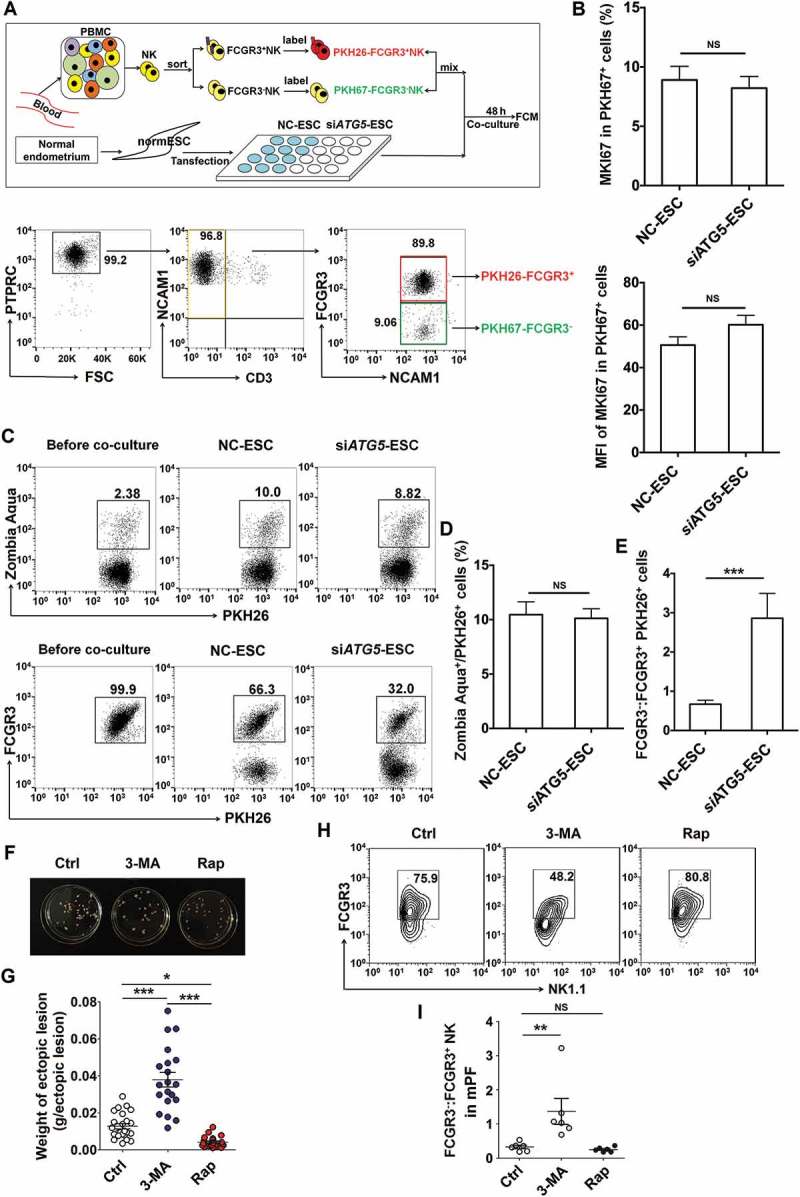
The low autophagy of ESCs triggers more differentiation of FCGR3− NK cells from FCGR3+ NK cells. (a,e) FCGR3+NK and FCGR3− NK cells were sorting from peripheral blood NK cells, and then labeling with PKH26 and PKH67, respectively. Then, PKH26-FCGR3+ NK and PKH67-FCGR3− NK cells (n = 6) were mixed and further cocultured with NC-ESC and siATG5-ESC (a). After 48 h, the positive ratio and MIF of MKI67 in PKH67+ NK cells (b), Zombie Aqua+ dead PKH26+ NK (c,d), and the differentiation of PKH26-FCGR3+ NK to PKH26-FCGR3− NK cells (c,e) were analyzed by FCM (Student t test). (f,g) The size and weight of EMS-like lesions from the EMS mouse model treated with vehicle, 3-MA (100 mg/kg/d) or Rap (100 mg/kg/d) (n = 6 mice/group) were measured (one-way ANOVA). (h,i) The C57BL/6 EMS mouse model was constructed, and treated with vehicle, 3-MA (100 mg/kg/d) or rapamycin (Rap, 100 mg/kg/d) (n = 6 mice/group) on day 3 and day 10 after surgery. Then the ratio of FCGR3−:FCGR3+ NK cells in PF was analyzed by FCM (one-way ANOVA). Data are expressed as the mean± SEM. **P < 0.05, **P < 0.01 and ***P < 0.001. NS, no statistical difference.
Additionally, intraperitoneal injection with 3-MA showed an increased weight of ectopic lesion (Figure 3(f,g)) as well as a high level of FCGR3− NK in PF in a mouse EMS model (Figure 3(h,i)). Taken together, these data suggest that the suppression of the autophagy level in ectoESCs possibly mediated by high estrogen plays a crucial role in the regulation of FCGR3− NK differentiation and the growth of ectopic lesions.
The suppression of ESC autophagy upregulates FCGR3− NK cells by downregulation of HCK
To investigate the potential effect of ESCs autophagy on FCGR3− NK cells, a proteomic microarray was performed to evaluate the differential proteins of cell lysates and supernatants between Ctrl-ESC and 3-MA-ESC (Figure 4(a,b)). Furthermore, based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg), we obtained the highest relationship between the centrality of differential expressions of proteins (non-cytokines) and their interacting partners of NK-function molecules (e.g. FCGR3, KIR2DL1, KIR3DL1, NCR3, NCR2, NCR1, KLRK1, IFNG, PRF1 and GZMB) in the autophagy-NK cell signaling pathway. Then some proteins (such as HCK, hematopoietic cell kinase) came to our attention (Figure 4(a) right and 4B right). HCK is a member of the SFKs (SRC family of cytoplasmic tyrosine kinases) and is mainly expressed in cells of the myeloid and B-lymphocyte cell lineages [27]. Compared to normESC, and eutopic ESCs (eutoESC), HCK expression in ectoESC was significantly decreased, which was consistent with the autophagy level of these ESCs (Figure 4(c)). As autophagy regulators, both 3-MA and rapamycin regulated the expression of HCK in ESCs and lesions in vitro and in vivo (Figure 4(d,e)). Similarly, siATG5-ESC had low levels of autophagy (low level of ATG5, BECN1, LC3B and high level of SQSTM1/p62) and HCK compared with NC-ESC (Figure 4F,G), which suggests the low level of autophagy in ectoESCs leads to a decrease in HCK.
Figure 4.
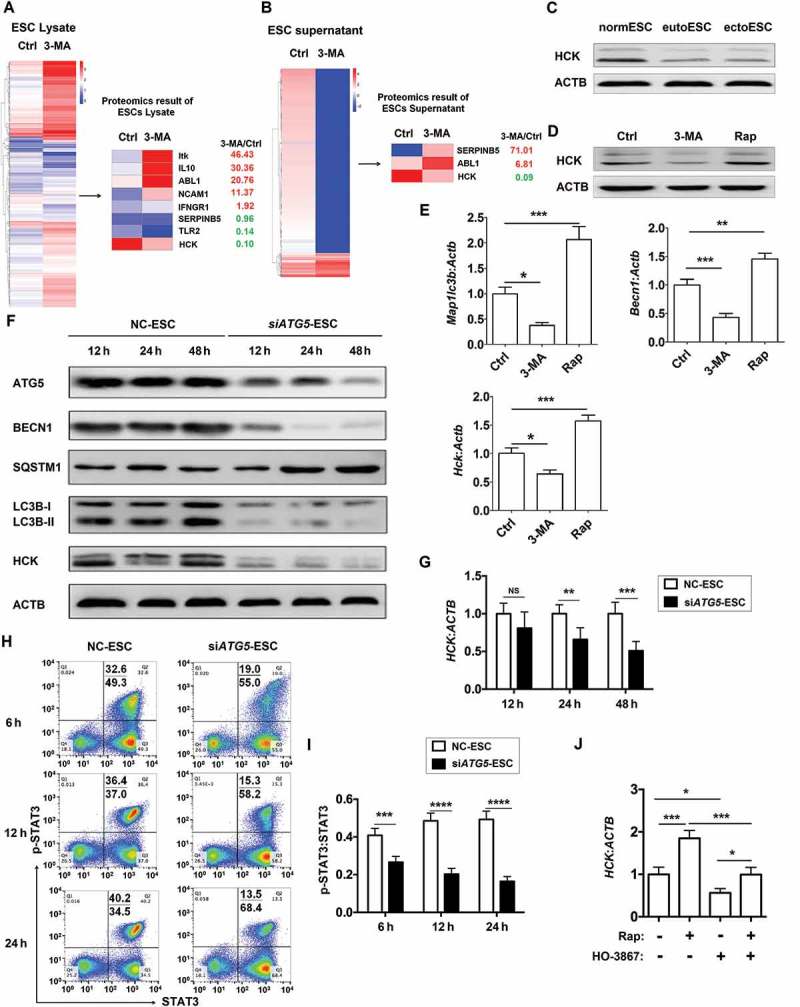
ESC autophagy upregulates expression of HCK by activating STAT3 signaling pathway. (a,b) The proteomic microarray was performed to evaluate the different proteins of cell lysate (A, left) and supernatant (B, left) between Ctrl-ESC and 3-MA-ESC. KEGG database-derived bioinformatics analysis was used to look for the differential expressions of proteins (non-cytokines) (A and B, right), which has the relationship with NK function molecules (FCGR3, KIR2DL1, KIR3DL1, NCR3, NCR2, NCR1, KLRK1, IFNG, PRF1 and GZMB). (c) The expression of HCK in normESC (n = 6), eutoESC (n = 6) and ectoESC (n = 6) was analyzed by western blotting. (d) The expression of HCK in Ctrl-ESC (n = 6), 3-MA-ESC (n = 6) or Rap-ESC (n = 6) was analyzed by western blotting. (e) The C57BL/6 EMS mice were treated with vehicle, 3-MA or Rap (n = 6 mice/group) on day 3 and day 10 after surgery. Then the transcriptional level of Map1lc3b, Becn1 and Hck in EMS-like lesions was detected by RT-PCR (one-way ANOVA). (f,g) NC-ESC and siATG5-ESC were stimulated with Rap (1 mM) for 4 h, and then cultured for another 12, 24, or 48 h, and the protein level of ATG5, BECN1, SQSTM1, LC3B and HCK, and the mRNA level of HCK was detected by western blotting and real-time PCR. (h,i) NC-ESC and siATG5-ESC were stimulated with Rap (1 mM) for 4 h, and then cultured for another 6, 12, or 24 h, and the level of phosphorylated and total STAT3 was analyzed by FCM. (j) Ctrl-ESC was treated with Rap (1 mM) for 4 h, and incubated with the selective inhibitor of STAT3 (HO-3867, 10 μM) for another 4 h, and then the mRNA level of HCK was analyzed by real-time PCR. eutoESC, ESCs of eutopic endometrium from women with EMS. Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.001.
It has been reported that there is a close link between autophagy, STAT3 (signal transducer and activator of transcription 3) and HCK [28–31]. Therefore, we further investigated the mechanism of autophagy on HCK protein level. As shown, silencing ATG5 led to the decrease of autophagy, HCK expression and STAT3 phosphorylation in ESCs (Figure 4(f to i). In addition, the upregulation of HCK induced by rapamycin was partly inhibited by a selective STAT3 inhibitor (HO-3867) (Figure 4(j)). These data suggest that autophagy upregulates the expression of HCK by activation of STAT3.
Further analysis showed that silencing of HCK (siHCK) and overexpression of HCK (HCK [OE]) (Fig. S2) amplified and impaired the stimulatory effects of ESCs on FCGR3− NK differentiation and expression of functional molecules (KIR2DL1, KIR3DL1, NCR3, NCR2, IFNG, PRF1 and GZMB), respectively (Figure 5(a,b) and Fig. S3). Subsequently, an endometrial fragment from the hck knockout (KO) mouse (hck−/-) was injected intraperitoneally into a wild-type mouse (WT) for constructing a hck special KO-EMS mouse model (Figure 5(c). Consistent with 3-MA, the absence of HCK gave rise to the upregulation of the MKI67/Ki-67 level and the weight of mouse ectopic lesions (Figure 5(d)), which suggests the low autophagy and HCK levels result in the growth of ectopic lesions. In addition, the ratio of FCGR3− NK cells significantly increased, but PRF1+, GZMB+ and IFNG+ NK cells were decreased in hck−/- EMS mice (Figure 5(e,f). These data above suggest that the downregulation of autophagy and HCK in ESCs increases the ratio of FCGR3− NK cells in ELM.
Figure 5.
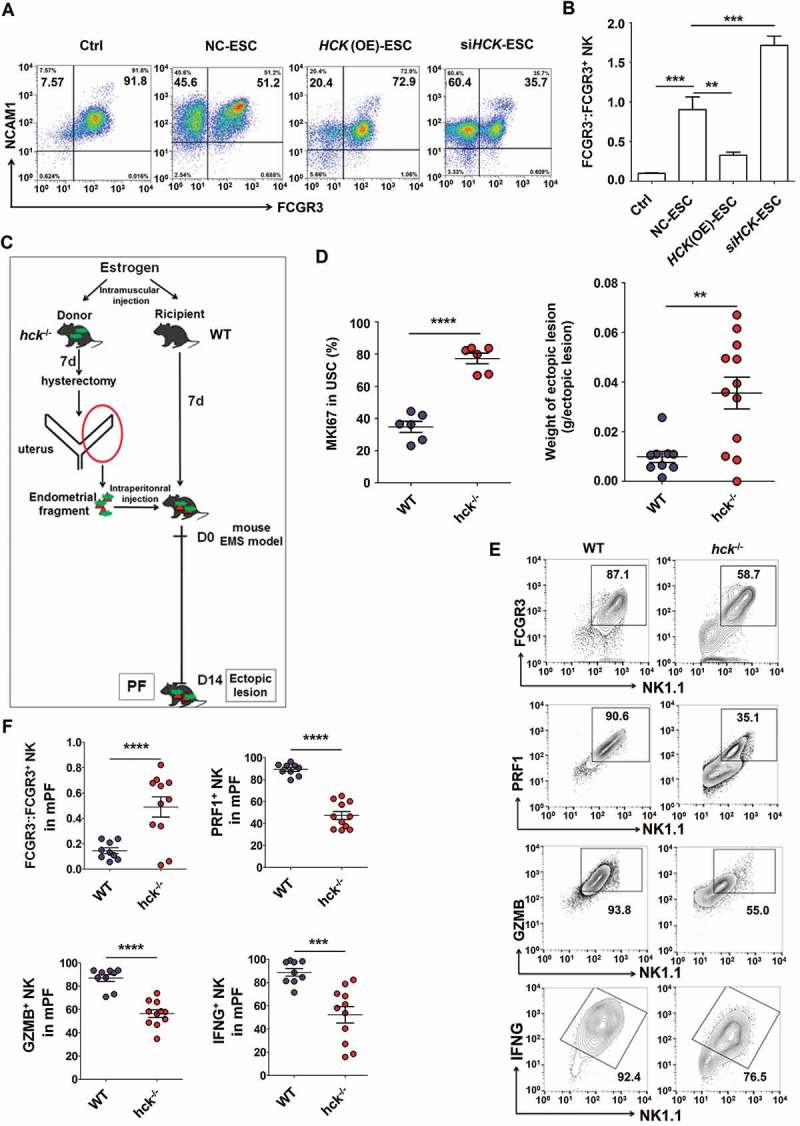
Hck downregulates FCGR3− NK cells. (a,b) The ratio of FCGR3−:FCGR3+ NK cells were analyzed after coculture with or without NC-ESC, HCK (OE)-ESC or siHCK-ESC by FCM (Student t test). (c) The endometrial fragment from Hck knockout (KO) mice (hck−/-) was intraperitoneally injected to wild-type mice (WT) for contrast with a hck special KO-EMS mouse model. (d) The level of MKI67 in USCs (left), and the weight of EMS-like lesions (right) from WT and hck−/- (n = 6 mice/group) EMS mice by FCM (Student t test). (e,f) The levels of PRF1+ NK, GZMB+ NK and IFNG+ NK cells were analyzed in PF from WT (n = 9 mice/group) and hck−/- (n = 10 mice/group) EMS mice by FCM (Student t test). NC-ESC, control ESCs transfected with GV230-vector plasmid; HCK (OE), HCK-overexpressing ESCs transfected with the GV230-HCK plasmid; WT: EMS mice (donor mice, WT mice); hck−/-, hck special KO-EMS mice (donor mice, hck−/- mice); USCs, mouse uterus stromal cells. Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.001.
The HCK-CXCL8/IL8-IL23A signal axis of ESCs regulates FCGR3− NK cell differentiation
According to the differential expression of cytokines (the differential fold >10 in ESC lysate and supernatants) (Figure 6(a,b)) and KEGG database-derived signal net analysis, differentially expressed cytokines IL1A, CXCL8 and IL23A were predicted to be downstream of HCK (Figure 6(c)). HCK (OE) or siHCK as well as stimulation with rapamycin or 3-MA resulted in the decrease and increase of CXCL8 and IL23A levels respectively, from ESCs in vitro, not IL1A (Figure 6(d) and Fig. S4A). Compared to the control group, the level of IL23A was elevated in ectopic lesions or PF from a hck−/- EMS mouse or 3-MA treatment group (Figure 6(e) and Fig. S4B). These results indicate that the low level of autophagy and HCK signaling stimulates the secretion of CXCL8 and IL23A by ESCs.
Figure 6.
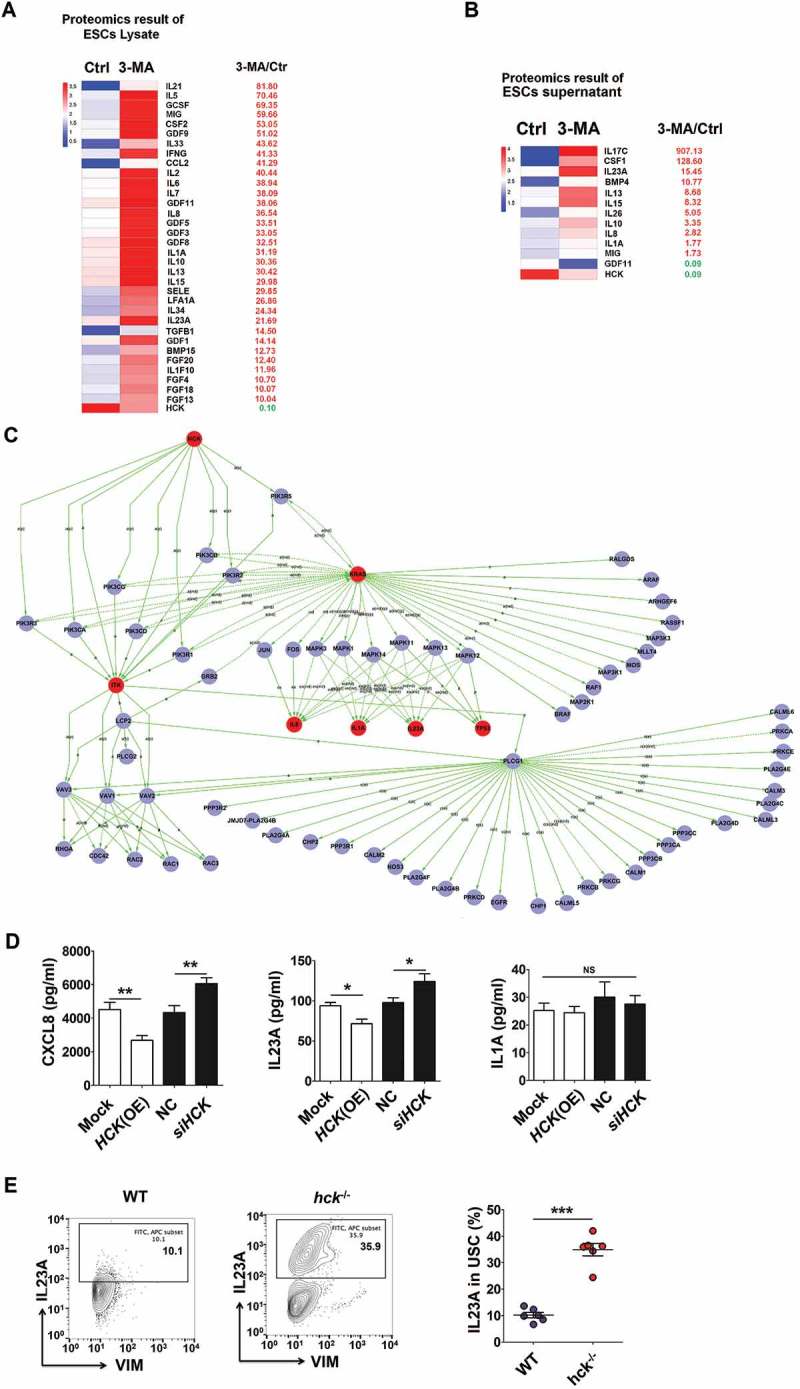
The low level of autophagy and HCK signaling stimulates the secretion of CXCL8 and IL23A by ESCs. (a,b) The differential expressions of cytokines (the differential fold > 10) in ESC lysate (a) and supernatants (b) by the proteomic microarray. (c) The differentially expressed cytokines IL1A, CXCL8 and IL23A were predicted as the downstream of HCK, according to KEGG database-derived signal net analysis. (d) The secretion levels of CXCL8, IL1A and IL23A from mock ESC (n = 6), HCK (OE)-ESC (n = 6), NC-ESC (n = 6) and siHCK-ESC (n = 6) were evaluated by ELISA (Student t test). (e) The levels of IL23A were evaluated in USCs of EMS-like lesions from WT (n = 6 mice/group) and hck−/- (n = 6 mice/group) EMS mice by FCM. (Student t test) mock, control ESCs transfected with GV230-vector plasmid; HCK (OE), HCK-overexpressing ESCs transfected with the GV230-HCK plasmid; Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001.
Based on KEGG database-derived bioinformatics analysis about the relationship between HCK, downstream cytokines (CXCL8, IL23A and IL1A), and Fc receptors FCGR3A and FCGR3B (Figure 7(a)), we next sought to confirm these relationships. As shown, recombinant human CXCL8 protein and IL23A overexpression (IL23A (OE), and neutralization antibodies for CXCL8/IL8 (anti-CXCL8) and siIL23A reversed the roles of HCK (OE) and siHCK in the regulation of FCGR3− NK differentiation and expression of functional molecules (e.g. IFNG, PRF1 and GZMB) in a coculture system, respectively (Figure 7(b) and Fig. S3). Blocking IL23A led to the decreased weight of ectopic lesions in EMS mice (Figure 7(c,d)). Of note, both PF and ESCs from EMS (stage III-IV) had higher level of CXCL8 and IL23A compared with healthy controls (Figure 7(e,f). Collectively, these data suggest that the HCK-CXCL8-IL23A signal axis of ESCs is involved in regulating FCGR3− NK differentiation and cytotoxicity.
Figure 7.
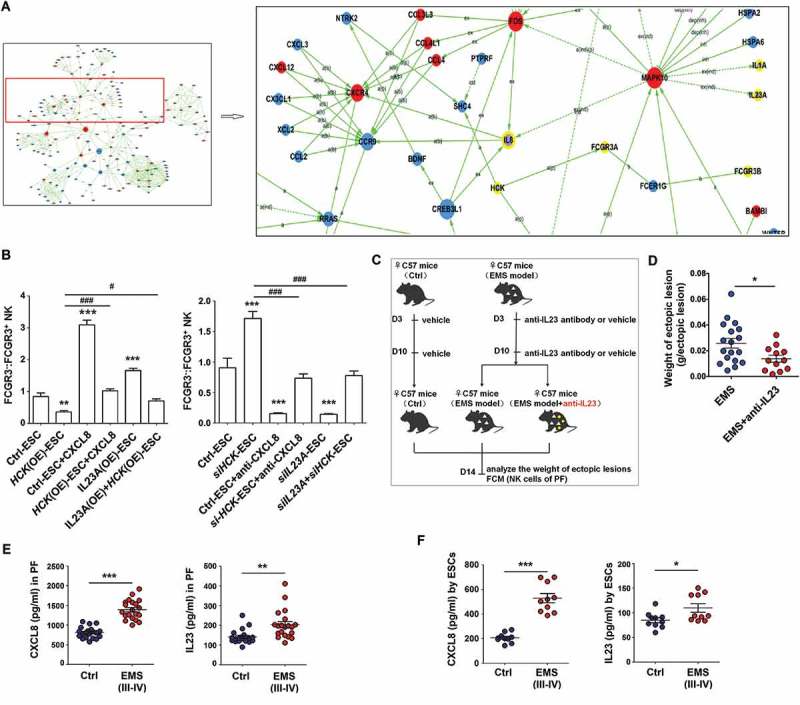
The HCK-CXCL8-IL23A signal axis of ESCs regulates FCGR3− NK cells differentiation. (a) KEGG database-derived bioinformatics analysis about the relationship between HCK downstream cytokines (CXCL8, IL23A and IL1A), Fc receptors FCGR3A and FCGR3B. (b) (left) After coculture with Ctrl-ESC, HCK (OE)-ESC, CXCL8 protein (100ng/ml) and/or IL23A (OE)-ESC for 48 h; (right) After coculture with Ctrl-ESC, siHCK-ESC, anti-CXCL8 (2 ug/ml) and/or siIL23A-ESC for 48 h, the ratio of FCGR3−:FCGR3+ NK cells (n = 6, right) were analyzed by FCM (one-way ANOVA). (c,d) The weights of EMS-like lesions from C57BL/6 EMS mice (n = 8 mice/group) were counted after treatment with or without intraperitoneal injection of anti-mouse IL23A neutralizing antibody (anti-IL23A, 0.1 mg/kg/d) on day 3 and day 10 (Student t test). (e,f) The concentration of CXCL8 and IL23A in PF from healthy control (n = 20) and EMS patients (stage III and IV, n = 20), supernatants of ESCs from healthy control (n = 10) and EMS patients (stage III and IV, n = 10) were detected by ELISA (Student t test). CXCL8, recombinant human CXCL8 protein; anti-CXCL8, anti-human CXCL8 neutralizing antibody. Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01and ***P < 0.001 (compared to Ctrl-ESC); #P < 0.05 and ###P < 0.001 (compared to HCK (OE)-ESC or siHCK-ESC group).
The FCGR3− NK cell differentiation in ELM is negatively regulated by MIR1185-1-3p
A variety of microRNAs have been found to be involved in the regulation of NK cell differentiation, maturation, and function, such as Mir155 [16], and MIR150/Mir150 [17]. However, the key microRNA and target genes for regulating the differentiation of FCGR3− NK cells remain largely unknown. Therefore, microRNA 4.0 and Human Transcriptome Array (HTA 2.0) microarrays were performed to analyze the differential miRNAs and genes in NK cells cocultured with Ctrl-ESC and 3-MA-ESC. Among these differential miRNAs (Figure 8(a)), the changes in 9 MIRs were beyond 3-fold. Further experiments confirmed that 3-MA-ESC or siHCK-ESC only decreased the level of MIR1185-1-3p in cocultured NK cells (Figure 8(b)). In contrast, MIR1185-1-3p in NK cells was perceptibly upregulated after the cells were cocultured with rapamycin-stimulated-ESCs (Rap-ESC) or HCK (OE)-ESC (Figure 8(b)). Meanwhile, CXCL8 and IL23A downregulated the MIR1185-1-3p level of NK cells in the coculture system (Figure 8(c,d)). Compared with healthy controls, NK cells in PF from EMS had a low level of MIR1185-1-3p (Figure 8(e)).
Figure 8.
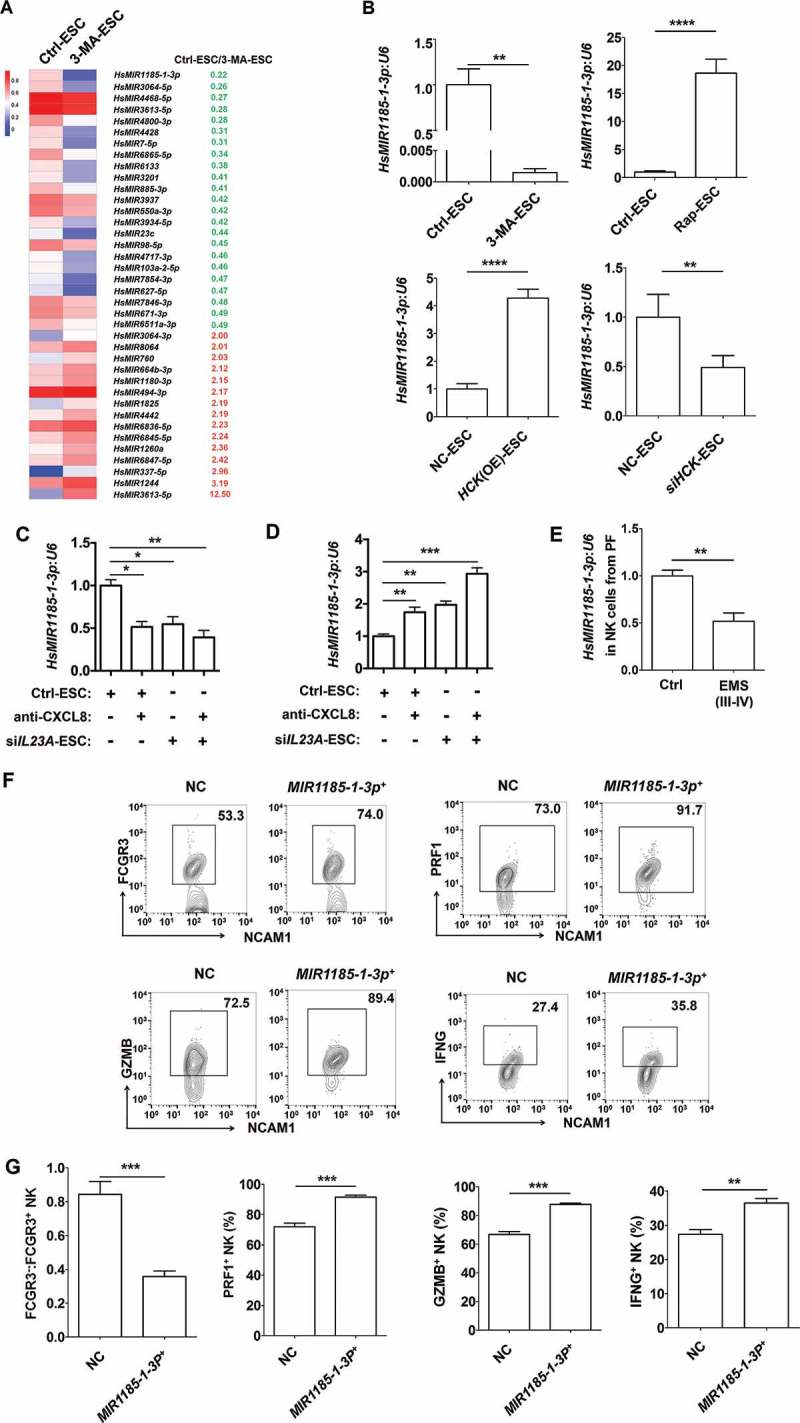
The FCGR3− NK cell differentiation in ELM is negatively regulated by MIR1185–1-3p. (a) The differential MicroRNAs (> 3-fold) in NK cells cocultured with Ctrl-ESC and 3-MA-ESC by microRNA 4.0 array. (b) The transcriptional levels of HsMIR1185-1-3p in NK cells (n = 6) were detected by RT-PCR, after coculture with Ctrl-ESC, 3-MA-ESC or Rap-ESC. In addition, the transcriptional levels of HsMIR1185-1-3p in NK cells (n = 6) cocultured with NC-ESC, HCK (OE)-ESC or siHCK-ESC were detected by RT-PCR (Student t test). (c,d) After coculture with Ctrl-ESC, IL23A (OE)-ESC, Ctrl-ESC plus CXCL8 protein (100 ng/ml) or IL23A (OE)-ESC plus CXCL8 protein (100 ng/ml), Ctrl-ESC, siIL23A-ESC, Ctrl-ESC plus anti-CXCL8 (2 µg/ml), or siIL23A-ESC plus anti-CXCL8 (2 µg/ml) for 48 h, the transcriptional levels of HsMIR1185-1-3p in NK cells (n = 6) were detected by RT-PCR (one-way ANOVA). (e) The transcriptional levels of HsMIR1185-1-3p in NK cells from healthy control (n = 6) and EMS patients (stage III and IV, n = 6) were detected by RT-PCR (Student t test). (f,g) FCM analysis of FCGR3, PRF1, GZMB and IFNG in NC NK cells (n = 6) and MIR1185-1-3p+ NK cells (n = 6) after coculture with normal ESCs (Student t test). NC, Ctrl NK cells transfected with control miRNA lentivirus; MIR1185-1-3p+, HsMIR1185-1-3p-overexpressing NK cells transfected with Mir-1185-1-3p mimic lentivirus. Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
We then investigated the role of MIR1185-1-3p in FCGR3− NK differentiation and molecule expression in MIR1185-1-3p overexpressing (MIR1185-1-3p+) or control (NC) NK cells (Fig. S5A) that were cocultured with normESCs. Compared to the NC group, NK cells in MIR1185-1-3p+ group had less FCGR3, PRF1, GZMB and IFNG (Figure 8(f,g) and Fig. S5B). In addition, overexpression of MIR1185-1-3p partly abrogated the effect of 3-MA-ESC on FCGR3− NK differentiation and functional molecules (Fig. S5B). Collectively, these results suggest that MIR1185-1-3p regulated by autophagy and the HCK level in ESCs should be an important suppressor for FCGR3− PRF1low GZMBlow IFNGlow NK differentiation.
The PTGS2 targeted by Mir1185-1-3p upregulates FCGR3− NK cells
We only found an intersection gene (PTGS2) based on the intersection analysis between differential genes in NK cells (coculture with Ctrl-ESC or 3-MA-ESC) and predicted target genes (i.e. EGR2, EREG and PTGS2) of MIR1185-1-3p (Figure 9(a,b). Meanwhile, the results of bioinformatics analysis (KEGG database-derived Signal net) told us that PTGS2/COX-2/COX2 was predicted as downstream of FCGR3/FcγR3 and was associated with CXCL8 and IL23A (Figure 9(c)). As shown, MIR1185-1-3p could target to regulate the transcription of PTGS2 (Figure 9(d,e)). Subsequently, we observed that MIR1185-1-3p+ suppressed the PTGS2 level in NK cells (Figure 9(f,g) and partly abolished the stimulatory effects of siHCK-ESC and 3-MA-ESC on PTGS2 expression in NK cells (Figure 9(f,g). In addition, PTGS2+ NK cells in PF of hck−/- EMS mice were significantly higher compared to WT EMS mouse cells (Figure 9(h)). Blocking IL23A decreased the ratio of PTGS2+ NK cells and FCGR3− NK cells in PF of the mouse EMS model (Fig. S6). Overall, these data indicate that the autophagy and HCK signaling of ESCs is involved in regulating the PTGS2 level in NK cells by MIR1185-1-3p.
Figure 9.
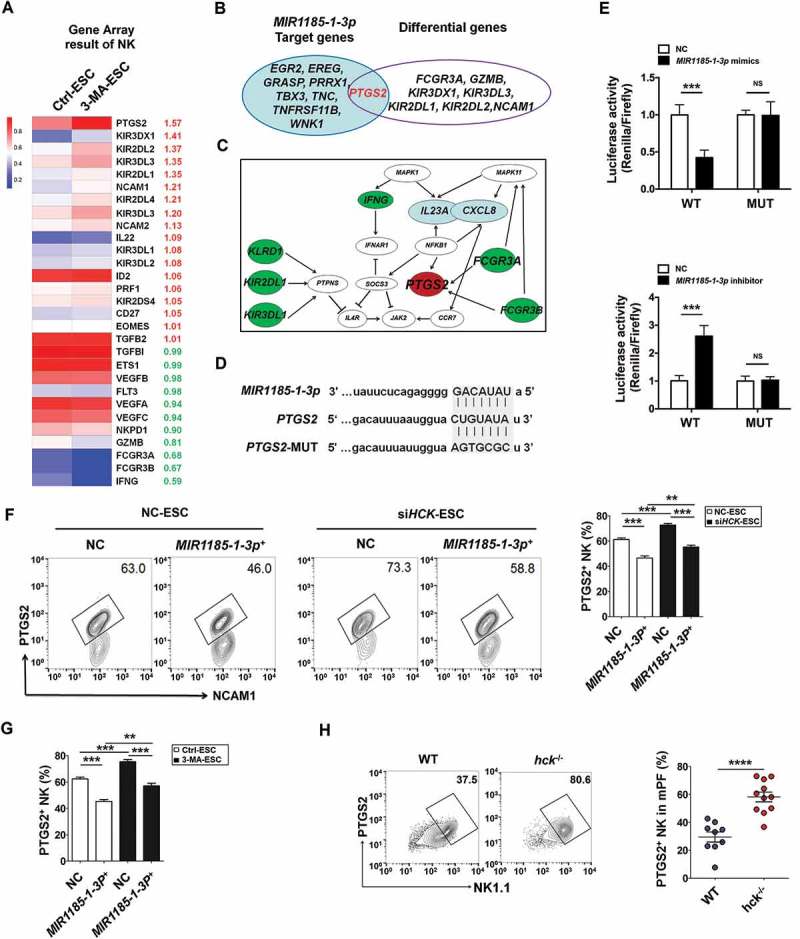
The autophagy and HCK signaling of ESCs are involved in regulating the PTGS2 level in NK cells by MIR1185-1-3p. . (a) The NK function-related genes in NK cells cocultured with Ctrl-ESC and 3-MA-ESC by Human Gene 2.0 ST Array. (b) The intersection analysis between differential genes in NK cells (coculture with Ctrl-ESC or 3-MA-ESC) and predicted target genes (i.e. EGR2, EREG and PTGS2) of MIR1185-1-3p. (c) The bioinformatics analysis (KEGG database-derived Signal net) about the relationship between PTGS2 (COX-2), FCGR3, CXCL8 and IL23A. (d,e) The relative luciferase activity of PTGS2 in HEK-293T cells was analyzed by a dual luciferase reporter assay, after cotransfection with plasmids (NC, MIR1185-1-3p mimics, MIR1185-1-3p inhibitor) and luciferase reporter plasmids (PTGS2-Luc [WT], PTGS2 mutation-Luc [MUT]) (Student t test). (f) FCM analysis of PTGS2 level in NC NK cells (n = 6) and MIR1185-1-3p + NK cells (n = 6) after coculture with Ctrl-ESC or siHCK-ESC (one-way ANOVA). (g) FCM analysis of PTGS2 level in NC NK cells (n = 6) and MIR1185-1-3p +NK cells (n = 6) after coculture with NC ESC or 3-MA-ESC (one-way ANOVA). (h) FCM analysis of PTGS2 level in NK cells of PF from WT (n = 9 mice/group) and hck−/- (n = 10 mice/group) EMS mice by FCM (Student t test). MIR1185-1-3p mimics, HsMIR1185-1-3p mimics; MIR1185-1-3p inhibitor, HsMIR1185-1-3p inhibitor. Data are expressed as the mean± SEM. **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Prior to the confirmation test about the role of PTGS2 in FCGR3− NK cell production, we analyzed the phenotype between PTGS2+ NK cells and PTGS2− NK cells in PF of WT EMS mice. As shown, PTGS2+ NK cells poorly expressed FCGR3, PRF1 and IFNG compared with PTGS2− NK cells (Figure 10(a)). Additionally, there were more FCGR3+ PRF1+ GZMB+ IFNG+ NK cells in PF and the uterus of ptgs2−/- mice (Figure 10(b-d) and Fig. S7A).
Figure 10.
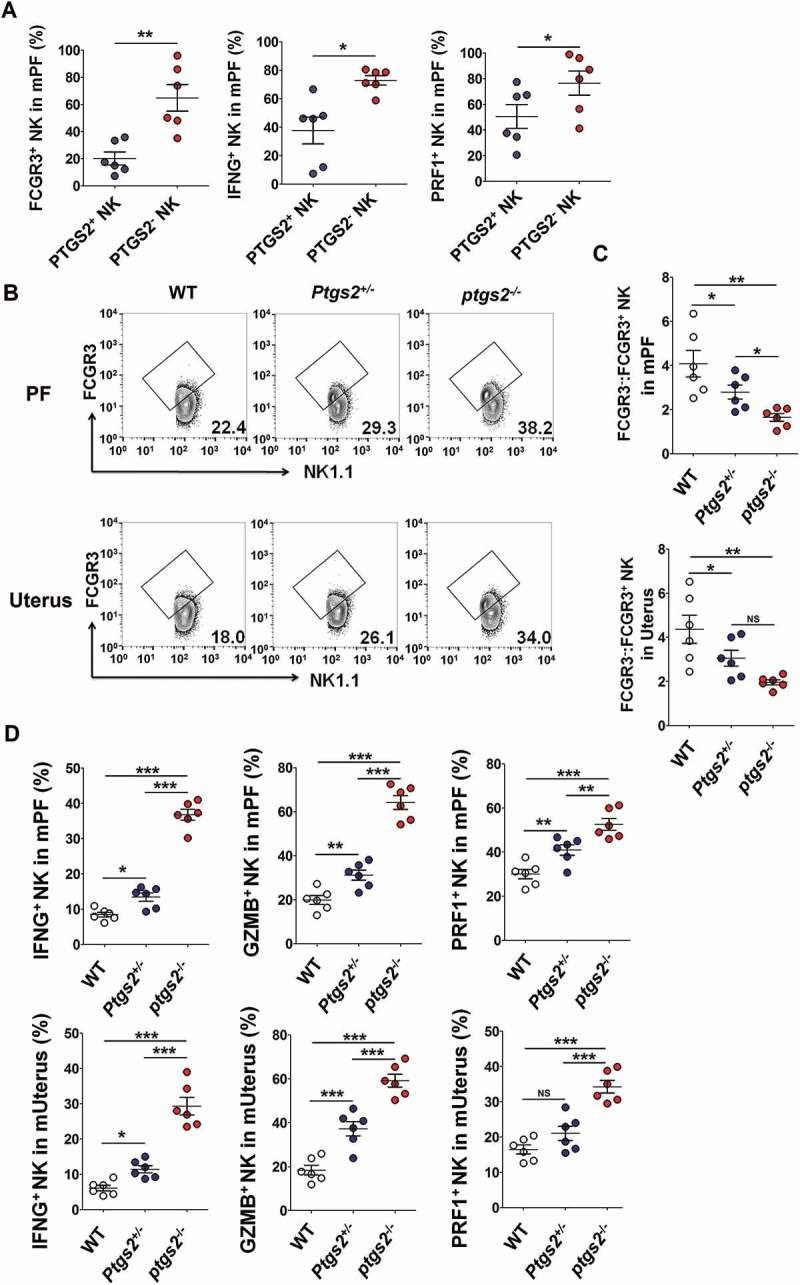
There are more FCGR3+ PRF1+ GZMB+ IFNG+ NK cells in PF and the uterus of ptgs2−/- mice. (a) FCM analysis of FCGR3, IFNG and PRF1 in PTGS2+ NK and PTGS2− NK cells of PF from C57BL/6 mice (n = 6 mice/group) (Student t test). (b,c) The ratio of FCGR3−:FCGR3+ NK cells were analyzed in PF and uterus from WT, ptgs± and ptgs−/- mice (n = 6 mice/group) by FCM (one-way ANOVA). (d) FCM analysis of PRF1, GZMB and IFNG in NK cells of PF and uterus from WT, ptgs2+/- and ptgs2−/- mice (n = 6 mice/group) by FCM (one-way ANOVA). Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001.
PTGS2+ FCGR3− NK cells present low levels of PRF1, GZMB and IFNG
Interestingly, blocking FCGR3 with a neutralizing antibody (anti-FCGR3) led to an increase of PTGS2 in NK cells cocultured with ESCs (Figure 11(a)), and the results of fcgr3−/- mice (Figure 11(b,c) and Fig.S7B) echoed this phenomenon. In contrast to ptgs2−/- mice, the PF and uterus of fcgr3−/- mice presented fewer PRF1+GZMB+ IFNG+ NK cells (Figure 11(d and Fig. S7B to e). Intraperitoneal injection with anti-FCGR3 and celecoxib (a PTGS2-specific inhibitor) led to completely opposite effects on PTGS2, GZMB and IFNG in NK cells (Figure 11(e,f). In addition, the ratio of PTGS2+ FCGR3− to PTGS2− FCGR3+ NK cells in PF from EMS was significantly increased (Figure 11(g,h)), and PTGS2+ FCGR3− NK cells had lower level of PRF1, GZMB and IFNG (Fig. S8A,B). Therefore, it can be concluded that PTGS2 triggers FCGR3−PRFlow GZMBlow IFNGlow NK differentiation, and there is a negative feedback regulation loop between PTGS2 and FCGR3 in NK cells, which may be important for homeostasis regulation of NK cell differentiation and function.
Figure 11.
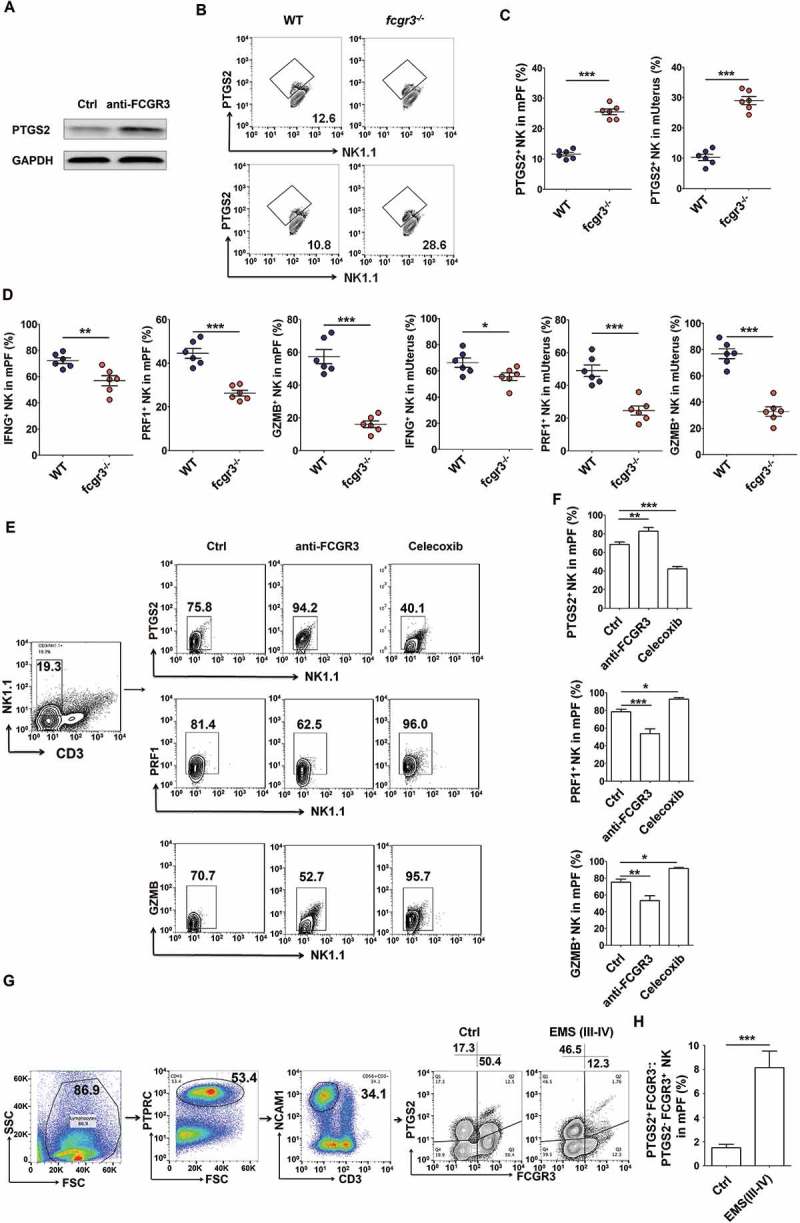
PTGS2+ FCGR3− NK cells present low levels of PRF1, GZMB and IFNG. (a) The expression of PTGS2 (n = 5) in NK cells cocultured with ESCs and treated with or without anti-FCGR3 (10 µg/ml) for 24 h was analyzed by western blotting. (b-d) FCM analysis of PTGS2, PRF1, GZMB and IFNG in NK cells of PF and uterus from WT and fcgr3−/- mice (n = 6 mice/group) by FCM (Student t test). (e,f) The C57BL/6 EMS mice were treated with vehicle, anti-FCGR3 (0.25 mg/kg/d) or celecoxib (5 mg/kg/d) (n = 6 mice/group) on day 3 and day 10 after surgery. Then the levels of PTGS2, GZMB and IFNG in NK cells of PF were detected by FCM (one-way ANOVA). (g) The ratio of PTGS2+ FCGR3− to PTGS2− FCGR3+ NK cells was analyzed in PF from healthy controls (n = 10) and EMS patients (stage III and IV, n = 10) by FCM (Student t test). Anti-FCGR3, anti-FCGR3 neutralizing antibody. Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001.
The ptgs2high FCGR3− NK cells accelerate the growth of ectopic lesions and progression of EMS
Our previous work shows that IL10 [24,25] and TGFB [258] can promote the growth and invasion of ESCs in EMS. Owing to the higher level of IL10 and TGFB in PTGS2+ FCGR3− NK cells (Figure 12(a,b), we next investigate the feedback effect of PTGS2high FCGR3− NK cells (as the downstream product of autophagy and HCK signaling) on ESCs biological behaviors, we collected NK cells from fcgr3−/- or ptgs2−/- mice and found that fcgr3−/- NK cells stimulated and restricted the viability and apoptosis of mouse uterus stromal cells (USCs) in vitro. However, the effects of ptgs2−/- NK cells were diametrically opposed (Figure 12(c,d). In vivo trials further verified these results because blocking FCGR3 and PTGS2 led to high and low levels of BIRC5/survivin (baculoviral IAP repeat containing 5) and PCNA (proliferating cell nuclear antigen) and weight of ectopic lesions, respectively (Figure 12(e-g). Compared with NK cells from WT mice, NK cells from ptgs2−/- mice decreased the weight of ectopic lesions in NK-depleted EMS mice (Figure 12(h,i), suggesting that PTGS2+ NK cells promote the growth of ectopic lesions in vivo. Taken together, when driven by an impaired autophagy and HCK signaling, PTGS2high FCGR3− NK cells, in turn, promote the growth of ectopic lesions and development of EMS.
Figure 12.
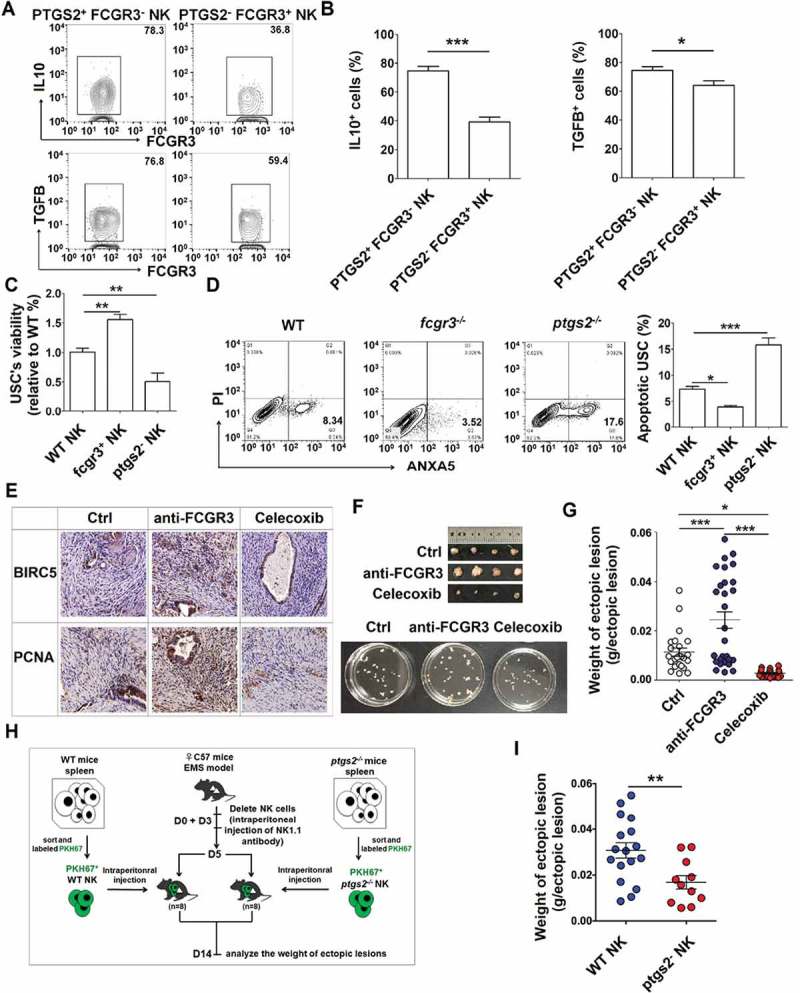
PTGS2highFCGR3− NK cells accelerate the growth of ectopic lesion and progression of EMS. (a,b) The expression of IL10 and TGFB in PTGS2+ FCGR3− NK cells and PTGS2− FCGR3+ NK cells in PF from patients with EMS (stage III and IV) (n = 10) was analyzed by FCM (Student t test). (c,d) The NK cells from WT, fcgr3−/- or ptgs2−/- mice were cocultured with mouse uterus stromal cells (USCs) for 48 h, and then the viability and apoptosis of USCs in vitro were analyzed by CCK8 assay (c) and apoptosis assay (d) (one-way ANOVA). (e) Immunohistochemistry analysis for the expression of BIRC5/survivin and PCNA in EMS-like lesions, in the EMS mouse model treated with vehicle, anti-FCGR3 (0.25 mg/kg/d) or celecoxib (5 mg/kg/d) (n = 6 mice/group). Original magnification: × 200. (f,g) The weight of EMS-like lesions in the EMS mouse model treated with vehicle, anti-FCGR3 or celecoxib (n = 6 mice/group) (one-way ANOVA). (h,i) The weight of EMS-like lesions in the NK-depleted EMS mouse model (anti-NK1.1 antibodies on days 0 and 3) treated with NK cells (day 5) from WT or ptgs2−/- mice (n = 8 mice/group) (Student t test). Data are expressed as the mean± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Discussion
Autophagy affects various physiological and pathological processes, including cell death, development, immunity, and tumorigenesis. As a double-edged sword, autophagy has both tumor-suppressive and tumor-promoting functions in tumorigenesis and metastasis [32]. Autophagy has also been recognized as an effector and regulator of innate and adaptive immunity, and there is a broad variety of studies that have focused on its role in the response against intracellular pathogens [33]. However, the role and mechanism of non-immune cell autophagy in regulation of immune cell differentiation and function are almost unknown. We found in this study that there was a low autophagy level of ESCs in ectopic lesions from patients with EMS, and this abnormality is associated with a local high concentration of estrogen. Interestingly, the decrease in autophagy promoted FCGR3− NK cells differentiation and impaired NK cell cytotoxicity (such as low levels of LAMP1, GZMB, PRF1, and IFNG) induced by ESCs, which was mediated by HCK.
HCK is a member of the highly conserved Src-family of cytoplasmic protein tyrosine kinases (SRC, FYN, YES, LCK, HCK, FGR, LYN, and BLK) that transduce a variety of extracellular signals, which ultimately affects cellular processes such as proliferation, migration and differentiation [34]. HCK is specifically expressed in myeloid cells, such as neutrophils, eosinophils, monocytes and macrophages and dendritic cells (DCs), which are considered as key regulators for controlling gene expression in alternatively activated monocytes and macrophages and FCGR-mediated phagocytosis of macrophages [35]. Prior investigations have reported that particular subsets of stromal cells have the capacity to function as non-professional APCs (antigen presenting cells) with constitutive or stimulation-induced expression of MHC class II molecules. For example, decidual stromal cells (DSCs) in the uterus differentiated from ESCs driven by pregnancy-related hormones, have been shown to express MHC class II molecules and display an ability to regulate allogenic T cells in vitro [36]. In our present study, we found that HCK is highly expressed in normESC. However, there is abnormally low HCK expression in ectoESC. It is noteworthy that HCK is positively controlled by ESC autophagy and STAT3. Further experiments should clarify the molecular mechanism of the estrogen and autophagy-STAT3 axis on HCK expression in ESCs.
HCK, which is rapidly activated by LPS signaling in neutrophils and macrophages usually mediates host defense and inflammatory responses (e.g., stimulates nitric oxide and TNF production, chemotaxis, and degranulation) [34]. Exposure with 3-MA stimulates CXCL8 and IL23A production from ESCs. The bioinformatics analysis and subsequent experiments in the current study confirm that HCK inhibits the secretion of CXCL8 and IL23A by ESCs, not IL1A. These effects may be associated with downstream PIK3R5 (phosphoinositide-3-kinase regulatory subunit 5)-KRAS-MAPKs (mitogen-activated protein kinases) [37], as well as JUN and FOS signals, which need further research.
Both siHCK and 3-MA-ESCs induce high levels of FCGR3−PTGS2high NK cells with impaired cytotoxic activity (low levels of GZMB, PRF1, IFNG, NCR3, and NCR2, high levels of KIR2DL1A and KIR3DL1) in a coculture system, and in vivo trials in the hck−/- EMS mouse model show similar results. Interestingly, both blocking CXCL8 and IL23A in ESCs or overexpressing MIR1185-1-3p in NK cells can partially reverse these effects, which suggests that autophagy, as well as HCK-CXCL8 and IL23A signals in ESCs regulate the differentiation of FCGR3−PTGS2high NK cells by MIR1185-1-3p. Blocking IL23A decreased the differentiation FCGR3−PTGS2high NK cells of PF and weight of ectopic lesions in the mouse EMS model. Limited evidence has shown that Mir155 and Mir15/16 appear to be essential for normal NK cell maturation [38], and MIR182 augments NK-cell cytotoxicity against liver cancer by modulating KLRK1 and KLRC1/NKG2A expression [39]. In fact, the posttranscriptional regulation of NK cells development by MIRs is largely unknown. Although this study was the first to discover the potential role of MIR1185-1-3p in regulating NK cell cytotoxic function, we could not further validate it in vivo because of the absence of mouse-derived Mir1185-1-3p. Fortunately, the role of its target gene (PTGS2) in FCGR3− IFNGlow PRF1low GZMBlow NK cells differentiation in ELM was confirmed.
It has been reported that many transcription factors (e.g., ID2 [40,41], TBET [42], EOMES [40,42] and NFIL3 [40]) are involved in the regulation of early and late stages of NK cell development. The results of the gene array indicate the low autophagy of ESCs induced by 3-MA does not change the levels of ID2 and EOMES expression, but upregulates levels of PTGS2/COX-2 and KIRs, and downregulates the levels of GZMB, FCGR3A, FCGR3B and IFNG in cocultured NK cells. Therefore, instead of maturation and development of NK cells, ESC autophagy just regulates the differentiation of impaired cytotoxic FCGR3−PTGS2high NK cells. Extensive research has shown high PTGS2 in ectopic ESCs from patients with EMS, and thus overproduction of prostaglandin E2 (PGE2) is involved in the development of EMS [43,44]. In this study, we have found that NK cells in PF and the uterus from ptgs2−/- mice show an IFNGhigh PRF1high GZMBhigh FCGR3high phenotype. In contrast, NK cells of fcgr3−/- mice tend to exhibit an IFNGlow PRF1low GZMBlow FCGR3low phenotype. Further analysis reveals that there is a negative feedback regulation loop between PTGS2 and FCGR3 in NK cells, which may be important for homeostasis regulation of NK cell function. It has been reported that PGE2 participates in the suppression of NK cell functions in breast cancer, which is generated largely through the elevated activity of PTGS2 that is common to many epithelial tumors [45,46]. Therefore, further research is needed to determine whether the regulation of PTGS2 on FCGR3 in NK cells depends on the production of PGE2, and the mechanism of bidirectional negative regulation between FCGR3 and PTGS2.
As impaired cytotoxic NK cells, ptgs2−/- NK cells restrict the apoptosis and enhance the viability of mouse USCs in vitro, while fcgr3−/- NK cells promote the opposite effects. Similarly, PTGS2+ NK cells promote the growth of ectopic lesions in vivo. Blocking FCGR3 or PTGS2 in vivo regulates the growth-related molecules BIRC5 and PCNA and leads to the development of EMS-like lesions. These effects may be associated with IL10 and TGF-β. Therefore, FCGR3−PTGS2high IFNGlow PRF1low GZMBlow NK cells induced by ESCs with low levels of autophagy and HCK stimulate the growth and accelerate the progression of EMS.
Collectively, as shown in Fig. S9, the aberrant low level of HCK in ectopic ESCs triggered by the decrease of ESC autophagy leads to the increase of CXCL8 and IL23A secretion, and further results in the differentiation of FCGR3−PTGS2high IFNGlow PRF1low GZMBlow NK cells by downregulating MIR1185-1-3p. On the one hand, these less cytotoxic NK cells mediate the immune escape of ectopic ESCs. On the other hand, these cells promote ectopic growth. With the progression of disease, the growth of ectopic ESCs may induce more FCGR3−PTGS2high IFNGlow PRF1low GZMBlow NK cells in ELM through autophagy and HCK signals. These processes will form a vicious positive feedback circle and finally accelerate the development of EMS.
There are several treatment options for EMS, including hormonal and surgical treatment. However, these approaches do not provide long-term relief. The high recurrence rate makes EMS intractable in clinical situations. It has been reported that a PTGS2 inhibitor can effectively inhibit the growth of ectopic lesion in mouse and rat EMS models [47]. Clinically, this treatment is widely used to relieve abdominal pain in patients with EMS. Based on this study, future treatment of EMS will likely combine both a PTGS2 inhibitor and hormonal treatment. In addition, COX inhibitors relieve MHC-mediated inhibition of NK cytotoxicity, which leads to recognition and lysis of metastatic tumor cell [48]. Therefore, these studies indicate that PTGS2 inhibitors may have a potential therapeutic value for treating diseases related to NK cell cytotoxic activity.
Materials and methods
Patients and sample collection
The protocol for this study was approved by the Human Research Ethics Committee of Obstetrics and Gynecology Hospital, Fudan University, and written informed consent was obtained from all participants. All of the eutopic endometrial and endometriotic tissues were obtained by laparoscopy from 75 patients with EMS (mean age 38.5 years; range 33 to 45 years) at the Obstetrics and Gynecology Hospital of Fudan University. The patients were classified according to the revised American Fertility Society (AFS) classification: 10 patients were in early stages (stage I and II), and 65 patients were in advanced stages (stage III and IV) of the disease. Normal endometrium was obtained through hysterectomy from patients with leiomyoma (168 cases) as healthy control samples. None of the included patient experienced complications related to pelvic inflammatory disease and no patients took any medications or received hormonal therapy within 6 months prior to surgery. All of the samples were obtained in the secretory phase of the cycle, which was confirmed histologically according to established criteria.
Collection and preparation of PF
PF was aspirated from the cul de sac at the beginning of the standard laparoscopic procedure under general anesthesia. Samples of PF contaminated by blood were excluded from the study. The mononuclear cells were isolated from the PF using lymphocyte separation medium and density-gradient centrifugation at 300 × g for 20 min at 4°C. The cells were collected, washed in ice-cold phosphate-buffered saline (PBS; GENOM Co., Ltd, Hangzhou, China, GNM-20,012) and suspended in antibody staining buffer (BioLegend, 926,002) at the desired concentration for flow cytometry.
Mice
Fcgr3 (003171) and Ptgs2 (002476) heterozygous mice were obtained from The Jackson Laboratories, Hck heterozygous mice were bred by Shanghai Model Organisms Center, Inc. (Shanghai, China) and all mice were maintained in the Laboratory Animal Facility of Fudan University (Shanghai, China). Fcgr3 knockout (fcgr3−/-), ptgs2−/- and hck−/- mice and wild-type littermates (WT) were obtained by mating of male and female Fcgr3, Ptgs2, or Hck heterozygous mice, respectively. A group of adult female C57BL/6 mice were purchased from the Laboratory Animal Facility of Fudan University and used for this study. The mice were usually maintained for 2 wk in the animal facility before the experiments. The Animal Care and Use Committee of the Obstetrics and Gynecology Hospital, Fudan University approved all of the animal protocols.
Antibodies for flow cytometry (FCM)
To identify and evaluate the NK cells in mononuclear cells from peritoneal fluid, the cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-human PTPRC/CD45 antibody (304,006); phycoerythrin-cyanine 7 (PE-Cy7)-conjugated anti-human CD3 antibody (300,420); Brilliant Violet 421TM (BV421) anti-human NCAM1/CD56 antibody (318,328) and allophycocyanin (APC)-conjugated anti-human FCGR3 antibody (302,012) (all from BioLegend). Human NK cells cocultured with ESCs were stained with PE-Cy7 or BV510-conjugated anti-human NCAM1/CD56 (318,318, 318,340), PE-Cy7 or APC-conjugated anti-human FCGR3 (302,012, 302,016), PE-Cy7 anti-human LAMP1/CD107a (328,618), APC anti-human MKI67/Ki-67 (350,514), BV510-conjugated anti-human IFNG (502,544), BV421-conjugated anti-human PRF1 (308,122), FITC-conjugated anti-human/mouse GZMB (515,403), APC-conjugated anti-human IL10 (501,410), APC-conjugated anti-human TGFB1 (349,608) (all from Biolegend) and PE-conjugated anti-human/mouse PTGS2/COX2 (Cell Signaling Technology, 13,314). Dead NK cells were analyzed by using Zombie Aqua Fixable Viability Kit (BioLegend, 423,102). Human ESCs were stained with APC-conjugated anti-human PVR/CD155 (337,618), APC-conjugated anti-human MICA/MICB (320,907), APC-conjugated anti-human HLA-E (342,605), PE APC anti-human STAT3 (371,804), and Alexa Fluor® 647-conjugated anti-human phospho-STAT3 antibodies (651,008) (all from BioLegend).
Mouse NK cells were stained with FITC-conjugated anti-mouse PTPRC/CD45 (103,108), percp/cy5.5-conjugated anti-mouse CD3 (100,218), PE-Cy7-conjugated anti-mouse NK1.1 (108,714), APC-Cy7-conjugated anti-mouse FCGR3/CD32 (101,327), BV421-conjugated anti-mouse IFNG (505,829), Alexa Fluor® 647-conjugated anti-human/mouse GZMB (515,405) (all from BioLegend), and APC-conjugated anti-mouse PRF1 (eBioscience, 17–9392). The mouse uterus stromal cells (USCs) were stained with a FITC-conjugated anti-mouse VIM/vimentin antibody (MyBioSource, MBS570210), an eFluor® 660-conjugated anti-mouse IL23A antibody (eBioscience, 50–7023) and am APC-conjugated anti-mouse MKI67/Ki-67 antibody (BioLegend, 652,406).
Western blotting
Cells were washed in PBS, detached with a cell scraper and centrifuged for 20 min at 10,000 × g at 4°C. The pellet was resuspended in high efficiency cell tissue rapid lysis buffer (RIPA; Beyotime, P0013J) containing 1% phenylmethanesulfonylfluoride (PMSF; Beyotime, P1008) proteinase and 1% phosphatase inhibitors (Roche Diagnostics, 04 693 132 001). Cell lysates were boiled for 10 min at 95°C and then were stored at −80°C. Protein concentrations were quantified using the BCA protein assay kit (Beyotime, P0012). Total proteins (20 µg) were electrophoresed in SDS-PAGE gels (Epizyme Biotech, LK102) using a Miniprotein III system (Bio-Rad, USA) and were transferred to PVDF membranes (Millipore, USA) for 2 h, followed by overnight incubation with primary antibody against LC3B (1:500; Cell Signaling Technology, 3868), BECN1 (1:500; Cell Signaling Technology, 3495), HCK (1:500; Cell Signaling Technology, 14,643), ATG5 (1:1000; Cell Signaling Technology, 12,994), SQSTM1 (1:1000; Cell Signaling Technology, 5114) and ACTB (1:1000; Cell Signaling Technology, 4970) at 4°C. Then PVDF membranes were washed three times with PBST solution (Beyotime, P0222) and were incubated at room temperature for 1 h in peroxidase-conjugated goat anti-rabbit IgG secondary antibodies (1:5000; Bioworld Technology, Co. Ltd., Louis Park, MN). Thereafter the membrane was washed three times and processed for chemiluminescence using the Immobilon Western Chemiluminescent HRP Substrate Kit (Millipore).
Isolation and culture of human ESCs and mouse USCs
We isolated human ESCs from the endometrium of healthy control, eutopic endometrium and or ectopic lesion from women with EMS according to a previously described method [24]. This method supplied more than 98% VIM+ KRT7/CK7− ESCs, which were confirmed by FCM analysis. The separation of USCs was similar to ESCs.
Purification of NK cells
PBMCs were isolated from healthy fertile women (n = 101). PF was collected from healthy control and EMS patients (stage III and IV). Human NK cells were isolated from PBMCs or PF using magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany, 130–096-892) for in vitro experiments. The FCGR3+ NK and FCGR3− NK cells were sorted from PBMC by fluorescence-activated cell sorting (FACS), and then these cells were labeled with PKH26 (red fluorescence dye; Sigma, PKH26GL) and PKH67 (green fluorescence dye; Sigma, PKH67GL), respectively. In addition, mouse NK cells were isolated from PF of fcgr3−/- mice, ptgs2−/- mice or WT mice through fluorescence-activated cell sorting (FACS) for in vitro experiments.
HCK-overexpressing ESCs, HCK-silenced ESCs, ATG5-silenced ESCs, IL23A-silenced ESCs and HsMIR1185-1-3p-overexpressing NK cells
We obtained the HCK-overexpressing ESCs (HCK (OE)), IL23A (OE) and control ESCs (mock) through transfection with the GV230-HCK plasmid, GV230-IL23A plasmid and GV230-vector plasmid (all from GeneChem Co., Ltd); the HCK-silenced ESCs (siHCK), siIL23A and control ESCs (NC) through transfection with HCK-siRNA, IL23A-siRNA and negative control siRNA (all from GeneChem Co., Ltd) and were confirmed by western blotting analysis. In addition, the ATG5 lentivirus and its corresponding control lentivirus, the HsMIR1185-1-3p mimic lentivirus (MIR1185-1-3p-mimics) and its corresponding control MIR lentivirus (MIR-NC, negative ctrl) were constructed by GenePharma. Control ESCs (NC-ESC), ATG5-silenced ESCs (siATG5-ESC), HsMIR1185-1-3p-overexpression (MIR1185-1-3p+) and the corresponding control (NC) of NK cells was then established. The efficiency of overexpression was verified by QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific, Inc., Waltham, MA, USA, 4,485,694).
Protein microarray, microrna array and gene array
After stimulation with 3-MA (10 mM) or not, the lysate and supernatant from Ctrl-ESC (secretory phase, n = 10) and 3-MA-ESC (secretory phase, n = 10) were collected and analyzed using the proteomic microarray RayBio® L-Series Human Antibody Array 1000 (RayBiotech Inc.). For protein microarray: After culture of cells, the supernatants were centrifuged at 1,000 × g for 10 min. The attached cells were washed by PBS twice and lysed with Cell Lysis Buffer (Invitrogen; Thermo Fisher Scientific, Inc., FNN0021). Then the lysates were gently rocked at for 30 min and centrifuged at 10,000 × g for 10 min at 2 to 8°C. And cells were normalized between arrays by determining cell lylate concentration using a total protein assay (BCA Protein Assay Kit; Pierce, 23,252). Then the supernatants and lysates were hybridized to RayBio® L-Series Human Antibody Array 1000 glass slide, and incubated and washed as the manufacturer’s instructions. Then the slide was scanned by HiLyte Plus™ Fluor 532, and analyzed by the RayBio® Analysis Tool software (RayBiotech Inc.).
After coculture with Ctrl-ESC and 3-MA-ESC, NK cells (n = 10) were collected and characterized by Affymetrix GeneChip® Human Gene 2.0 ST Array (Affymetrix Inc.) and Affymetrix GeneChip® microRNA 4.0 Array (Affymetrix Inc.). For microRNA array: For Affymetrix microarray profiling, total RNA was isolated with TRIzol® reagent (Invitrogen, 10,290–010) and purified using RNeasy Mini Kit (Qiagen, 74,106), including a DNase digestion treatment. Ensure that the purification method retains low molecular weight (LMW) RNA. RNA concentrations were determined by the absorbance at 260 nm and quality control standards were A260:A280 = 1.8–2.1, using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). RNA was labeled using FlashTag® Biotin HSR labeling kit as per the manufacturer’s instructions (Genisphere, HSR30FTA). Labeled RNA was hybridized to Affymetrix GeneChip® microRNA 4.0 array (Affymetrix Inc.) according to the User Manuals. Affymetrix® Expression Console Software (version 1.3.1) was used for microarray analysis, including data normalization, summarization, and quality control assessment. Median-centric normalization was used for the custom microRNA oligonucleotide chips. Affymetrix chips were normalized using the robust multichip analysis (RMA) procedure. For gene array: The Affymetrix GeneChip® Human Gene 2.0 ST Array (Affymetrix Inc.) is composed of lncRNAs and protein-coding mRNAs from the human genome. We collected 11,086 lincRNAs, 9,066 ncRNAs and 40,716 mRNAs from authoritative data sources including NCBI RefSeq (release 51), Ensembl (release 65), lncRNA db, Broad Institute, Human Body Map lincRNAs and TUCP (transcripts of uncertain coding potential) catalog. Each Human Gene 2.0 ST Array is composed of 1.35 million distinct probes (25-mers) and each transcript is represented by 21 probes (median) to enhance statistical confidence. Each transcript is represented by a specific exon or intron probe to identify individual transcripts accurately. The microarray hybridization and bioinformatics analysis were performed by Genminix, Shanghai, China.
Cytotoxicity trials of NK cells to ESC (LDH release-based)
The cytotoxicity of NK in response to ESC (normal ESC, ectopic ESC, NC-ESC, siATG5-ESC) was analyzed using a LDH (lactate dehydrogenase) release assay according to our previous procedure [49]. ESC (2,500 to 5,000 cells/well) were plated, and the next day NK cells were added at various ratios (the ratio of ESC to NK was 100:1, 10:1, 1:1, 1:3, or 1:10) (all samples in triplicate). After 4 h of coculture, an aliquot of 50 μl media was used in the LDH cytotoxic assay using the CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega, G1780). The value of corrected experimental LDH release was calculated by subtracting the value of spontaneous LDH release from effector cells at corresponding dilutions. NK cytotoxicity was defined as %Cytotoxicity = (Experimental value − Effector Cells Spontaneous Control − Target Cells Spontaneous Control)/(Target Cell Maximum Control − Target Cells Spontaneous Control) × 100.
Enzyme-linked immunosorbent assay (ELISA)
The supernatants of ctrl ESCs, ESCs from patients with EMS (stage III and IV), mock ESC, HCK (OE) ESC, NC ESC and siHCK ESC were collected, and cytokine concentrations (CXCL8, IL23A and IL1A) in the supernatants were measured using ELISA kits (R&D, D8000C, D2300B, DLA50).
The transcription of ATG5, HCK, Map1lc3b, Becn1, Hck and MIR1185-1-3p
The transcription levels of ATG5 and HCK in ESCs, Map1lc3b, Becn1 and Hck in mouse ectopic lesions and the levels of MIR1185-1-3p in NK cells were analyzed by QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific, 4,485,694) according to standard protocols. Human MIR1185-1-3p (CD201-T) and U6 (CD201-0145) primers were designed and synthesized by TIANGEN. Other primer sequences were designed and synthesized by TaKaRa Biotechnology Co., Ltd as described in Table S1.
Dual luciferase reporter assay
The NC, HsMIR1185-1-3p mimics, HsMIR1185-1-3p inhibitor, PTGS2, PTGS2 mutation luciferase reporter plasmids were constructed by RiboBio Co., Ltd. These plasmids were transfected into HEK-293T cells by Effectene Transfection Reagent (QIAGEN, 301,425) according to our previous procedure [24]. The relative luciferase activity was analyzed by the Dual-Luciferase® Reporter (DLR™) Assay (Promega, E1910).
Cell viability and apoptosis assays
ESCs were resuspended in DMEM/F-12 with 10% FBS, and seeded at a density of 1 × 104 cells/well in 96-well flat-bottom microplates (for the CCK-8 assay), or 1 × 105 cells/well in 24-well flat-bottom microplates (for ANXA5/Annexin V-FITC assay). After attaining 70 to 80% confluence, the cells were starved with DMEM containing 1% FBS for 12 h before treatment. The medium was removed once again, and the cells were treated with NK cells from WT, fcgr3−/-mice or ptgs2−/- mice, respectively. Then the Cell Counting Kit-8 (CCK-8) (Dojindo, CK04) assay and ANXA5-FITC (eBioscience, BMS500FI-100) assay were applied to evaluate the effects of NK cells on cell viability and apoptosis of ESCs, according to our previous procedure [24].
Intraperitoneal EMS model
We divided female C57BL/6 mice (age: 8 weeks old, weight: 20 to 23g) into 3 groups, including control, 3-MA and rapamycin (Rap) groups as well as control, anti-FCGR3 and celecoxib groups by using a table of random numbers organized by body weight, age and family. This study was an unblinded trial.
For WT C57BL/6 mice, intraperitoneal EMS-like lesions were induced surgically by suturing uterine tissue samples to the abdominal wall. For autologous transplantation, the left uterine area of the recipient animal was divided into 4 equal parts and sewn into 4 quadrants of the peritoneum. EMS-like lesions developed along the transplanted uterine tissue samples. The mice were treated with vehicle, 3-MA (100 mg/kg/d, Sigma, M9281), Rap (100 mg/kg/d, Sigma, V900930), anti-FCGR3 (0.25 mg/kg/d, R&D, AF1960), celecoxib (5 mg/kg/d, Sigma, Y0001445) or anti-mouse IL23A neutralizing antibody (0.1 mg/kg/d, R&D, AF1619) on days 3 and 10 after surgery. After 2 weeks, the sizes of the EMS-like lesions were measured. In addition, the EMS-like lesions and PF were collected and detected. The relative molecule expression of NK cells in PF was analyzed by flow cytometry. In addition, immunohistochemistry was used to analyze the expression of BIRC5 and PCNA in the endometriosis-like lesions.
For immunohistochemistry, paraffin sections (5 µM) of the endometriosis-like lesions were dehydrated in graded ethanol, then incubated with hydrogen peroxide and 1% bovine serum albumin (BSA)/TBS to block endogenous peroxidase. The samples were then incubated with rabbit anti-mouse BIRC5/survivin (1:200; CST, 2808), rabbit anti-mouse PCNA (1:200; CST, 13,110) or rabbit IgG isotype overnight at 4°C in a humid chamber. After washing 3 times with TBS, the sections were overlaid with peroxidase-conjugated goat anti-mouse IgG, and the reaction was developed with 3,3-diaminobenzidine (DAB) and counterstained with hematoxylin.
For the hck special KO-EMS mouse model, an endometrial fragment from hck−/- mice was injected intraperitoneally into wild-type mice (WT) for constructing the hck special KO-EMS mouse model.
For depletion of NK cells, C57BL/6 EMS mice were injected with 100 ml of 0.5% FBS in PBS containing 300 mg of anti-NK1.1 (BioLegend, clone PK136) or isotype control antibodies on days 0 and 3 according to Christopher et al. report [50]. The NK cells were sorted from spleen of WT mice or ptgs2−/- mice by fluorescence-activated cell sorting (FACS), and then these cells were labeled with PKH67. These NK cells were then intraperitoneally injected to NK depleted EMS mice on day 5.
Statistics
The continuous variable is shown as the mean ± SEM. Continuous variables were analyzed using a Student t test for 2 groups and one-way ANOVA using the Tukey post-hoc test for multiple groups. All analyses were conducted using the SPSS 19.0 Statistical Package for the Social Sciences software. The results were considered statistically significant at P < 0.05.
Funding Statement
This work was supported by the National Natural Science Foundation of China (NSFC) [91542108, 81471513, 81601354, 81490744, 81471548, 31671200, 31600735, 81370730, 81571512, 81730039]; National Basic Research Program of China [2015CB943300]; Shanghai Rising-Star Program [16QA1400800]; Development Fund of Shanghai Talents [201557]; Program of Shanghai Outstanding Academic Leader [15XD1500900]; Program for Zhuoxue of Fudan University [/]; Nationan Science of Jiangsu Province [BK20160128]; Fundamental Research Funds for the Central Universities [021414380180]; Program for Shanghai leaders [/]; Oriented Project of Science and Technology Innovation from Key Lab. of Reproduction Regulation of NPFPC [CX2017-2].
Acknowledgments
We thank Dr. Wen-Jing Zhao from Genminix Informatics Ltd., Co for help with bioinformatics analysis, and Dr. Yi-Qin Wang in the Department of Pathology, Hospital of Obstetrics and Gynecology, Shanghai Medical School, Fudan University for helping with histological analysis. We are grateful to Dr. Jie Duan from Department of Obstetrics and Gynecology, Hospital of Obstetrics and Gynecology, Shanghai Medical School, Fudan University for her helps with in vivo experiments. This study supported by the National Basic Research Program of China (No. 2015CB943300), and the National Natural Science Foundation of China (NSFC) (No. 91542108, 81471513, 81601354, 81490744, 81471548, 31671200, 31600735, 81370730, 81571512, 81730039 and 81671460), the Shanghai Rising-Star Program (No. 16QA1400800), the Development Fund of Shanghai Talents (No. 201557), the Innovation-oriented Science and Technology Grant from NPFPC Key Laboratory of Reproduction Regulation (CX2017-2), the Program for Zhuoxue of Fudan University, the National Science Foundation of Jiangsu Province (No. BK20160128), the Fundamental Research Funds for the Central Universities (No. 021414380180), the Open Project Program of Shanghai Key Laboratory of Female Reproductive Endocrine-Related Diseases (No. 14DZ2271700), the Program for Shanghai leaders, and the Program of Shanghai Outstanding Academic Leader (15XD1500900).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplymentary materials
Supplymentary materials can be accessed here
References
- [1].Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol. 2008;8:259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cooper MA, Fehniger TA, Caligiuri MA.. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. [DOI] [PubMed] [Google Scholar]
- [3].Cooper MA, Fehniger TA, Turner SC, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56 (bright) subset. Blood. 2001;97:3146–3151. [DOI] [PubMed] [Google Scholar]
- [4].Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–227. [DOI] [PubMed] [Google Scholar]
- [5].Vivier E, Tomasello E, Baratin M, et al. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. [DOI] [PubMed] [Google Scholar]
- [6].Yokoyama WM. Mistaken notions about natural killer cells. Nat Immunol. 2008;9:481–485. [DOI] [PubMed] [Google Scholar]
- [7].Ferlazzo G, Thomas D, Lin SL, et al. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J Immunol. 2004;172:1455–1462. [DOI] [PubMed] [Google Scholar]
- [8].Bulun SE. Endometriosis. N Engl J Med. 2009;360:268–279. [DOI] [PubMed] [Google Scholar]
- [9].Berkkanoglu M, Arici A. Immunology and endometriosis. Am J Reprod Immunol. 2003;50:48–59. [DOI] [PubMed] [Google Scholar]
- [10].Eisenberg VH, Zolti M, Soriano D. Is there an association between autoimmunity and endometriosis? Autoimmun Rev. 2012;11:806–814. [DOI] [PubMed] [Google Scholar]
- [11].Osuga Y, Koga K, Hirota Y, et al. Lymphocytes in endometriosis. Am J Reprod Immunol. 2011;65:1–10. [DOI] [PubMed] [Google Scholar]
- [12].Oosterlynck DJ, Cornillie FJ, Waer M, et al. Women with endometriosis show a defect in natural killer activity resulting in a decreased cytotoxicity to autologous endometrium. Fertil Steril. 1991;56:45–51. [DOI] [PubMed] [Google Scholar]
- [13].Funamizu A, Fukui A, Kamoi M, et al. Expression of natural cytotoxicity receptors on peritoneal fluid natural killer cell and cytokine production by peritoneal fluid natural killer cell in women with endometriosis. Am J Reprod Immunol. 2014;71:359–367. [DOI] [PubMed] [Google Scholar]
- [14].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- [15].Leong JW, Sullivan RP, Fehniger TA. microRNA management of NK-cell developmental and functional programs. Eur J Immunol. 2014;44:2862–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zawislak CL, Beaulieu AM, Loeb GB, et al. Stage-specific regulation of natural killer cell homeostasis and response against viral infection by microRNA-155. Proc Natl Acad Sci U S A. 2013;110:6967–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim N, Kim M, Yun S, et al. MicroRNA-150 regulates the cytotoxicity of natural killers by targeting perforin-1. J Allergy Clin Immunol. 2014;134:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Berezikov E, van Tetering G, Verheul M, et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res. 2006;16:1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Deng H, Chu X, Song Z, et al. MicroRNA-1185 induces endothelial cell apoptosis by targeting UVRAG and KRIT1. Cell Physiol Biochem. 2017;41:2171–2182. [DOI] [PubMed] [Google Scholar]
- [20].Deng H, Song Z, Xu H, et al. MicroRNA-1185 promotes arterial stiffness though modulating VCAM-1 and E-Selectin expression. Cell Physiol Biochem. 2017;41:2183–2193. [DOI] [PubMed] [Google Scholar]
- [21].Auberger P, Puissant A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 2017;129:547–552. [DOI] [PubMed] [Google Scholar]
- [22].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mei J, Zhu XY, Jin LP, et al. Estrogen promotes the survival of human secretory phase endometrial stromal cells via CXCL12/CXCR4 up-regulation-mediated autophagy inhibition. Hum Reprod. 2015;30:1677–1689. [DOI] [PubMed] [Google Scholar]
- [24].Chang KK, Liu LB, Jin LP, et al. IL-27 triggers IL-10 production in Th17 cells via a c-Maf/RORγt/Blimp-1 signal to promote the progression of endometriosis. Cell Death Dis. 2017;8:e2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li MQ, Wang Y, Chang KK, et al. CD4+Foxp3+ regulatory T cell differentiation mediated by endometrial stromal cell-derived TECK promotes the growth and invasion of endometriotic lesions. Cell Death Dis. 2014;5:e1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wei CY, Mei J, Tang LL, et al. 1-Methyl-tryptophan attenuates regulatory T cells differentiation due to the inhibition of estrogen-IDO1-MRC2 axis in endometriosis. Cell Death Dis. 2016;7:e2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kovács M, Németh T, Jakus Z, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med. 2014;211:1993–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Noman MZ, Janji B, Berchem G, et al. Hypoxia-induced autophagy: a new player in cancer immunotherapy? Autophagy. 2012;8:704–706. [DOI] [PubMed] [Google Scholar]
- [29].Noman MZ, Janji B, Kaminska B, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011;71:5976–5986. [DOI] [PubMed] [Google Scholar]
- [30].Portillo JC, Muniz-Feliciano L, Lopez Corcino Y, et al. Toxoplasma gondii induces FAK-Src-STAT3 signaling during infection of host cells that prevents parasite targeting by autophagy. PLoS Pathog. 2017;13:e1006671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schreiner SJ, Schiavone AP, Smithgall TE. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem. 2002;277:45680–45687. [DOI] [PubMed] [Google Scholar]
- [32].White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Erns M, Inglese M, Scholz GM, et al. Constitutive activation of the SRC family kinase Hck results in spontaneous pulmonary inflammation and an enhanced innate immune response. J Exp Med. 2002;196:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Suzuki T, Kono H, Hirose N, et al. Differential involvement of Src family kinases in Fc gamma receptor-mediated phagocytosis. J Immunol. 2000;165:473–482. [DOI] [PubMed] [Google Scholar]
- [36].Nagamatsu T, Schust DJ, Sugimoto J, et al. Human decidual stromal cells suppress cytokine secretion by allogenic CD4+ T cells via PD-1 ligand interactions. Hum Reprod. 2009;24:3160–3171. [DOI] [PubMed] [Google Scholar]
- [37].Mócsai A, Jakus Z, Vántus T, et al. Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol. 2000;164:4321–4331. [DOI] [PubMed] [Google Scholar]
- [38].Rp S, Jw L, Se S, et al. microRNA-15/16 antagonizes Myb to control NK cell maturation. J Immunol. 2015;195:2806–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Abdelrahman MM, Fawzy IO, Bassiouni AA, et al. Enhancing NK cell cytotoxicity by miR-182 in hepatocellular carcinoma. Hum Immunol. 2016;77:667–673. [DOI] [PubMed] [Google Scholar]
- [40].Male V, Nisoli I, Kostrzewski T, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med. 2014;211:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Delconte RB, Shi W, Sathe P, et al. The helix-loop-helix protein ID2 governs NK cell fate by tuning their sensitivity to interleukin-15. Immunity. 2016;44:103–115. [DOI] [PubMed] [Google Scholar]
- [42].Gordon SM, Chaix J, Rupp LJ, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wu MH, Lin SC, Hsiao KY, et al. Hypoxia-inhibited dual-specificity phosphatase-2 expression in endometriotic cells regulates cyclooxygenase-2 expression. J Pathol. 2011;225:390–400. [DOI] [PubMed] [Google Scholar]
- [44].Mei J, Lp J, Ding D, et al. Inhibition of IDO1 suppresses cyclooxygenase-2 and matrix metalloproteinase-9 expression and decreases proliferation, adhesion and invasion of endometrial stromal cells. Mol Hum Reprod. 2012;18:467–476. [DOI] [PubMed] [Google Scholar]
- [45].Ma X, Holt D, Kundu N, et al. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology. 2013;2:e22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mamessier E, Sylvain A, Thibult ML, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. 2011;121:3609–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kundu N, Walser TC, Ma X, et al. Cyclooxygenase inhibitors modulate NK activities that control metastatic disease. Cancer Immunol Immunother. 2005;54:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Machado DE, Berardo PT, Landgraf RG, et al. A selective cyclooxygenase-2 inhibitor suppresses the growth of endometriosis with an antiangiogenic effect in a rat model. Fertil Steril. 2010;93:2674–2679. [DOI] [PubMed] [Google Scholar]
- [49].Zhou WJ, Chang KK, Wu K, et al. Rapamycin synergizes with cisplatin in antiendometrial cancer activation by improving IL-27-stimulated cytotoxicity of NK Cells. Neoplasia. 2018;20:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Andoniou CE, van Dommelen SL, Voigt V, et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6:1011–1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.