

Abstract
γδNKT cells are neonatal-derived γδ T lymphocytes that are grouped together with iNKT (invariant natural killer T) cells based on their shared innate-like developmental program characterized by the transcription factor PLZF (Promyelocytic leukemia zinc finger). Previous studies have demonstrated that the population size of γδNKT cells is tightly controlled by Id3-mediated inhibition of E-protein activity in mice. However, how E proteins promote γδNKT cell development and expansion remains to be determined. Here, we report that the transcription factor, Egr2, which also activates PLZF expression in iNKT cells, is essential for regulating γδNKT cell expansion. We observed a higher expression of Egr family genes in γδNKT cells compared to the conventional γδ T cell population. Loss of function of Id3 caused an expansion of γδNKT cells, which is accompanied by further up-regulation of Egr family genes as well as PLZF. Deletion of Egr2 in Id3-deficient γδNKT cells prevented cell expansion and blocked PLZF upregulation. We further show that this Egr2 mediated- γδNKT cell expansion is dependent on c-Myc. c-Myc knockdown attenuated the proliferation of Id3-deficient γδNKT cells, while c-Myc overexpression enhanced the proliferation of Id3/Egr2-double deficient γδNKT cells. Therefore, our data reveals a regulatory circuit involving Egr2-Id3-E2A, which normally restricts the population size of γδNKT cells by adjusting Egr2 dosage and c-Myc expression.
Keywords: Id3, γδNKT Cells, TCR signaling, Egr2, c-Myc
Introduction
γδ T cells are generated from fetal development through adult life in series of developmental waves (1, 2). Each wave of γδ T cells preferentially migrates to particular anatomical locations and serves site-specific immune regulatory functions ranging from tissue homeostasis to immunosurveillance (1, 3-5). The Vγ1.1Vδ6.3 γδ T cells represent the late fetal wave that expands during the neonatal window. These γδ T cells were first described by Pablo Pereira in 1997(6) and later referred to as NKTγδ T cells (7, 8) or γδNKT cells(9, 10). This population develops in the neonatal thymus and migrates to peripheral sites such as the spleen, lymph nodes and most prominently, the liver(6). γδNKT cells are considered an innate-like lineage based on expression of the innate signature transcription factor PLZF(8, 11). Mature γδNKT cells are phenotypically CD24lowCD44highNK1.1+ and are capable of secreting various cytokines of IFNγ, IL-4 and IL-13 upon TCR stimulation(8, 12-16). These innate features endow γδNKT cells with important functions in immune protection(17-19), as well as in immune disorders such as Sjögren’s Syndrome(14), dermatitis(8), asthma(15, 20).
The population size of γδNKT cells is tightly regulated during thymic development and typically represents 10% of total γδ T cells in C57Bl/6 mice. γδNKT cells undergo a dramatic expansion in Id3−/− mice, particularly on the background where Id2 expression is also partially compromised (11, 21-24). This phenomenon can be explained by elevated activity of E-proteins, since deletion of E2A in Id3-deficient mice attenuates γδNKT cell expansion(21, 23, 24). Furthermore, a number of labs have observed an enhanced development of γδNKT cells in mouse strains with deficiencies or impairments in TCR downstream molecules such as SLP-76(11, 25) and Interleukin-2-inducible T-cell kinase (Itk)(10, 15). Consistently, Dok1, as an inhibitor of ZAP-70, LAT, SLP-76, Akt and Erk1/2(26-29), promotes γδNKT cell development and expansion(28). Since Id3 is the most downstream factor in the TCR signaling pathway, it is possible that defects in these upstream signaling molecules may prevent proper activation of Id3, which consequently results in elevated E protein activity. Therefore a common mechanism underpinning the γδNKT expansion reported in these studies may be E protein mediated. Such a regulatory mechanism of γδNKT development and expansion has not yet been explored.
Members of the Egr family (Egr1, Egr2, and Egr3) sense TCR signaling through phosphorylated ERK (pERK) and transmit TCR signals through the upregulation of Id3, which in turn inhibits E-protein activity. TCR signal strength and duration of Egr activity are considered major factors distinguishing αβ from γδ T lineage at the preTCR checkpoint, with a weaker signal instructing αβ T lineage and a stronger signal supporting γδ T lineage development(30). Egr family members can also regulate the proliferation and survival of lymphocytes(31-33). A recent study has also shown that sustained induction of Egr2 in response to TCR signaling in iNKT cells results in upregulation of PLZF, which is crucial for an innate developmental program(34). Whether the same mechanism is involved in γδNKT cell development remains to be determined. Furthermore, the relationship between the Egr2-PLZF pathway and E2A-mediated γδNKT cell development and expansion has not been evaluated.
Here we have examined the role of Egr proteins, namely Egr1, Egr2, and Egr3, in γδNKT cell development, and found that Egr2 plays a prominent role in γδNKT cell expansion. We demonstrate that Id3 deletion results in upregulation of Egr family genes and PLZF among the expanded γδNKT cells. Deletion of Egr2 ameliorated the expansion of Id3-deficient γδNKT cells and prevented PLZF upregulation. We further show that Egr2 promotes γδNKT cell proliferation through c-Myc. Collectively, we revealed a regulation loop of Egr2-Id3-E2A in regulating γδNKT cell development and expansion.
Materials and Methods
Mice and reagents
Mice with Id3 conditional knockout(35) and LckCre transgenic(36) alleles have been described previously. Egr1−/−, Egr2f/f and Egr3−/− mice were as previously reported(37). All the strains are on B6 background. Animals were bred and maintained in the SPF facility managed by Duke University Division of Laboratory Animal Research. Animal procedures were approved by the Duke University Institutional Animal Care and Use Committee.
The antibodies used were as follows: FITC anti-mouse TCRδ (GL3), PE/Cy7 anti-mouse TCRδ (GL3), PE/Cy5 anti-mouse NK1.1 (PK136), PE/Cy7 anti-mouse/human CD44 (IM7), APC anti-mouse CD24 (M1/69), PE-Cy7 anti-mouse IFNγ (XMG1.2), and Pacific Blue™ anti-mouse IL-17 (TC11-18H10.1) were purchased from Biolegend; PE anti-mouse Vδ6.3/2 (8F4H7B7) APC anti-mouse PLZF (9E12) and APC BrdU Flow Kit were purchased from BD Biosciences. Rabbit anti-mouse Egr2 (EPR4004) and Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488, ab150077) were purchased from abcam.
Flow Cytometry
For cell surface analysis, single cells from the thymus were suspended in cold FACS buffer (1×PBS supplemented with 5% bovine calf serum). A total of 5×106 cells were stained with antibodies in the dark at 4°C for 30 min. After washing with cold FACS buffer, cells were analyzed using a FACSCanto II flow cytometer (BD Biosciences). FlowJo software (tree Star) was used for data analysis. In some experiments, γδ T cells were sorted with MoFlo XDP cell sorter.
For cytokine analysis, thymocytes were stimulated in vitro with PMA/Ionomycin in the presence of Brefeldin A and monensin for 4 hours. Cells were washed and stained with anti-TCRδ and Vδ6.3/2 antibodies. After a 30-minute incubation, cells were fixed and permeabilized according to BD Cytofix/Cytoperm™ Fixation/Permeabilization Kit protocol, followed by IFNγ and IL-17 analysis with FACS.
Real-time PCR analysis
Total RNA was extracted from purified Vδ6.3− and Vδ6.3+ γδ T cells with RNAqueous micro kit (Life Technologies). Reverse transcription was performed with Moloney murine leukemia virus reverse transcriptase (Life Technologies). SYBR-based real-time PCR was performed to quantitatively compare gene expression, with results normalized by β-actin expression. Quantitative PCR primer sequences are shown below.
Egr1 forward primer: 5’-AGCGCCTTCAATCCTCAAG-3’, Egr1 reverse primer: 5’-TTTGGCTGGGATAACTCGTC-3’; Egr2 forward primer: 5’-TTGACCAGATGAACGGAGTG-3’, Egr2 reverse primer: 5’-TGCCCATGTAAGTGAAGGTC-3’; Egr3 forward primer: 5’-TGCCCCAACCGCCGCTTACTCTCA-3’, Egr3 forward primer: 5’-GGCGCACCCCCTTTCTCCGACTTC-3’; PLZF forward primer: 5’-CCACCTTCGCTCACATACAG-3’, PLZF reverse primer: 5’-CACAGCCATTACACTCATAGGG-3’; c-Myc forward primer: 5’-GCTGTTTGAAGGCTGGATTTC-3’, c-Myc reverse primer: 5’-GATGAAATAGGGCTGTACGGAG-3’.
BrdU incorporation
For in vivo experiments, 50 μl (100μg) BrdU was injected intraperitoneally to neonatal mice for 4 hours of pulse labeling. For in vitro experiments, cultured γδ T cells were incubated with BrdU at a final concentration of 10 μM for 30 min. Single cells from thymus (in vivo) or culture (in vitro) were stained with anti-TCRδ, Vδ6.3/2, and BrdU antibodies according to the manufacturer’s instructions (BD Biosciences). The stained cells were analyzed on a FACSCanto II flow cytometer.
Retrovirus transduction
Vδ6.3+ γδ T cells from the thymi of Id3 deficient or Id3-Egr2 double deficient neonatal mice were sorted by FACS and cultured overnight with 5μg/ml anti-CD3 and anti-CD28 Abs in the presence of a cocktail of cytokines (5ng/ml IL-7, 1ng/ml IL-2, and 5ng/ml IL-15). Id3-deficient or Id3/Egr2 double deficient γδ T cells were infected with c-Myc shRNA and c-Myc overexpression virus, respectively. Forty-eight hours later, cells were collected for RNA or BrdU analysis. The shRNA (TGTAAGCTTCAGCCATAATTT) was designed to target the 3’UTR of c-Myc, and cloned into a vector carrying a miR30-based backbone(38).
Statistical analysis
The data were compared using Student t-test, with a p<0.05 considered significant, and p<0.01 and p<0.001 considered highly significant.
Results
Egr2 controls the expansion of Id3−/− γδ NKT cells
Previous studies have shown that Id3-deficiency causes a dramatic expansion of γδNKT cells over conventional γδ T cells in the neonatal thymus. We hypothesized that the loss function of Id3 leads to activation of an innate-associated transcription program. We therefore decided to search for transcription factors with critical roles in regulating innate-like T cell development. Egr2 has been shown to support γδ lineage differentiation by mediating the TCR signal(34, 37, 39). We noted that Egr2 expression was also higher in γδNKT cells than in any other subtypes of γδ T cells, as reported in Immgen. This prompted us to test the possibility that Egr2 may be involved in differentially regulating γδNKT cells vs. other γδ T cells.
To test whether Egr2 plays a necessary role in supporting γδNKT cell expansion in Id3-deficient mice, we first verified the reported Egr2 expression patterns in γδNKT cells (Vγ1.1+Vδ6.3+) and conventional γδ T cells (Vγ1.1+Vδ6.3−) purified from thymi of WT neonatal mice. Indeed, Egr2 expression was higher among γδNKT cells than in conventional γδ T cells (Fig.1A-1C). Egr3, but not Egr1, also displayed relatively higher level of transcripts in γδNKT cells than the control γδ population. Within the γδNKT cell lineage, Id3 deletion resulted in upregulation of all three Egr genes at mRNA levels (Fig.1A-1C). We further confirmed an upregulation of Egr2 protein level specifically among Id3 deficient γδNKT cells (Supplemental Fig.1). We then tested if any of the Egr genes are required for γδNKT cell development and/or expansion in Id3-deficient mice. Deletion of Egr2, but not Egr1 and Egr3, significantly reduced numbers of γδNKT cells in Id3-deficient mice. Deletion of both Egr2 and Egr3 reduced the population size of γδNKT cells in Id3-deficient mice to wild type levels, indicating that Egr2 and Egr3 work together to promote γδNKT cell expansion in response to elevated E-protein activity (Fig. 1D and 1E). Moreover, because Egr2/3-deficiency returns the number of γδNKT cells generated in Id3/Egr2/Egr3 triple knockout mice to wild type levels without completely eliminating them, Egr2 and 3 do not appear to be required for the generation of γδNKT cells. We then further tested the involvement of Egr2 in γδNKT cell proliferation using in vivo BrdU pulse labeling. We have shown in previous publications that Id3 deficiency results in vigorous proliferation of neonatal γδNKT cells(22). Egr2/Id3 double deficiency reduced the proliferation rate of γδNKT cells relative to that in Id3-deficient mice (Fig. 1F-1H). These findings demonstrate that Egr2 expression is essential for regulating the expansion of γδNKT cells in Id3-deficient mice. Additionally, Egr3 collaborates with Egr2 in promoting γδNKT cell expansion.
Fig.1. Egr2 is essential to γδNKT cell development in Id3−/− mice.
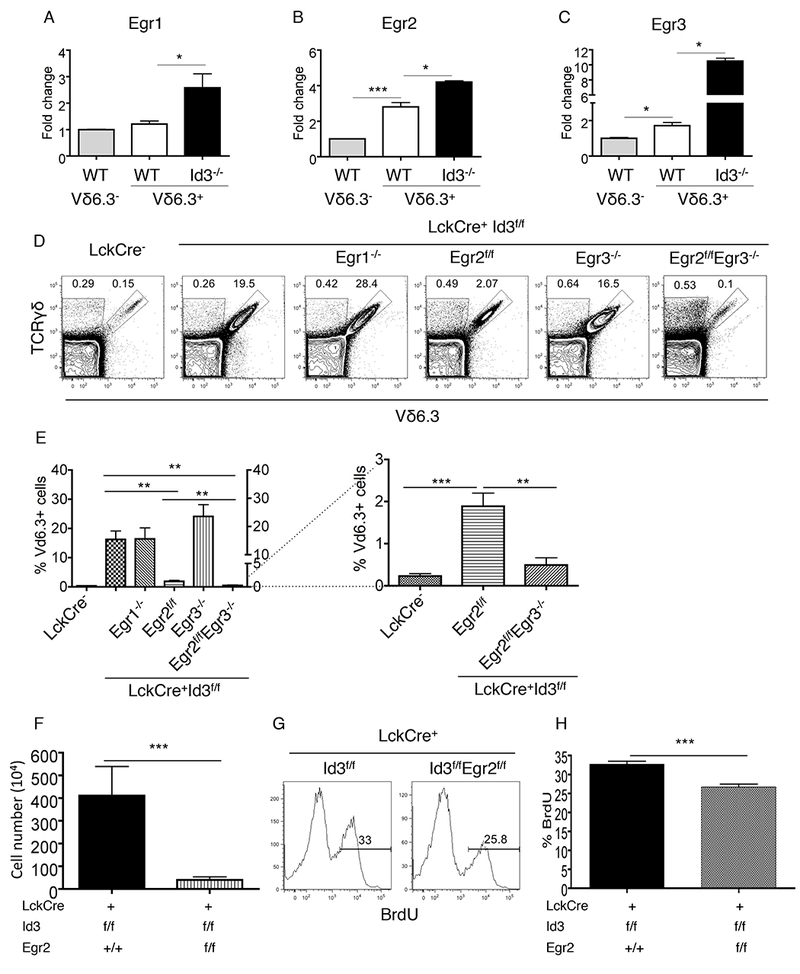
(A)-(C) Transcription analysis of Egr1, Egr2 and Egr3 in conventional and γδNKT cells from indicated strains. (D) Percentage of γδNKT cells in the thymus of indicated mouse strains. (E) Statistical summary for (D). (F) Absolute cell number of γδNKT cells in the thymus of indicated mouse strains. (G) Representative FACS plots for BrdU incorporation of thymic γδNKT cells from mouse strains. (H) Summary for the percentage of BrdU+ γδNKT cells in the thymus from mouse strains. The data are representative for 3 independent experiments.
Egr2 significantly affected maturation but not effector characteristics of Id3-deficient γδNKT cells
Maturation and effector differentiation are two major steps in γδ T cell development. CD24 down regulation has been well defined as a critical step in T cell maturation. We found that the majority of γδNKT cells in Id3-deficient mice are CD24 negative (Fig.2A). This phenotype remains unchanged upon further deletion of Egr2 on Id3-deficient background. Once γδNKT cells become matured, they typically enter effector phase expressing surface makers such as CD44 and NK1.1, and secreting cytokines such as IFNγ (10, 40). We found a similar expression pattern of CD44 and NK1.1 expression between WT and Id3−/− γδNKT cells (Fig. 2B). Deletion of Egr2 augmented the percentage, but not absolute numbers, of CD44+NK1.1− and CD44+NK1.1+ γδNKT cells (Fig. 2B-2D). Furthermore, we found that loss of Id3 increased the frequency of IL-17 producers but not IFNγ producers among total γδNKT cells (Fig. 2E and 2F). This effector pattern was not affected by further deletion of Egr2. Our data demonstrated that functions of Egr2 in γδNKT cells promote the maturation without dramatically influencing the effector features of these cells.
Fig.2. Effect of Egr2 on the maturation and effector feature of Id3−/− γδNKT cells.
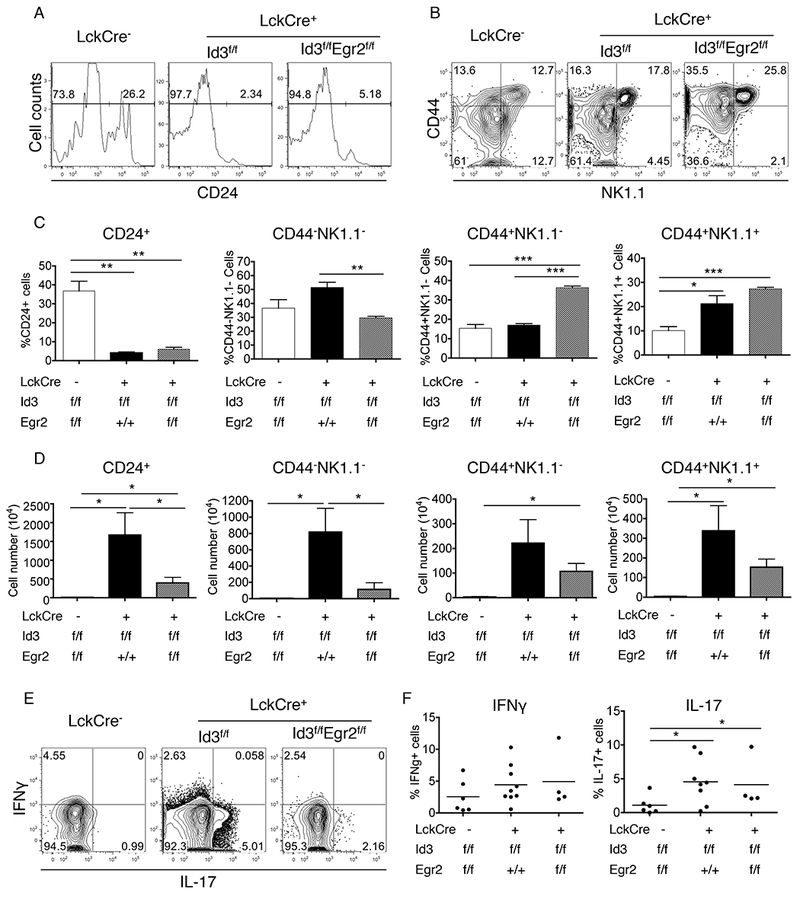
(A) CD24 expression in γδNKT cells from the indicated mouse strains. (B) CD44 and NK1.1 expression in γδNKT cells from the indicated mouse strains. (C) Statistical summary for (A) and (B). (D) The absolute number of γδNKT cells in indicated stages. (E) Representative FACS plots for IFNγ and IL-17 expression in γδNKT cells of thymus. (F) Summary for the percentage of IFNγ+ and IL-17+ cells in thymic γδNKT cells. The data are representative for 3 independent experiments.
Egr2 is required for increased PLZF expression in Id3−/− γδ T cells
PLZF is a key transcription factor that controls the differentiation of multiple innate populations, including γδNKT and iNKT cells (11, 41, 42). PLZF has been shown to be a direct target of Egr1/2 in iNKT cells(34). To address whether Egr2 can control PLZF expression in Id3−/− cells, we compared RNA and protein levels of PLZF in Id3−/− and Id3−/−Egr2−/− γδNKT cells. We found a 3-fold increase of PLZF transcription in Id3−/− γδNKT cells compared to WT γδNKT cells (Fig.3A). Egr2 deficiency returned PLZF transcription in Id3−/− γδNKT cells to WT levels. Likewise, intracellular staining for PLZF protein revealed that Id3 deficiency markedly increased the PLZF protein levels in γδNKT compared to WT γδNKT cells, and this was reversed by Egr2 deficiency (Fig.3B). This suggested a common role for Egr2 in regulating expression levels of PLZF in both iNKT cells and γδNKT cells. So far, our data demonstrated that in γδNKT cells, Egr2 lies upstream of PLZF, but is also controlled by a feedback regulation of the Id3/E2A axis.
Fig.3. PLZF expression controlled by Egr2 in Id3−/− γδNKT cells.
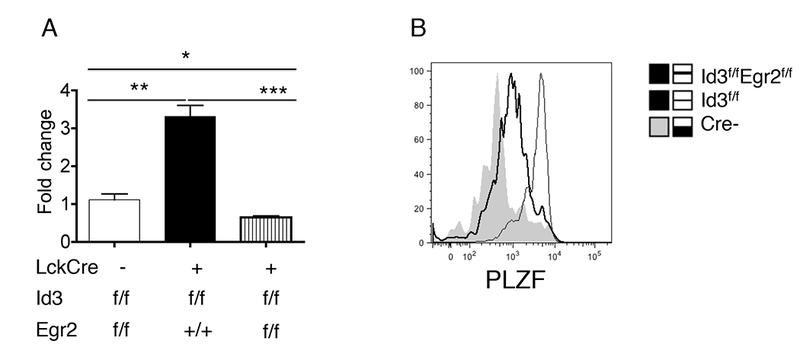
(A) The transcription of PLZF in thymic γδNKT cells from indicated mouse strains. (B) FACS analysis of PLZF protein expression in thymic γδNKT cells from indicated mouse strains. The data are representative for 3 independent mice.
c-Myc is responsive to the Egr2-Id3-E2A axis and required for Id3−/− γδNKT cell proliferation
c-Myc plays a critical role in cell cycle regulation(43), and has been reported to be an E2A target in a lymphoma cell line(44). Here, we explored whether c-Myc is involved in the Egr2-driven Id3−/− γδNKT cell proliferation. We found that Id3−/− γδNKT cells exhibited a significant increase in c-Myc transcription compared to WT cells (Fig. 4A). Egr2 deficiency mildly, but significantly, decreased c-Myc transcription. However, knockdown of c-Myc by shRNA did not affect Egr2 transcription (Fig. 4B and 4C). This data demonstrates that loss of Id3 enhances c-Myc transcription partially through the upregulation of Egr2. However, elevation of E protein activity in Id3-deficient γδNKT cells also appears to contribute to c-Myc induction in ways that are independent of Egr2.
Fig. 4. c-Myc is responsible to Egr2 and required for γδNKT cell proliferation.

(A) The transcription of c-Myc in γδNKT cells of indicated mouse strains. (B) Knockdown of c-Myc with retrovirus based shRNA. (C) Effect of c-Myc knockdown on Egr2 transcription. (D) Histogram of BrdU incorporation in Id3−/− γδNKT cells transduced with Mock or shRNA retrovirus. (E) Statistical summary for (D). (F) Histogram of BrdU incorporation in Id3−/−Egr2−/− γδNKT cells transduced with Mock or c-Myc overexpressing retrovirus. (G) Statistical summary for (F). Each data represents 3 independent experiments. The data are representative for 3 independent experiments.
To determine whether c-Myc contributes to Id3−/− γδNKT cell proliferation, we purified γδNKT cells from the thymus of neonatal Id3−/− mice, stimulated them with anti-CD3 and anti-CD28 antibodies in the presence of cytokine cocktails, and then transduced them with either mock or c-Myc shRNA for 48h. We found that compared to the mock control, knockdown of c-Myc significantly inhibited BrdU incorporation (Fig. 4D and 4E). Conversely, overexpression of c-Myc in Id3−/−Egr2−/− γδNKT cells, significantly enhanced proliferation, as evidenced by increased BrdU incorporation (Fig. 4F and 4G). Therefore, c-Myc is indeed involved in the proliferation of Id3−/− γδNKT cells, and controlled by high level of E-proteins and partially through Egr2.
Discussion
γδNKT cells, which exclusively express a Vγ1.1-Vδ6.3 TCR, have been considered as innate-like γδ lineage, distinct from the conventional γδ T cell compartment because of their uniformly high expression of PLZF and effector molecules. The mechanisms that control γδNKT cell population size are still unclear. Our study suggests that an Egr2-Id3-E2A feedback loop controls γδNKT cell lineage expansion. Normally, γδ TCR signaling activates a cascade that leads to phosphorylation of ERK and Egr1/2/3 induction, which then represses E protein activity through Id3, ultimately driving conventional γδ lineage development. However, here we report that elevated E protein activity in the absence of Id3 leads to upregulation of Egr2, PLZF, and c-Myc through a positive feedback loop, which then preferentially supports γδNKT lineage expansion.
Id3 deficiency not only increased the proportion, but also the absolute number of γδNKT cells. This suggests that a significant proliferation of γδNKT cells occurs in Id3−/− mice, which is also found in other mutant strains mention above. Indeed, BrdU incorporation experiments showed that Id3−/− γδNKT cells from neonatal, not young adult, mice had a significantly increased percentage of BrdU+ cells compared to WT(22). Consistent with this observation, adoptive transfer experiments employing adult bone marrow from WT and Id3−/− mice failed to replicate the expansion phenotype of γδNKT cells in Id3−/− mice(24). Furthermore, by analyzing TCR rearrangements in Id3−/− γδNKT cells, Verykokakis et al.,(24) showed that the junction sequences of Vg1.1-Jg4 and Vd6-Jd1 segments were highly narrowed and conserved. Additionally, the rearrangements exhibit the neonatal characteristic of lacking N nucleotide additions. Collectively, these evidences demonstrate that this population entered a robust proliferation primarily at the neonatal, but not the adult stage. The basis for this stage-restricted proliferation phenotype remains unclear.
TCR signaling strength has been shown to correlate with lineage commitment to the αβ or γδ T cell fate, such that strong TCR signaling favoring γδ T cell lineage differentiation(37, 45-47). There is also evidence in support of strong TCR signaling promoting the innate lineage differentiation of γδNKT. However, there are quite a few controversial studies supporting that dysfunction in TCR downstream molecules, such as Id3, KLF1, SLP-76, and Itk(10, 15), instead enhanced expansion of γδNKT cells(11, 21-25). At least in Id3−/− mice, γδNKT cell expansion appears to be supported by a feedback loop comprising increased Egr2 and PLZF expression, which is consistent with the strong TCR-signaling-induced elevation and sustainability of Egr1/Egr2 enhanced NKT cell lineage development(34). It seems that the dysfunction in TCR downstream molecules could reinforce the innate developmental pathway and allow an increase of innate lineage proliferation. γδNKT cells express a high level of CD5(8), an activation marker that is induced in proportion with TCR signal strength. This idea is in line with the view that γδNKT cells are auto-reactive and contribute to the occurrence of autoimmune diseases such as Sjögren’s Syndrome(14), and would therefore normally be eliminated by negative selection due to strong TCR signaling. Enforced expression of Vγ1.1/Vδ6.3 TCR has also been shown to be sufficient to support the generation of PLZF+ γδNKT cells, indicating that TCR signaling indeed plays a selective role in γδNKT lineage differentiation (8). Given the complex relationship between the Vγ1.1/Vδ6.3 TCR and its downstream signaling events, further investigation is needed to determine the signaling strength and quality generated by the stereotypic Vγ1.1/Vδ6.3 TCR.
The expansion of γδNKT cells in Id3−/− mice relies on high levels of E-proteins due to a release of Id3 inhibition(21, 23). Although we found that Egr2 is responsive to high levels of E-proteins and required for γδNKT cell expansion, a luciferase assay did not show evidence of E2A directly binding to the promoter region controlling Egr2 expression (data not shown). c-Myc, as a cell cycle regulator in multiple cell types(43, 48), has been demonstrated as one of E2A’s targets in a lymphoma cell line(44). We confirmed that c-Myc is responsive to high levels of E-proteins, contributes to γδNKT cell proliferation, and is partially dependent on Egr2 expression. Since Egr2-deficiency only partially reduced c-Myc expression, and not to WT levels, it is likely that the elevated levels of E-protein activity also regulate c-Myc in an Egr2 independent manner. In addition to Myc, it is also of interest to assess the relationship between the Id3/E2A axis and other pathways, such as SAP and Lin28, which have also been shown to be required for the development of γδNKT cells(11, 24, 49). However, these signals, including TCR, are not sufficient for such a dramatic expansion of γδNKT cells. We speculate that yet to be identified factors unique to the neonatal environment, such as specific antigens, nutrients, or signals, are essential to the expansion of γδNKT cells.
Overall, our study revealed that the level of Egr2 expression controls the population size of γδNKT cells. Id3, as a downstream target of Egr2, is involved in a feedback regulation to restrict the activity of Egr2 and thus to suppress the expansion of γδNKT cells. This mechanism may explain the general difficulty of detecting γδNKT development in the thymus. It remains to be discovered how such a repressive mechanism is alleviated exclusively in the neonatal thymus in the wild type mice.
Supplementary Material
Fig. 5. The feedback regulation loop of Egr2-Id3-E2A controls the development and expansion of γδNKT cells.
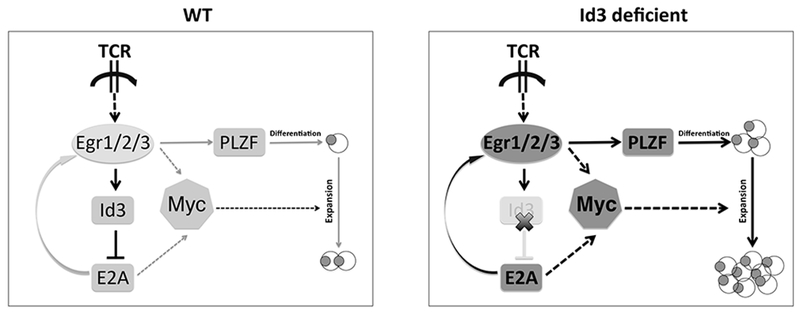
Sensing proximal TCR signaling, a cassette of downstream signaling is activated including pErk, Egr, Id3 and E2A molecules. The dosage of E-proteins is critical for γδ T cell lineage development. On the left side, normally pErk can activate Egr genes including Egr1, Egr2 and Egr3, then in turn to induce Id3 expression to suppress E-protein activity. The low dose of E-proteins could induce medium levels of Egr and PLZF gene expression and lead to the development of conventional γδ T cells and a very small population (1/10 in total γδ T cells) of γδNKT cells. On the right side, when Id3 is missing, the inhibition of E-protein activity is released, which strongly induces Egr2, Egr3 and PLZF expression to enhance γδNKT cell differentiation. Meanwhile, strong E-protein activity and Egr2 expression could elevate c-Myc expression to cause a dramatic expansion of γδNKT cells. Therefore, the regulation loop of Egr2-Id3-E2A controls γδNKT cell lineage differentiation and population size.
Acknowledgments
We thank the Flow Cytometry Facility of Duke Comprehensive Cancer Center for cell sorting, and thank Sumedha Roy, Ariana Mihai and Qun Wang for comments.
The funding supports are from NIH R01GM-059638 (Y.Z.), NIH P01AI102853 (Y.Z.), the National Basic Research Program of China 81771673 (B.Z.), and 1000-young talent program (B.Z.).
Reference
- 1.Carding SR and Egan PJ. 2002. Gammadelta T cells: functional plasticity and heterogeneity. Nature reviews. Immunology. 2:336–345. [DOI] [PubMed] [Google Scholar]
- 2.Xiong N and Raulet DH. 2007. Development and selection of gammadelta T cells. Immunological reviews. 215:15–31. [DOI] [PubMed] [Google Scholar]
- 3.Bonneville M, O’Brien RL, and Born WK. 2010. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nature reviews. Immunology. 10:467–478. [DOI] [PubMed] [Google Scholar]
- 4.Hayday A and Tigelaar R. 2003. Immunoregulation in the tissues by gammadelta T cells. Nature reviews. Immunology. 3:233–242. [DOI] [PubMed] [Google Scholar]
- 5.Jameson J, Ugarte K, Chen N, Yachi P, Fuchs E, Boismenu R, and Havran WL. 2002. A role for skin gammadelta T cells in wound repair. Science. 296:747–749. [DOI] [PubMed] [Google Scholar]
- 6.Azuara V, Levraud JP, Lembezat MP, and Pereira P. 1997. A novel subset of adult gamma delta thymocytes that secretes a distinct pattern of cytokines and expresses a very restricted T cell receptor repertoire. European journal of immunology. 27:544–553. [DOI] [PubMed] [Google Scholar]
- 7.Pereira P, Berthault C, Burlen-Defranoux O, and Boucontet L. 2013. Critical role of TCR specificity in the development of Vgamma1Vdelta6.3+ innate NKTgammadelta cells. Journal of immunology. 191:1716–1723. [DOI] [PubMed] [Google Scholar]
- 8.Kreslavsky T, Savage AK, Hobbs R, Gounari F, Bronson R, Pereira P, Pandolfi PP, Bendelac A, and von Boehmer H. 2009. TCR-inducible PLZF transcription factor required for innate phenotype of a subset of gammadelta T cells with restricted TCR diversity. Proceedings of the National Academy of Sciences of the United States of America. 106:12453–12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lees RK, Ferrero I, and MacDonald HR. 2001. Tissue-specific segregation of TCRgamma delta+ NKT cells according to phenotype TCR repertoire and activation status: parallels with TCR alphabeta+NKT cells. European journal of immunology. 31:2901–2909. [DOI] [PubMed] [Google Scholar]
- 10.Yin CC, Cho OH, Sylvia KE, Narayan K, Prince AL, Evans JW, Kang J, and Berg LJ. 2013. The Tec kinase ITK regulates thymic expansion, emigration, and maturation of gammadelta NKT cells. Journal of immunology. 190:2659–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alonzo ES, Gottschalk RA, Das J, Egawa T, Hobbs RM, Pandolfi PP, Pereira P, Nichols KE, Koretzky GA, Jordan MS, and Sant’Angelo DB. 2010. Development of promyelocytic zinc finger and ThPOK-expressing innate gamma delta T cells is controlled by strength of TCR signaling and Id3. Journal of immunology. 184:1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grigoriadou K, Boucontet L, and Pereira P. 2003. Most IL-4-producing gamma delta thymocytes of adult mice originate from fetal precursors. Journal of immunology. 171:2413–2420. [DOI] [PubMed] [Google Scholar]
- 13.Alonzo ES and Sant’Angelo DB. 2011. Development of PLZF-expressing innate T cells. Current opinion in immunology. 23:220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belle I, Mahlios J, McKenzie A, and Zhuang Y. 2014. Aberrant production of IL-13 by T cells promotes exocrinopathy in Id3 knockout mice. Cytokine. 69:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felices M, Yin CC, Kosaka Y, Kang J, and Berg LJ. 2009. Tec kinase Itk in gammadeltaT cells is pivotal for controlling IgE production in vivo. Proceedings of the National Academy of Sciences of the United States of America. 106:8308–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi Q, Xia M, Hu J, Hicks E, Iyer A, Xiong N, and August A. 2009. Enhanced development of CD4+ gammadelta T cells in the absence of Itk results in elevated IgE production. Blood. 114:564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belles C, Kuhl AK, Donoghue AJ, Sano Y, O’Brien RL, Born W, Bottomly K, and Carding SR. 1996. Bias in the gamma delta T cell response to Listeria monocytogenes. V delta 6.3+ cells are a major component of the gamma delta T cell response to Listeria monocytogenes. Journal of immunology. 156:4280–4289. [PubMed] [Google Scholar]
- 18.Matsuzaki G, Hiromatsu K, Yoshikai Y, Muramori K, and Nomoto K. 1993. Characterization of T-cell receptor gamma delta T cells appearing at the early phase of murine Listeria monocytogenes infection. Immunology. 78:22–27. [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura T, Matsuzaki G, and Nomoto K. 1999. The protective role of T-cell receptor Vgamma1+ T cells in primary infection with Listeria monocytogenes. Immunology. 96:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller C and August A. 2003. Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. Journal of immunology. 170:5056–5063. [DOI] [PubMed] [Google Scholar]
- 21.Ueda-Hayakawa I, Mahlios J, and Zhuang Y. 2009. Id3 restricts the developmental potential of gamma delta lineage during thymopoiesis. Journal of immunology. 182:5306–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang B, Dai M, Li QJ, and Zhuang Y. 2013. Tracking proliferative history in lymphocyte development with cre-mediated sister chromatid recombination. PLoS genetics. 9:e1003887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, Lin YY, Dai M, and Zhuang Y. 2014. Id3 and Id2 act as a dual safety mechanism in regulating the development and population size of innate-like gammadelta T cells. Journal of immunology. 192:1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verykokakis M, Boos MD, Bendelac A, Adams EJ, Pereira P, and Kee BL. 2010. Inhibitor of DNA binding 3 limits development of murine slam-associated adaptor protein-dependent “innate” gammadelta T cells. PloS one. 5:e9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan MS, Smith JE, Burns JC, Austin JE, Nichols KE, Aschenbrenner AC, and Koretzky GA. 2008. Complementation in trans of altered thymocyte development in mice expressing mutant forms of the adaptor molecule SLP76. Immunity. 28:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yasuda T, Bundo K, Hino A, Honda K, Inoue A, Shirakata M, Osawa M, Tamura T, Nariuchi H, Oda H, Yamamoto T, and Yamanashi Y. 2007. Dok-1 and Dok-2 are negative regulators of T cell receptor signaling. International immunology. 19:487–495. [DOI] [PubMed] [Google Scholar]
- 27.Dong S, Corre B, Foulon E, Dufour E, Veillette A, Acuto O, and Michel F. 2006. T cell receptor for antigen induces linker for activation of T cell-dependent activation of a negative signaling complex involving Dok-2, SHIP-1, and Grb-2. The Journal of experimental medicine. 203:2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Besin G, Yousefi M, Saba I, Klinck R, Pandolfi PP, and Duplay P. 2012. Dok-1 overexpression promotes development of gammadelta natural killer T cells. European journal of immunology. 42:2491–2504. [DOI] [PubMed] [Google Scholar]
- 29.Mashima R, Hishida Y, Tezuka T, and Yamanashi Y. 2009. The roles of Dok family adapters in immunoreceptor signaling. Immunological reviews. 232:273–285. [DOI] [PubMed] [Google Scholar]
- 30.Lee SY, Coffey F, Fahl SP, Peri S, Rhodes M, Cai KQ, Carleton M, Hedrick SM, Fehling HJ, Zuniga-Pflucker JC, Kappes DJ, and Wiest DL. 2014. Noncanonical mode of ERK action controls alternative alphabeta and gammadelta T cell lineage fates. Immunity. 41:934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marzec M, Halasa K, Liu X, Wang HY, Cheng M, Baldwin D, Tobias JW, Schuster SJ, Woetmann A, Zhang Q, Turner SD, Odum N, and Wasik MA. 2013. Malignant transformation of CD4+ T lymphocytes mediated by oncogenic kinase NPM/ALK recapitulates IL-2-induced cell signaling and gene expression reprogramming. Journal of immunology. 191:6200–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter JH, Lefebvre JM, Wiest DL, and Tourtellotte WG. 2007. Redundant role for early growth response transcriptional regulators in thymocyte differentiation and survival. Journal of immunology. 178:6796–6805. [DOI] [PubMed] [Google Scholar]
- 33.Lauritsen JP, Kurella S, Lee SY, Lefebvre JM, Rhodes M, Alberola-Ila J, and Wiest DL. 2008. Egr2 is required for Bcl-2 induction during positive selection. Journal of immunology. 181:7778–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seiler MP, Mathew R, Liszewski MK, Spooner CJ, Barr K, Meng F, Singh H, and Bendelac A. 2012. Elevated and sustained expression of the transcription factors Egr1 and Egr2 controls NKT lineage differentiation in response to TCR signaling. Nature immunology. 13:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Z, Li H, Han M, Xu T, Wu X, and Zhuang Y. 2011. Modeling Sjogren’s syndrome with Id3 conditional knockout mice. Immunology letters. 135:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hennet T, Hagen FK, Tabak LA, and Marth JD. 1995. T-cell-specific deletion of a polypeptide N-acetylgalactosaminyl-transferase gene by site-directed recombination. Proceedings of the National Academy of Sciences of the United States of America. 92:12070–12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haks MC, Lefebvre JM, Lauritsen JP, Carleton M, Rhodes M, Miyazaki T, Kappes DJ, and Wiest DL. 2005. Attenuation of gammadeltaTCR signaling efficiently diverts thymocytes to the alphabeta lineage. Immunity. 22:595–606. [DOI] [PubMed] [Google Scholar]
- 38.Zeng Y, Wagner EJ, and Cullen BR. 2002. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Molecular cell. 9:1327–1333. [DOI] [PubMed] [Google Scholar]
- 39.Lauritsen JP, Wong GW, Lee SY, Lefebvre JM, Ciofani M, Rhodes M, Kappes DJ, Zuniga-Pflucker JC, and Wiest DL. 2009. Marked induction of the helix-loop-helix protein Id3 promotes the gammadelta T cell fate and renders their functional maturation Notch independent. Immunity. 31:565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narayan K, Sylvia KE, Malhotra N, Yin CC, Martens G, Vallerskog T, Kornfeld H, Xiong N, Cohen NR, Brenner MB, Berg LJ, Kang J, and Immunological Genome Project C. 2012. Intrathymic programming of effector fates in three molecularly distinct gammadelta T cell subtypes. Nature immunology. 13:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, Chua K, Eidson M, Kim HJ, Im JS, Pandolfi PP, and Sant’Angelo DB. 2008. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nature immunology. 9:1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, Lantz O, and Bendelac A. 2008. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 29:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer N and Penn LZ. 2008. Reflecting on 25 years with MYC. Nature reviews. Cancer. 8:976–990. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, and Murre C. 2006. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proceedings of the National Academy of Sciences of the United States of America. 103:9976–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ciofani M, Knowles GC, Wiest DL, von Boehmer H, and Zuniga-Pflucker JC. 2006. Stage-specific and differential notch dependency at the alphabeta and gammadelta T lineage bifurcation. Immunity. 25:105–116. [DOI] [PubMed] [Google Scholar]
- 46.Hayes SM, Li L, and Love PE. 2005. TCR signal strength influences alphabeta/gammadelta lineage fate. Immunity. 22:583–593. [DOI] [PubMed] [Google Scholar]
- 47.Hayes SM, Shores EW, and Love PE. 2003. An architectural perspective on signaling by the pre-, alphabeta and gammadelta T cell receptors. Immunological reviews. 191:28–37. [DOI] [PubMed] [Google Scholar]
- 48.Hoffman B, Amanullah A, Shafarenko M, and Liebermann DA. 2002. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene. 21:3414–3421. [DOI] [PubMed] [Google Scholar]
- 49.Yuan J, Nguyen CK, Liu X, Kanellopoulou C, and Muljo SA. 2012. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science. 335:1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.