

Abstract
Significance: The two foremost concepts of aging are the mechanistic free radical theory (FRT) of how we age and the evolutionary antagonistic pleiotropy theory (APT) of why we age. Both date from the late 1950s. The FRT holds that reactive oxygen species (ROS) are the principal contributors to the lifelong cumulative damage suffered by cells, whereas the APT is generally understood as positing that genes that are good for young organisms can take over a population even if they are bad for the old organisms.
Recent Advances: Here, we provide a common ground for the two theories by showing how aging can result from the inherent chemical reactivity of many biomolecules, not just ROS, which imposes a fundamental constraint on biological evolution. Chemically reactive metabolites spontaneously modify slowly renewable macromolecules in a continuous way over time; the resulting buildup of damage wrought by the genes coding for enzymes that generate such small molecules eventually masquerades as late-acting pleiotropic effects. In aerobic organisms, ROS are major agents of this damage but they are far from alone.
Critical Issues: Being related to two sides of the same phenomenon, these theories should be compatible. However, the interface between them is obscured by the FRT mistaking a subset of damaging processes for the whole, and the APT mistaking a cumulative quantitative process for a qualitative switch.
Future Directions: The manifestations of ROS-mediated cumulative chemical damage at the population level may include the often-observed negative correlation between fitness and the rate of its decline with increasing age, further linking FRT and APT. Antioxid. Redox Signal. 29, 1003–1017.
Keywords: : aging, evolution, oxygen, reactive oxygen species, metabolism, chemistry, theory
“So I decided to look at the aging problem. I felt…that there had to be some common, some basic cause which is killing everything … a chemical process of some kind. Since we're dealing with molecules, chemicals … This is the simple basis in which we all exist.”—Denham Harman (73)“Any such small number of primary physiological factors is a logical impossibility if the assumptions made in the present study are valid… Such conclusions are always disappointing, but they have the desirable consequences of channeling research in directions that are likely to be fruitful.”—George Williams (136)
Introduction
Studies of aging (senescence) necessarily address two fundamental questions: how do we age and why? (20, 70, 78). Remarkably, the two most influential theories that address these questions are close age mates (Fig. 1). The mechanistic free radical theory (FRT) was published by Harman in 1956 (51), and the article by Williams presenting his evolutionary theory of aging based on gene pleiotropy appeared in 1957 (136). It was later termed antagonistic pleiotropy theory (APT) (104, 105).
FIG. 1.

Some milestones on the parallel tracks of FRT and APT of aging with focus on topics discussed in this article. FRT events are above and APT events are below the timeline. APT, antagonistic pleiotropy theory; FRT, free radical theory. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Scientific information did not circulate as rapidly and widely 60 years ago as it does now. The two theories were utterly independent, and there are no indications that their authors were even aware of each other. Had they been, the course of events associated with developing a coherent general theory of aging might have been different.
A noteworthy feature of both seminal publications is that neither provided an explicit definition of the phenomenon they sought to explain. Harman's approach in 1956 was very vague: “The phenomenon of growth, decline and death–aging.” Williams in 1957 preferred the term “senescence,” which corresponds rather to the second component of the Harman's triad already given. Williams emphatically warned against confusing the senescence of living bodies with the wear-and-tear of manufactured objects, basing his caution on the ability of living things, as distinct from inanimate things, to renew themselves; he regarded senescence as a decline manifested primarily in “decreasing probability of reproduction with increasing adult age.” Almost five decades later, Harman defined aging as “the accumulation of changes that increase the risk of death” (53). The latter definition is quite attributable to wear-and-tear, which increases the risk of malfunction or wreck. At the same time, the risk of death certainly decreases the probability of self-reproduction, of which manufactured objects are incapable. Therefore, the approach of Williams seems more general with regard to living things and more specific in a broader context due to differentiating living things from inanimate things. That said, both authors seem to have avoided defining aging (senescence) with a few words and, instead, each preferred to express his understanding in a more general way.
The two theories, running on parallel tracks, dramatically influenced research and conceptual advances in the aging field. However, their experimental analyses eventually turned inconclusive and even their theoretical bases came to be questioned [e.g., Gladyshev (34)].
Being related to essentially the same phenomenon, the theories, to be relevant, have to be mutually compatible. Their compatibility with each other and with concepts related to further aspects of aging are explored hereunder.
Antagonistic Pleiotropy Theory
The concept of antagonistic pleiotropy (AP) is rooted in population genetics and in evolutionary theory and consequently carries connotations that are not necessarily related to aging (86). Pleiotropic genes are those that influence several distinctive aspects of fitness. Antagonistically pleiotropic genes reinforce certain aspects of fitness and at the same time compromise other aspects. This phenomenon was first recognized at the population level to explain the persistence of alleles that produce negative effects on fitness in homozygotes, for example, the mutation responsible for sickle cell disease persists because it confers resistance to malaria in heterozygotes (11). Williams extended this notion to individual organisms by invoking pleiotropic genes “that have opposite effects on fitness at different ages or, more accurately, in different somatic environments” (136) to explain the pervasiveness of aging despite its negative effects on life-long fitness. Today, the “antagonistic pleiotropy” concept (or hypothesis) is almost unanimously attributed to the article published in 1957 (136); however, the term “antagonistic” is nowhere used in it. It was about two decades later that the phrase “antagonistic pleiotropy” was introduced by Rose (104) and was recognized as applicable to the concept put forward by Williams.
To give a mechanistic example of what he means, Williams considered the hypothetical possibility that a gene that enhances bone calcification might be advantageous in early life but associated with increased blood vessel wall calcification and hence deleterious in later life. Artery wall calcification is a hallmark of Mönckeberg's arteriosclerosis whose prevalence increases with aging. Ever since, aging-related AP has been sought mainly among genes implicated in late-onset diseases (13, 103). One of the genes suggested to show this behavior is indeed implicated in calcium homeostasis, although in a reverse manner: ALOX15 has a variant that enhances bone calcification early in life but increases the risk of postmenopausal osteoporosis (85). Other cases interpreted as exhibiting late-acting AP include (i) the p53 gene involved in protection from cancer on one hand and in cell death by apoptosis and cell senescence on the other (130); (ii) the apoE gene, whose apoE4 variant is both a factor for enhanced fertility and cognitive performance in young adults, and a risk factor for sporadic late-onset Alzheimer's disease (49, 63); and (iii) the Huntington gene with expanded series of CAG repeats, which is associated with both higher fecundity at reproductive age and with the late-onset Huntington's chorea (25). A reverse case is a mutation in a growth hormone receptor gene that results in the form of dwarfism known as Laron syndrome and is associated with decreased risks of late-onset diseases such as cancer and diabetes (77).
However, not everyone develops Alzheimer's or Huntington's disease, or cancer, or overt diabetes, yet everybody experiences age-associated decreases in glucose tolerance, muscular contractility, cardiac performance, nerve conductance velocity, and other functional deficits. Moreover, organisms quite different from humans and not subject to the mentioned late-onset human conditions nevertheless experience similar age-related decline. Even bacterial cells show aging when their interdivision periods are sufficiently extended (45, 93), as discussed hereunder in more detail. Therefore, if AP is responsible for all this, it should be sought among genes common to all forms of life, that is, the genes implicated in core metabolic and other vital functions. How can AP be related to such genes?
Another problem is that age-associated functional deficits do not appear all at once after a certain age or one after another in a defined order, but gradually develop in a parallel manner. How can this be reconciled with the commonly held view that AP effects, as distinct from beneficially pleiotropic effects, are manifested at postreproductive ages? This view is expressed, for example, in the following quotation: “Senescence can develop because some genes have nonseparable, but typically different or opposite, functions in reproductive-age and in old individuals (AP; Williams, 1957). Such genes, selected according to their “youthful” function, may thus impose a distinct senescent phenotype in old age” (141); “mutations that are damaging for the organism later in life (and hence contribute to senescence) could actually be favoured by natural selection if they are advantageous early in life, resulting in increased reproductive success of their carriers” (103). What then switches on the late effects?
Free Radical Theory
The FRT of aging is rooted in chemistry and radiobiology (73). The observations that the effects of irradiation mimic many manifestations of aging and may be mediated by hydroxyl radicals (•OH) prompted Harman to suggest that endogenous oxygen-derived free radicals are the main cause of aging (51). Harman further proposed the use of antioxidants to slow aging and extend lifespan. Initial experiments with β-mercaptoethylamine administration to mice were encouraging (52). However, subsequent elaborations of FRT [mitochondrial theory, oxidative stress theory, etc. (23, 53, 72)] led to much controversy (10, 33, 76, 84, 90, 119), and epidemiological studies provided no evidence that antioxidant preparations, including vitamins and food additives, increase human lifespan (106, 131). The recognition of the physiological signaling functions of reactive oxygen species (ROS) [reviewed in refs. (29, 56, 112)] added to the mounting confusion.
The molecules covered by the commonly used umbrella term “reactive oxygen species” include superoxide (•O2−), •OH, hydrogen peroxide (H2O2), and singlet oxygen 1O2. The basic chemical relationships between them, which in living cells usually involve transition valence metal (Me) ions, can be outlined as follows:

The chemical properties of ROS have been thoroughly studied and reviewed (56, 59, 112), largely for their involvement, first, in diseases and aging and, second, in signaling and, therefore, will not be detailed here. It is also increasingly recognized that various ROS readily react with nitric oxide, hydrogen sulfide, carbon monoxide, their derivatives, and probably other small messenger species capable of redox interactions to form a tightly integrated regulatory system, whose conditions change with aging and may influence its development as discussed in recent reviews (19, 65, 66).
All in all, it emerges that ROS are required for vitality and, at the same time, produce adverse effects implicated in aging. Is this not a mechanistic basis for AP? If it is, how may it be associated with genes, and what are these genes?
FRT and APT Meet Each Other
With regard to the relevance of ROS to AP, recent research has focused particularly on genes coding for the NOX (NADPH oxidase) family of enzymes (75). These enzymes catalyze the reduction of molecular oxygen by transferring one electron from NADPH to O2, producing superoxide anion radical •O2−:
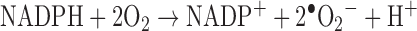 |
Several types of NOX enzymes are expressed in various cells in animals ranging from insects to primates. The best-known function of NOX enzymes is in phagocytes that produce oxidants to kill bacteria, fungi, and other pathogens. However, the oxidants invariably damage everything in their vicinity, including the intercellular matrix and its resident cells, of which possible delayed consequences are inflammation and autoimmune reactions. This is indeed an obvious case of AP: saving life and the associated ability to contribute to reproduction comes at the price of later decrease in vitality.
Relatively well studied is the role of NOX in insulin signaling, which involves a range of changes in the redox states of stress-activated protein kinases, insulin receptor substrate-1, protein tyrosine phosphatase B, PTEN, FOXO, and other signaling cascade components that may differentially depend on H2O2 levels (116). NOX involvement in these and other (97) cellular functions may contribute to the development of diverse pathologies of late life, most notably fibrosis, hypertension, insulin resistance, cancer, and neurodegenerative diseases (2, 54, 75, 135).
If an NOX enzyme gene is antagonistically pleiotropic because the useful product of the enzyme, that is, •O2−, reacts chemically with other biomolecules in ways that lead to diseases or compromise vital functions, why shouldn't this apply to other genes whose metabolic products have adverse chemical activities?
The list of known metabolites that can react in damaging ways with other metabolites, including those incorporated in macromolecules as their building blocks, is expanding (40, 41, 50, 83). Some representative examples of such metabolites are shown in Table 1. A more comprehensive listing is available in the Chemical Damage-MINE database (79).
Table 1.
Examples of Chemically Reactive Metabolites Implicated in Aging and Diseases
| Metabolite | Reactive derivative in fast equilibrium with its source | Target | Final reaction product | Relevance to aging and/or disease (reference) |
|---|---|---|---|---|
| d-Glucopyranose | d-Glucose | Arginine + lysine | Glucosepane | Protein crosslinking (88) |
| Phosphatidyl-ethanolamine | Fructosyl phosphatidylethanolamine | Membrane damage (91) | ||
| d-Ribofuranose-5-phosphate | d-Ribose-5-phosphate | Adenosine | N2-carboxyethyl-2′-deoxyadenosine | DNA damage (4) |
| Glyceraldehyde-3-phosphate | Methylglyoxal | Guanine | 6,7-Dihydro-6,7-dihydroxy-6-methyl-imidazo-[2,3-b]-guanine-9-one | DNA damage (100) |
| Urea | Isocyanate | Lysine | Homocitrulline | Protein damage, autoimmunity, renal failure (46) |
| Acyl-CoA | — | Lysine | ɛ-Acyllysine | Mitochondrial protein modifications implicated in aging (57) |
| Pyruvate | — | Tryptophan or tryptamine | Carboline derivatives | Neurotoxicity (55) |
| Water | — | Deoxyadenosine | Adenine and deoxyribose | Depurination of DNA (82) |
| Dopamine | — | Cysteine | 5-S-cysteinyldopamine | Protein damage, neurotoxicity (36) |
It should be stressed that the inherently reactive metabolites in Table 1 do not need molecular oxygen or free radicals for the initiation of their damaging effects, although under aerobic conditions these effects may be significantly modified by oxygen and ROS, as exemplified in Figure 2. Still, these metabolites would have been able to do their damaging work before aerobic life began.
FIG. 2.

Oxygen interference with an anaerobic pathway of protein damage by glycation. (I) Reversible Schiff base formation; (II) Irreversible Amadori rearrangement; (III) Enolization; (IV) Double bond migration; (V) Dehydration; (VI) Condensation of α-dicarbonyl with arginine; (VII) Enediol oxidation (associated with the generation of reactive oxygen species, including hydrogen peroxide); (VIII) Oxidative cleavage of α-dicarbonyl. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
ROS and the Evolutionary Origin of Aging
Did senescence first emerge when cells acquired the ability to make metabolic use of oxygen (with the associated ROS production), or did it emerge earlier, when cells started generating oxygen as a byproduct of photosynthesis (Fig. 3)? Should APT be extended to the genes whose products are responsible for the endogenous generation or use of oxygen (and, by implication, for its reactive forms)?
FIG. 3.
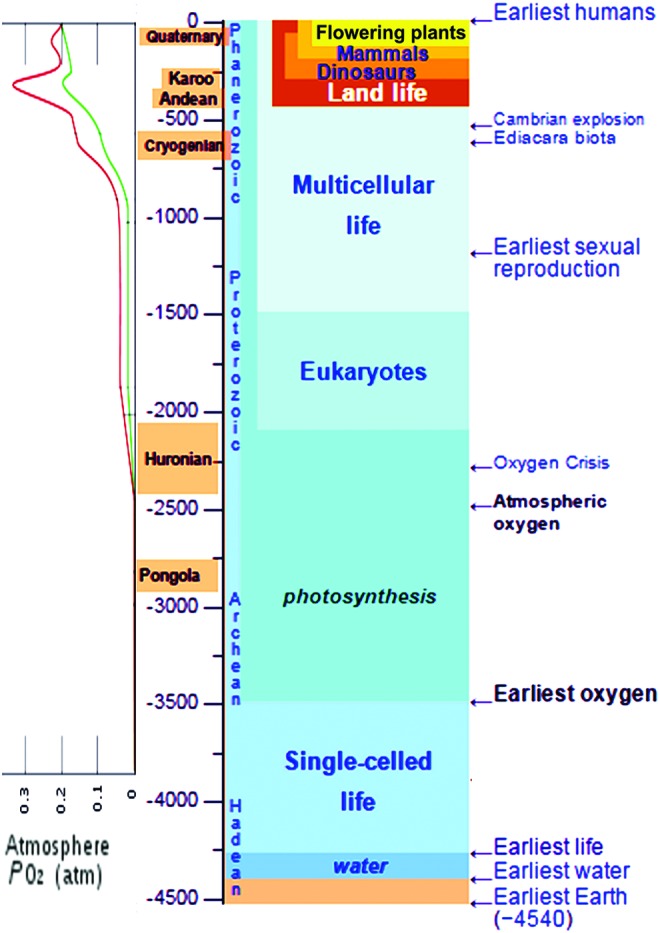
The history of molecular oxygen and life on the Earth. The red and green curves on the left show the highest and lowest PO2 estimates, respectively (based on https://en.wikipedia.org/wiki/Geological_history_of_oxygen). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In this regard, it is important that the photosynthetic machinery that introduced oxygen into life's environment most probably evolved under selection pressures defined by (i) the need to gain energy as reducing molecules were depleted and (ii) the need to protect cells from ultraviolet (UV) when there was no ozone layer, with the pigment molecules initially used as UV screens being subsequently recruited as light acceptors in photosynthesis (14). Much of the UV-induced damage to cells is mediated not only by singlet oxygen and ozone but also by free radicals and the resulting chain reactions, which can be initiated by photosensitizers such as tryptophan and tyrosine, available in primordial cells. The availability of free iron (which was not yet precipitated in the form of oxides during earth's early history) would have added to the initiation and propagation of free radical reactions. Moreover, H2O2 is hypothesized to have been present right from the dawn of life (6). ROS generation in cells even before the emergence of the aerobic life is, therefore, plausible. Indeed, phylogenetic studies indicate that peroxidases could have been present in the last universal common ancestor (LUCA) (35, 114, 115). Molecular oxygen could thus have been generated endogenously in primordial anaerobic cells by the detoxification of peroxides (115).
Because metabolites other than ROS can damage macromolecules (Table 1), it is hard to assess the contribution of ROS to the overall endogenous damage suffered by LUCA cells. Most likely, it was far less significant than it is now. In any case, could molecular damage give rise to anything resembling aging in LUCA? To consider this issue, it is necessary first to decide whether the concept of aging, at least in the sense already outlined in the introductory section, can be applied to unicellular organisms that multiply by division only.
Obviously, any damage that accumulates in slowly turning-over macromolecules, including proteins and DNA, will be diluted by increasing the normal biomass produced by cell growth and division. More time between successive cell divisions will provide more opportunities for damage to accumulate in a cell. Can an increase in interdivision time lead to damage accumulation to a level sufficient to compromise cell viability? A recent experiment indicates that it may be so. Tracking single Escherichia coli cells in microfluidic chambers revealed negative correlations between the rates of cell proliferation and cell death (67). Furthermore, it is known that metabolite damage products can accumulate with time in bacteria (45, 93).
The fact that damage can accumulate in unicellular organisms up to levels that compromise viability is confirmed by the existence of special mechanisms “molded” by evolution to protect cell structures from endogenous damage and to repair damaged structures (1, 102), to excrete damaged macromolecules (17), and to ensure an uneven partitioning of damage between daughter cells (16, 47), the progeny of one being destined to die out from damage overload, leaving the other rejuvenated and able to produce damage-free progeny.
Taken together, the mentioned considerations suggest that unicellular prokaryotes can age under conditions resulting in an extension of their interdivision time sufficient for endogenous damage to accumulate to levels compromising cell vitality. LUCA was most probably subject to this rule. The aging experienced by primordial unicells could have resulted from the intrinsic chemical properties of metabolites, such as those shown in Table 1, including those incorporated in macromolecules. These properties already suffice to cause aging, regardless of the role of other factors. Therefore, the assertion that aging is “molded” by natural selection (48) is not fully correct because the propensity for aging has been an independent factor to be coped with by natural selection ever since life emerged from inanimate matter (38, 40). Molded indeed were species-specific adaptive traits on which the ever present “prototypic driving force of aging” (40) could act to produce species-specific senescent phenotypes.
From the dawn of life onward, aging could gradually increase the probability of death of unicellular organisms, depending on the time elapsed from their birth by mother cell division, and, therefore, must have been a factor in determining the overall cell loss from the population. The equilibration of cell loss rate with cell proliferation rate can establish a stationary level of damage in a cell population. The level of damage can be kept within tolerable limits either (i) by increasing the rate of cell division or (ii) by intensifying the protection of macromolecules from damage and/or the repair/clearance of damage. Both the protection and the repair options consume resources and thus come at a cost. In particular, protection enzymes are expensive to build and some of them consume energy/metabolic currency, for example, the consumption of NAD(P)H in the reduction of reactive carbonyl compounds. Protection and repair become more important under conditions that reduce the rate of proliferation and thus increase the time available for damage to accumulate.
This simple reasoning is sufficient to suggest that tradeoffs between reproduction and protection from endogenous damage probably existed long before the emergence of the dichotomy between germ cells and soma, which is the basis of another influential evolutionary concept of aging, the disposable soma theory (DST) (22, 69, 70).
The contribution of oxygen and ROS to the aging of LUCA cells should not be discounted, for otherwise there would have been no need for peroxidases; however, other factors such as methylglyoxal and other electrophiles (Table 1) were probably much more significant before the emergence of photosynthetic O2 production. The same must have been true for the first cells that started to produce molecular oxygen as a byproduct of photosynthesis. This is where AP related to O2 and associated ROS production and handling begins to enter the picture. However, the estimates of O2 levels at the sites of its generation based on possible relationships between the rates of O2 production, diffusion, and dissipation in the environment suggest that O2 levels were initially very low (68). It took about 1 billion years to increase environmental O2 (and corresponding intracellular O2) to levels associated with the so-called “oxygen crisis,” aka “the Great Oxygenation Event” (GOE) (Fig. 3).
One consequence of the GOE was that most then-existing forms of life were poisoned by O2 and driven to extinction, except for those that managed to enhance their preexisting antioxidant protection mechanisms and/or to develop novel mechanisms (14, 28). Some of those that succeeded in protecting themselves from oxygen even managed to make use of it as the final acceptor for electrons taken from nutritional substrates and carried to oxygen via a series of intermediate acceptors. The resulting redox energy gradient was coupled in the respiratory chain with the transmembrane gradient of proton concentration, which could be used to produce high-energy compounds. The surplus source of chemical energy started another story, which proceeded via the emergence of eukaryotes, sexual versus clonal mode of proliferation, multicellular organisms with the associated dichotomy between germ cells and soma, and, finally, nonrenewable structures used in the soma to protect the germ cells. As far as we know, all this would have been impossible without the giant leap in energy availability provided by oxygen reduction (14, 101, 138).
ROS and the Evolution of Aging in Metazoans
Simple mathematical modeling (33) shows that if the emergence of a new function increases the chance that its host will survive to reproductive age, then this function will enhance the overall reproductive output of the population even if its operation gradually decreases individual reproductive fitness because of damage accumulation in nonrenewable structures. One such “new function” is ROS generation by NOX enzymes, which appeared very early in the phylogeny of metazoans (60).
There are, however, some metazoan species that, although aerobic, show no evidence of aging, at least in the sense that their mortality rate does not increase and fecundity does not decrease over time. One example is the cnidarian Hydra vulgaris (87, 109, 128). Such species are distinctive in that they can regenerate themselves from virtually any part of their bodies. All of their cell populations are renewable, as well as the small amounts of intercellular matrix materials secreted by the cells; therefore, even if each of their individual cells can produce and accumulate damage and is thus subject to aging in the sense applicable to unicellular organisms, the overall damage in their multicellular bodies can be kept at a tolerable stationary level by the turnover of cell populations, just as in a clone of a unicellular organism.
Other taxa that might contain nonaging organisms (i.e., organisms that are potentially immortal if protected from all external causes of death) include sponges, flatworms, and echinoderms (9, 98, 99, 134) (Fig. 4). Yet, their regenerating potencies constitute only necessary but not sufficient conditions for nonaging. A recent study of the flatworm Macrostomum lignano showed that this organism does age (89). Perhaps the most interesting case is placozoans, because they are likely to be the closest to the common root of all metazoans (99, 120). However, they are also least studied with regard to aging, and it is still not clear whether the early phylogenetic pathway to chordates passed through ancient placozoans. As to the other species whose entire bodies are renewable, they belong to taxonomic dead ends that clearly diverge from the route leading to chordates and could have evolved to occupy very specific aquatic niches (9). In particular, all of these organisms consume oxygen through their entire surface, that is, need no breathing and specialized oxygen adsorbing and transporting apparatuses.
FIG. 4.

Evolutionary relationship between the ability for total regeneration and aging. Dashed phylogenetic branches indicate uncertain positions. Note that most metazoan phyla have not been studied with regard to aging. Based on Telford et al. (127) and Bely (7). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Because only nonrenewable structures, such as the endo- and exoskeleton, the extracellular matrix, and the brain, provide place and time for accumulation of damage produced by ROS and other reactive endogenous metabolites, the emergence of such structures is what made aging such a prominent characteristic of complex organisms. Since the reduction of molecular oxygen is the only plausible source of energy required for the emergence of nonrenewable structures (14), O2 with its associated reactive forms is indeed indispensable for the evolutionary origin of senescence, in the sense that without O2 there would have been no metazoans elaborate enough to senesce.
Can the use of oxygen for energy production by mitochondria be associated with AP attributable to a specific gene of a multicellular organism? Evidence on this point comes, for instance, from knockout and knockdown strains of the roundworm Caenorhabditis elegans, whose genes coding for the respiratory chain proteins were targeted. An analysis of several dozen cases (21) led to the conclusion that in no individual case can the observed changes in lifespan and/or aging rate (see hereunder a discussion of differences between these parameters) be unequivocally attributed to changes in energy supply for vital functions or changes in ROS production or action. There were always significant confounding factors, such as compensatory changes in downstream, upstream, regulatory, or competing pathways and mechanisms. However, taken together, the data suggest that gains in lifespan are generally associated with decreases in damage associated with ROS generation, and that these gains usually come at the cost of compromised fecundity, motility, or other components of fitness (21).
There are at least two approaches that explain this cost. According to one approach, less vigor is associated with less oxygen consumed to produce energy and less ROS generated to produce damage. This approach is generally consistent with the rate-of-living theory “live fast, die young” [see Muller et al. (90)]. According to the other approach, lesser vigor spares more resources that may be allocated to protection from ROS and thus to the maintenance of vigor. This is generally consistent with DST, whose main emphasis, however, is on the tradeoff between allocation of resources for soma protection from damage (somatic maintenance) and germ cells' propagation to progeny (proliferation) (69, 70, 132). The DST can be regarded as extending or complementing the APT (31). Nevertheless, as already noted, the split between soma and germ cells, which is implicit in DST, is not necessary for the emergence of tradeoffs between maintenance and reproduction; such tradeoffs may well operate in unicells that multiply by division. Something that foreshadows disposable soma may be recognized only in the cases of uneven partitions of damage between daughter cells.
Are There Populational Manifestations of AP, and How Do They Relate to ROS?
Extending the mentioned considerations to higher organisms requires still more caveats. First, changes in body properties resulting in an increase in the mean (or maximal, median, 95th percentile, etc.) lifespan can be achieved in two ways: by increasing the initial vitality (vigor) or by decreasing the rate of its decline. The difference between the two options is captured by the Gompertz–Makeham law of mortality (30, 37, 38, 71, 95):
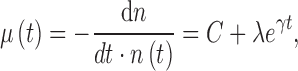 |
where μ is the rate of decrease in the number of objects n having age t (age-specific mortality rate), λ is inversely proportional to the initial vitality (at t = 0), and γ reflects the rate of its decline, whereas C reflects the contribution of factors that do not depend on age to the overall mortality. Highly idealized illustrative plots showing changes in lifespan distributions as they depend on the different parameters of the Gompertz–Makeham law are shown in Figure 5.
FIG. 5.

Illustrative plots of changes in lifespan depending on changes in the parameters of the Gompertz–Makeham law. Upper row: survivorship curves, n(t); middle row: age-at-death (lifespan) distributions, −dn/dt; lower row: logarithms of mortality force ln[−dn/(dt × n(t))]. Lifespan may increase because (A) the age-independent mortality C decreases; (B) the rate of aging γ decreases; or (C) the initial vigor (1/λ) increases. The column (D) shows a case when γ and λ are linked according to the SM correlation: greater initial vigor is associated with faster aging rate; for example, an increase in the availability or use of aerobic energy is associated with a greater rate of damage to macromolecules. On the semilogarithmic plots of μ versus t (the lower row), increases in C make the plots increasingly curved, changes in γ are manifested in plot slopes, and changes in λ in the parallel shifts of the plots, whereas negatively correlated changes in λ and γ make the plots converge. Note that when C is small, its changes may mimic patterns that correspond to the SM correlation. SM, Strehler–Mildvan.
Parenthetically, data-derived μ(t) plots related to postmaturation age periods usually contain no special points that may be attributed to the ages when gene effects switch from beneficial to antagonistic. This is consistent with the idea that AP effects are not switched on late in life but are accumulating lifelong (see the ROS and the Evolutionary Origin of Aging section). There are also no points suggesting a defined limit to the longevity of an individual. This means that aging does not lead to death at a certain species-specific age; it “only” increases the probability to die within a certain interval of time (in other words, increases the probability density of death). The increase starts long before the last members of successive cohorts of a population die; therefore, the ages at death of individuals belonging to a defined species may be very different. However, because the increase in mortality rate is exponential, mortality eventually becomes so high that most members of any of the successive finite cohorts of a given species will die within a narrow interval of time since their birth, the interval being close to the time when the entire finite cohort is exhausted by mortality, that is, close to the age-at-death of the last member of the cohort. Somewhat counterintuitively, this means that the maximal observed lifespan must correlate with the size of the population where it is observed. Indeed, such effects can easily be modeled and have been observed (26, 137). An implication of that mentioned is that the parameters of mortality kinetics (γ and λ and C in the Gompertz–Makeham model), not of lifespan, are what must really matter.
A meta-analysis of available evidence for strains and mutants of C. elegans and Mus musculus suggests that γ is more variable than λ (Fig. 5B) in C. elegans, whereas the opposite is true (Fig. 5C) for mice (58). This means that genetic interventions influence primarily the rate of aging in roundworms and the initial vitality in mice. The same is generally true for other interventions, except for calorie restriction, which in rodents appears to decelerate aging (to decrease γ) quite robustly (113).
In the analysis of studies wherein survivorship data were sufficient to reveal relationships between γ and λ, these parameters were found to show a negative correlation (111, 113) consistent with the equation lnλ = A − Bγ (Fig. 5D). To extend this observation further, it may be noted that the roughly twofold increase in human lifespan over the last century is almost completely attributable to a decrease in C (Fig. 5A), whereas small changes in λ and γ are such that a decrease in λ is associated with an increase in γ, making these parameters negatively correlated [(30, 38, 42, 124) and references therein].
This effect is known as the Strehler–Mildvan (SM) correlation, or as the compensational effect of mortality (30, 123). It may mean that, within a given species, a gain in vitality is achieved at the expense of its accelerated decline. The significance, implications, and even existence of the SM correlation are debated (11, 27, 80, 140). Some authors regard it as a mathematical artifact (11, 126). However, there are reasons to believe that it has a real biological basis (38).
To express this basis in APT terms, one may say that a change in the (epi)genetic makeup associated with an increase in early fitness (1/λ) is supported by natural selection even if it is associated with a greater decrease in fitness at a later time because of the acceleration of the decrease (of the rate of aging, γ).
To express the basis in terms of relationships between aerobic energy supply and ROS-mediated damage to living bodies, one may say that an enhancement of vital functions made possible by an increase in the available aerobic energy is associated with the accelerated attenuation of this and other functions because of damage caused by ROS. This is somewhat reminiscent of the rate-of-living theory of aging. The difference is that the correlation between vitality and the rate of its decline is based not so much on simple stoichiometric relationships and balances as on numerous superimposed nonlinear regulatory interactions involving molecular mechanisms, which are becoming known in increasing detail.
To start with, a textbook notion is that increasing ATP consumption for performing this or that function is associated with increases in the levels of ADP and AMP. ADP is known to be a positive allosteric regulator of rate-limiting Krebs cycle enzymes, most notably, NAD+-dependent isocitrate dehydrogenase. This alone may result in increased NADH production and, thus, in “pushing” electrons to the respiratory chain resulting in its overall reduction, which predisposes to •O2− generation.
Moreover, AMP activates the AMP-dependent protein kinase (AMPK), a sensor of energy deficit, which activates via a number of signal transduction pathways, the mechanisms that facilitate energy generation, such as glucose transmembrane transport and fatty acid β-oxidation (64), thus reinforcing respiratory chain feeding with electrons derived from oxidized substrates.
Moreover, some Krebs cycle enzymes, most notably the FAD-dependent pyruvate and α-ketoglutarate dehydrogenases, have recently been shown to generate •O2− and H2O2 at rates comparable with that attributed to Complex I of the respiratory chain [reviewed in ref. (86)]. Irrespective of the possible signaling functions of H2O2 generated in this way, including the inhibition of α-ketoglutarate dehydrogenase itself and of aconitase, the net result is that increasing metabolic flux through the Krebs cycle must be associated with increasing ROS generation. Relationships responsible for this are thus far beyond stoichiometric balances and the law of mass action.
Still more, increased oxygen consumption by mitochondria in response to functional demands has been shown in very different cell types, that is, muscular (94), neuroendocrine (139), and pancreatic (108), to decrease O2 level to extents associated with the activation of hypoxia-inducible factor (HIF-1), which traditionally is considered in the context of changes, usually decrements, in oxygen supply rather than consumption. HIF-1, a constitutively expressed transcription factor, is destabilized when its proline residues are modified by dedicated dioxygenases, which split oxygen for hydroxylation of proline in HIF-1 and for concomitant oxidative decarboxylation of α-ketoglutarate. As a result, reduced oxygen leads to HIF-1 stabilization and increased stationary levels and to the resulting upregulation of the expression of numerous genes controlled by HIF-1, which are implicated in facilitating oxygen supply to cells, their effects being realized at the tissue (neovascularization) and systemic (erythropoiesis, etc.) levels.
Another facet of HIF-1 activity, which is better known because of its involvement in protecting cells from hypoxia/reoxygenation, is activation of genes encoding ROS-detoxifying enzymes. This is likely to moderate increases in extramitochondrial ROS-mediated damage associated with stimulation of aerobic functional capacities. In contrast, HIF-1 is implicated in stimulation of intramitochondrial ROS generation, probably for signaling purposes, including AMPK stimulation (117).
Yet another facet of the whole situation is the metabolic cost of the repair of DNA damage caused by ROS. Single strand break (SSB) repair requires NAD+ for poly(ADP-ribose) formation by poly(ADP-ribose)polymerases (PARP). Thus, increasing SSB is associated with NAD+ depletion (5), which makes a case of competition between reparative and functional activities for common resources and, at the same time, a case of functional activity as a cause of ROS-mediated damage, which requires channeling a part of the resources away from this same functional activity. PARP also compete with sirtuins for their common substrate NAD+ (133). Other aspects of PARP interference with energy metabolism and associated ROS generation are discussed in Bal et al. (5).
All of that mentioned should be regarded as only parts of a bigger picture because HIF-1 and AMPK are much discussed in current literature with regard to their relationships with other hubs of functional networks implicated in aging, such as sirtuins, mTOR, and FOXO (64, 96, 107), each of which is more or less relevant to ROS in aging. The molecular aspects to be taken into account are so numerous and tangled that any inferences from an inevitably limited set thereof should be correlated with observations at the higher levels to which the inferences are intended to refer.
So what, if any, is the physiological evidence for ROS-related tradeoffs between vitality and the rate of its decline? This question has been tackled by ecological physiology or functional ecology studies. Caveats in interpreting the results of such studies were discussed in Cohen et al. (18), the conclusion being that “free radicals can have beneficial roles in signaling as well as causing damage and should not be interpreted out of context” and “despite many approaches for manipulating oxidative stress, it seems to have little effect on ageing. One hypothesis is that antioxidant defenses are already optimized, and thus ROS are not a causal mechanism of ageing, even if they can be disrupted during ageing (as a secondary effect) and in particular diseases (as the drivers of disease).”
Still, there are notable observations with regard to the SM correlation as a manifestation of AP mediated by ROS. Some illustrative examples are presented hereunder.
A meta-analysis of studies that address the relationship between growth rate and oxidative stress concluded that, within a particular species, higher growth rates are associated with greater oxidative damage to tissues (118). Increases in the levels of markers of damage occur despite their possible dilution by increasing biomass. No consistent effects of antioxidants were found. No straightforward extrapolations of these observations to humans are possible because greater body sizes (apart from adiposity) are achievable not because of more available calories but because of better overall conditions of development. It is true that increases in life expectancy over the last century were paralleled by increases in body size (92). However, there is no evidence of associated decreases in the rates of demographic aging. Rather, increases in the initial vitality correlate with increases in demographic aging rate (the SM correlation, see above).
Another notable topic related to the role of ROS in physiological tradeoffs is the predator–prey relationship. For example, it has been shown that in water-dwelling insect (damselfly) larvae under predation risk and the associated increased energy expenditure for fight-or-flight responses, •O2− levels and lipid peroxidation were increased, growth rate was decreased, and the subsequent responses to risk were blunted (60, 61).
An ever-hot topic in this area is the relevance of oxidative stress to the cost of reproduction. Some authors have concluded that litter size positively correlates with subsequently measured parameters of oxidative stress and that oxidative stress plays a key role in life history evolution (121). In a recent large-scale epidemiological study in humans, parity and associated lactation have been found to correlate with oxidative stress markers (142). An independent study (110) has shown that elevated markers of “derivatives of reactive oxygen metabolites” correlate with all-cause mortality, mainly attributed to cardiovascular diseases and cancer. Taken together these findings suggest that ROS are involved in the association of higher parity with higher subsequent mortality and thus, the aging rate, at least in the demographic sense.
Together, these studies support the idea that ROS are particularly significant contributors to the processes that drive aging and are responsible for tradeoffs interpretable in terms of APT. However, the aggregate contribution of all other drivers must be no less significant. Such drivers include, among others:
Damage to macromolecules, which is caused by excessive chemical potencies of metabolites other than oxygen and ROS, such as carbonyl-containing metabolites, acyl phosphates, or even water, which can damage DNA by spontaneous hydrolysis (40, 41, 50, 79). Nonenzymatic reactions caused solely by the chemical properties of their participants (and the respective theoretical approach to aging) have been termed parametabolic (38, 41).
Protein misfolding, which can be spontaneous, but can be enhanced by ROS-related and other parametabolic reactions (3, 24, 74, 125).
Telomere attrition, which can be accelerated by ROS (44, 62, 90, 122).
Cell senescence, which is partially attributable to telomere attrition and can be accelerated by ROS (8, 15), but can also take place irrespective of telomere attrition and ROS effects because of the inherent stochasticity of the molecular processes underlying cell proliferation and differentiation (39, 43).
DNA damage associated with transcription (12) and replication (81, 129).
The aggregate result of operation of all these and other factors, ROS included, is cumulative (Fig. 6) and is conceptualized as the deleteriome, whose composition becomes increasingly complex and whose impact on fitness becomes increasingly significant as aging advances (34).
FIG. 6.

Relationships between pleiotropic beneficial and antagonistic effects of two hypothetical genes. Gene A codes for the enzyme that makes metabolite M2, which can give rise spontaneously to a damaging product D. D can directly damage DNA, thus making Gene A antagonistically pleiotropic, and can induce the aggregation of the protein product of Gene B, thus making Gene B antagonistically pleiotropic. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In summary, although oxygen is indispensable for the emergence of organisms having nonrenewable structures for which aging may become significant, ROS are not mechanistically necessary for aging, although they may be—and usually are—important contributors to it. In the course of evolution, ROS were superimposed on preexisting endogenous chemical factors of aging, which continue working in the background, and their relative contribution may increase when ROS-mediated damage is attenuated. This could be partly because ROS-related and -unrelated damaging factors compete for common targets, so that attenuating one type of damage opens the way for the other type to occur.
Concluding Comments
This review offers a birds-eye view of the evidence and the reasoning relevant to the role of ROS in aging. The evidence ranges from pure chemistry to natural selection, metabolism, biological demography, and ecological physiology. Much of the reasoning resides at the interface of the mechanistic FRT of aging and the evolutionary APT. We suggest that these theories, although incomplete, are deeply compatible with each other, as well as with other relevant aspects of aging. Our arguments run as follows:
Aging has not been “molded” by natural selection to optimize ecological or population-genetic tradeoffs, and it is not the consequence of some unfortunate frozen accidents missed by selection. Instead, aging is the outworking of inescapable constraints imposed on natural selection by the chemical properties of biomolecules. These constraints have been operating since life sprang from inanimate matter, and thus they contribute independently to tradeoffs important for the evolution of life histories.
ROS are only one component of the mentioned constraints and were added to its preexisting components when aerobic metabolism emerged. In the extant aerobic forms of life, ROS are particularly significant drivers of aging; however, the aggregate contribution of all other driving forces is no less significant.
AP does not involve late-acting switches from good to bad effects, but results from lifelong cumulative damaging effects, which are not limited to just a few genes whose action is considered at different points in lifespan. ROS-mediated damage fits this approach to APT well enough to make the two concepts, APT and FRT, mutually consistent.
Although the historical primacy of FRT is indisputable, logically it is only a specific embodiment of more general concepts of why and how we age, that is, the closely related concepts of metabolite damage (50, 79, 83), parametabolic damage (38, 40, 41), and imperfectness culminating in the deleteriome (32, 34).
Abbreviations Used
- AMPK
AMP-dependent protein kinase
- APT
antagonistic pleiotropy theory
- DST
disposable soma theory
- FRT
free radical theory
- GOE
great oxygenation event
- HIF-1
hypoxia-inducible factor
- LUCA
last universal common ancestor
- NOX
NADPH oxidase
- PARP
poly(ADP-ribose)polymerases
- ROS
reactive oxygen species
- SM
Strehler–Mildvan
- SSB
single strand break
- UV
ultraviolet
References
- 1.Abdallah J, Mihoub M, Gautier V, and Richarme G. The DJ-1 superfamily members YhbO and YajL from Escherichia coli repair proteins from glycation by methylglyoxal and glyoxal. Biochem Biophys Res Commun 470: 282–286, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Rahman EA, Mahmoud AM, Aaliya A, Radwan Y, Yasseen B, Al-Okda A, Atwa A, Elhanafy E, Habashy M, and Ali SS. Resolving contributions of oxygen-consuming and ROS-generating enzymes at the synapse. Oxid Med Cell Longev 2016: 7, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adrover M, Marino L, Sanchis P, Pauwels K, Kraan Y, Lebrun P, Vilanova B, Munoz F, Broersen K, and Donoso J. Mechanistic insights in glycation-induced protein aggregation. Biomacromolecules 15: 3449–3462, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Akhter F, Salman Khan M, Shahab U, and Moinuddin AS. Bio-physical characterization of ribose induced glycation: a mechanistic study on DNA perturbations. Int J Biol Macromol 58: 206–210, 2013 [DOI] [PubMed] [Google Scholar]
- 5.Bai P, Nagy L, Fodor T, Liaudet L, and Pacher P. Poly(ADP-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol Metab 26: 75–83, 2015 [DOI] [PubMed] [Google Scholar]
- 6.Ball R. and Brindley J. The life story of hydrogen peroxide III: chirality and physical effects at the dawn of life. Orig Life Evol Biosph 46: 81–93, 2016 [DOI] [PubMed] [Google Scholar]
- 7.Bely AE. Evolutionary loss of animal regeneration: pattern and process. Integr Comp Biol 50: 515–527, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, and Csoka AB. Cellular senescence as the causal nexus of aging. Front Genet 7: 13, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bilinski T, Bylak A, and Zadrag-Tecza R. Principles of alternative gerontology. Aging (Albany NY) 8: 589–602, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buffenstein R, Edrey YH, Yang T, and Mele J. The oxidative stress theory of aging: embattled or invincible? Insights from non-traditional model organisms. Age 30: 99–109, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burger O. and Missov TI. Evolutionary theory of ageing and the problem of correlated Gompertz parameters. J Theor Biol 408: 34–41, 2016 [DOI] [PubMed] [Google Scholar]
- 12.Callegari AJ. Does transcription-associated DNA damage limit lifespan? DNA Repair (Amst) 41: 1–7, 2016 [DOI] [PubMed] [Google Scholar]
- 13.Carter AJ. and Nguyen AQ. Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med Genet 12: 160, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catling DC, Glein CR, Zahnle KJ, and McKay CP. Why O2 is required by complex life on habitable planets and the concept of planetary “oxygenation time”. Astrobiology 5: 415–438, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Chandrasekaran A, Idelchik MdPS, and Melendez JA. Redox control of senescence and age-related disease. Redox Biol 11: 91–102, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chao L, Rang CU, Proenca AM, and Chao JU. Asymmetrical damage partitioning in bacteria: a model for the evolution of stochasticity, determinism, and genetic assimilation. PLoS Comput Biol 12: e1004700, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen-Or I, Katz C, and Ron EZ. AGEs secreted by bacteria are involved in the inflammatory response. PLoS One 6: e17974, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen AA, de Magalhães JP, and Gohil K. Ecological, biomedical and epidemiological approaches to understanding oxidative balance and ageing: what they can teach each other. Funct Ecol 24: 997–1006, 2010 [Google Scholar]
- 19.Cortese-Krott MM, Koning A, Kuhnle GG, Nagy P, Bianco C, Pasch A, Wink DA, Fukuto J, Jackson AA, van Goor H, Olson KR, and Feelisch M. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxid Redox Signal 27: 684–712, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Costa JP, Vitorino R, Silva GM, Vogel C, Duarte AC, and Rocha-Santos T. A synopsis on aging—theories, mechanisms and future prospects. Ageing Res Rev 29: 90–112, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dancy BM, Sedensky MM, and Morgan PG. Effects of the mitochondrial respiratory chain on longevity in C. elegans. Exp Gerontol 56: 245–255, 2014 [DOI] [PubMed] [Google Scholar]
- 22.Drenos F. and Kirkwood TB. Modelling the disposable soma theory of ageing. Mech Ageing Dev 126: 99–103, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Droge W. Oxidative stress and aging. Adv Exp Med Biol 543: 191–200, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Eftekharzadeh B, Hyman BT, and Wegmann S. Structural studies on the mechanism of protein aggregation in age related neurodegenerative diseases. Mech Ageing Dev 156: 1–13, 2016 [DOI] [PubMed] [Google Scholar]
- 25.Eskenazi BR, Wilson-Rich NS, and Starks PT. A Darwinian approach to Huntington's disease: subtle health benefits of a neurological disorder. Med Hypotheses 69: 1183–1189, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Finch CE. and Pike MC. Maximum life span predictions from the Gompertz mortality model. J Gerontol A Biol Sci Med Sci 51: B183–B194, 1996 [DOI] [PubMed] [Google Scholar]
- 27.Finkelstein M. Discussing the Strehler-Mildvan model of mortality. Demogr Res 26: 191–206, 2012 [Google Scholar]
- 28.Fischer WW, Hemp J, and Valentine JS. How did life survive Earth's great oxygenation? Curr Opin Chem Biol 31: 166–178, 2016 [DOI] [PubMed] [Google Scholar]
- 29.Forman HJ. Redox signaling: an evolution from free radicals to aging. Free Radic Biol Med 97: 398–407, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gavrilov LA. and Gavrilova NS. The Biology of life Span: A Quantitative Approach. New York: Harwood Academic Publisher, 1991 [Google Scholar]
- 31.Gavrilov LA. and Gavrilova NS. Theoretical perspectives on biodemography of aging and longevity. In: Handbook of Theories of Aging, edited by Bengtson VL. and Richard AS. New York, NY: Springer Publishing LLC, 2016, pp. 643–667 [Google Scholar]
- 32.Gladyshev VN. On the cause of aging and control of lifespan. BioEssays 34: 925–929, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gladyshev VN. The free radical theory of aging is dead. Long live the damage theory! Antioxid Redox Signal 20: 727–731, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gladyshev VN. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15: 594–602, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glansdorff N, Xu Y, and Labedan B. The last universal common ancestor: emergence, constitution and genetic legacy of an elusive forerunner. Biol Direct 3: 29, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldstein DS, Kopin IJ, and Sharabi Y. Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol Ther 144: 268–282, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golubev A. Does Makeham make sense? Biogerontology 5: 159–167, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Golubev A. How could the Gompertz-Makeham law evolve. J Theor Biol 258: 1–17, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Golubev A. Applications and implications of the exponentially modified gamma distribution as a model for time variabilities related to cell proliferation and gene expression. J Theor Biol 393: 203–217, 2016 [DOI] [PubMed] [Google Scholar]
- 40.Golubev A, Hanson AD, and Gladyshev VN. Non-enzymatic molecular damage as a prototypic driver of aging. J Biol Chem 292: 6029–6038, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golubev AG. The other side of metabolism: a review. Biochem Mosc 61: 1443–1460, 1996 [PubMed] [Google Scholar]
- 42.Golubev AG. The issue of the feasibility of a general theory of aging. III. Theory and practice of aging. Adv Gerontol 2: 109–119, 2012 [Google Scholar]
- 43.Golubev AG, Khrustalev S, and Butov AA. An in silico investigation into the causes of telomere length heterogeneity and its implications for the Hayflick limit. J Theor Biol 225: 153–170, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Gomes NMV, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR, Austad SN, Venditti C, Pagel M, Shay JW, and Wright WE. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 10: 761–768, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonidakis S. and Longo VD. Assessing chronological aging in bacteria. Methods Mol Biol 965: 421–437, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorisse L, Pietrement C, Vuiblet V, Schmelzer CEH, Köhler M, Duca L, Debelle L, Fornès P, Jaisson S, and Gillery P. Protein carbamylation is a hallmark of aging. Proc Natl Acad Sci U S A 113: 1191–1196, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta A, Lloyd-Price J, and Ribeiro AS. In silico analysis of division times of Escherichia coli populations as a function of the partitioning scheme of non-functional proteins. In Silico Biol 12: 9–21, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamilton WD. The moulding of senescence by natural selection. J Theor Biol 12: 12–45, 1966 [DOI] [PubMed] [Google Scholar]
- 49.Han SD. and Tuminello ER. The apolipoprotein e antagonistic pleiotropy hypothesis: review and recommendations. Int J Alzheim Dis 2011:726197, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanson AD, Henry CS, Fiehn O, and Crécy-Lagard Vd. Metabolite damage and metabolite damage control in plants. Annu Rev Plant Biol 67: 131–152, 2016 [DOI] [PubMed] [Google Scholar]
- 51.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956 [DOI] [PubMed] [Google Scholar]
- 52.Harman D. Free radical theory of aging: effect of free radical reaction inhibitors on the mortality rate of male LAF mice. J Gerontol 23: 476–482, 1968 [DOI] [PubMed] [Google Scholar]
- 53.Harman D. The free radical theory of aging. Antioxid Redox Signal 5: 557–561, 2003 [DOI] [PubMed] [Google Scholar]
- 54.Hecker L. and Thannickal VJ. Getting to the core of fibrosis: targeting redox imbalance in aging. Ann Transl Med 4: 93, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herraiz T. and Galisteo J. Naturally-occurring tetrahydro-beta-carboline alkaloids derived from tryptophan are oxidized to bioactive beta-carboline alkaloids by heme peroxidases. Biochem Biophys Res Comm 451: 42–47, 2014 [DOI] [PubMed] [Google Scholar]
- 56.Holmstrom KM. and Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15: 411–421, 2014 [DOI] [PubMed] [Google Scholar]
- 57.Hong SY, Ng LT, Ng LF, Inoue T, Tolwinski NS, Hagen T, and Gruber J. The role of mitochondrial non-enzymatic protein acylation in ageing. PLoS One 11: e0168752, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes BG. and Hekimi S. Different mechanisms of longevity in long-lived mouse and Caenorhabditis elegans mutants revealed by statistical analysis of mortality rates. Genetics 204: 905–920, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huie RE. and Neta P. Chemistry of reactive oxygen species. In: Reactive Oxygen Species in Biological Systems: An Interdisciplinary Approach. Boston, MA: Springer, 2002, pp. 33–73. [Google Scholar]
- 60.Janssens L. and Stoks R. Predation risk causes oxidative damage in prey. Biol Lett 9, 20130350, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Janssens L. and Stoks R. Chronic predation risk reduces escape speed by increasing oxidative damage: a deadly cost of an adaptive antipredator response. PLoS One 9: e101273, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jarman SN, Polanowski AM, Faux CE, Robbins J, De Paoli-Iseppi R, Bravington M, and Deagle BE. Molecular biomarkers for chronological age in animal ecology. Mol Ecol 24: 4826–4847, 2015 [DOI] [PubMed] [Google Scholar]
- 63.Jasienska G, Ellison PT, Galbarczyk A, Jasienski M, Kalemba-Drozdz M, Kapiszewska M, Nenko I, Thune I, and Ziomkiewicz A. Apolipoprotein E (ApoE) polymorphism is related to differences in potential fertility in women: a case of antagonistic pleiotropy? Proc Biol Sci 282: 20142395, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med 48: e245, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones DP. Redox theory of aging. Redox Biol 5: 71–79, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jones DP. and Sies H. The redox code. Antioxid Redox Signal 23: 734–746, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jouvet L, Rodriguez-Rojas A, and Steiner UK. Demographic variability and heterogeneity among individuals within and among clonal bacteria strains. bioRxiv 105353, 2017. DOI: 10.1101/105353 [DOI]
- 68.Kihara S, Hartzler DA, and Savikhin S. Oxygen concentration inside a functioning photosynthetic cell. Biophys J 106: 1882–1889, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kirkwood TB. Evolution of ageing. Nature 270: 301–304, 1977 [DOI] [PubMed] [Google Scholar]
- 70.Kirkwood TB. and Austad SN. Why do we age? Nature 408: 233–238, 2000 [DOI] [PubMed] [Google Scholar]
- 71.Kirkwood TBL. Deciphering death: a commentary on Gompertz (1825) ‘On the nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies. Philos Trans R Soc Lond B Biol Sci 370: 20140379, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kirkwood TBL. and Kowald A. The free-radical theory of ageing–older, wiser and still alive. BioEssays 34: 692–700, 2012 [DOI] [PubMed] [Google Scholar]
- 73.Kitani K. and Ivy GO. “I thought, thought, thought for four months in vain and suddenly the idea came”–an interview with Denham and Helen Harman mitochondrial dysfunction and ageing. Biogerontology 4: 401–412, 2003 [DOI] [PubMed] [Google Scholar]
- 74.Labbadia J. and Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 43: 332–347, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lapointe J. and Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci 67: 1–8, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laron Z, Kauli R, Lapkina L, and Werner H. IGF-I deficiency, longevity and cancer protection of patients with Laron syndrome. Mutat Res 772: 123–133, 2016 [DOI] [PubMed] [Google Scholar]
- 78.Le Bourg E. Evolutionary theories of aging can explain why we age. Interdiscip Top Gerontol 39: 8–23, 2014 [DOI] [PubMed] [Google Scholar]
- 79.Lerma-Ortiz C, Jeffryes James G, Cooper Arthur JL, Niehaus Thomas D, Thamm Antje MK, Frelin O, Aunins T, Fiehn O, de Crécy-Lagard V, Henry Christopher S, and Hanson Andrew D. ‘Nothing of chemistry disappears in biology’: the top 30 damage-prone endogenous metabolites. Biochem Soc Trans 44: 961–971, 2016 [DOI] [PubMed] [Google Scholar]
- 80.Li T. and Anderson JJ. The Strehler–Mildvan correlation from the perspective of a two-process vitality model. Popul Stud (Camb) 69: 91–104, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lim B, Mun J, and Kim SY. Intrinsic molecular processes: impact on mutagenesis. Trends Cancer 3: 357–371, 2017 [DOI] [PubMed] [Google Scholar]
- 82.Lindahl T. Instability and decay of the primary structure of DNA. Nature 362: 709–715, 1993 [DOI] [PubMed] [Google Scholar]
- 83.Linster CL, Van Schaftingen E, and Hanson AD. Metabolite damage and its repair or pre-emption. Nat Chem Biol 9: 72–80, 2013 [DOI] [PubMed] [Google Scholar]
- 84.Liochev SI. Reflections on the theories of aging, of oxidative stress, and of science in general. Is it time to abandon the free radical (oxidative stress) theory of aging? Antioxid Redox Signal 23: 187–207, 2014 [DOI] [PubMed] [Google Scholar]
- 85.Madimenos FC. An evolutionary and life-history perspective on osteoporosis. Annu Rev Anthropol 44: 189–206, 2015 [Google Scholar]
- 86.Mailloux RJ. Still at the center of it all; novel functions of the oxidative krebs cycle. Bioenergetics 4: 122, 2015 [Google Scholar]
- 87.Martinez DE. Mortality patterns suggest lack of senescence in Hydra. Exp Gerontol 33: 217–225, 1998 [DOI] [PubMed] [Google Scholar]
- 88.Monnier VM, Mustata GT, Biemel KL, Reihl O, Lederer MO, Zhenyu DAI, and Sell DR. Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on “a Puzzle Nearing Resolution.” Ann N Y Acad Sci 1043: 533–544, 2005 [DOI] [PubMed] [Google Scholar]
- 89.Mouton S, Willems M, Back P, Braeckman BP, and Borgonie G. Demographic analysis reveals gradual senescence in the flatworm Macrostomum lignano. Front Zool 6: 15, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muller FL, Lustgarten MS, Jang Y, Richardson A, and Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med 43: 477–503, 2007 [DOI] [PubMed] [Google Scholar]
- 91.Naudí A, Jové M, Ayala V, Cabré R, Portero-Otín M, and Pamplona R. Non-enzymatic modification of aminophospholipids by carbonyl-amine reactions. Int J Mol Sci 14: 3285–3313, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.NCD Risk Factor Collabration. A century of trends in adult human height. eLife 5: e13410, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nystrom T. The free-radical hypothesis of aging goes prokaryotic. Cell Mol Life Sci 60: 1333–1341, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O'agan KA, Cocchiglia S, Zhdanov AV, Tambuwala MM, Cummins EP, Monfared M, Agbor TA, Garvey JF, Papkovsky DB, Taylor CT, and Allan BB. PGC-1α is coupled to HIF-1α-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci U S A 106: 2188–2193, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olshansky SJ. and Carnes BA. Ever since Gompertz. Demography 34: 1–15, 1997 [PubMed] [Google Scholar]
- 96.Pan H. and Finkel T. Key proteins and pathways that regulate lifespan. J Biol Chem 292: 6452–6460, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pereira EJ, Smolko CM, and Janes KA. Computational models of reactive oxygen species as metabolic byproducts and signal-transduction modulators. Front Pharmacol 7: 457, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Perrigue PM, Najbauer J, Jozwiak AA, Barciszewski J, Aboody KS, and Barish ME. Planarians as a model of aging to study the interaction between stem cells and senescent cells in vivo. Pathobiol Aging Age Relat Dis 5: 30052, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Petralia RS, Mattson MP, and Yao PJ. Aging and longevity in the simplest animals and the quest for immortality. Ageing Res Rev 16: 66–82, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rabbani N. and Thornalley PJ. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem Soc Trans 42: 425–432, 2014 [DOI] [PubMed] [Google Scholar]
- 101.Reinhard CT, Planavsky NJ, Olson SL, Lyons TW, and Erwin DH. Earth's oxygen cycle and the evolution of animal life. Proc Natl Acad Sci U S A 113: 8933–8938, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Richarme G, Liu C, Mihoub M, Abdallah J, Leger T, Joly N, Liebart JC, Jurkunas UV, Nadal M, Bouloc P, Dairou J, and Lamouri A. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 357: 208–211, 2017 [DOI] [PubMed] [Google Scholar]
- 103.Rodríguez JA, Marigorta UM, Hughes DA, Spataro N, Bosch E, and Navarro A. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat Ecol Evol 1: 0055, 2017 [DOI] [PubMed] [Google Scholar]
- 104.Rose MR. Life history evolution with antagonistic pleiotropy and overlapping generations. Theor Popul Biol 28: 342–358, 1985 [Google Scholar]
- 105.Rose MR. and Graves JL., Jr. What evolutionary biology can do for gerontology. J Gerontol 44: B27–B29, 1989 [DOI] [PubMed] [Google Scholar]
- 106.Sadowska-Bartosz I. and Bartosz G. Effect of antioxidants supplementation on aging and longevity. BioMed Res Int 2014: 404680, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salminen A, Kaarniranta K, and Kauppinen A. AMPK and HIF signaling pathways regulate both longevity and cancer growth: the good news and the bad news about survival mechanisms. Biogerontology 17: 655–680, 2016 [DOI] [PubMed] [Google Scholar]
- 108.Sato Y, Endo H, Okuyama H, Takeda T, Iwahashi H, Imagawa A, Yamagata K, Shimomura I, and Inoue M. Cellular hypoxia of pancreatic β-cells due to high levels of oxygen consumption for insulin secretion in vitro. J Biol Chem 286: 12524–12532, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schaible R, Scheuerlein A, Dańko MJ, Gampe J, Martínez DE, and Vaupel JW. Constant mortality and fertility over age in Hydra. Proc Natl Acad Sci U S A 112: 15701–15706, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schöttker B, Brenner H, Jansen EH, Gardiner J, Peasey A, Kubínová R, Pająk A, Topor-Madry R, Tamosiunas A, Saum KU, Holleczek B, Pikhart H, and Bobak M. Evidence for the free radical/oxidative stress theory of ageing from the CHANCES consortium: a meta-analysis of individual participant data. BMC Med 13: 300, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shen J, Landis GN, and Tower J. Multiple metazoan life-span interventions exhibit a sex-specific Strehler–Mildvan inverse relationship between initial mortality rate and age-dependent mortality rate acceleration. J Gerontol A Biol Sci Med Sci 72: 44–53, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sies H, Berndt C, and Jones DP. Oxidative stress. Annu Rev Biochem 86: 715–748, 2017 [DOI] [PubMed] [Google Scholar]
- 113.Simons MJ, Koch W, and Verhulst S. Dietary restriction of rodents decreases aging rate without affecting initial mortality rate—a meta-analysis. Aging Cell 12: 410–414, 2013 [DOI] [PubMed] [Google Scholar]
- 114.Ślesak I, Ślesak H, and Kruk J. Oxygen and hydrogen peroxide in the early evolution of life on earth: in silico comparative analysis of biochemical pathways. Astrobiology 12: 775–784, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ślesak I, Ślesak H, Zimak-Piekarczyk P, and Rozpądek P. Enzymatic antioxidant systems in early anaerobes: theoretical considerations. Astrobiology 16: 348–358, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smith GR. and Shanley DP. Computational modelling of the regulation of Insulin signalling by oxidative stress. BMC Syst Biol 7: 41, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smith KA, Waypa GB, and Schumacker PT. Redox signaling during hypoxia in mammalian cells. Redox Biol 13: 228–234, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smith SM, Nager RG, and Costantini D. Meta-analysis indicates that oxidative stress is both a constraint on and a cost of growth. Ecol Evol 6: 2833–2842, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Speakman JR. and Selman C. The free-radical damage theory: accumulating evidence against a simple link of oxidative stress to ageing and lifespan. BioEssays 33: 255–259, 2011 [DOI] [PubMed] [Google Scholar]
- 120.Srivastava M, Begovic E, Chapman J, Putnam NH, Hellsten U, Kawashima T, Kuo A, Mitros T, Salamov A, Carpenter ML, Signorovitch AY, Moreno MA, Kamm K, Grimwood J, Schmutz J, Shapiro H, Grigoriev IV, Buss LW, Schierwater B, Dellaporta SL, and Rokhsar DS. The Trichoplax genome and the nature of placozoans. Nature 454: 955–960, 2008 [DOI] [PubMed] [Google Scholar]
- 121.Stier A, Reichert S, Massemin S, Bize P, and Criscuolo F. Constraint and cost of oxidative stress on reproduction: correlative evidence in laboratory mice and review of the literature. Front Zool 9: 37, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stone RC, Horvath K, Kark JD, Susser E, Tishkoff SA, and Aviv A. Telomere length and the cancer–atherosclerosis trade-off. PLoS Genet 12: e1006144, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Strehler BL. and Mildvan AS. General theory of mortality and aging. Science 132: 14–21, 1960 [DOI] [PubMed] [Google Scholar]
- 124.Strulik H. and Vollmer S. Long-run trends of human aging and longevity. J Popul Econ 26: 1303–1323, 2013 [Google Scholar]
- 125.Tanase M, Urbanska AM, Zolla V, Clement CC, Huang L, Morozova K, Follo C, Goldberg M, Roda B, Reschiglian P, and Santambrogio L. Role of carbonyl modifications on aging-associated protein aggregation. Sci Rep 6: 19311, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tarkhov AE, Menshikov LI, and Fedichev PO. Strehler-Mildvan correlation is a degenerate manifold of Gompertz fit. J Theor Biol 416: 180–189, 2017 [DOI] [PubMed] [Google Scholar]
- 127.Telford MJ, Budd GE, and Philippe H. Phylogenomic insights into animal evolution. Curr Biol 25: R876–R887, 2015 [DOI] [PubMed] [Google Scholar]
- 128.Tomczyk S, Fischer K, Austad S, and Galliot B. Hydra, a powerful model for aging studies. Invertebr Reprod Dev 59: 11–16, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tubbs A. and Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ungewitter E. and Scrable H. Antagonistic pleiotropy and p53. Mech Ageing Dev 130: 10–17, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vaiserman AM, Lushchak V, and Koliada AK. Anti-aging pharmacology: promises and pitfalls. Ageing Res Rev 31: 9–35, 2016 [DOI] [PubMed] [Google Scholar]
- 132.van den Heuvel J, English S, and Uller T. Disposable soma theory and the evolution of maternal effects on ageing. PLoS One 11: e0145544, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science 350: 1208–1213, 2015 [DOI] [PubMed] [Google Scholar]
- 134.Watanabe H, Hoang VT, Mättner R, and Holstein TW. Immortality and the base of multicellular life: lessons from cnidarian stem cells. Semin Cell Dev Biol 20: 1114–1125, 2009 [DOI] [PubMed] [Google Scholar]
- 135.Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Al Ghuzlan A, Bidart JM, Schlumberger M, and Dupuy C. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 31: 1117–1129, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution 11: 398–411, 1957 [Google Scholar]
- 137.Wilmoth JR, Deegan LJ, Lundström H, and Horiuchi S. Increase of maximum life-span in Sweden, 1861–1999. Science 289: 2366–2368, 2000 [DOI] [PubMed] [Google Scholar]
- 138.Zhang X. and Cui L. Oxygen requirements for the Cambrian explosion. J Earth Sci 27: 187–195, 2016 [Google Scholar]
- 139.Zhdanov AV, Ward MW, Prehn JHM, and Papkovsky DB. Dynamics of intracellular oxygen in PC12 cells upon stimulation of neurotransmission. J Biol Chem 283: 5650–5661, 2008 [DOI] [PubMed] [Google Scholar]
- 140.Zheng H, Yang Y, and Land K. Heterogeneity in the Strehler-Mildvan general theory of mortality and aging. Demography 48: 267–290, 2012 [DOI] [PubMed] [Google Scholar]
- 141.Zimniak P. What is the proximal cause of aging? Front Genet 3: 189, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ziomkiewicz A, Sancilio A, Galbarczyk A, Klimek M, Jasienska G, and Bribiescas RG. Evidence for the cost of reproduction in humans: high lifetime reproductive effort is associated with greater oxidative stress in post-menopausal women. PLoS One 11: e0145753, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]