

ABSTRACT
PTEN phosphorylation at its C-terminal (C-tail) serine/threonine cluster negatively regulates its tumor suppressor function. However, the consequence of such inhibition and its downstream effects in driving lung cancer remain unexplored. Herein, we ascertain the molecular mechanisms by which phosphorylation compromises PTEN function, contributing to lung cancer. Replacement of the serine/threonine residues with alanine generated PTEN-4A, a phosphorylation-deficient PTEN mutant, which suppressed lung cancer cell proliferation and migration. PTEN-4A preferentially localized to the nucleus where it suppressed E2F1-mediated transcription of cell cycle genes. PTEN-4A physically interacted with the transcription factor E2F1 and associated with chromatin at gene promoters with E2F1 DNA-binding sites, a likely mechanism for its transcriptional suppression function. Deletion analysis revealed that the C2 domain of PTEN was indispensable for suppression of E2F1-mediated transcription. Further, we uncovered cancer-associated C2 domain mutant proteins that had lost their ability to suppress E2F1-mediated transcription, supporting the concept that these mutations are oncogenic in patients. Consistent with these findings, we observed increased PTEN phosphorylation and reduced nuclear PTEN levels in lung cancer patient samples establishing phosphorylation as a bona fide inactivation mechanism for PTEN in lung cancer. Thus, use of small molecule inhibitors that hinder PTEN phosphorylation is a plausible approach to activate PTEN function in the treatment of lung cancer.
Abbreviations
- AKT
V-Akt Murine Thymoma Viral Oncogene
- CA
Cancer adjacent
- CDK1
Cyclin dependent kinase 1
- CENPC-C
Centromere Protein C
- ChIP
Chromatin Immunoprecipitation
- co-IP
Co-immunoprecipitation
- COSMIC
Catalog of Somatic Mutations In Cancer
- CREB
cAMP Responsive Element Binding Protein
- C-tail
Carboxy terminal tail
- E2F1
E2F Transcription Factor 1
- ECIS
Electric Cell-substrate Impedance Sensing
- EGFR
Epidermal Growth Factor Receptor
- GSI
Gamma Secretase Inhibitor
- HDAC1
Histone Deacetylase 1
- HP1
Heterochromatin protein 1
- KAP1/TRIM28
KRAB-Associated Protein 1/Tripartite Motif Containing 28
- MAF1
Repressor of RNA polymerase III transcription MAF1 homolog
- MCM2
Minichromosome Maintenance Complex Component 2
- miRNA
micro RNA
- MTF1
Metal-Regulatory Transcription Factor 1
- PARP
Poly(ADP-Ribose) Polymerase
- PD-1
Programmed Cell Death 1
- PD-L1
Programmed Cell Death 1 Ligand 1
- PI3K
Phosphatidylinositol-4,5-Bisphosphate 3-Kinase
- PLK
Polo-like Kinase
- pPTEN
Phosphorylated PTEN
- PTEN
Phosphatase and Tensin Homolog deleted on chromosome ten
- PTM
Post Translational Modification
- Rad51
RAD51 Recombinase
- Rad52
RAD52 Recombinase
- RPA1
Replication protein A
- SILAC
Stable Isotope Labeling with Amino Acids in Cell Culture
- SRF
Serum Response Factor
- TKI
Tyrosine Kinase inhbitors
- TMA
Tissue Microarray
- TOP2A
DNA Topoisomerase 2A
KEYWORDS: E2F1, lung cancer, phosphorylation, PTEN, transcription
Introduction
PTEN (phosphatase and tension homolog deleted on chromosome 10) is a dual lipid and protein phosphatase that functions as a major tumor suppressor, exerting its function through several AKT dependent and independent pathways [1]. PTEN plays an important role in lung development, while PTEN loss in the lung epithelium causes hyperplasia and expansion of the cancer stem cell population in mouse models [2,3]. Cigarette smoke, which is the most prominent risk factor for lung cancer, reduces PTEN levels in the airway epithelial cells [4]. Indeed, pre-neoplastic lesions in the lung are characterized by low PTEN and high phospho-AKT levels. Despite its role in maintaining lung epithelial cell homeostasis, PTEN is only mutated in <10% of lung tumors [5]. However, lung tumors frequently show aberrant PI3K/AKT signaling which is associated with chemo-resistance and poor prognosis [6,7]. This may be attributed to reduced PTEN levels, which are routinely observed in lung cancer, and are associated with poor prognosis [8–12]. Several genomic and non-genomic mechanisms such as promoter hyper-methylation, degradation of PTEN mRNA by miRNAs and post translational modifications regulate PTEN levels in several cancers, including lung cancer [13]. Given the role PTEN plays in determining sensitivity to tyrosine kinase inhibitors (TKIs), which are widely used for lung cancer treatment, it is important to characterize mechanisms and downstream consequences of PTEN inactivation [14].
Phosphorylation of PTEN at its C-tail region (Ser380, Thr382, Thr383 and Ser385) causes an intramolecular association of the PTEN C-terminal tail (C-tail) with the rest of the PTEN body resulting in a closed/inactive conformation of PTEN that has reduced catalytic activity and membrane binding [15–17]. Upon PTEN dephosphorylation, the intramolecular association is disrupted which enhances PTEN catalytic activity and promotes its dual localization to the cell membrane and nucleus [18]. Given the emerging role of PTEN in the nucleus, it is imperative to determine how PTEN phosphorylation and the ensuing conformational changes could alter its nuclear function.
In the nucleus, PTEN is known to influence the cell cycle, modulate the function of transcription factors and participate in the maintenance of chromosomal stability. PTEN interacts with CENP-C, MCM2, TOP2A and RPA1 amongst other proteins to preserve genomic stability [19–21]. PTEN also associates with DNA damage repair proteins such as Rad51 and Rad52 in response to DNA damage [19,22–26]. While PTEN is not known to bind DNA, it associates with several transcription factors such as MTF1, SRF, CREB, MAF1, histones and chromatin remodelers to control the expression of selected genes [27–32]. PTEN exerts an inhibitory effect on the cell cycle [33–36], however the effect of PTEN phosphorylation on different phases of the cell cycle is only recently emerging. Phosphorylation of PTEN contributes to its effect on the G2/M phase of the cell cycle. Dephosphorylated PTEN induces prometaphase arrest by interacting with and enhancing the nuclear retention of the cyclin B1/CDK1 complex [37]. Further, PTEN when phosphorylated at Ser380 by Plk1 associates with chromatin and is required for progression of cells through the mitotic phase [38]. Consequently, a PTEN phosphorylation-deficient mutant causes mitotic exit. However, the effect of PTEN phosphorylation on the G1 phase of the cell cycle remains unexplored.
In the present work, we demonstrate that a phosphorylation-deficient PTEN mutant, PTEN-4A, selectively inhibits the transcription of E2F1-regulated cell cycle genes in the G1 phase. PTEN-4A physically binds to E2F1 and associates with native promoter regions of E2F1-regulated genes in the chromatin context in the nucleus. The C2 domain of PTEN is required for its transcriptional functions such that disease associated PTEN mutations in the C2 domain abrogate its ability to suppress E2F1-mediated transcription. Finally, we observe that PTEN expression levels are reduced in lung cancer and residual PTEN observed in the tumors remains phosphorylated, establishing PTEN-phosphorylation as an inhibitory mechanism for PTEN function in lung cancers.
Results
PTEN-4A is a more Potent Tumor Suppressor than PTEN-WT
To study the effect of PTEN C-terminal phosphorylation on its tumor suppressor function, a C-tail phosphorylation-deficient mutant, PTEN-4A, which remains in the open/active form, was used (Figure 1A). PTEN-4A, was derived by replacement of the serine-threonine cluster (S380, T382, T383, S385) in the PTEN C-tail by alanine residues [39]. A lung adenocarcinoma cell line (NCI-H1299) expressing low levels of PTEN was used for all experiments. Lentiviral transduction was used to produce stable cell lines that either expressed Flag-tagged wild type form of PTEN (PTEN-WT) or the Flag-tagged phosphorylation deficient mutant of PTEN (PTEN-4A). Both forms of PTEN were stably expressed at similar levels in H1299 cells (Figure 1B). Proliferation assays were performed on H1299 parent cell line, PTEN-WT stable and PTEN-4A stable cell lines using colorimetric assays (Figure 1C) and the ECIS (Electric Cell-substrate Impedance Sensing) method (Figure 1D). PTEN-4A suppressed cell proliferation to a greater extent than PTEN-WT (Figure 1C (i) and 1D). The stable cell lines were treated with the cytokine leptin, a known activator of the PI3K/AKT pathway, which induces lung cancer cell proliferation [40]. Leptin is also elevated in mouse lung tumors deficient in PTEN [40]. Likewise, high levels of leptin are detected in patient lung tumors [41,42]. Further, leptin is also known to induce PTEN C-tail phosphorylation, thereby inactivating it [43]. Upon leptin treatment, we observed that cells expressing PTEN-4A proliferated at a lower rate as compared to PTEN-WT and parent H1299 cells, indicating that PTEN-4A can counter the proliferative effects of a cytokine like leptin (Figure 1C (ii)). Our migration assays in the stable cell lines revealed that the PTEN-4A expressing cells have the least migratory capacity compared to the PTEN-WT or parent H1299 cells (Figure 1E). Therefore, the phosphorylation deficient PTEN mutant, PTEN-4A, is a more potent tumor suppressor compared to PTEN-WT.
Figure 1.
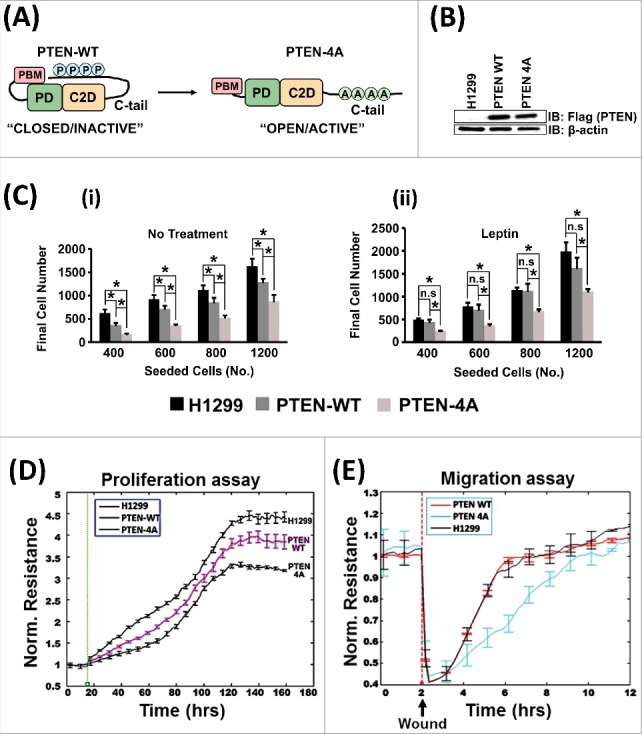
PTEN-4A is a more Potent Tumor Suppressor than PTEN-WT. (A) In the PTEN-4A mutant protein, the serine-threonine cluster (S380, T382, T383, S385) in the PTEN C-tail is replaced by alanine residues, converting it into an open/active conformer. (B) Lentiviral transduction, followed by puromycin selection, was used to stably express Flag PTEN-WT and Flag PTEN-4A proteins in PTEN deficient NSCLC cell line H1299. The expression levels of both PTEN-WT and PTEN-4A proteins were similar. (C) PTEN-4A inhibited cell proliferation significantly more than PTEN-WT in a standard cell proliferation assays (i). Even in the presence of a proliferative signal such as leptin, PTEN-4A remained a potent inhibitor of cell proliferation (ii). Data are derived from experiments performed in triplicates ± S.E. (n = 3, *p<0.05). (D) Cell proliferation assays on H1299, PTEN-WT and PTEN-4A cells measured by the ECIS method also revealed that PTEN-4A (black line) significantly suppressed cell proliferation as compared to PTEN-WT (pink line). (E) Likewise, PTEN-4A (blue line) significantly inhibited the migratory potential of H1299 lung cancer cells as compared to PTEN-WT (red line).
PTEN-4A Preferentially Localizes to the Nucleus
Previous reports indicate that PTEN-4A exhibits increased membrane localization [16,44,45]. However, given the emerging role of PTEN in the nucleus, we wanted to determine whether PTEN-4A exhibited a differential nuclear localization pattern compared to PTEN-WT. Therefore, we isolated nuclear protein from the PTEN-WT and PTEN-4A expressing stable cell lines and performed immunofluorescence experiments in 293T cells. We found that PTEN-4A preferentially localized to the nucleus (Figure 2A-C), consistent with recent findings in various cell types [18,44–47].
Figure 2.
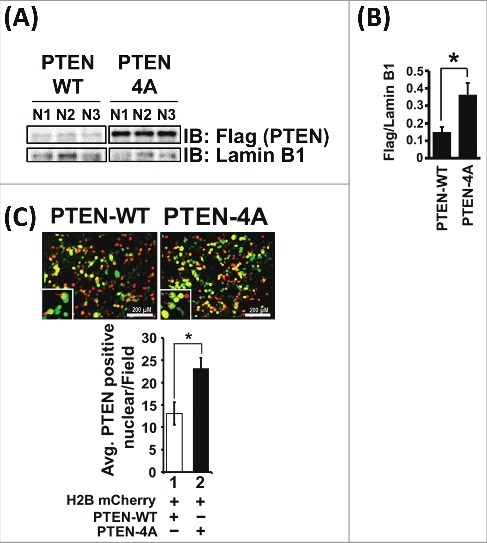
PTEN-4A Preferentially Localizes to the Nucleus. (A) Total nuclear proteins isolated from H1299 cells stably expressing Flag-tagged PTEN-WT and PTEN-4A revealed that PTEN-4A preferentially localized to the nucleus as compared to PTEN-WT, as examined by immunoblotting with Flag antibodies. (B) Lamin B1, an exclusively nuclear protein, was used as a loading control and to normalize densitometric values obtained for Flag-tagged PTEN-WT and PTEN-4A protein levels in the nucleus. Levels of PTEN-4A was approximately 2.5-fold higher in the nucleus than PTEN-WT. (C) Preferential nuclear localization for PTEN-4A detected by immunofluorescence signals of GFP-tagged PTEN-WT and GFP-tagged PTEN-4A protein expression in 293T cells. Expression of H2B mCherry stained the nucleus red. Data are derived from three independent experiments ± S.E. (n = 3, *p<0.05).
PTEN-4A Preferentially Inhibits E2F1-mediated Transcription
PTEN-4A selectively inhibited the transcriptional potential of several well established E2F1 regulated gene promoters that are involved in the cell cycle (Figure 3A-E). The effect of PTEN-WT and PTEN-4A on the transcriptional potential of the E2F1-regulated gene promoters: the artificial E2F1 reporter, native E2F1 promoter, cyclin E1 and cyclin D1 were assessed using luciferase-based promoter-reporter plasmids in H1299 cells and PTEN-null PC3 cells (Figure 3A-E and Suppl Figure 2). The artificial E2F1 reporter, E2F-Luc, was used for all further experiments to ascertain the mechanistic role of PTEN in suppressing E2F1-mediated transcription.
Figure 3.
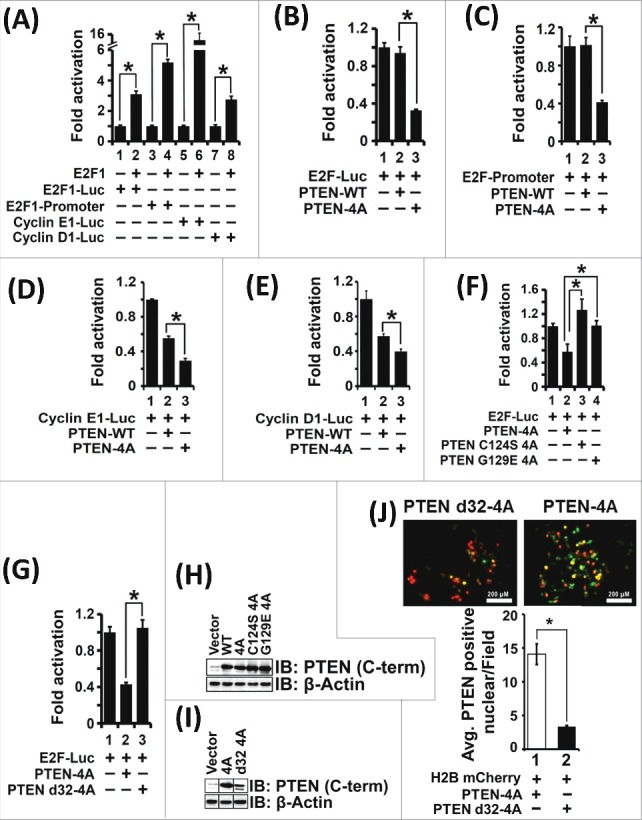
PTEN-4A Preferentially Inhibits E2F1-mediated Transcription. (A) Transcriptional assays on four E2F1-responsive promoter-luciferase reporter plasmids co-transfected with E2F1 expression plasmid in H1299 cancer cells indicate that the four promoters are all activated by E2F1 expression. (B) PTEN-4A preferentially suppresses (∼3 fold) the transcription from an artificial E2F1 reporter (E2F-Luc) compared to PTEN-WT. (C) PTEN-4A suppresses transcription mediated by the native E2F1 promoter 2.5 fold more than PTEN-WT. (D) Both PTEN-WT and PTEN-4A likewise suppress transcription mediated by the cyclin E1 promoter. However, PTEN-4A suppresses transcription by the cyclin E1-promoter approximately 2 fold more than PTEN-WT. (E) Both PTEN-WT and PTEN-4A suppress cyclin D1-transcription. However, PTEN-4A suppresses transcription by the cyclin D1-promoter 1.5 fold more than PTEN-WT. (F) As compared to PTEN-4A (lane 2), PTEN catalytic mutants, PTEN C124S 4A (lane 3) and PTEN G129E 4A (lane 4) cannot suppress E2F1-mediated transcription as detected by E2F1-Luc reporter activity in H1299 cells. (G) A PTEN-4A mutant lacking the nuclear localization sequence of PTEN (PTEN d32-4A) cannot suppress E2F1-mediated transcription (lanes 2 and 3), compared to PTEN-4A. (H and I) PTEN-WT, PTEN-4A, PTEN C124S-4A, PTEN G129E-4A and PTEN d32-4A containing expression plasmids stably express the proteins upon transient transfection. (J) Expression of GFP-tagged PTEN-4A and PTEN d32-4A followed by immunofluorescence detection indicate that PTEN d32-4A is excluded from the nucleus. The nucleus is stained red by using a H2B mCherry construct. Positive co-localization (yellow color) was counted independently by two researchers using ImageJ after merging Texas Red and GFP field images. Ten fields/transfection were recorded by each of the two researchers and the data obtained are represented from three independent experiments carried out in triplicate, represented as mean ± S.E. (*p value ≤0.05).
PTEN inhibits the PI3K/AKT pathway through its lipid phosphatase activity [48]. Therefore, we sought to determine whether the effect of PTEN on E2F1-mediated transcription is dependent on its lipid phosphatase activity using two PTEN mutants: PTEN(C124S)4A (dual lipid and protein phosphatase dead mutant) and PTEN(G129E)4A (lipid phosphatase dead mutant) [49]. We observed that the lipid-phosphatase activity of PTEN contributed to E2F1-mediated transcriptional suppression (Figure 3F and 3H). These results indicate an AKT-dependent mechanism of PTEN's effect on E2F1-mediated transcription.
Next, we wanted to determine the relationship between the nuclear localization of PTEN-4A and its effect on transcriptional suppression of E2F1-regulated genes. PTEN does not possess a canonical nuclear localization sequence. However, the first 32 amino acids of PTEN are critical for its nuclear transport [50]. A PTEN-4A mutant lacking the first 32 amino acids, PTEN d32-4A, displayed defective nuclear localization and was unable to suppress E2F1-mediated transcription (Figure 3G, 3I and 3J), indicating that nuclear localization of PTEN-4A is a prerequisite for its effect on E2F1-mediated transcription. Our results indicate that it is essential for PTEN-4A to be localized to the nucleus to suppress E2F1-mediated transcription in a phosphatase-dependent manner.
The PTEN C2 Domain is Required for E2F1-mediated Transcriptional Suppression
To determine PTEN protein domains critical for its transcriptional suppressive function, we cloned and expressed various PTEN deletion mutants and tested their transcriptional activities in luciferase-based transcriptional assays on E2F-Luc reporter (Figure 4A). We observed that the C2 domain (C2D) is required for E2F1-mediated transcriptional suppression. Deletion of the C2 domain, as seen with the deletion mutant PBM-PD (Figure 4B, lane 4), resulted in loss of transcriptional suppression. Even though the C2 domain of PTEN lacks the canonical nuclear localization sequence, it does contain several nuclear localization-like sequences [51]. Indeed, immunofluorescence experiments indicate that the C2 domain does truly localize to the nucleus (Figure 4C). Our results are consistent with recent findings that the C2 domain of PTEN contains a chromatin localization sequence and is responsible for several nuclear functions of PTEN [22,32]. Thus, results from Figure 3F and Figure 4B indicate that both phosphatase-dependent and -independent mechanisms are utilized by PTEN to regulate E2F1-mediated transcription.
Figure 4.
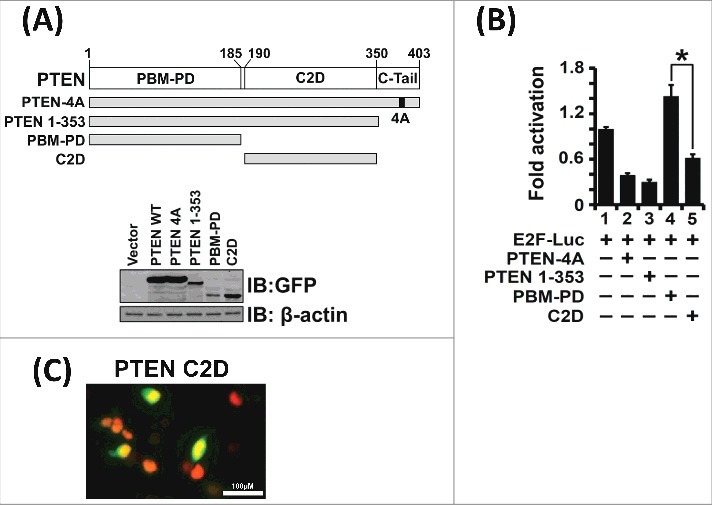
The PTEN C2 Domain is Required for E2F1-mediated Transcriptional Suppression. (A) The PTEN deletion mutants, PTEN 1–353, PBM-PD (AA 1–185) and C2 domain (C2D, AA 190–350) are stably expressed in transfection assays. (B) Expression of PTEN-4A, PTEN 1–353 and the PTEN C2 domain alone suppressed E2F1-mediated transcription as assessed from the E2F-Luc reporter activity (lanes 2, 3 and 5). However, the PTEN phosphatase domain alone (PBM-PD), which lacks the C2 domain, failed to suppress transcriptional activity (lane 4). These results indicate that the PTEN C2 domain is required for E2F1-mediated transcriptional suppression. (C) Expression of GFP-tagged C2 domain (C2D) as detected by immunofluorescence demonstrate that the PTEN C2D localize to the nucleus (yellow fluorescence). The nucleus is stained red upon expression of histone-H2B mCherry. Nuclear co-localization of GFP-tagged-C2D and histone-H2B mCherry is depicted by yellow fluorescence.
Several disease-associated mutations occur in the PTEN C2 domain as seen in the COSMIC database [52]. To investigate whether these mutations affect transcriptional functions of PTEN, we cloned and expressed four point mutants (P246L, F271S, D331G and P339L) and four truncation mutants (Q214*, R233*, K260*, R335*) (Figure 5D-G). To prevent any confounding results due to conformational changes modulated by the C-tail region, the point mutants were cloned and expressed without the PTEN C-tail region i.e, in the PTEN 1–353 background (Figure 4A). The transcriptional activities of these four point mutants were compared to the activity of PTEN 1–353 protein (Figure 5A and Figure 5C). Our transcriptional assays revealed that the two point mutants PTEN F271S and PTEN P339L cannot suppress E2F1 mediated transcription. Further, all the truncation mutants tested failed to suppress transcription of E2F1-regulated genes (Figure 5B). All these truncation mutants lack a loop and β-sheet encompassing amino acids 336–353 (Figure 5F, indicated in red), which may likely be required for the effect of PTEN on E2F1-mediated transcription. In summary, we identified disease (cancer)-associated PTEN mutants that have lost their ability to suppress E2F1-mediated transcription involved in driving the cell cycle.
Figure 5.
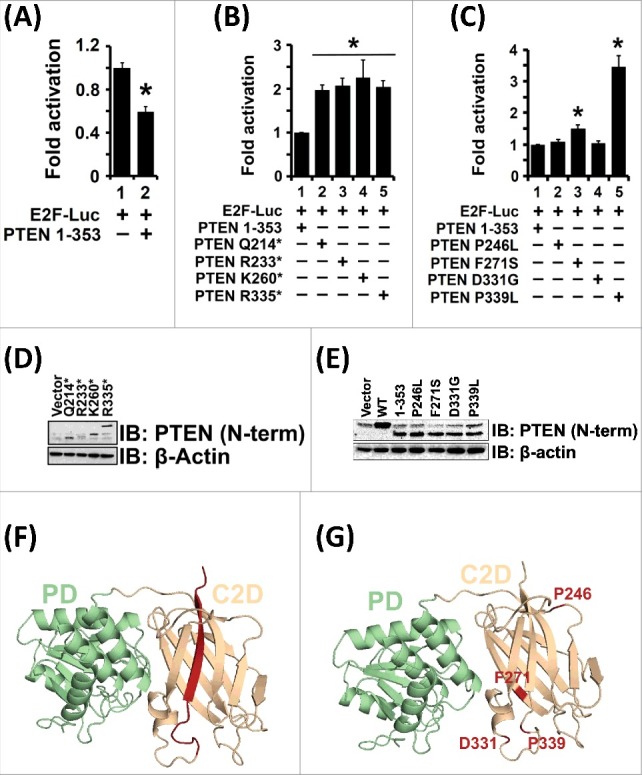
Pathogenic PTEN C2 Domain Mutants Cannot Suppress E2F1-mediated Transcription. (A) PTEN 1–353 suppressed E2F1-mediated transcription. Therefore, all subsequent C2 domain mutants were made in the PTEN 1–353 context to avoid confounding influence of variable phosphorylation and intramolecular conformational closure of PTEN via its C-tail in all mutants tested. (B) All of the analyzed PTEN truncation mutants lost their ability to suppress E2F1-mediated transcription as compared to the PTEN 1–353 protein (compare lane 1 to lanes 2, 3, 4 and 5). (C) Expression of PTEN C2 domain point mutant proteins: F271S and P339L, could not inhibit E2F1-mediated transcription when compared to PTEN 1–353 protein (compare lane 1 to lanes 3 and 5). The expression of PTEN proteins with point mutations: P246L and D331G, however, retained their ability to suppress E2F1-mediated transcription (compare lane 1 to lanes 2 and 4). (D and E) All PTEN C2 domain truncations and point mutations maintained protein stability following transient transfection. (F) All PTEN C2 domain truncation mutants lack a loop and β-sheet (indicated in red). (G) Spatial location of PTEN C2 domain point mutants (indicated in red) on the PTEN crystal structure. All transfection data are represented from three independent experiments carried out in triplicate ± S.E. (*p value ≤0.05).
PTEN-4A Physically Interacts with E2F1 Protein and at E2F1 DNA Binding Sites on Cell Cycle Gene Promoters on Chromatin
Association of PTEN with several transcription factors, including Sp1, MTF1, CREB in the nucleus is known to regulate the expression of multiple genes [27,29,53]. Therefore, we tested whether PTEN physically associates with the E2F1 protein via co-immunoprecipitation (co-IP) experiments. Flag-tagged PTEN-WT or Flag-tagged PTEN-4A and HA-tagged E2F1 plasmids were co-expressed in 293T cells. Reciprocal immunoprecipitation (IP) experiments using HA and Flag co-IP revealed that E2F1 binds to both PTEN-WT and PTEN-4A (Figure 6A). Further, in 293T cell nuclear extracts, endogenous PTEN and E2F1interaction was detected (Figure 6B). To determine whether PTEN-4A can associate with E2F1-binding sites on cell cycle gene promoter regions in the native chromatin context, we performed chromatin immunoprecipitation (ChIP) assays in lung cancer cell line H1299 stably expressing PTEN-4A. Our results indicate the presence of both E2F1 and PTEN-4A at E2F1-binding sites on the native promoters of cyclin D1 and cyclin E1 in the nucleus (Figure 6C). Taken together, PTEN-4A physically interacted with the E2F1 protein and at E2F1-binding sites in the chromatin context in the nucleus.
Figure 6.
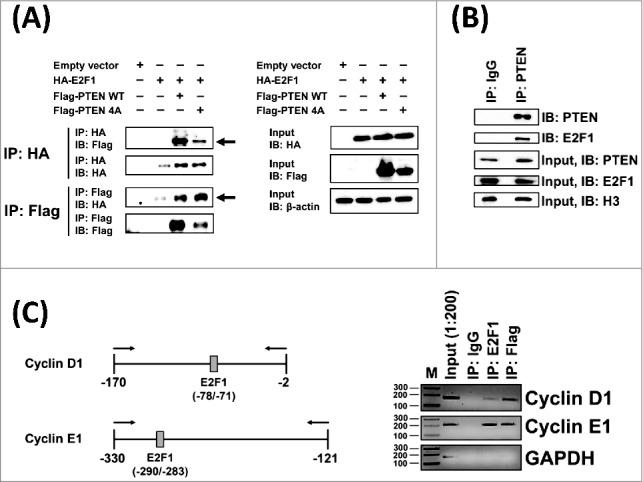
PTEN-4A Physically Interacts with E2F1 Protein and at E2F1 DNA Binding Sites on Chromatin. (A) Both Flag-PTEN-WT and Flag-PTEN-4A proteins physically associated with the HA-E2F1 containing protein complex in co-immunoprecipitation assays performed on cell extracts derived from 293T cells (indicated by arrows). (B) Endogenous PTEN and E2F1 proteins interact in 293T cell nuclear extracts. (C) Chromatin-immunoprecipitation assays indicate that both PTEN-4A and E2F1 associated with E2F1 DNA-binding sites on the chromatin at the native cyclin D1 and cyclin E1 promoters in the PTEN-4A stable cell line.
PTEN Expression Levels are Reduced in Lung Cancer
Staining of a lung cancer tissue microarray (TMA) with a PTEN antibody revealed that lung tumors had a decreased level of PTEN compared to cancer-adjacent (CA) or normal lung tissue. Intriguingly, cancer-adjacent tissues had greater PTEN expression compared to normal lung tissue (Figure 7A). However, most of the PTEN in cancer-adjacent tissues remains phosphorylated, suggesting compromised PTEN function (see Figure 8). Both non-small cell (adenocarcinoma and squamous carcinoma) and small-cell lung cancer tissues had reduced PTEN expression as compared to normal and cancer-adjacent tissues (Figure 7B). Further, the expression of PTEN was inversely correlated with tumor grade (Figure 7C).
Figure 7.
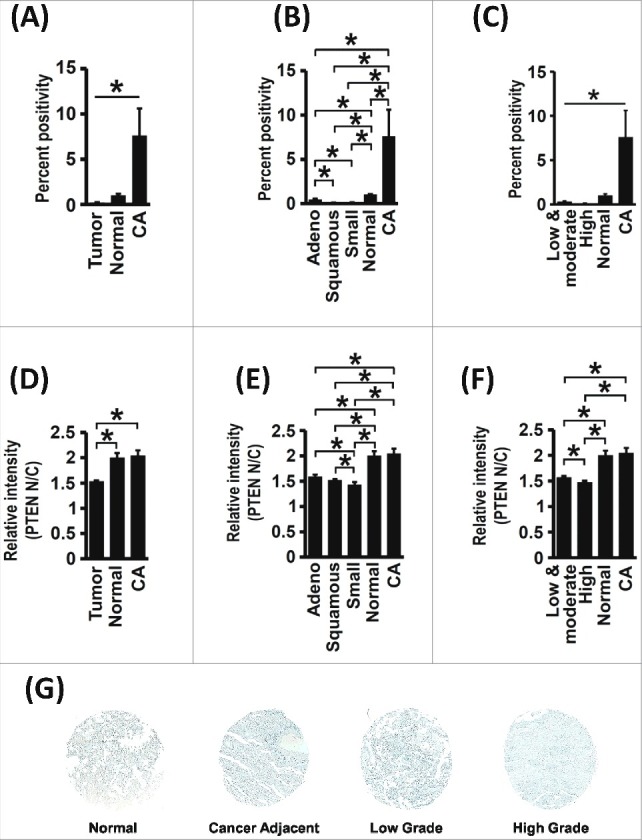
Total PTEN and Nuclear PTEN levels are Reduced in Human Lung Cancer Samples. (A) Total PTEN expression levels are reduced in lung tumor tissue when compared with normal and cancer adjacent (CA) lung tissue, as quantitatively examined by histological staining of a tissue microarray (No. of samples: 137 (tumor), 16 (normal), 13 (cancer-adjacent), *p<0.05). (B) Total PTEN expression levels are reduced in non-small cell lung tumors (adenocarcinoma and squamous cell carcinomas) and in small-cell lung tumors (No. of samples: 60 (adenocarcinoma), 57 (squamous carcinoma), 18 (small-cell lung cancer), 16 (normal), 13 (cancer-adjacent), *p<0.05). (C) Total PTEN levels decreased with increasing tumor grade (No. of samples: 87 (low and moderate grade), 31 (high grade), 16 (normal), 13 (cancer-adjacent), *p<0.05). (D) The nuclear to cytoplasmic ratio (N/C ratio) of PTEN levels is decreased in tumor tissue when compared with normal and cancer adjacent lung tissue (No. of samples: 160 (tumor), 20 (normal), 10 (cancer-adjacent), *p<0.05). (E) PTEN N/C ratios are reduced in non-small cell lung tumors (adenocarcinoma and squamous cell carcinomas) and in small-cell lung tumors (No. of samples: 66 (adenocarcinoma), 71 (squamous carcinoma), 22 (small cell lung cancer), 20 (normal), 10 (cancer-adjacent), *p<0.05). (F) PTEN N/C ratios decreased with increasing tumor grade (No. of samples: 96 (low and moderate grade), 37 (high grade), 20 (normal), 10 (cancer-adjacent), *p<0.05). (G) Representative images showing total PTEN levels in patient lung tumor samples.
Figure 8.
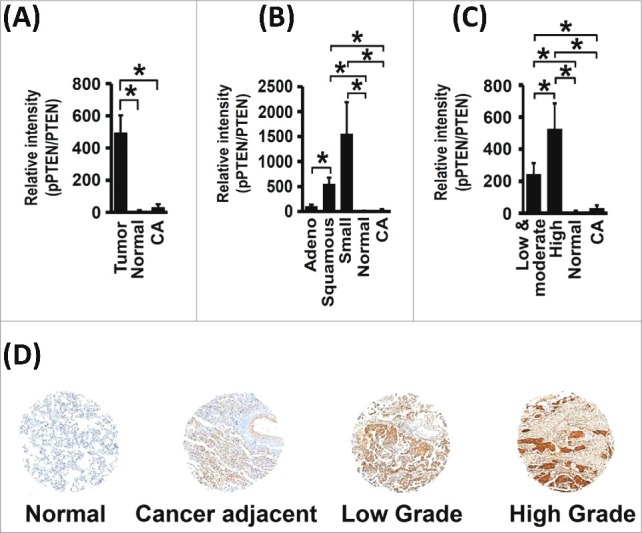
PTEN C-tail Phosphorylation is Increased in Human Lung Cancer. (A) Relative pPTEN expression levels are increased in tumor tissue when compared with normal and cancer adjacent lung tissue (No. of samples: 136 (tumor), 14 (normal), 13 (cancer-adjacent), *p<0.05). (B) Relative pPTEN expression levels are increased in non-small cell lung tumors (adenocarcinoma and squamous cell carcinomas) and in small-cell lung tumors (No. of samples: 59 (adenocarcinoma), 57 (squamous carcinoma), 18 (small-cell lung cancer), 14 (normal), 13 (cancer-adjacent), *p<0.05). (C and D) Relative pPTEN expression levels increased with increasing tumor grade (No. of samples: 87 (low and moderate grade), 31 (high grade), 14 (normal), 13 (cancer-adjacent, *p<0.05).
Nuclear PTEN Levels are Reduced in Lung Cancer
Nuclear and cytoplasmic levels of PTEN were determined using the tissue microarray (TMA) stained with the PTEN antibody. Lung tumor tissues showed a lower PTEN nuclear-to-cytoplasmic (N/C) ratio compared to cancer-adjacent or normal lung tissue (Figure 7D). The PTEN N/C ratio was reduced across all histological subtypes of lung cancer analyzed (Figure 7E) and decreased with increasing tumor grade (Figure 7F). These results are consistent with the critical role of PTEN in the nucleus, as identified by us and others [21,22,27,32,37,38,54–60]
PTEN C-tail Phosphorylation is Increased in Lung Cancer
Our cell-based assays indicated that phosphorylation of PTEN at the C-tail compromised its function as a tumor suppressor. To validate our findings in lung cancer patients, we stained the lung cancer TMA using a phospho-specific PTEN antibody, which recognizes C-tail phosphorylation. Levels of pPTEN (phosphorylated PTEN) were normalized to total PTEN to account for lower PTEN expression in tumor samples. We observed that the relative pPTEN levels were elevated in tumor tissue (Figure 8A), across all histological subtypes (Figure 8B) and increased with the grade of the tumor (Figure 8C and Figure 8D). These results indicate that PTEN C-tail phosphorylation is a bona fide inactivation mechanism for PTEN in human lung cancer and is positively correlated with a higher grade, indicative of its role in advanced stage lung cancers.
Discussion
In spite of its functional versatility, PTEN mutations are infrequent in lung cancer. However, a majority of patient lung tumors exhibit hyper-activation of PI3K/AKT signaling despite the relative low frequency of PTEN mutations [6,7]. Loss of PTEN expression, as shown by us (Figure 7) and others [8–12]. could explain, in part, the aberrant activation of the PI3K/AKT pathway observed in patient lung tumors. Further, several non-genomic mechanisms contribute to PTEN inactivation resulting in oncogenic transformation and progression. PTEN function and subcellular localization is frequently modulated by post-translational modifications, particularly phosphorylation and ubiquitination [16,18,47,61,62]. Indeed, PTEN phosphorylation has increasingly been observed to compromise PTEN function in several cancers [63–66] and other non-malignant diseases [67–69].
Using a phosphorylation deficient mutant of PTEN, PTEN-4A, we have investigated the effect of PTEN phosphorylation on its nuclear function, particularly focusing on its role in the transcription of cell cycle genes. We reveal that PTEN-4A preferentially localized to the nucleus where it inhibited the transcription of E2F1-regulated genes in H1299 lung cancer cells and PC3 PTEN-null prostate cancer cells, indicating a unique function across multiple cell types. PTEN-4A also physically associated with the E2F1 protein and at E2F1 DNA binding sites on the native cyclin D1 and cyclin E1 promoters in the chromatin context in the nucleus.
The preferential nuclear localization of PTEN-4A has been shown by other research groups in several different cell models [16,18,37,39]. Nguyen et. al have shown that the Lys13 residue of PTEN is exposed, when dephosphorylated, and ubiquitination of this Lys13 residue promotes its nuclear accumulation [18]. Further, it may be possible that PTEN-4A preferentially binds to nuclear import proteins [50]. thereby enhancing its nuclear levels.
The exact mechanism by which PTEN inhibits E2F1-mediated transcription remains to be determined. It is possible that transcriptional suppression by PTEN of E2F1-responsive cell cycle genes is, in part, a reflection of the global effect of PTEN in facilitating chromatin condensation via its interaction with histone H1 [32]. Whether PTEN can directly bind to DNA remains to be determined; although it's binding to chromatin and transcriptional repressor activity may suffice its role as a transcriptional modulator.
Our mechanistic studies revealed that the effect of PTEN on E2F1-mediated transcription occurs through both phosphatase dependent and independent effects. We observed that PTEN catalytic domain mutants were unable to repress transcription of E2F1-mediated genes indicating an AKT-dependent mechanism. Further, deletion analysis of PTEN domains revealed that the C2 domain is critical for its transcriptional function, revealing a phosphatase-independent mechanism. Indeed, several of our disease associated C2 domain PTEN mutants lose their ability to suppress E2F1-mediated transcription. Although the exact mechanism of this loss of function is not clear, it is possible that these PTEN mutants have altered nuclear localization patterns, or have impaired E2F1 binding capabilities, which remains to be elucidated. The recent findings of PTEN dimerization [70] and existence of multiple PTEN proteoforms [71] supports the notion that the PTEN mutations in the C2 domain may engage in aberrant PTEN homo/hetero-dimerization, leading to loss of PTEN function. In summary, our transcriptional studies indicate that both PTEN-WT and PTEN-4A can inhibit the transcription of E2F1 regulated genes and that the tumor suppressive effect of PTEN-4A is more pronounced due to its enhanced lipid-phosphatase activity, increased nuclear localization and accessibility of the C2 domain.
In patient samples, PTEN levels were reduced in lung tumors and had an inverse correlation with tumor grade. The nuclear levels of PTEN were also reduced in the tumors, indicating that deficiency of nuclear PTEN may contribute to lung cancer progression. Further, the relative levels of pPTEN (normalized to total PTEN levels) were significantly higher in tumor tissue and had a direct correlation with the tumor grade. However, we did not observe a correlation between nuclear levels of pPTEN in tumor and normal tissue. Perhaps, additional PTMs such as monoubiquitination or sumoylation may have a more profound effect on PTEN nuclear transport compared to PTEN phosphorylation [54,61]. Further, since non-phosphorylated PTEN preferentially localizes to the nucleus it may be more pertinent to stain the TMA using a non-phospho specific PTEN antibody.
While it is still not clear whether loss of PTEN is a driving factor for development or progression of lung cancer, PTEN levels/activity frequently influence tumor drug sensitivity [72]. Loss of PTEN is associated with resistance to TKI therapy, particularly in Epidermal Growth Factor Receptor (EGFR) mutant lung tumors [14]. PTEN loss also causes resistance to gamma-secretase inhibitors (GSIs) [73] and T-cell mediated immunotherapy in melanoma [74]. two therapeutic strategies that are gaining popularity in lung cancer, particularly with the emergence of PD-1/PD-L1 based immunotherapies [75]. Further, loss of PTEN renders several different tumor types susceptible to certain drugs, allowing the use of synthetic lethal combination therapies. For example, PTEN-deficient cancers are found to be particularly susceptible to treatment with PARP inhibitors [76] and PI3K/p110β-specific inhibitors [77–79]. Therefore, PTEN status could potentially be exploited to tailor therapeutic regimens for lung cancer patients with PTEN-deficient tumors or patients whose lung tumors have high levels of phosphorylated PTEN.
Materials and Methods
Plasmids
The 800 pSG5L-HA-PTEN WT, 977 pSG5L-HA-PTEN A4, 811 pSG5L-HA-PTEN C124S, 882 pSG5L-HA-PTEN G129E, 878 pSG5L-HA-PTEN 1–353. 809 pCDNA3 GFP PTEN WT, and pCDNA3 GFP PTEN A4 plasmids were a gift from Dr. William Sellers (Addgene plasmid #10750, #10753, #10744, #10746, #10765, #10759 and #10760) [15,80]. The 10–4 cyclin E promoter was a gift from Dr. Robert Weinberg (Addgene plasmid #8458) [81]. -1748 human cyclin D1 promoter cloned in pGL3 basic luciferase reporter was a gift from Dr. Frank McCormick (Addgene plasmid #32726) [82], pGL2-AN was a gift from Dr. William Kaelin (Addgene plasmid #20950) [83]. E2F-Luc and HA-E2F1 plasmids were a gift from Dr. Srikumar Chellappan (H. Lee Moffitt Cancer Center and Research Institute) [84,85]. H2B mCherry plasmid was a gift from Dr. Robert Benezra (Addgene plasmid #20972) [86], pIRES-Flag-PTEN WT was a gift from Dr. Lih-Yuan Lin (National Tsing Hua University, Taiwan) [29]. pIRES-Flag-PTEN 4A was cloned by PCR amplification from the pIRES-Flag-PTEN WT plasmid. pSG5L-HA PTEN C124S 4A, pSG5L-HA PTEN G129E 4A and pSG5L-HA PTEN d32 4A were engineered by PCR amplification from the parent plasmid pSG5L-HA-PTEN A4. pIRES-Flag PTEN WT and pIRES-Flag PTEN 4A were sub-cloned into the pLVX-tdTomato-C1 vector (Takara Bio USA Inc., #632564) for assembly into lentiviruses. For PTEN deletion mutants (PBM-PD and C2 domain), the various deletions were PCR amplified and cloned into the pSG5L-HA vector. The deletion mutants were then sub-cloned into the pcDNA3-GFP vector (Life Technologies). PTEN truncation mutants Q214*, R233*, K260* and R335* were cloned in the pSG5L-HA vector using standard PCR amplification. PTEN point mutants P246L, F271S, D331G and P339L were cloned in the pSG5L-HA vector using nested PCR [87]. All plasmids were verified by sequencing. Please see Table 1 for primer sequences.
Table 1.
Primer Sequences for Cloning and ChIP Assays.
 |
Cell culture and Stable Cell Line Generation
NCI-H1299 cells (ATCC® CRL-5803™) and 293T cells (ATCC® CRL-3216™), a gift from Dr. Mary Zhang (Wayne State University), were cultured in DMEM (Dulbecco's Modified Eagle's medium, Sigma Aldrich, #D5796) supplemented with 10% Fetal Bovine Serum (FBS), Antibiotic-antimycotic solution (final concentration 200 units/ml penicillin G, 200 µg/ml streptomycin sulfate and 0.5 µg/ml amphotericin B, Sigma Aldrich,#A9909) and Plasmocin (1.25 µg/ml, Invivogen). The cells were maintained in a humidified incubator with 5% CO2 at 37°C.
The pLVX-TdTomato-C1-Flag-PTEN-WT and pLVX-TdTomato-C1-Flag-PTEN-4A plasmids were assembled into viruses at the Viral Vector Core (University of Iowa). H1299 cells were infected with the lentiviruses at a MOI (Multiplicity of Infection) of 0.5 for a period of 8 hours. Polybrene was used at a concentration of 12 µg/ml to facilitate viral infection. 72 hours post-infection, the cells were treated with increasing concentrations of puromycin to identify the stable transformants. The cells were maintained at 5 µg/ml of puromycin.
Luciferase-based Reporter and Immunofluorescence Assays
Transient transfection with luciferase-based promoter-reporter plasmids and expression plasmids was performed in H1299, 293T or PC3 cells using the PEI (Polyethylenimine) method [40,88]. Briefly, 6-well plates at 50–60% cell confluence were co-transfected with 1 or 2µg of the luciferase-reporter plasmids and varying amounts of different PTEN (or mutant PTEN) expression plasmids. The total amount of DNA was kept constant across wells using corresponding empty vector controls. 48 hours after transfection, luciferase activity was measured using the Synergy H4 Hybrid Reader (BioTek). The transfection efficiency was normalized for each well using 62.5 ng of pCMV β-galactosidase plasmid (Takara Bio USA Inc.), co-expressed in each experiment, as described previously [89]. Data obtained represent the average of three transfection experiments, each carried out in triplicate and depicted as means ± S.E. unless stated otherwise. Transient transfection with H2B mCherry, as a nuclear marker, and GFP-tagged PTEN expression plasmids was performed in 293T cells using the PEI method. Briefly, cells were grown to ∼50% confluence and were co-transfected with H2B mCherry and indicated GFP-tagged PTEN construct. Immunofluorescence images were obtained at 20X and 40X magnification using Texas Red and GFP filters per each field on an Olympus IX51 microscope. Positive co-localization (yellow color) was counted independently by two researchers using ImageJ after merging Texas Red and GFP field images. Ten fields/transfection were recorded by each of the two researchers and the data obtained is represented as mean ± S.E. unless stated otherwise.
Protein Isolation, Co-immunoprecipitation Assays and Western Blots
For quantitation of relative protein expression or for immunoprecipitation experiments, whole cell extracts, were prepared using Pierce IP Lysis Buffer (Thermo Fisher Scientific; 25 mM Tris.HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) supplemented with phosphatase and protease inhibitors (Sigma Aldrich). Protein concentration was determined using Bradford Dye Reagent (BioRad, #5000006). Nuclear proteins were isolated without using detergents as described previously [90]. For immunoprecipitation (IP) assays, 300 µg of protein was incubated with the desired antibody/beads overnight on a rotator. The beads/immuno-complex was then washed 3 times with the Lysis Buffer and boiled in 100 µl of 2X Laemmli Buffer containing 5% β-mercaptoethanol. For endogenous IP assays, 1 mg of nuclear protein was incubated with beads overnight on a rotator. The beads were then washed 6 times with 1X TBS (20 mM Tris, 150 mM NaCl, pH 7.6) and boiled in 50 µl of 2X Laemmli Buffer containing 5% β-mercaptoethanol. The co-immunoprecipitated (co-IPed) proteins released were analyzed by immunoblotting. Briefly, proteins were separated on a 12% SDS-PAGE gel and electroblotted to nitrocellulose membranes (0.45 μm; GE Healthcare). Blots were blocked with 5% nonfat dry milk or bovine serum albumin in TBST buffer (20 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.6) and incubated with corresponding primary antibodies. Peroxidase conjugated secondary antibodies were used at dilution of 1:10,000. Blots were developed by chemiluminescence (Thermo Fisher Scientific) on X-ray films or developed on a ChemidocMP System (BioRad). The list of antibodies used, with their concentrations, is outlined in Table 2.
Table 2.
Details of antibodies used.
 |
Chromatin Immunoprecipitation Assays (ChIP)
Chromatin was isolated from the PTEN-4A stable cell line using ChIP IT Express Magnetic Chromatin Immunoprecipitation kit (Active Motif, #53009) as per the manufacturer's instructions. Briefly, the cells were fixed with formaldehyde, the chromatin was isolated and digested with micrococcal nuclease. The DNA was purified from an aliquot of the digestion mix to verify optimal chromatin digestion. DNA present in the digestion mix was extracted and purified using a phenol:chloroform:isoamyl alcohol mixture (25:24:1, v/v). The DNA was precipitated and washed with 100% and 70% ethanol respectively, before dissolving it in 30 µl of water. 1 to 3 µgs of the purified DNA was run on a 3% agarose gel in 1X TAE (40mM Tris (pH 7.6), 20mM acetic acid, 1mM EDTA) to verify that optimal micrococcal nuclease digestion had occurred, resulting in DNA fragments ranging between 150–300 base-pairs. Once the chromatin digestion was verified, the original micrococcal nuclease digestion mix containing cross-linked chromatin was immunoprecipitated overnight using either Rabbit IgG, E2F1 or Flag antibodies (for PTEN ChIP) bound to Protein G Magnetic beads. The immuno-precipitates were washed, reverse-cross linked and subjected to Proteinase K digestion. The released DNA was used to PCR-amplify GAPDH, cyclin D1 and cyclin E1 genomic DNA derived from defined promoter regions. GAPDH promoter region served as a negative control. The primer sequences are listed in Table 1.
Proliferation and Migration Assays
Cell proliferation assays were performed using Cell Counting Kit-8 (Dojindo Molecular Technologies Inc., #CK04-05). Parent H1299 cells, PTEN-WT and PTEN-4A stable cells were plated in 96-well plates at increasing density ranging from 400 to 1200 cells/well and cultured in DMEM growth medium as described above. The cells were then serum starved for 24 hours following which they were replenished with serum free medium alone or serum free medium containing 100 ng/ml of human recombinant leptin (R&D Systems, #398-LP-01M). 24 hours following leptin treatment, the final cell numbers were assessed by measuring absorbance at 450 nm.
Parent H1299, PTEN-WT and PTEN-4A stable cells were grown on electric cell substrate impedance sensing (ECIS) 8-well plate arrays (8W1E; Applied Biophysics) in above growth media. For proliferation assays, the impedance was measured using the ECIS system (Applied Biophysics) at 1000 Hz for 7 days until the cells were completely confluent. For migration assays, the cells were wounded using an elevated field pulse of 1,400 mA at 32,000 Hz applied for 20 seconds, producing a uniform circular lesion of 250 mm in size, and wounds were tracked over a period of 12 hours. The impedance (Z) was measured at 16,000 Hz, normalized to its value at the initiation of data acquisition, and plotted as a function of time. Assays were performed in triplicates and reported as mean ± S.E. unless stated otherwise (p value ≤ 0.05).
Immunohistochemistry
A TMA comprising of multiple types of lung cancer tissue (of varying grades), cancer adjacent and normal lung tissue was obtained from US Biomax (LC-2085a). The TMAs were stained with either an antibody to PTEN (6H.2, BioCare) or phospho-PTEN (pSer380/pThr382/pThr383, SAB4300044, Sigma Aldrich). Red chromogen or DAB was used as substrates for the PTEN and phospho-PTEN antibodies, respectively. Mayer's hematoxylin was used as a counter stain. All staining was performed at the Tissue Core at H. Lee Moffitt Cancer Center and Research Institute (Tampa, FL).
Tissue Microarray Image and Statistical Analysis
The stained tissue cores in the TMA were analyzed to calculate percent positivity (percentage of the core that showed any and all expression of the protein). Quantitation of nuclear and cytoplasmic PTEN levels was performed using an algorithm that extends the cells from the nucleus (identified by the hematoxylin stain) in a fixed distance in all directions. The TMAs were analyzed at the Analytical Microscopy Core at H. Lee Moffitt Cancer Center and Research Institute (Tampa, FL). Statistical analysis for the analyzed data was done using the IBM SPSS Software Suite. Outliers were removed using Tukey's Test [91,92] and significance was determined using Mann-Whitney Tests.
Supplementary Material
Funding Statement
American Heart Association (AHA) [grant number AHA-SDG-0830101N]; Moffitt Cancer Center Lung SPORE [grant number P50-CA119997]; National Institute of Health [grant number NIH-1R21AG047473-01A1].
Acknowledgements
This work was supported by the Moffitt Cancer Center Lung SPORE Career Development grant # P50-CA119997 (V.D.), USF-COM Start-up Fund (V.D.), AHA-SDG-0830101N (V.D.), and NIH-1R21AG047473-01A1 (V.D.).
Disclosure of Interest
The authors report no conflict of interest.
References
- [1].Blanco-Aparicio C, Renner O, Leal JFM, et al. . PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386. doi: 10.1093/carcin/bgm052. PMID:17341655 [DOI] [PubMed] [Google Scholar]
- [2].Yanagi S, Kishimoto H, Kawahara K, et al. . Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. The Journal of Clinical Investigation 2007;117:2929–2940. doi: 10.1172/JCI31854. PMID:17909629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dave V, Wert SE, Tanner T, et al. . Conditional deletion of Pten causes bronchiolar hyperplasia. American Journal of Respiratory Cell and Molecular Biology. 2008;38:337–345. doi: 10.1165/rcmb.2007-0182OC. PMID:17921358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shaykhiev R, Otaki F, Bonsu Y, et al. . Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. PMID:20820852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Forgacs E, Biesterveld EJ, Sekido Y, et al. . Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557–1565. doi: 10.1038/sj.onc.1202070. PMID:9794233 [DOI] [PubMed] [Google Scholar]
- [6].Fumarola C, Bonelli MA, Petronini PG, et al. . Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90:197–207. doi: 10.1016/j.bcp.2014.05.011. PMID:24863259 [DOI] [PubMed] [Google Scholar]
- [7].Sarris EG, Saif MW, Syrigos KN. The Biological role of PI3K pathway in lung cancer. Pharmaceuticals. 2012;5:1236–1264. doi: 10.3390/ph5111236. PMID:24281308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Marsit CJ, Zheng S, Aldape K, et al. . PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Human Pathol. 2005;36:768–776. doi: 10.1016/j.humpath.2005.05.006. PMID:16084946 [DOI] [PubMed] [Google Scholar]
- [9].Tang JM, He QY, Guo RX, et al. . Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer. 2006;51:181–191. doi: 10.1016/j.lungcan.2005.10.003. PMID:16324768 [DOI] [PubMed] [Google Scholar]
- [10].Cetin Z, Ozbilim G, Erdogan A, et al. . Evaluation of PTEN and Mcl-1 expressions in NSCLC expressing wild-type or mutated EGFR. Medical Oncol. 2010;27:853–860. doi: 10.1007/s12032-009-9296-7. [DOI] [PubMed] [Google Scholar]
- [11].Yanagawa N, Leduc C, Kohler D, et al. . Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J Thorac Oncol. 2012;7:1513–1521. doi: 10.1097/JTO.0b013e3182641d4f. PMID:22982652 [DOI] [PubMed] [Google Scholar]
- [12].Panagiotou I, Tsiambas E, Lazaris AC, et al. . PTEN expression in non small cell lung carcinoma based on digitized image analysis. J Buon. 2012;17:719–723. PMID:23335531 [PubMed] [Google Scholar]
- [13].Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature Reviews Molecular Cell Biol. 2012;13:283–296. PMID:22473468 [DOI] [PubMed] [Google Scholar]
- [14].Sos ML, Koker M, Weir BA, et al. . PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. PMID:19351834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vazquez F, Grossman SR, Takahashi Y, et al. . Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627–48630. doi: 10.1074/jbc.C100556200. PMID:11707428 [DOI] [PubMed] [Google Scholar]
- [16].Rahdar M, Inoue T, Meyer T, et al. . A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. P Natl Acad Sci USA. 2009;106:480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Odriozola L, Singh G, Hoang T, et al. . Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282:23306–23315. doi: 10.1074/jbc.M611240200. PMID:17565999 [DOI] [PubMed] [Google Scholar]
- [18].Nguyen HN, Yang JM, Miyamoto T, et al. . Opening the conformation is a master switch for the dual localization and phosphatase activity of PTEN. Sci Rep-Uk. 2015;5:12600. doi: 10.1038/srep12600. PMID:26216063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen WH, Balajee AS, Wang J, et al. . Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. PMID:17218262 [DOI] [PubMed] [Google Scholar]
- [20].Kang X, Song C, Du X, et al. . PTEN stabilizes TOP2A and regulates the DNA decatenation. Sci Rep. 2015;5:17873. doi: 10.1038/srep17873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Feng JW, Liang J, Li JJ, et al. . PTEN Controls the DNA Replication Process through MCM2 in Response to Replicative Stress. Cell Reports. 2015;13:1295–303. doi: 10.1016/j.celrep.2015.10.016. PMID:26549452 [DOI] [PubMed] [Google Scholar]
- [22].Choi BH, Chen Y, Dai W. Chromatin PTEN is involved in DNA damage response partly through regulating Rad52 sumoylation. Cell Cycle. 2013;12:3442–3447. doi: 10.4161/cc.26465. PMID:24047694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen JH, Zhang P, Chen WD, et al. . ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy. 2015;11:239–252. doi: 10.1080/15548627.2015.1009767. PMID:25701194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ming M, Feng L, Shea CR, et al. . PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011;71:5287–5295. doi: 10.1158/0008-5472.CAN-10-4614. PMID:21771908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vuono EA, Mukherjee A, Vierra DA, et al. . The PTEN phosphatase functions cooperatively with the Fanconi anemia proteins in DNA crosslink repair. Sci Rep. 2016;6:36439. doi: 10.1038/srep36439. PMID:27819275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guan J, Zhao Q, Mao W. Nuclear PTEN interferes with binding of Ku70 at double-strand breaks through post-translational poly(ADP-ribosyl)ation. Biochim Biophys Acta. 2016;1863:3106–3115. doi: 10.1016/j.bbamcr.2016.10.003. PMID:27741411 [DOI] [PubMed] [Google Scholar]
- [27].Gu TT, Zhang Z, Wang JL, et al. . CREB Is a Novel Nuclear Target of PTEN Phosphatase. Cancer Res. 2011;71:2821–2825. doi: 10.1158/0008-5472.CAN-10-3399. PMID:21385900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Horita H, Wysoczynski CL, Walker LA, et al. . Nuclear PTEN functions as an essential regulator of SRF-dependent transcription to control smooth muscle differentiation. Nat Commun. 2016;7:10830. doi: 10.1038/ncomms10830. PMID:26940659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lin MC, Liu YC, Tam MF, et al. . PTEN interacts with metal-responsive transcription factor 1 and stimulates its transcriptional activity. Biochem J. 2012;441:367–377. doi: 10.1042/BJ20111257. PMID:21883094 [DOI] [PubMed] [Google Scholar]
- [30].Palian BM, Rohira AD, Johnson SAS, et al. . Maf1 is a novel target of PTEN and PI3K signaling that negatively regulates oncogenesis and lipid metabolism. Plos Genet. 2014;10(12):e1004789. doi: 10.1371/journal.pgen.1004789. PMID:25502566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fan C, He L, Kapoor A, et al. . PTEN inhibits BMI1 function independently of its phosphatase activity. Mol Cancer. 2009;8:98. doi: 10.1186/1476-4598-8-98. PMID:19903340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen ZH, Zhu M, Yang J, et al. . PTEN interacts with histone H1 and controls chromatin condensation. Cell Reports. 2014;8:2003–2014. doi: 10.1016/j.celrep.2014.08.008. PMID:25199838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G(0)-G(1) in MCF-7 cells. Cancer Res. 2003;63:282–286. PMID:12543774 [PubMed] [Google Scholar]
- [34].Chung JH, Ostrowski MC, Romigh T, et al. . The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum Mol Genet. 2006;15:2553–2559. doi: 10.1093/hmg/ddl177. PMID:16849370 [DOI] [PubMed] [Google Scholar]
- [35].Jacob AI, Romigh T, Waite KA, et al. . Nuclear PTEN levels and G2 progression in melanoma cells. Melanoma research. 2009;19:203–210. doi: 10.1097/CMR.0b013e32832ccd6e. PMID:19478684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Howitt J, Low LH, Putz U, et al. . Ndfip1 represses cell proliferation by controlling Pten localization and signaling specificity. J Mol Cell Biol. 2015;7:119–131. doi: 10.1093/jmcb/mjv020. PMID:25801959 [DOI] [PubMed] [Google Scholar]
- [37].Kim SJ, Lee HW, Baek JH, et al. . Activation of nuclear PTEN by inhibition of Notch signaling induces G2/M cell cycle arrest in gastric cancer. Oncogene. 2016;35:251–260. doi: 10.1038/onc.2015.80. PMID:25823029 [DOI] [PubMed] [Google Scholar]
- [38].Choi BH, Pagano M, Dai W. Plk1 Protein Phosphorylates Phosphatase and Tensin Homolog (PTEN) and Regulates Its Mitotic Activity during the Cell Cycle. J Biol Chem. 2014;289:14066–14074. doi: 10.1074/jbc.M114.558155. PMID:24706748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vazquez F, Ramaswamy S, Nakamura N, et al. . Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/MCB.20.14.5010-5018.2000. PMID:10866658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pathak RR, Grover A, Malaney P, et al. . Loss of phosphatase and tensin homolog (PTEN) induces leptin-mediated leptin gene expression: feed-forward loop operating in the lung. J Biol Chem. 2013;288:29821–29835. doi: 10.1074/jbc.M113.481523. PMID:23963458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Terzidis A, Sergentanis TN, Antonopoulos G, et al. . Elevated serum leptin levels: a risk factor for non-small-cell lung cancer? Oncology. 2009;76:19–25. doi: 10.1159/000177952. PMID:19033693 [DOI] [PubMed] [Google Scholar]
- [42].Song C-H, Liao J, Deng Z-H, et al. . Is leptin a predictive factor in patients with lung cancer? Clinical Biochemistry. 2014;47:230–232. doi: 10.1016/j.clinbiochem.2013.12.003. PMID:24355691 [DOI] [PubMed] [Google Scholar]
- [43].Ning K, Miller LC, Laidlaw HA, et al. . A novel leptin signalling pathway via PTEN inhibition in hypothalamic cell lines and pancreatic beta-cells. EMBO Journal. 2006;25:2377–2387. doi: 10.1038/sj.emboj.7601118. PMID:16675953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nguyen HN, Afkari Y, Senoo H, et al. . Mechanism of human PTEN localization revealed by heterologous expression in Dictyostelium. Oncogene. 2014;33:5688–5696. doi: 10.1038/onc.2013.507. PMID:24292679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nguyen HN, Yang JM, Afkari Y, et al. . Engineering ePTEN, an enhanced PTEN with increased tumor suppressor activities. P Natl Acad Sci USA. 2014;111:E2684–E2693. doi: 10.1073/pnas.1409433111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nguyen HN, Yang JM, Rahdar M, et al. . A new class of cancer-associated PTEN mutations defined by membrane translocation defects. Oncogene. 2015;34:3737–3743. doi: 10.1038/onc.2014.293. PMID:25263454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stumpf M, Blokzijl-Franke S, den Hertog J. Fine-tuning of Pten localization and phosphatase activity is essential for zebrafish angiogenesis. Plos One. 2016;11(5):e0154771. doi: 10.1371/journal.pone.0154771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sun H, Lesche R, Li DM, et al. . PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. PMID:10339565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Myers MP, Pass I, Batty IH, et al. . The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. PMID:9811831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gil A, Andres-Pons A, Fernandez E, et al. . Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: Involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell. 2006;17:4002–4013. doi: 10.1091/mbc.E06-05-0380. PMID:16807353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chung JH, Ginn-Pease ME, Eng C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res. 2005;65:4108–4116. doi: 10.1158/0008-5472.CAN-05-0124. PMID:15899801 [DOI] [PubMed] [Google Scholar]
- [52].Forbes SA, Beare D, Gunasekaran P, et al. . COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. PMID:25355519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rankin SL, Guy CS, Mearow KM. PTEN downregulates p75NTR expression by decreasing DNA-binding activity of Sp1. Biochem Bioph Res Co. 2009;379:721–725. doi: 10.1016/j.bbrc.2008.12.075. [DOI] [PubMed] [Google Scholar]
- [54].Bassi C, Ho J, Srikumar T, et al. . Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–399. doi: 10.1126/science.1236188. PMID:23888040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chang CJ, Mulholland DJ, Valamehr B, et al. . PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol. 2008;28:3281–3289. doi: 10.1128/MCB.00310-08. PMID:18332125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].He JX, Kang X, Yin YX, et al. . PTEN regulates DNA replication progression and stalled fork recovery. Nat Commun. 2015;6:7620. doi: 10.1038/ncomms8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang GX, Li Y, Wang P, et al. . PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 2015;25:1189–1204. doi: 10.1038/cr.2015.115. PMID:26403191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ming M, He YY. PTEN in DNA damage repair. Cancer Lett. 2012;319(2):125–129. doi: 10.1016/j.canlet.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gong LL, Govan JM, Evans EB, et al. . Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle. 2015;14:2323–2332. doi: 10.1080/15384101.2015.1044174. PMID:25946202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Misra S, Mukherjee A, Karmakar P. Phosphorylation of PTEN at STT motif is associated with DNA damage response. Mutat Res-Fund Mol M. 2014;770:112–119. doi: 10.1016/j.mrfmmm.2014.08.008. [DOI] [PubMed] [Google Scholar]
- [61].Trotman LC, Wang X, Alimonti A, et al. . Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. PMID:17218261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lee MS, Jeong MH, Lee HW, et al. . PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun. 2015;6:7769. doi: 10.1038/ncomms8769. PMID:26183061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kechagioglou P, Papi RM, Provatopoulou X, et al. . Tumor suppressor PTEN in breast cancer: heterozygosity, mutations and protein expression. Anticancer Res. 2014;34:1387–1400. PMID:24596386 [PubMed] [Google Scholar]
- [64].Nakahata S, Ichikawa T, Maneesaay P, et al. . Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat Commun. 2014;5:3393. doi: 10.1038/ncomms4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yang Z, Yuan XG, Chen J, et al. . Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: A novel mechanism of PTEN inactivation. Clin Res Hepatol Gas. 2013;37:72–79. doi: 10.1016/j.clinre.2012.03.002. [DOI] [PubMed] [Google Scholar]
- [66].Roy D, Dittmer DP. Phosphatase and tensin homolog on chromosome 10 is phosphorylated in primary effusion lymphoma and kaposi's sarcoma. Am J Pathol. 2011;179:2108–2119. doi: 10.1016/j.ajpath.2011.06.017. PMID:21819957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lee EJ, Kim N, Kang KH, et al. . Phosphorylation/inactivation of PTEN by Akt-independent PI3K signaling in retinal pigment epithelium. Biochem Bioph Res Co. 2011;414:384–389. doi: 10.1016/j.bbrc.2011.09.083. [DOI] [PubMed] [Google Scholar]
- [68].Hua F, Ha TZ, Ma J, et al. . Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J Immunol. 2007;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. PMID:17513782 [DOI] [PubMed] [Google Scholar]
- [69].Horita H, Furgeson SB, Ostriker A, et al. . Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. J Am Heart Assoc. 2013;2(3):e000188. doi: 10.1161/JAHA.113.000188. PMID:23727701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Papa A, Wan L, Bonora M, et al. . Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157:595–610. doi: 10.1016/j.cell.2014.03.027. PMID:24766807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Malaney P, Uversky VN, Dave V. PTEN proteoforms in biology and disease. Cell Mol Life Sci. 2017;74:2783–2794. doi: 10.1007/s00018-017-2500-6. PMID:28289760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Current drug targets. 2014;15:65–79. doi: 10.2174/1389450114666140106100909. PMID:24387334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nair JS, Sheikh T, Ho AL, et al. . PTEN regulates sensitivity of melanoma cells to RO4929097, the gamma-secretase inhibitor. Anticancer Res. 2013;33:1307–1316. PMID:23564767 [PubMed] [Google Scholar]
- [74].Peng WY, Chen JQ, Liu CW, et al. . Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. PMID:26645196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jing W, Li M, Zhang Y, et al. . PD-1/PD-L1 blockades in non-small-cell lung cancer therapy. OncoTargets and Therapy. 2016;9:489–502. doi: 10.2147/OTT.S94993. PMID:26889087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mendes-Pereira AM, Martin SA, Brough R, et al. . Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. Embo Mol Med 2009;1:315–322. doi: 10.1002/emmm.200900041. PMID:20049735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ni J, Liu Q, Xie S, et al. . Functional characterization of an isoform-selective inhibitor of PI3K-p110beta as a potential anticancer agent. Cancer Discov. 2012;2:425–433. doi: 10.1158/2159-8290.CD-12-0003. PMID:22588880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Edgar KA, Wallin JJ, Berry M, et al. . Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res 2010; 70:1164–1172. doi: 10.1158/0008-5472.CAN-09-2525. PMID:20103642 [DOI] [PubMed] [Google Scholar]
- [79].Chen H, Mei L, Zhou L, et al. . PTEN restoration and PIK3CB knockdown synergistically suppress glioblastoma growth in vitro and in xenografts. Journal of Neuro-Oncol. 2011;104:155–167. doi: 10.1007/s11060-010-0492-2. PMID:21188471 [DOI] [PubMed] [Google Scholar]
- [80].Ramaswamy S, Nakamura N, Vazquez F, et al. . Regulation of G(1) progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. P Natl Acad Sci USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Geng Y, Eaton EN, Picon M, et al. . Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. PMID:8649818 [PubMed] [Google Scholar]
- [82].Tetsu O, McCormick F. beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. PMID:10201372 [DOI] [PubMed] [Google Scholar]
- [83].Neuman E, Flemington EK, Sellers WR, et al. . Transcription of the E2f-1 gene is rendered cell-cycle dependent by E2f DNA-binding sites within its promoter. Mol Cell Biol. 1994;14:6607–6615. doi: 10.1128/MCB.14.10.6607. PMID:7935380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang S, Nath N, Fusaro G, et al. . Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals. Mol Cell Biol. 1999;19:7447–7460. doi: 10.1128/MCB.19.11.7447. PMID:10523633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rizwani W, Schaal C, Kunigal S, et al. . Mammalian lysine histone demethylase KDM2A regulates E2F1-mediated gene transcription in breast cancer cells. Plos One. 2014;9(7):e100888. doi: 10.1371/journal.pone.0100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nam HS, Benezra R. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem Cell. 2009;5:515–526. doi: 10.1016/j.stem.2009.08.017. PMID:19896442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ho SN, Hunt HD, Horton RM, et al. . Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. PMID:2744487 [DOI] [PubMed] [Google Scholar]
- [88].Boussif O, Lezoualch F, Zanta MA, et al. . A versatile vector for gene and oligonucleotide transfer into cells in culture and in-vivo – polyethylenimine. P Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].V D, Childs T, Whitsett JA. Nuclear factor of activated T cells regulates transcription of the surfactant protein D gene (Sftpd) via direct interaction with thyroid transcription factor-1 in lung epithelial cells. J Biol Chem. 2004;279:34578–34588. doi: 10.1074/jbc.M404296200. PMID:15173172 [DOI] [PubMed] [Google Scholar]
- [90].Brunner E, Ahrens CH, Mohanty S, et al. . A high-quality catalog of the Drosophila melanogaster proteome. Nature Biotechnology. 2007;25:576–583. doi: 10.1038/nbt1300. PMID:17450130 [DOI] [PubMed] [Google Scholar]
- [91].Hoaglin DC, Iglewicz B, Tukey JW. Performance of some resistant rules for outlier labeling. J Am Stat Assoc. 1986;81:991–999. doi: 10.1080/01621459.1986.10478363. [DOI] [Google Scholar]
- [92].Hoaglin DC, Iglewicz B. Fine-tuning some resistant rules for outlier labeling. J Am Stat Assoc. 1987;82:1147–1149. doi: 10.1080/01621459.1987. PMID:10478551.10478551 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.